Lack of the Cell-Cycle Inhibitor p27Kip1 Results in Selective Increase of Transit-Amplifying Cells for Adult Neurogenesis (original) (raw)
Abstract
The subventricular zone (SVZ) is the largest germinal layer in the adult mammalian brain and comprises stem cells, transit-amplifying progenitors, and committed neuroblasts. Although the SVZ contains the highest concentration of dividing cells in the adult brain, the intracellular mechanisms controlling their proliferation have not been elucidated. We show here that loss of the cyclin-dependent kinase inhibitor p27Kip1 has very specific effects on a population of CNS progenitors responsible for adult neurogenesis. Using bromodeoxyuridine and [3H]thymidine incorporation to label cells in S phase and cell-specific markers and electron microscopy to identify distinct cell types, we compared the SVZ structure and proliferation characteristics of wild-type and p27Kip1-null mice. Loss of p27Kip1 had no effect on the number of stem cells but selectively increased the number of the transit-amplifying progenitors concomitantly with a reduction in the number of neuroblasts. We conclude that cell-cycle regulation of SVZ adult progenitors is remarkably cell-type specific, with p27Kip1 being a key regulator of the cell division of the transit-amplifying progenitors.
Keywords: cell division, differentiation, neural progenitors, stem cells, CNS, proliferation
The adult mouse subventricular zone (SVZ) is the richest source of stem cells in the adult brain and constantly generates new neurons destined for the olfactory bulb. The SVZ is composed of a thin layer of dividing cells extending along the length of the lateral wall of the lateral ventricle (Steindler et al., 1996; Temple and Alvarez-Buylla, 1999). On the basis of cell-cycle length and ultrastructural characteristics, four distinct cell types can be identified in this region: slowly dividing SVZ astrocytes (type B cells), rapidly dividing progenitors (type C cells), proliferating migratory neuroblasts (type A cells), and ependymal cells, which line the lateral ventricle (Lois and Alvarez-Buylla, 1994; Doetsch et al., 1997; Peretto et al., 1999). Recently a lineage progression from B→C→A cell has been proposed (Doetsch et al., 1999a,b), with type B cells being the primary precursors (stem cells) that generate migratory neuroblasts via the highly proliferative transit-amplifying progenitor (type C cell). Cells within this lineage possess the capability of undergoing proliferation, differentiation, or apoptosis (Morshead and van der Kooy, 1992; Lois and Alvarez-Buylla, 1994; Biebl et al., 2000). Recent studies have suggested that cell death may be one of the potential mechanisms for lineage restriction in the postnatal SVZ (Levison et al., 2000).
The SVZ of adult mice has a characteristic architecture. The migrating neuroblasts form elongated aggregates called “chains” that are ensheathed by GFAP-positive B cells. Type C cells are found as clusters interspersed among the chains (Doetsch et al., 1997). The chains of neuroblasts form an extensive network of interconnected pathways that converge at the anterior tip of the SVZ to join the rostral migratory stream (RMS), which terminates in the olfactory bulb (Lois and Alvarez-Buylla, 1994; Doetsch and Alvarez-Buylla, 1996).
Several studies on cell-cycle length in the neonatal rat brain (Schultze and Korr, 1981; Menezes et al., 1995, 1998; Thomaidou et al., 1997; Smith and Luskin, 1998) and adult mouse (Schultze and Korr, 1981;Morshead and van der Kooy, 1992) have indicated the existence of distinct populations with different cell-cycle times within the SVZ and RMS. It has been reported, for instance, that cells in the anterior SVZ have a faster cell-cycle time than the migratory cells in the RMS (Smith and Luskin, 1998).
The length of the cell-cycle transit time during neurogenesis depends primarily on the duration of the G1 transition (Caviness et al., 1999) and is modulated by the enzymatic activity of cyclin-dependent kinases (CDKs). Two main enzymatic activities have been described: CDK4, acting in early to mid-G1, and CDK2, acting in late G1, very close to the entry into the S replicative phase. These two activities differ in terms of substrate specificity and modality of regulation. CDK4, for instance, is positively regulated by cyclin D and can be inhibited by binding to members of the inhibitors of CDK4 (INK4) family, which act by preventing association of the CDK4 catalytic subunit to the positive regulator cyclin D1. Inhibitors of the kinase inhibitory protein (Kip) family can also bind CDK4/cyclin D complexes, although with lower affinity than the INKs, but this event does not result in efficient functional inhibition of enzymatic activity (Sherr and Roberts, 1995). CDK2, in contrast, is positively regulated by cyclin E and negatively regulated by the Kips. Three members of the Kip family have been identified: p21Waf1, p27Kip1, and p57Kip2. It has been shown that p27Kip1 effectively binds to preformed CDK2/cyclin E complexes and stoichiometrically inhibits their enzymatic activity, thus affecting the duration of the G1 phase of the cell cycle (Sherr and Roberts, 1995). Interestingly, only two G1 regulators are expressed in the SVZ and in the RMS: p27Kip1 and p19Ink4d. P27Kip1 is expressed in the proximity of the ventricle (van Lookeren Campagne and Gill, 1998), whereas p19Ink4d expression is low close to the ventricle and highly enriched in the RMS (Coskun and Luskin, 2001).
We have demonstrated previously that loss of p27Kip1 function in mice results in a twofold increase in glial cell number as a result of impaired exit from the cell cycle of glial progenitors in the cortex and in the cerebellum (Casaccia-Bonnefil et al., 1997, 1999). The defective growth arrest was observed for both oligodendrocytes and astrocytes and resulted in expanded pools of glial cells (Casaccia-Bonnefil et al., 1997, 1999). The pattern of p27Kip1 expression in the SVZ (van Lookeren Campagne and Gill, 1998) and the consideration of differences in cell-cycle transit time in distinct neonatal SVZ subpopulations (Menezes et al., 1995, 1998) led us to hypothesize a role for p27Kip1 in growth arrest of SVZ progenitor cells. The detailed information available for the identification of the primary precursors for adult neurogenesis and their lineage progression makes the SVZ an ideal experimental system in which to investigate the cell types that are affected by lack of p27Kip1 function. We now characterize distinct cell populations in the SVZ of p27Kip1-null mice (Kiyokawa et al., 1996; Casaccia-Bonnefil et al., 1997, 1999) and show that p27Kip1 plays an important role in the regulation of type C cell number. The data presented indicate that CDK2 regulators may have a critical role in cell-cycle control of transit-amplifying progenitors and suggest that distinct cell-cycle inhibitors are used by the different SVZ cell types.
MATERIALS AND METHODS
Mouse genotyping. Null and wild-type mice were obtained from heterozygous p27Kip1 breeding pairs (in a mixed genetic background of C57BL/6 and B6SJL) (Kiyokawa et al., 1996). The genotype was determined by PCR analysis of genomic tail DNA. Briefly, tails were digested in lysis buffer (100 mm NaCl, 10 mm Tris, pH 8.0, 0.5% SDS, 25 mm EDTA, 148 μg/ml proteinase K stock) for 14–18 hr at 55°C. Genomic DNA was isolated and amplified by PCR using the following primers: 5′-CGCCCCGACTGCATCTGCGTGTTCGAA-3′ and 5′-TCAAACGTGAGAGTGTCTAACGG-3′ directed against the_p27Kip1_ gene and 5′-AGGGCTTATGATTCTGAAAGTCG-3′ corresponding to the neo sequence. After a 35 cycle reaction (denaturation at 94°C for 45 sec, annealing at 62°C for 1 min, extension at 72°C for 1 min), the wild-type allele yielded a 210 bp PCR product and the null allele yielded a longer 290 bp product because of neo insertion in the first exon.
