Neuronal Deficiency of Presenilin 1 Inhibits Amyloid Plaque Formation and Corrects Hippocampal Long-Term Potentiation But Not a Cognitive Defect of Amyloid Precursor Protein [V717I] Transgenic Mice (original) (raw)
Abstract
In the brain of Alzheimer's disease (AD) patients, neurotoxic amyloid peptides accumulate and are deposited as senile plaques. A major therapeutic strategy aims to decrease production of amyloid peptides by inhibition of γ-secretase. Presenilins are polytopic transmembrane proteins that are essential for γ-secretase activity during development and in amyloid production. By loxP/Cre-recombinase-mediated deletion, we generated mice with postnatal, neuron-specific presenilin-1 (PS1) deficiency, denoted PS1(n−/−), that were viable and fertile, with normal brain morphology. In adult PS1(n−/−) mice, levels of endogenous brain amyloid peptides were strongly decreased, concomitant with accumulation of amyloid precursor protein (APP) C-terminal fragments. In the cross of APP[V717I]xPS1 (n−/−) double transgenic mice, the neuronal absence of PS1 effectively prevented amyloid pathology, even in mice that were 18 months old. This contrasted sharply with APP[V717I] single transgenic mice that all develop amyloid pathology at the age of 10–12 months. In APP[V717I]xPS1 (n−/−) mice, long-term potentiation (LTP) was practically rescued at the end of the 2 hr observation period, again contrasting sharply with the strongly impaired LTP in APP[V717I] mice. The findings demonstrate the critical involvement of amyloid peptides in defective LTP in APP transgenic mice. Although these data open perspectives for therapy of AD by γ-secretase inhibition, the neuronal absence of PS1 failed to rescue the cognitive defect, assessed by the object recognition test, of the parent APP[V717I] transgenic mice. This points to potentially detrimental effects of accumulating APP C99 fragments and demands further study of the consequences of inhibition of γ-secretase activity. In addition, our data highlight the complex functional relation of APP and PS1 to cognition and neuronal plasticity in adult and aging brain.
Keywords: PS1, Alzheimer's disease, neuronal plasticity, cognition, amyloid pathology, mouse model
Accumulation and deposition of neurotoxic amyloid peptides in vasculature and brain parenchyma as senile plaques is an important pathological hallmark of Alzheimer's disease (AD). The amyloid peptides, Aβ40 and Aβ42, are generally believed to play a central but still poorly understood role in AD and are formed by sequential endoproteolytic cleavages of the amyloid precursor protein (APP) by β- and γ-secretases, hence representing important therapeutic targets (Vassar and Citron, 2000; Golde and Younkin, 2001; Selkoe, 2001).
Presenilin-1 (PS1) is critical for the generation of amyloid peptides from APP (De Strooper et al., 1998), either by exerting γ-secretase activity itself or by controlling trafficking of γ-secretase and its substrates (for review, see Esler and Wolfe, 2001). Presenilins are polytopic transmembrane proteins that are proteolytically processed into N- and C-terminal fragments that form biologically active complexes in combination with other proteins, e.g., nicastrin and others (Haass and De Strooper, 1999; Yu et al., 2000;Esler and Wolfe, 2001). PS1 is involved not only in APP processing, but also in the processing of other integral membrane proteins, among which Notch1 is most critical for embryonic development (Naruse et al., 1998;Song et al., 1999; Struhl and Greenwald, 1999). The pleomorphic effects of PS1 are best illustrated by the phenotype of the “classic” PS1 knock-out mice. Severely impaired mouse embryogenesis is characterized by disturbed somitogenesis, cranial hemorrhages, impaired neurogenesis with thinning of the ventricular zone, bilateral cerebral cavitations resulting in malformation of brain, and late embryonic lethality (Shen et al., 1997; Wong et al., 1997; Hartmann et al., 1999).
Although PS1 plays a critical role in the generation of amyloid peptides, its potential as a therapeutic target for AD remains to be scrutinized, especially regarding its role in adult brain. We have now generated mice with a restricted postnatal and neuron-specific deletion of PS1, to circumvent the embryonic lethality of the classically induced, complete PS1 deficiency. This approach demonstrated that PS1(n−/−) mice were viable and fertile and had normal brain morphology, indicating that neuronal deficiency of PS1 is well tolerated in adult mice.