_Whole-mount bromodeoxyuridine incorporation.Bromodeoxyuridine (BrdU, Sigma, St. Louis, MO) was injected intraperitoneally (100 μg/gm body weight) 1 hr before whole-mount dissection. Whole mounts were fixed overnight in 3% paraformaldehyde; washed in PBS; sequentially incubated in 100% methanol (30 min at −20°C) and 100% acetone (30 min at −20°C); washed three times in PBS/0.5% Triton X-100; incubated in 2N HCl at 37°C for 10 min; washed; blocked in 10% horse serum in PBS/0.5% Triton X-100 for 2 hr; incubated for 48–72 hr with 1:200 anti-BrdU antibodies (Dako, Carpinteria, CA) in blocking solution; and revealed with secondary antibodies, avidin–biotin complex (ABC) reaction (Vector Laboratories, Burlingame, CA), and 0.02% DAB (Sigma) and 0.01% H2O2 (Fisher, Houston, TX). The distribution and number of BrdU-positive cells were analyzed in whole-mount preparations of the lateral wall of the lateral ventricle using a computerized mapping system (Alvarez-Buylla and Vicario, 1988). Counts were also analyzed in three subregions of the SVZ, as defined by the anatomy of the ventricle: the anterior horn from +1.4 mm to 0 mm relative to bregma, the intermediate bridge from 0 mm to −1.5 mm, and the inferior horn from −1.5 mm to −2 mm (Mitro and Palkovits, 1981). Statistical analysis of the samples was performed using a two-tailed_t test distribution for samples of unequal variance.
Whole-mount terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining. Adult mice (2–3 months of age) were intracardially perfused with 0.9% saline, and the brains were removed. The lateral walls of the lateral ventricle were dissected, and the resulting whole mounts were fixed overnight in 3% paraformaldehyde. Whole mounts were immersed for several hours in PBS, followed by methanol and acetone, as described above, and then washed three times in PBS. For terminal deoxynucleotidyl transferase (TdT)-mediated biotinylated UTP nick end labeling (TUNEL) processing, whole mounts were treated with proteinase K (20 μg/ml in 10 mm Tris/HCl, pH 7.4–7.8) for 30 min at room temperature; washed three times in PBS; and immersed in TdT buffer (Boehringer Mannheim, Indianapolis, IN), 1.25 mmcobalt chloride, biotinylated dUTPs, and 0.25 U/μl TdT (Boehringer Mannheim) for 1.5 hr at 37 C. After washing, positive cells were revealed with Elite ABC (Vector Laboratories) and DAB as substrate. The distribution and number of TUNEL-positive cells were analyzed in whole-mount preparations of the lateral wall of the lateral ventricle using a computerized mapping system (Alvarez-Buylla and Vicario, 1988). For immunostaining in combination with TUNEL, the whole mounts were first stained with antibodies against polysialic acid (PSA)-neural cell adhesion molecule (NCAM) (1:1000; a gift from Dr. Rougon, University de Luminy, Marseille, France) and Dll (1:50; a gift from Dr. Panganiban, University of Wisconsin, Madison, WI), GFAP (1:200; Boehringer Mannheim), or Mac-1 (1:50; Boehringer Mannheim), followed by incubation in Cy-3- or Cy-5-conjugated secondary antibodies (1:500; Jackson ImmunoResearch Laboratories, West Grove, PA). The whole mounts were then processed for TUNEL, with the omission of the proteinase K step, using biotinylated dUTPs followed by avidin Cy-2 (1:1000; Jackson ImmunoResearch Laboratories).
_Frozen sections and immunohistochemistry on sections._Wild-type and p27−/− animals were anesthetized with avertin and perfused with 4% paraformaldehyde in PBS. After overnight post-fixation at 4°C in the same fixative, the tissue was incubated for 24 hr in 30% sucrose in 0.1 m phosphate buffer, pH 7.5, embedded in optimal cutting temperature (OCT) compound, and frozen in cryomolds in an 8-methylbutane bath on dry ice. Cryosections (10–15 μm thick) were collected on Superfrost glass slides (Fischer Scientific, Swanee, GA) and processed for immunofluorescence. Frozen sections were thawed and incubated for 1 hr in blocking solution (PGBA: 0.1 m PBS, 0.1% gelatin, 1% BSA, 0.002% sodium azide) with 10% NGS (Vector Laboratories). Primary antibodies were typically incubated overnight [Dll 1:50, PSA-NCAM 1:1000, GFAP (Dako, Carpinteria, CA) 1:500, or TuJ-1 (Sigma) 1:1000 in PGBA containing 10% NGS and 0.3% Triton X-100], followed by biotinylated secondary antibodies (1:200; Southern Biotechnology, Birmingham, AL) and avidin conjugated to distinct fluorophores (1:500; Amersham Biosciences, Arlington Heights, IL). For immunolabeling with anti-BrdU antibodies, sections were then treated with 2N HCl for 10 min at 37°C to denature DNA, followed by equilibration in 0.1 m sodium borate, pH 8.6, for 10 min. Primary antibody anti-BrdU was used at a 1:100 dilution in 0.1m PBS, 0.1% gelatin, 1% BSA, 0.002% sodium azide, and 0.5% Triton X-100 for at least 3 hr at room temperature, followed by Texas Red-conjugated secondary antiserum. Sections were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) (Molecular Probes, Eugene, OR) to visualize cell nuclei. Photomicrographs were obtained with a Hamamatsu (Bridgewater, NJ) CCD camera interfaced to a Leica (Nussloch, Germany) TMC fluorescence microscope. Double labeling was confirmed by scanning individual cells along the_z_-axis with a Leica TCS confocal microscope.
Electron microscopy, [3H]thymidine autoradiography, and cell counts. Four adult male wild-type mice and four p27Kip1-null littermates were injected 1 hr before they were killed with 50 μl of 1 mCi [3H]thymidine. Animals were deeply anesthetized with Nembutal and transcardially perfused with 0.9% saline followed by 100 ml of Karnovsky's fixative (2% paraformaldehyde and 2.5% glutaraldehyde). The heads were removed and post-fixed in the same fixative overnight. The brains were then removed from the skull and washed in 0.1 m PBS for 2 hr. Transverse 200 μm sections were cut on a Vibratome, post-fixed in 2% osmium for 2 hr, rinsed, dehydrated, and embedded in araldite (Durcupan; Fluka, Buchs, Switzerland). Tritiated thymidine autoradiography was performed as described in detail by Doetsch et al. (1997). For the identification of individual cell types, ultrathin (0.05 μm) sections were cut with a diamond knife, stained with lead citrate, and examined under a Jeol (Peabody, MA) 100CX electron microscope. The number of profiles corresponding to the different cell types along the ventricular wall of the anterior horn was quantified in a fixed region in six to eight ultrathin sections for each genotype under the electron microscope (Doetsch et al., 1997). The dorsolateral corner of the SVZ was not included in the analysis, because the high density of cells precludes an accurate quantification in this area. Cells with only small fragments of cytoplasm or nucleus in a given section were classified as unidentified. All quantifications were performed blind as to the genotype. Total cell counts obtained by single-section analysis are comparable with those in three-dimensional reconstructions, indicating that our counts are not biased by cell size (Doetsch et al., 1997). For [3H]thymidine counts, the dorsoventral extent of four semithin sections each from wild-type and p27Kip1-null mice were photographed in their entirety, then re-embedded for electron microscopy. Labeled and unlabeled cells were then identified and quantified with an electron microscope.
Neurosphere cultures, BrdU labeling, and staining. Adult wild-type and knock-out mice were killed, and the SVZ was dissected and collected in Petri dishes containing PIPES buffer (in mm: 20 PIPES, 25 glucose, 120 NaCl, 0.5 KCl), pH 7.4. After digestion with papain and DNase as described previously (Doetsch et al., 1999), cells were dissociated by trituration, using a fire-polished Pasteur pipette and resuspended in DMEM-F12 supplemented with N2, 2 mm glutamine, 20 μg/ml insulin, and 15 mm HEPES. The cells were counted, and equal numbers of cells were plated for wild-type and knock-out mice in the medium described above supplemented with 20 ng/ml epidermal growth factor (EGF). After 5 d in culture, the number of neurospheres from wild-type and knock-out mice was counted. For differentiation of neurospheres, cells were dissociated by gentle trituration and then plated onto laminin-coated LabTek chambers
RESULTS
Increased incorporation of BrdU in the SVZ of p27Kip1-null mice
Because p27Kip1 is the only member of the Kip family to be expressed in the SVZ (van Lookeren Campagne and Gill, 1998), we asked whether loss of p27Kip1 affects proliferation in this area of postnatal neurogenesis in the rodent brain. To address this question, 9-week-old wild-type and p27Kip1-null littermates were pulsed with BrdU for 1 hr_in vivo_. The incorporation of BrdU into SVZ cells was then evaluated by immunohistochemistry in whole-mount preparations of the walls of the lateral ventricles. Labeled nuclei were counted, and each BrdU-positive cell was recorded and its location displayed within the outline of the entire ventricular wall as shown in Figure1. In p27Kip1-null mice (n = 8), we observed a statistically significant increase (p = 0.0028) in the number of proliferating cells compared with wild-type mice (n = 5). The total number of BrdU-positive cells counted in p27Kip1-null mice was 4499.5 ± 579 (mean ± SEM; n = 8); the total number in wild-type littermates was 3756 ± 185 (mean ± SEM; n = 5). Interestingly, the increased number of BrdU-positive cells was not evenly distributed throughout the SVZ but rather was localized in the anterior horn, with no differences in the intermediate bridge or in the posterior horn (Fig. 1). This pattern of increased BrdU labeling in the mutant mice parallels the distribution of type A and C cells, whose incidence is higher in the anterior horn of the lateral ventricles (Doetsch et al., 1997).
Fig. 1.
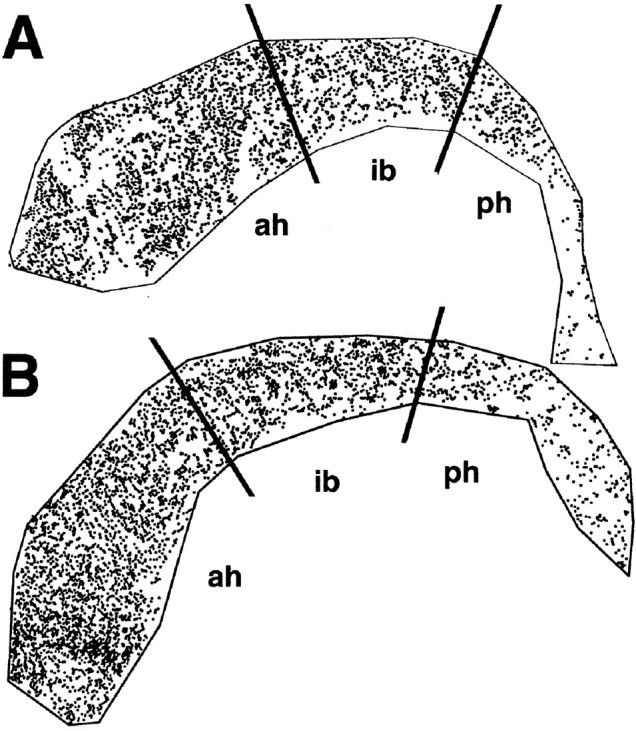
Increased BrdU incorporation in the SVZ of adult p27Kip1-null mice. A computer-generated map of BrdU-labeled cells in the SVZ of adult wild-type (A) and knock-out (B) mice is shown. Nine-week-old mice received a single pulse of BrdU and were killed after 1 hr. The number of proliferating cells was determined by staining SVZ whole mounts with anti-BrdU antibodies. Each dot represents the nucleus of a BrdU-positive cell. The black lines indicate arbitrary anatomical subdivisions of the SVZ, as described in Materials and Methods. Note that the increased number of BrdU-positive cells in the p27Kip1-null mice is localized predominantly to the anterior horn (ah) rather than the intermediate bridge (ib) or the posterior horn (ph). The topographic location of the BrdU-positive cells parallels the distribution of type A and C cells in the adult SVZ.
Loss of p27Kip1 results in selective increase of type C cells in the adult SVZ
To identify the cell types that were affected by loss of p27Kip1 function, we performed electron microscopic analysis of the anterior horn of the SVZ. Six to eight ultrathin transverse sections of the ventricular lateral wall were obtained from wild-type (n = 4) and p27Kip1-null (n = 4) mice, and distinct cell types were identified according to established ultrastructural criteria using an electron microscope (Doetsch et al., 1997). Briefly, type A cells (neuroblasts) were identified on the basis of their elongated morphology, scant dark cytoplasm, abundant free ribosomes, and microtubules oriented along the longitudinal axis of the cells. Type B cells (SVZ astrocytes) were characterized by the presence of multiple processes intercalating among other cells, thick bundles of intermediate filaments, glycogen granules, and dense bodies in the cytoplasm and gap junctions. Type C cells (transit-amplifying progenitors) were more electron-dense than type B cells and more electron-lucent than type A neuroblasts. They were identified on the basis of their large size, deeply invaginated nuclei, the presence of free ribosomes in the cytoplasm, and lack of the intermediate filaments typical of type B cells.