The PS1(n−/−) mice were further crossed with our APP[V717I] mice, which display some phenotypic changes that are highly reminiscent for AD and shared by other APP transgenic models (Games et al., 1995; Hsiao et al., 1996, 1998; Holcomb et al., 1998, 1999; Calhoun et al., 1999;Moechars et al., 1999; Dewachter et al., 2000; Van Dorpe et al., 2000;Duff and Rao, 2001), including impaired long-term potentiation (LTP) and cognitive deficits with progressive development with age of typical amyloid plaques and vascular pathology.
In this model we studied whether phenotypic changes in APP[V717I] mice could be alleviated by neuronal deficiency of PS1, not only because this is highly relevant to validate therapeutic strategies based on γ-secretase inhibition, but also because it is very informative for our fundamental understanding of the etiology of AD. The exact contribution of the different biochemical APP derivatives, i.e., the amyloid peptides 40/42 and the C-terminal stubs, still remains to be established, as well as in which form, i.e., soluble or precipitated, they exert or cause neurotoxicity (Nalbantoglu et al., 1997; Suh, 1997; Chapman et al., 1999; Holcomb et al., 1999; Hsia et al., 1999; Dewachter et al., 2000; Suh et al., 2000).
The results that are presented highlight the complex functional relation of APP and PS1 and of their metabolites to cognition and neuronal plasticity. This includes data supporting a critical role for amyloid peptides in impairing LTP in APP transgenic mice. In addition, our data demonstrate that neuronal deficiency of PS1 inhibited amyloid peptide formation, prevented plaque formation, and corrected deficits in LTP in APP transgenic mice, hence supporting the use of γ-secretase inhibition as therapy for AD. However, the current data also underline the potential detrimental effects of the accumulation of APP β-stubs or C99 fragments, which necessitate further detailed study of the use of therapeutic strategies aimed at inhibition of γ-secretase activity.
MATERIALS AND METHODS
Generation of transgenic mice. A PS1 allele was constructed to contain three loxP sites and a neomycin resistance cassette (neoR) by gene targeting in embryonic stem cells. This allele contains two loxP sites flanking exon 7 and a third loxP site downstream of neoR (see Fig. 1). Thy-1 Cre-recombinase transgenic mice were generated essentially as described for other neuron-specific expression constructs (Moechars et al., 1996, 1999; Dewachter et al., 2000; Spittaels et al., 2000; Tesseur et al., 2000). Southern blotting and PCR analysis of DNA isolated from tail biopt was used for genotyping of all mice. Thy-1-driven Cre-recombinase activity was assessed by crossing with LacZ reporter mice, demonstrating prominent β-gal activity in neurons of the cortex and hippocampus of adult mice (see Fig. 1), consistent with our previous observations in transgenic mice overexpressing transgenes under control of the mouse thy-1 gene promoter.
Fig. 1.

Generation of PS1(n−/−) mice. A, Overall strategy to generate mice with a conditional inactivation of PS1, denoted PS1(n−/−), and APP transgenic mice with a neuronal PS1 deficiency. B, Schematic representation of the targeting vector, the wild-type, and the targeted PS1 gene. Lox P sites are represented by open triangles flanking the neomycin cassette and exon 7. Arrowheads represent synthetic primers used for genotyping by PCR. C, β-Galactosidase staining of brain section of the selected thy1-Cre-recombinase transgenic mouse line after crossing with the LacZ reporter mouse (Akagi et al., 1997) demonstrating neuronal staining in cortex and hippocampus. D, Genotyping by PCR analysis of DNA isolated from tail biopts using primers NE277 and NE278 (B) to identify the wild-type and targeted PS1 genes in mice with the indicated genotypes. E, In situ hybridization of brain sections of PS1(n−/−) mice (left panel) and of mice with a floxed but active PS1 gene (right panel) with RNA probes specific for PS1 exon 7, which is deleted by Cre-recombinase. F, Western blotting for the C-terminal fragment of mouse PS1 (∼20 kDa) in total brain extracts from four individual PS1(n−/−) mice [lanes marked _PS1(n−/−)_] and from four individual mice with floxed but active PS1 genes (lanes marked Control), and diluted sample (1, 0.5, 0.25). Quantitation revealed levels of 100.0 ± 5.5 and 16.0 ± 0.7% in control mice and PS1(n−/−) mice, respectively. Western blotting with antibodies against APP and LRP was used as a loading control.