The total number of cells counted with the electron microscope was 1751 for p27Kip1-null mice and 1536 for wild-type littermates (Table1). The most striking difference between the two genotypes was the increased number of type C cells in the p27Kip1-null mice (Table 1). On average, 19 ± 4 type C cells were counted in a wild-type mouse SVZ per 1000 μm length, whereas 38 ± 7 C cells were counted in p27Kip1-null mice. Interestingly, no difference was observed either in the number of ependymal cells (wild-type, 80 ± 8; p27Kip1-null, 74±18) or in the number of type B stem cells (wild-type, 63 ± 11; p27Kip1-null, 58 ±16). The mutant mice also showed a twofold reduction in the number of type A cells (p27Kip1-null, 29 ± 6) compared with wild-type mice (56 ± 12).
Table 1.
Average number of distinct cell types in sections of the anterior horn of the SVZ
| Cell type | Wild type | p27Kip1-null | p27Kip1-null/wild type | |||
|---|---|---|---|---|---|---|
| Cells/1000 μm | SEM | Cells/1000 μm | SEM | Relative change | p value | |
| A | 56 | 12 | 29 | 6 | Twofold decrease | 0.001 |
| B | 43 | 3 | 45 | 7 | No change | NS |
| C | 19 | 4 | 38 | 7 | Twofold increase | 0.0001 |
| E | 58 | 5 | 64 | 12 | No change | NS |
| Neurons | 2 | 1 | 2 | 1 | No change | NS |
| Mitosis | 0.7 | 0.7 | 1 | 0.8 | No change | NS |
| Unidentified | 6 | 2 | 5 | 2 | No change | NS |
| Microglia | 0 | 0 | 2 | 0.8 | 6.6-fold increase | <0.0001 |
| Total | 1536 | 1751 |
The increased number of C cells combined with the decrease in type A cells resulted in the disruption of the characteristic architecture of the SVZ in p27Kip1-null mice (Figs. 2,3). In the wild-type mice, B cells were organized in gliotubes surrounding chains of migratory type A neuroblasts (Fig. 2). Type C cells, the precursors of type A, cells were distributed in clusters interspersed among the chains, as described previously (Doetsch et al., 1997). In contrast, in the p27Kip1-null mice, type A neuroblasts were sometimes observed at unusual locations (Figs. 3,4B,F), whereas the more abundant type C cells were closer to the ventricle and scattered in distribution, primarily interspersed among type B cells (Figs. 3, 4D,F). Microglia were also more common in the p27Kip1-null mice than in wild-type mice (see Fig. 6). These data indicate that loss of p27Kip1 function selectively increases the number of type C cells and that modification of the relative proportion of each cell type disrupts the orderly structure of the SVZ.
Fig. 2.
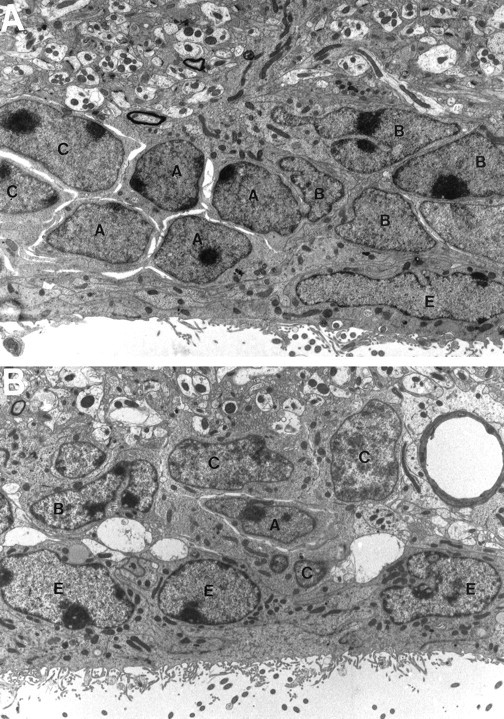
Loss of p27Kip1 function results in increased C-cell number and disorganization of the SVZ. A, Photomicrograph of the typical structure of the adult mouse SVZ as observed in wild-type animals. The migrating neuroblasts (A cells), identified by scant dark cytoplasm and dense heterochromatin, are surrounded by type B cells (B), characterized by light cytoplasm with several inclusions and irregular contours. Type C cells (C) are interspersed in clusters, whereas ependymal cells (E) line the ventricular wall (4812× magnification). B, Photomicrograph of the SVZ structure in p27Kip1-null littermates. There are significantly more type C cells, whereas type B cells and ependymal cells are constant and type A cells are reduced in number (4156× magnification).
Fig. 3.

Displaced A cells in the p27Kip1-null mice. Electron micrograph of the anterior horn of the SVZ of mutant mice showing the ependymal cells (E) and the transit amplifying progenitors (C) close to the ventricular lumen (left). Note the unusual location of the migratory A cells (arrow) away from the lumen of the lateral ventricle.
Fig. 4.
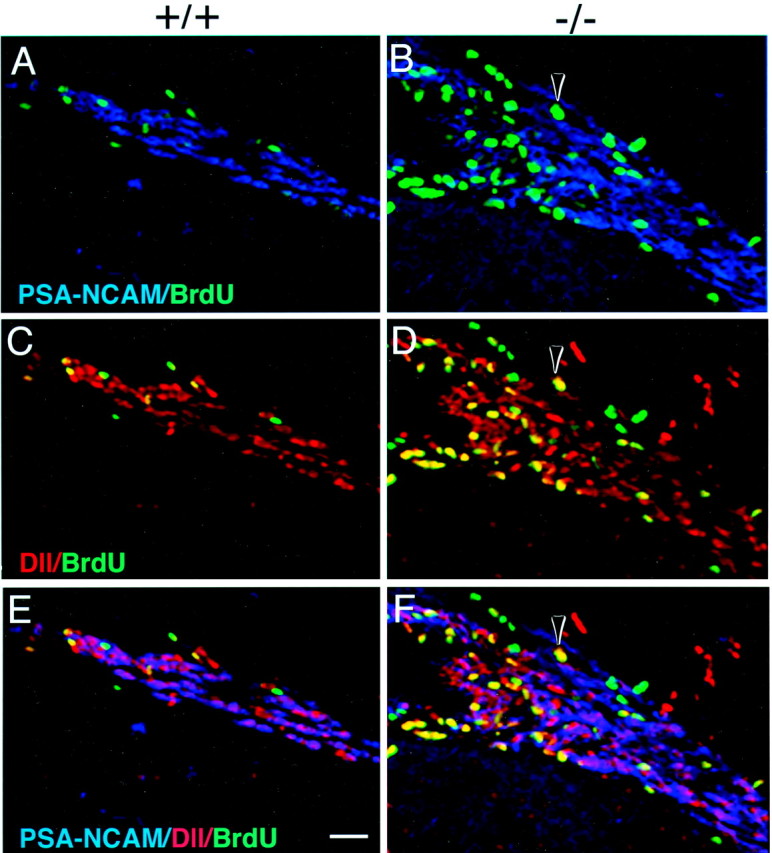
Identification of proliferating type C cells in p27Kip1-null mice. Triple immunofluorescence of frozen coronal sections obtained from the SVZ of wild-type (+/+, A, C, E) and p27Kip1-null (−/−, B, D, F) mice and stained for BrdU (green nuclei), PSA-NCAM (blue immunofluorescence), and Dll (red_immunofluorescence). The arrowhead indicates a cell that is BrdU+/PSA-NCAM− (B, F) but Dll+ (D, F) and therefore identified as a proliferating type C cell. Note that in the mutant, the accumulation of proliferating C cells close to the ventricular lumen (on the_left of each panel) displaces the PSA-NCAM+ type A cells laterally. Scale bar, 100 μm.