Analysis of behavior and cognition. The object recognition task was essentially performed as described (Tang et al., 1999; Rampon et al., 2000). Briefly, the mice were habituated for 1 hr to a Plexiglas open-field box (52 × 52 × 40 cm) with black vertical walls and a translucent floor dimly illuminated by a lamp placed underneath the box. The next day the animals were placed in the same box and submitted to a 10 min acquisition trial. During this trial mice were placed individually in the open field in the presence of object A (marble or dice), and the time spent exploring object A (when the animal's snout was directed toward the object at a distance <1 cm) was measured. During a 10 min retention trial (second trial), which was performed 3 hr later, a novel object (object B: marble or dice) was placed together with the familiar object (object A) in the open field. The time (_t_A and_t_B) the animal spent exploring the two objects was recorded. The recognition index (RI), defined as the ratio of the time spent exploring the novel object over the time spent exploring both objects [(_t_B/(_t_A+ _t_B)) × 100] was used to measure nonspatial memory. Statistical analysis was done by using ANOVA single factor as described (Moechars et al., 1996, 1999).
Western blotting analysis. Biochemical analysis of APP-processing intermediates has been described (Dewachter et al., 2000). Briefly, mouse brains were homogenized in 6.5 vol of ice-cold buffer containing 20 mm Tris-HCl, pH 8.5, and a mixture of proteinase inhibitors (Roche, Darmstadt, Germany). After centrifugation at 135,000 × g at 4°C for 1 hr, the supernatant was centrifuged again for 2 hr at 200,000 ×g before analysis of soluble amyloid peptides by specified ELISA. The pellets from the first centrifugation were resuspended in TBS with proteinase inhibitors and used for analysis of membrane-bound proteins. Biochemical analysis of intact membrane-bound APP and of the C-stubs was done as described (Moechars et al., 1999; Dewachter et al., 2000). β-C-stubs derived from human APP in APP transgenic mice and APPxPS1(n−/−) mice were measured by immunoblotting with antibody WO2 (Ida et al., 1996); C-stubs derived from murine APP were measured by immunoblotting with polyclonal B10/4. For detection of PS1 and PS2 protein, brains were homogenized in sucrose buffer [5 mm Tris, 250 mm sucrose, 1 mm EGTA, pH 7.4, and a mixture of proteinase inhibitors (Roche, Darmstadt, Germany)] with a potter homogenizer, spun at 12,000 × g at 4°C for 10 min. The supernatant was taken and further centrifuged for 30 min at 100,000 × g at 4°C. Proteins were denatured and reduced in sample buffer containing a final concentration of 2% SDS, 1% 2-ME separated on 4–12% Nu-Page gels (Novex, San Diego, CA). PS1 and PS2 were detected with the mouse monoclonal antibody Ab 5232 (Chemicon, Temecula, CA), recognizing C-terminal fragments of PS1, and the goat polyclonal antibody sc-1456 (Santa Cruz Biotechnology, Santa Cruz, CA), recognizing C-terminal fragments of PS2. After incubation with appropriate secondary antibodies, all Western blots were developed with the ECL detection system and photographically recorded. Densitometric scanning of films and calculation and normalization were performed as described (Moechars et al., 1999; Dewachter et al., 2000) using a flatbed optical density scanner and dedicated software for analysis and measurement (Image Master; Pharmacia, Uppsala, Sweden).