Fig. 6.
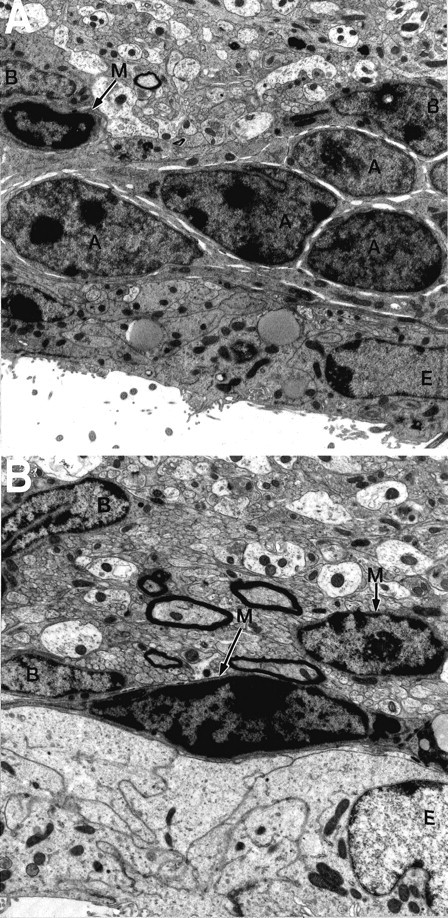
Microglial cells in the p27Kip1-null mouse SVZ.A, Photomicrograph of the adult mouse SVZ, as observed in wild-type animals. Occasional microglial cells (M,arrows) can be observed (4812× magnification).B, A typical section from p27Kip1-null mice reveals an increased number of microglial cells (M,arrows) (6453× magnification). A, A cells; B, B cells; E, ependymal cells.
Proliferation in p27Kip1-null mice
To determine the effect of loss of p27Kip1 on cell proliferation in the SVZ, we used [3H]thymidine and BrdU incorporation to label cells in S phase and electron microscopic analysis and immunohistochemistry to identify labeled cells in the SVZ. Electron microscopic analysis of brain sections labeled for 1 hr with [3H]thymidine revealed that the relative contribution of each population to the total number of proliferating cells was completely altered in p27Kip1-null mice compared with controls (Table 2). Although the proportion of labeled type B cells did not change significantly between the two genotypes (Table 2), there was an increase in proliferating transit-amplifying type C cells and a decrease in proliferating type A neuroblasts in the anterior horn of the SVZ. Type A cells, for instance, which account for 26% of the total number of proliferating cells in wild-type mice (8 of 31 cells), represent only 7% of the total number of cells in S phase in p27Kip1-null mice (2 of 28 cells). Conversely, type C cells accounted for 64% (20 of 31 cells) of the total number of [3H]thymidine-labeled cells in wild-type animals and 86% (24 of 28 cells) of the proliferating population in the p27Kip1-null mice (Table 2). Proliferation of type B cells did not change (Table 2). From these data we conclude that the loss of p27Kip1 does not equally affect the cell-cycle regulation of distinct subpopulations in the SVZ. Loss of this cell-cycle inhibitor increases the number of type C cells and decreases the number of type A cells in S phase.
Table 2.
Relative contribution of distinct cell types to total proliferation in the anterior horn of the SVZ
| Cell type | Wild type | P27Kip-1-null | ||||
|---|---|---|---|---|---|---|
| n (cells in each population) | n (thy + cells) | % relative to Thy + cells | n (cells in each population) | n (thy + cells) | % relative to Thy + cells | |
| A | 98 | 8 | 26 | 50 | 2 | 7 |
| B | 75 | 3 | 10 | 68 | 2 | 7 |
| C | 28 | 20 | 64 | 78 | 24 | 86 |
| Pykno | 0 | 0 | 0 | 2 | 0 | 0 |
| Micro | 1 | 0 | 0 | 2 | 0 | 0 |
| Unidentified | 4 | 0 | 0 | 6 | 0 | 0 |
| Total | 206 | 31 | 100 | 206 | 28 | 100 |
To confirm the data obtained from the electron microscopic analysis, we performed immunohistochemistry on frozen sections, using BrdU to label cells in S phase and cell-specific markers to identify distinct cell types. Type A and C cells can be distinguished by double-immunostaining for Dlx-2 and PSA-NCAM. Type A cells express both PSA-NCAM and Dlx-2, whereas type C cells express only Dlx-2 (F. Doetsch and A. Alvarez-Buylla, unpublished results). Thus, triple immunostaining for Dll (which recognizes all forms of the Dlx family, including Dlx-2) (Panganiban et al., 1995), PSA-NCAM, and BrdU allowed us to distinguish BrdU-positive type C cells (Dll+/PSA-NCAM−) from BrdU-positive type A cells (Dll+/PSA-NCAM+). Consistent with the results of the [3H]thymidine labeling, immunohistochemical analysis of the SVZ of wild-type and p27Kip1-null mice injected with BrdU revealed that the majority of the BrdU-labeled cells in p27Kip1-null mice were PSA-NCAM− and Dll+ type C cells (Fig.4).
Loss of p27Kip1 function results in increased apoptosis in the SVZ
Because type C cells are increased in number and represent the transit-amplifying population of progenitors for type A neuroblasts, we investigated why the number of A cells was significantly reduced in the mutant animals. We therefore performed TUNEL and analyzed the distribution and number of TUNEL-positive cells in whole-mount preparations of the SVZ to evaluate cell death in the mutant mice. In p27Kip1-null mice, we counted a total of 568 ± 64 (mean ± SEM, n = 6) TUNEL-positive cells, compared with the 290 ± 18 (mean ± SEM, n = 5) apoptotic cells in wild-type animals (Fig. 5). Consistent with an increase in apoptosis in the mutant mice, a large number of microglial cells (which are typically involved in phagocytosis of dead cell fragments) were observed in the SVZ of mutant mice (Fig. 6). To identify the dying cells, we performed TUNEL in combination with immunohistochemistry for cell-specific antigens (Figs. 7, 8). When triple immunostaining was performed for PSA-NCAM, Dll, and TUNEL, we very rarely observed labeled A cells (TUNEL+/PSA-NCAM+/Dlx2+) and occasionally found labeled type C cells (TUNEL+/PSA-NCAM−/Dlx2+) (Fig.7). Colabeling for TUNEL and specific antibodies for type B cells (i.e., GFAP) or microglia (i.e., Mac-1) also revealed few double-labeled cells. The majority of TUNEL-positive cells did not stain with any cellular markers, suggesting that apoptotic cells may downregulate expression of markers before death.
Fig. 5.