ELISA of amyloid peptides. Protein extracts were applied on reversed-phase columns (C18-Sep-pack cartridges; Waters Corporation, Milford, MA) and washed with increasing concentrations of acetonitrile (5, 25, and 50%) containing 0.1% trifluoroacetic acid. The last fraction contained the amyloid peptides and was dried in vacuo overnight and dissolved for measurements in ELISA. Sandwich ELISA for human Aβ40 and Aβ42 peptides was performed using the capture antiserum JRF/cAβ40/14 and 21F12, respectively, and they were developed with monoclonal antibodies JRFcAβtot/14 and 3D6, respectively (Vanderstichele et al., 2000). Sandwich ELISA for murine Aβ40 and Aβ42 peptides was performed using the capture antibodies JRF/cAβ40/14 and JRF/cAβ42/17, respectively, and they were developed with the HRP-coupled monoclonal antibody JRF/Aβ1–15/2.
Northern blotting. Total RNA was isolated from mouse brain, separated by electrophoresis, and transferred by capillary transfer to nylon membranes. Filters were prehybridized for 6 hr at 42°C in SSPE (150 mm sodium chloride, 20 mm sodium phosphate, 5 mm EDTA), Denhardt's solution, 0.5% SDS, 50% deionized formamide, 100 μg/ml denatured sperm DNA, and 50 μg/ml heparin and hybridized in the same solution supplemented with 10% dextran sulfate at 42°C overnight with addition of a radiolabeled probe. Membranes were washed in 0.3× SSPE, 0.5% SDS at 60°C for 1 hr. Specified DNA probes were generated by PCR using mouse brain cDNA as template, with specified primers as follows: for Hes5, 5′-CCAAGTACCGTGGCGGTGGA and 5′-GAGATGGCCGTCAGCTACCT; for Dll1, 5′-CTCCTTCAGCCTGCCTGATG and 5′-AGGCACCTCACTGTGGGAGA.
Histology and quantitative analysis. Brains were dissected, and the left cerebral hemisphere was snap frozen and stored at −70°C. The right cerebral hemisphere was immersion fixed in 4% paraformaldehyde in PBS overnight and used for histological and quantitative analyses. Coronal vibratome sections (40 μm) were cut from the occipital two-thirds of the right hemisphere. Thioflavin-S staining was performed on vibratome sections according to standard protocols. Immunohistochemistry with monoclonal antibody JRF/AβN25 specific for Aβ was done on free-floating vibratome sections according to previously published protocols, using diaminobenzidine as chromogen (Van Dorpe et al., 2000). Quantitative analysis of amyloid plaque load in the brain was performed on Thioflavin-S-stained and immunostained coronal vibratome sections. Well defined coronal sections at bregma −2.54 mm were selected for quantification of amyloid load (Franklin et al., 1997). For quantification of amyloid plaque load in the subiculum, two serial sections were used for Thioflavin-S staining and immunostaining with JRF/AβN25, respectively. Images (200× magnification) from these sections were collected from a 3CCD color video camera and analyzed with appropriate software (AIS/C; Imaging Research, St. Catherine, Ontario, Canada). The surface of individual amyloid deposits and the surface of the subiculum were measured. The total amyloid plaque load was expressed as a percentage of the total surface of the subiculum. Classic cresyl violet staining was performed on paraffin sections prepared as described (Dewachter et al., 2000).
In situ hybridization. In situ hybridization was performed on paraffin sections (6 μm) of mouse brain placed on silylated glass slides, dewaxed, and rehydrated through an ethanol series. Sections were digested with proteinase K (20 μg/ml), post-fixed in 4% paraformaldehyde, and treated with 0.25% acetic anhydride in 0.1 m triethanolamine. Sections were hybridized overnight in 50% deionized formamide, 0.3m NaCl, 20 mm Tris-HCl, and 5 mm EDTA, pH 8.0, with 10% dextran sulfate, 1× Denhardt's solution, 0.5 mg/ml yeast RNA, and 10 mm dithiothreitol, supplemented with the appropriate radiolabeled riboprobe. After stringency washes and ribonuclease A treatment, sections were dehydrated and dipped in photographic emulsion (LM-1, Amersham Biosciences) and exposed for 1 week. For synthesis of the sense and antisense PS1 RNA, a 221 bp PCR product from exon 7 of the presenilin-1 gene was cloned in a pGEM-T vector (Promega, Madison, WI). The plasmid was linearized with either _Not_I or _Sph_I and transcribed with T7 and SP6RNA polymerase, respectively, in the presence of [33P]UTP. In brain of PS(+)/(+) mice, PS1 was readily detected in neurons from the pyramidal layers of the hippocampus and in neurons from the cortex. In neurons from PS1(n−/−) brain, staining was practically absent, comparable to staining intensity in control mice with a sense probe (results not shown).
Electrophysiology. Hippocampal slices were bathed in artificial CSF containing (in mm ): 124 NaCl, 5 KCl, 26 NaHCO3, 1.24 KH2PO4, 2.4 CaCl2, 1.3 MgSO4, 10 glucose, aerated with 95% O2 and 5% CO2. Mice were anesthetized with ether and decapitated, and transverse slices (400 μm thick) were cut in cold artificial CSF using a vibratome and kept at room temperature until placed in the interface recording chamber at 30°C. Electrophysiological recordings were begun not earlier than 3 hr after dissection to allow recovery of the slice. The chamber was perfused with artificial CSF (1 ml/min). Bipolar tungsten microelectrodes (World Precision Instruments, Sarasota, FL) were used to stimulate Schaffer's collaterals, although evoked field EPSPs (fEPSPs) were recorded in the stratum radiatum of the CA1 region with low resistance (2 MΩ) glass microelectrodes filled with 2 m NaCl. Test stimuli were 0.1 msec constant-voltage pulses delivered every 30 sec at an intensity sufficient to evoke an ∼33% maximal response. LTP was induced by an electrical high-frequency stimulation at an intensity evoking a 50% maximal response. The slope of the field EPSP (millivolts per millisecond) was measured from the average wave from four consecutive responses.
RESULTS
Generation of mice with postnatal neuron-specific ablation of PS1
Total inactivation of the PS1 gene severely impaired mouse embryogenesis by disturbed somitogenesis, cranial hemorrhage, impaired neurogenesis with thinning of the ventricular zone, bilateral cerebral cavitations resulting in malformation of brain, and late embryonic lethality (Shen et al., 1997; Wong et al., 1997; De Strooper et al., 1998; Hartmann et al., 1999). To circumvent the embryonic lethality, we have used the Cre/loxP gene targeting system to generate mice with a postnatal, neuron-specific deficiency of PS1, denoted PS1(n−/−).
These mice were generated by crossing two transgenic mouse strains: (1) mice with a targeted PS1 gene to introduce loxP sites (PS1-flox mice) and (2) mice expressing Cre-recombinase under control of the mouse thy1-gene promoter (Fig. 1). The characteristics of the thy1-gene promoter have been amply documented by us and by others to yield postnatal expression of the transgene in central neurons only (Moechars et al., 1996, 1999; Dewachter et al., 2000; Spittaels et al., 2000; Tesseur et al., 2000; Van Dorpe et al., 2000).
In addition, the specificity of the selected thy1-Cre-recombinase transgenic mouse strain was demonstrated in a cross with a reporter transgenic mouse strain that carries a constitutionally inactive reporter gene construct, i.e., an actin promoter and LacZ reporter gene separated by a “floxed” spacer (Akagi et al., 1997). Prominent β-galactosidase staining was evident in nearly all neurons in cortex and hippocampus of double transgenic mice, i.e., thy1-Cre recombinase x LacZ-reporter mice, proving the specific neuronal expression of the Cre-recombinase in our selected transgenic mouse strain (Fig.1C).
The resulting PS1(n−/−) mice were genotyped by four independent PCR reactions (Fig. 1D). The expression of PS1 was demonstrated to be decreased specifically in cortical and hippocampal neurons as assayed by in situ hybridization with RNA probes specific for PS1 exon 7 (Fig. 1E), as opposed to normal expression of PS1 in the parent transgenic mice with a “floxed” PS1 gene. These had normal expression of PS1 and were used throughout this study as “control” mice in all experiments, unless indicated otherwise. Western blotting established strongly decreased levels of PS1 protein in total brain extracts of PS1(n−/−) mice (84% decrease) (Fig. 1F), with the very low residual expression likely caused by glial and other non-neuronal cells, as judged from in situ hybridization.