Increased apoptosis in whole mounts of SVZ from p27Kip1-null mice. A computer-generated map of the SVZ of adult wild-type (A) and knock-out (B) mice whole mounts stained with TUNEL. Each_dot_ represents the nucleus of a TUNEL-positive cell, and the black lines indicate arbitrary subdivisions of the SVZ based on anatomical landmarks, as defined in Materials and Methods. Note that the increased number of TUNEL-positive cells in the p27Kip1-null mice is localized predominantly to the anterior horn (ah) of the SVZ rather than the intermediate bridge (ib) or the posterior horn (ph), which parallels the distribution of type A and C cells in the adult SVZ.
Fig. 7.
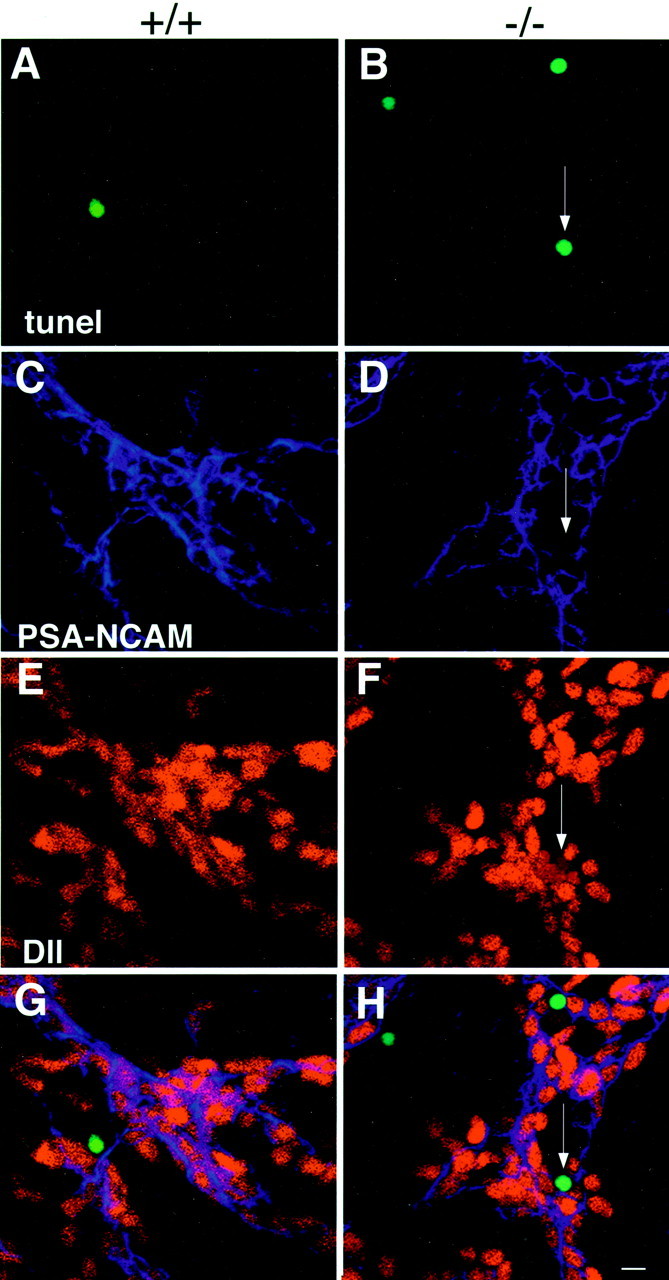
Identification of apoptotic cells in the SVZ of wild-type and p27Kip1-null mice using type A- and type C-specific cell markers. Triple immunofluorescence of whole mounts from wild-type mice (+/+, left column) and p27Kip1-null mice (−/−,right column) stained for TUNEL (green nuclei), PSA-NCAM (_blue_immunofluorescence), and Dll (red immunofluorescence). The arrow indicates the apoptotic nucleus (B) of a cell that is Dll-positive (F) and PSA-NCAM-negative (D) and therefore identified as a type C apoptotic cell in the mutant mice. Scale bar, 50 μm.
Fig. 8.

Identification of apoptotic cells in the SVZ of wild-type and p27Kip1-null mice using type B-specific (gfap) and microglial-specific (mac-1) cell markers. Double immunofluorescence of whole mounts from wild-type mice (+/+) and p27Kip1-null mice (−/−) stained for TUNEL (green nuclei), GFAP (red_immunofluorescence; A–F), or Mac-1 (red immunofluorescence; G–L). The_arrow indicates an apoptotic nucleus of a microglial cell in wild-type mice. The arrowheads indicate apoptotic cells that do not stain with Mac-1 in the mutant mice. Scale bar, 150 μm.
Loss of p27Kip1 function results in increased number of neurospheres
Single precursor cells isolated from the adult brain and cultured in suspension divide to generate clusters of cells called neurospheres in the presence of appropriate growth factors (Reynolds and Weiss, 1992; Morshead et al., 1994). The SVZ is the richest source of EGF-responsive neurospheres in the adult brain. We therefore dissociated the SVZ from wild-type and p27Kip1 littermates (four mice per experiment) and cultured the same number of cells in medium containing EGF (10 ng/ml) to determine the ability of p27Kip1-null mice to generate neurospheres. After 5 d, the total number of neurospheres was counted in wild-type and knock-out dishes. Interestingly, the average of three independent experiments revealed a significant increase in the number of neurospheres generated from p27Kip1-null mice (mean, 2077 ± 91) compared with wild-type mice (mean, 1461 ± 165). No difference was observed between wild-type and p27Kip1-null neurospheres in their ability to generate O4+ oligodendrocytes, GFAP+ astrocytes, and TuJ-1+ neurons (Fig.9). We conclude that loss of p27Kip1 results in increased numbers of neurospheres, although it does not affect their ability to generate neurons and glia.
Fig. 9.

Neurospheres isolated from p27Kip1-null mice maintain their ability to differentiate in vitro. Pluripotent cells were isolated from the SVZ of p27Kip1-null mice and grown in vitro to generate neurospheres. After dissociation and plating on laminin-coated dishes, these cells differentiate into glial cells (A) or into neurons (B). A, p27Kip1-null GFAP-positive astrocyte (_green_immunofluorescence). B, p27Kip1-null A cell stained with the neural marker TuJ-1 (red immunofluorescence). Individual nuclei are labeled by DAPI (_blue_immunofluorescence). Scale bar, 25 μm.
DISCUSSION
The number of stem cells, transit-amplifying progenitors, and neuroblasts at any given stage in the SVZ lineage is the result of a dynamic equilibrium between self-renewal, lineage progression, and death (Fig. 10). In this report, we have examined the consequences of eliminating one of the crucial G1 regulators of the cell cycle, the CDK inhibitor p27Kip1, on the cell types in the SVZ.
Fig. 10.

Schematic model of population dynamics in the anterior horn of the SVZ. A model of SVZ lineage progression is shown. Type B cells are the GFAP-positive primary precursors that give rise to type C transit-amplifying progenitors, which can further differentiate into type A migratory neuroblasts. The number of cells in each stage of the lineage is determined by self-renewal, differentiation, or death.