Decreased γ-secretase cleavage of APP in PS1(n−/−) mice
PS1 is critical for generating the amyloid peptides from APP in primary cultures of neurons (De Strooper et al., 1998), either by exerting γ-secretase activity itself or as an essential substrate binding adaptor protein or chaperone in the γ-secretase complex (for review, see Esler and Wolfe, 2001).
In the brain of PS1(n−/−) mice, the levels of murine Aβ40 and Aβ42 derived from endogenous APP were dramatically decreased, as expected (Fig. 2). Concomitantly, the C-terminal fragments of mouse APP accumulated, whereas the level of membrane-bound full-length mouse APP remained unchanged (Fig. 2).
Fig. 2.

APP processing in brain of PS1(n−/−) mice. Western blotting (left panel) of mouse brain extracts demonstrating very similar levels of membrane-bound APP (APPm; ∼110 kDa) (top) and accumulation of the APP C-terminal fragments (∼10–12 kDa) (bottom) in PS1(n−/−) mice. Results shown are from three individual PS1(n−/−) mice (three right lanes) and from three individual mice with floxed but active PS1 genes (control, three left lanes). Antiserum B10/4 was used that is specific for the C-terminal domain of human and mouse APP (Dewachter et al., 2000). Levels of soluble amyloid peptides (right panel) (nanograms per gram brain tissue) extracted from the brain of mice with a floxed but active PS1 gene (control) and from PS1(n−/−) mice, as measured with specific ELISA for murine Aβ40 (black bars) and Aβ42 (gray bars) (mean with SEM,n = 6 for each genotype).
The combined data proved that the neuronal deficiency of PS1 was reflected in the expected biochemical effects on APP-processing, i.e., decreased levels of amyloid peptides and accumulation of C-terminal fragments of APP. This established the pronounced inhibition of γ-secretase processing of APP in the brain of PS1(n−/−) transgenic mice.
Unaltered expression of Notch ligand Dll1 and Hes5, an effector gene downstream of Notch
PS1 is also involved in processing of other integral membrane proteins, among which Notch1 is crucial for embryonic development (Naruse et al., 1998; De Strooper et al., 1999; Song et al., 1999;Struhl and Greenwald, 1999). In mouse brain in vivo, the very low levels of the Notch1 intracellular domain are beyond detection limits, which precludes direct in vivo analysis of this metabolite of Notch cleavage. We therefore analyzed whether Notch signaling was affected in brain of adult PS1(n−/−) mice by analysis of the expression level of Hes5, a gene affected downstream of Notch (Ohtsuka et al., 1999; Handler et al., 2000). In addition, we measured the mRNA levels of the Notch ligand Dll1. Northern analysis demonstrated no significant differences in the mRNA levels of Hes5 and Dll1 in brain of PS1(n−/−) mice (Fig.3). This confirmed, in addition to the normal phenotype of the adult PS1(n−/−) mice, that Notch signaling is unaltered, in contrast to the severe embryonic phenotype of the complete PS1 knock-out, in which Hes5 levels were reduced and Dll1 expression was increased (Handler et al., 2000).
Fig. 3.

Analysis of Notch signaling and PS2 expression. Northern blotting analysis of RNA extracted from brain of mice with floxed but active PS1 genes (lanes 1–3) and of PS1(n−/−) mice (lanes 4–6) demonstrates no changes of mRNA levels of Hes5 (top) and Dll1 (middle). Western blotting for the C-terminal fragment of mouse PS2 (∼20 kDa) (bottom), in total brain extracts from four individual PS1(n−/−) mice [_lanes_marked _PS1(n−/−)] and from four individual mice with floxed but active PS1 genes (lanes marked_Control), and diluted sample (1, 0.5, 0.25). Quantitation revealed levels of 100 ± 2 and 127 ± 11% in control mice and PS1(n−/−) mice, respectively.