The main conclusion of this study is that loss of the G1 regulator p27Kip1 selectively increases the number of the transit-amplifying type C cells, concomitant with a decrease in the number of type A neuroblasts. This clearly indicates that cell-cycle regulation is not equivalent in the different cell populations of the SVZ. If the role of p27Kip1 was equivalent in distinct SVZ cell types, then its loss would equally affect cell division in the three cell populations and proportionally increase the number of cells in each population. In contrast, loss of p27Kip1 enhanced proliferation of type C cells and not B cells or A neuroblasts, as evidenced by the increased number of [3H]thymidine-positive and BrdU-positive C cells and by the increase in the total number of type C cells. Our results suggest that the different SVZ cell types rely on distinct molecules regulating the G1/S transition, which represents, for the majority of eukaryotic cells, the decisional point between exit or re-entry into the cell cycle. More specifically, we show here that the number of type C cells is regulated primarily by the p27Kip1 inhibitor acting at the G1/S checkpoint. P27Kip1 is expressed in the SVZ (van Lookeren Campagne and Gill, 1998) and has been shown to regulate the length of the G1 phase of the cell cycle in CNS progenitors (Mitsuhashi et al., 2001). It is likely that in the absence of p27Kip1, type C cells lose their inhibitory control and that this results in an untimely exit from the cell cycle, as reported in other cellular systems (Durand et al., 1998). Although our data do not directly rule out the possibility that loss of p27Kip1 may selectively increase the duration of the S phase in C cells and therefore result in an increased number of [3H]thymidine-positive and BrdU-positive cells, this is an unlikely scenario. P27Kip1 was originally identified as a regulator of the G1phase of the cell cycle (Polyak et al., 1994; Toyoshima and Hunter, 1994). Consistent with this role, transgenic mice overexpressing p27Kip1 in CNS progenitor cells show lengthening of the G1 phase of the cell cycle (Mitsuhashi et al., 2001). In addition, loss of p27Kip1 function in rodents (Casaccia-Bonnefil et al., 1997; Durand et al., 1998), in_Drosophila_ (de Nooij et al., 1996; Lane et al., 1996), or in_Caenorhabditis elegans_ (Hong et al., 1998) has been shown not to affect the length of the S phase but rather the G1 checkpoint of progenitor cells, which is crucial for determining the timing of cell-cycle exit before differentiation. We propose that the increased number of type C cells in the adult SVZ of p27Kip1-null mice is the consequence of additional rounds of cell division caused by loss of one of the crucial regulators of the G1 checkpoint.
The selectivity of the effect of p27Kip1 in regulating the G1 phase of type C cells is also supported by our finding that the number of type B cells is not changed and type A cells are actually decreased in number. The lower number of type A cells in S phase in the p27Kip1-null mice may be attributable to compensatory mechanisms of homeostasis. Recently, type A neuroblasts have been shown to express the G1 regulator p19Ink4d (Zindy et al., 1997; Coskun and Luskin, 2001). The increased number of type C cells in p27Kip1-null mice may elicit a feedback signal onto type A cells, which, through a p19Ink4d-dependent mechanism, results in type A cells undergoing fewer rounds of cell division. Distinct cell-cycle molecules, therefore, may have cell type-selective and stage-specific effects during the SVZ lineage progression. A second mechanism that could underlie the decreased number of type A cells is increased apoptosis (Fig. 10). In p27Kip1-null mice, increased numbers of TUNEL-positive cells were present. Triple immunohistochemistry for TUNEL, PSA-NCAM, and Dll revealed that some TUNEL-positive cells were type C cells, and very occasionally A cells, but that the majority of TUNEL-positive cells did not express cellular markers. This could be attributable to the downregulation of markers by dying cells. It is likely that increased apoptosis is an additional compensatory mechanism activated in the p27Kip1-null mice to maintain cell number in the SVZ.
The most likely explanation for the decreased number of type A cells is therefore a delay in cell-cycle exit by type C cells. Loss of the G1 checkpoint in p27Kip1-null mice likely causes type C cells to undergo extra rounds of cell division at the expense of lineage progression. The fact that C cells do eventually progress into A cells suggests that other cell-cycle inhibitors are involved in this decision. Because of the increased number of dividing C cells close to the ventricle and their delayed progression into the lineage, type A cells appear “dislocated” away from the ventricular lumen. As these C cells progress into type A neuroblasts, however, the cell-cycle control is taken over by the G1 inhibitor p19Ink4d (Coskun and Luskin, 2001). This possible compensatory mechanism results in decreased A cell proliferation, exit from the cell cycle, and aggregation of type A neuroblasts in chains that converge to form the RMS.
In conclusion, by taking advantage of the known lineage relationships in the adult SVZ, we have identified p27Kip1 as one of the main cell-cycle inhibitors regulating proliferation of the transit-amplifying progenitors, type C cells. In the absence of p27Kip1 function, control of proliferation is lost in a cell-specific manner, and this results in increased division of type C cells and in a consequent decrease of type A neuroblasts and altered histogenesis of the SVZ.
Footnotes
This paper was supported by National Multiple Sclerosis Society Grant RG 3154-A-2 (P.C.B.) and National Institutes of Health Grants HD23315 (M.V.C.) and HD32116 (A.A.-B.). We are extremely grateful to Dr. Pangagiban for the generous gift of the anti-Dll antibody; to Parawmen Navidad, and Michela Muggironi for technical help; and to Drs. Nowakowski and Di Cicco-Bloom for critical reading of this manuscript.
Correspondence should be addressed to Patrizia Casaccia-Bonnefil, Department of Neuroscience and Cell Biology, University of Medicine and Dentistry of New Jersey, 675 Hoes Lane, Piscataway, NJ 08854. E-mail: casaccpa@umdnj.edu.
REFERENCES
- 1.Alvarez-Buylla A, Vicario DS. Simple microcomputer system for mapping tissue sections with the light microscope. J Neurosci Methods. 1988;25:165–173. doi: 10.1016/0165-0270(88)90155-0. [DOI] [PubMed] [Google Scholar]
- 2.Biebl M, Cooper CM, Winkler J, Kuhn HG. Analysis of neurogenesis and programmed cell death reveals a self-renewing capacity in the adult rat brain. Neurosci Lett. 2000;291:17–20. doi: 10.1016/s0304-3940(00)01368-9. [DOI] [PubMed] [Google Scholar]
- 3.Casaccia-Bonnefil P, Kiyokawa H, Tikoo R, Friedrich V, Koff A, Chao MVC. Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27Kip1. Genes Dev. 1997;11:2335–2346. doi: 10.1101/gad.11.18.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casaccia-Bonnefil P, Hardy RJ, Teng KK, Levine JM, Koff A, Chao MV. Loss of p27Kip1 function results in increased proliferative capacity of oligodendrocyte progenitors but unaltered timing of differentiation. Development. 1999;126:4027–4037. doi: 10.1242/dev.126.18.4027. [DOI] [PubMed] [Google Scholar]
- 5.Caviness VS, Jr, Takahashi T, Nowakowki RS. The G1 restriction point as critical regulator of cortical neuronogenesis. Neurochem Res. 1999;24:497–506. doi: 10.1023/a:1022579712262. [DOI] [PubMed] [Google Scholar]
- 6.Coskun V, Luskin MB. The expression pattern of cell cycle inhibitor p19 ink4d by progenitor cells of the rat embryonic telencephalon and the neonatal anterior subventricular zone. J Neurosci. 2001;21:3092–3103. doi: 10.1523/JNEUROSCI.21-09-03092.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Nooij J, Letendre M, Hariharan I. A cyclin-dependent kinase inhibitor, Dacapo, is necessary for timely exit from the cell cycle during Drosophila embryogenesis. Cell. 1996;87:1237–1247. doi: 10.1016/s0092-8674(00)81819-x. [DOI] [PubMed] [Google Scholar]
- 8.Doetsch F, Alvarez-Buylla A. Network of tangential pathway for neural migration in adult mammalian brain. Proc Natl Acad Sci USA. 1996;93:14895–14900. doi: 10.1073/pnas.93.25.14895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci. 1997;17:5046–5061. doi: 10.1523/JNEUROSCI.17-13-05046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999a;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- 11.Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A. Regeneration of a germinal layer in the adult mammalian brain. Proc Natl Acad Sci USA. 1999b;96:11619–11624. doi: 10.1073/pnas.96.20.11619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durand B, Fero ML, Roberts JM, Raff MC. p27Kip1 alters the response of cells to mitogen and is part of a cell-intrinsic timer that arrests the cell cycle and initiates differentiation. Curr Biol. 1998;8:431–440. doi: 10.1016/s0960-9822(98)70177-0. [DOI] [PubMed] [Google Scholar]
- 13.Hong Y, Roy R, Ambros V. Developmental regulation of a cyclin-dependent kinase inhibitor controls cell cycle progression in Caenorhabditis elegans. Development. 1998;125:3585–3597. doi: 10.1242/dev.125.18.3585. [DOI] [PubMed] [Google Scholar]
- 14.Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 15.Lane M, Sauer K, Wallace K, Jan Y, Lehner C, Vaessin H. Dacapo, a cyclin-dependent kinase inhibitor, stops cell proliferation during Drosophila development. Cell. 1996;87:1225–1235. doi: 10.1016/s0092-8674(00)81818-8. [DOI] [PubMed] [Google Scholar]
- 16.Levison SW, Rothstein RP, Brazel CY, Young GM, Albrecht PJ. Selective apoptosis within the rat subependymal zone: a plausible mechanism for determining which lineages develop from neural stem cells. Dev Neurosci. 2000;22:106–115. doi: 10.1159/000017432. [DOI] [PubMed] [Google Scholar]
- 17.Lois C, Alvarez-Buylla A. Long distance neuronal migration in the adult mammalian brain. Science. 1994;264:1145–1148. doi: 10.1126/science.8178174. [DOI] [PubMed] [Google Scholar]
- 18.Menezes JR, Smith CM, Nelson KC, Luskin MB. The division of neuronal progenitor cells during migration in the neonatal mammalian forebrain. Mol Cell Neurosci. 1995;6:496–508. doi: 10.1006/mcne.1995.0002. [DOI] [PubMed] [Google Scholar]
- 19.Menezes JR, Dias F, Garson AV, Lent R. Restricted distribution of S-phase cells in the anterior subventricular zone of the postnatal mouse forebrain. Anat Embryol. 1998;198:205–211. doi: 10.1007/s004290050178. [DOI] [PubMed] [Google Scholar]
- 20.Mitro M, Palkovits M. Morphology of the rat brain ventricles, ependymal and periventricular structures. In: Lierse W, editor. Bibliotecha anatomica, No. 21. Karger; Basel: 1981. [PubMed] [Google Scholar]
- 21.Mitsuhashi T, Aoki Y, Eksaioglu YZ, Takahashi T, Bhide PG, Reeves SA, Caviness VSJ. Overexpression of p27kip1 lengthens the G1 phase in a mouse model that targets inducible gene expression to central nervous system progenitor cells. Proc Natl Acad Sci USA. 2001;98:6435–6440. doi: 10.1073/pnas.111051398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morshead CM, van der Kooy D. Postmitotic death is the fate of constitutively proliferating cells in the subependymal layer of the adult mouse brain. J Neurosci. 1992;12:249–256. doi: 10.1523/JNEUROSCI.12-01-00249.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morshead C, Reynolds BA, Craig CG, McBurney MW, Staines WA, Morassutti D, Weiss S, van der Kooy D. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron. 1994;13:1071–1082. doi: 10.1016/0896-6273(94)90046-9. [DOI] [PubMed] [Google Scholar]
- 24.Panganiban G, Sebring A, Nagy L, Carroll S. The development of crustacean limbs and the evolution of arthropods. Science. 1995;270:1363–1366. doi: 10.1126/science.270.5240.1363. [DOI] [PubMed] [Google Scholar]
- 25.Peretto P, Merighi A, Fasolo A, Bonfanti L. The subependymal layer in rodents: a site of structural plasticity and cell migration in the adult mammalian brain. Brain Res Bull. 1999;49:221–243. doi: 10.1016/s0361-9230(99)00037-4. [DOI] [PubMed] [Google Scholar]
- 26.Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massague J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 28.Schultze B, Korr H. Cell kinetic studies of different cell types in the developing and adult brain of the rat and mouse: a review. Cell Tissue Kinet. 1981;14:309–325. doi: 10.1111/j.1365-2184.1981.tb00535.x. [DOI] [PubMed] [Google Scholar]
- 29.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 30.Smith CM, Luskin MB. Cell cycle length of olfactory bulb neuronal progenitors in the rostral migratory stream. Dev Dyn. 1998;213:220–227. doi: 10.1002/(SICI)1097-0177(199810)213:2<220::AID-AJA7>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 31.Steindler DA, Kadrie T, Fillmore H, Thomaas LB. The subependymal zone: “brain marrow.”. Prog Brain Res. 1996;108:349–363. [PubMed] [Google Scholar]
- 32.Temple S, Alvarez-Buylla A. Stem cells in the adult mammalian central nervous system. Curr Opin Neurobiol. 1999;9:135–141. doi: 10.1016/s0959-4388(99)80017-8. [DOI] [PubMed] [Google Scholar]
- 33.Thomaidou D, Mione MC, Cavanagh JFR, Parnavelas JG. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J Neurosci. 1997;17:1075–1085. doi: 10.1523/JNEUROSCI.17-03-01075.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 35.van Lookeren Campagne M, Gill R. Tumor-suppressor p53 is expressed in proliferating and newly formed neurons of the embryonic and postnatal rat brain: comparison with expression of the cell cycle regulators p21waf1, p27Kip1, p57Kip2, p16Ink4a, cyclin G1, and the protooncogene Bax. J Comp Neurol. 1998;397:181–198. doi: 10.1002/(sici)1096-9861(19980727)397:2<181::aid-cne3>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 36.Zindy F, Soares H, Herzog K-H, Morgan J, Sherr CJ, Roussel MF. Expression of INK4 inhibitors of cyclin D-dependent kinases during mouse brain development. Cell Growth Differ. 1997;8:1139–1150. [PubMed] [Google Scholar]