We further measured the expression of PS2 mRNA and protein levels, demonstrating no change of PS2 mRNA levels, and a small increase (27%) in the levels of the PS2 C-terminal fragment in brain of PS1(n−/−) mice. Although this might compensate for the PS1 deficiency, further study is required to establish the actual effect of this minor change.
No effect of postnatal neuron-specific PS1 deletion on brain morphology and behavior
The strategy to generate mice deficient postnatally in PS1 in neurons only, as outlined in Figure 1, proved successful in circumventing developmental problems and the lethality of the complete PS1 deficiency. PS1(n−/−) mice were born at the expected mendelian frequency and were viable and fertile. They did not show any morphological anomaly in brain sections examined by cresyl violet staining (Fig. 4A) and by immunostaining with different antibodies, i.e., GAP43, MAP2, APP, and synaptophysin (Fig. 4; and results not shown). No cerebral hemorrhages, cavities, or tumors were found associated with the neuronal PS1 deficiency in mice up to 2 years old.
Fig. 4.
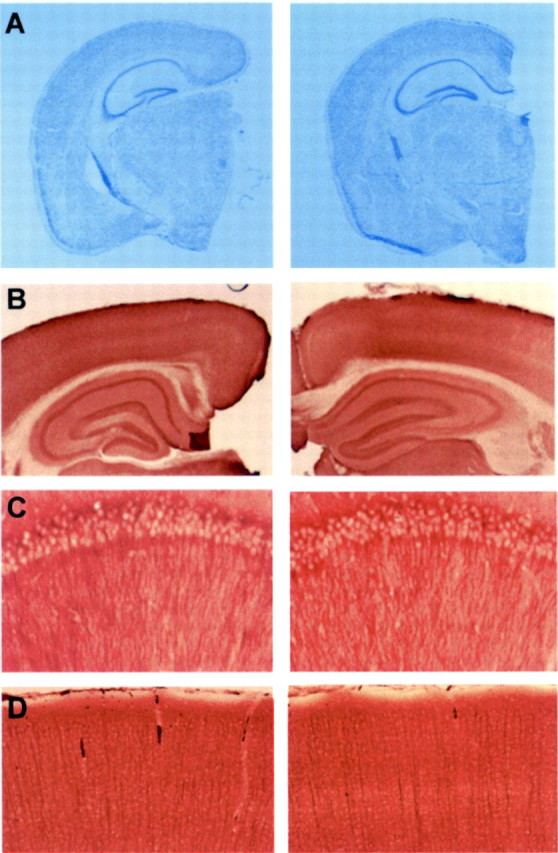
Morphological analysis of brain of PS1(n−/−) mice. A, Cresyl violet staining of paraffin-embedded sections from brain of mice with floxed but active PS1 genes (left panel) and from PS1(n−/−) mice (right panel), aged 6 months, demonstrating normal brain architecture in PS1(n−/−) mice. B, Immunohistochemical staining for GAP43 of vibratome sections from brain of mice with floxed but active PS1 genes (left panel) and PS1(n−/−) mice (right panel), demonstrating normal morphology of hippocampus.C, D, Higher magnification of neurons in hippocampal CA1 region (C) and cortex (D) after immunohistochemical staining for MAP2 of vibratome sections from brain of mice with floxed but active PS1 genes (left) and PS1(n−/−) mice (right).
PS1(n−/−) mice did not behave differently from wild-type mice during normal handling by caretakers and displayed no cognitive deficit in the object recognition test, as detailed further. Taken together, these data demonstrated that the postnatal neuronal deficiency of PS1 was tolerated very well, and we conclude therefore that neuronal PS1 is not essential for maintaining gross neuronal architecture and cognitive functioning in the object recognition test in adult or aging mouse brain.
Generation and characterization of APP[V717I]xPS1(n−/−) mice
The validation of the PS1(n−/−) transgenic mice by the functional and biochemical characteristics described, i.e., the effective inhibition of γ-secretase cleavage of APP, which drastically inhibited amyloid peptide formation without causing major abnormalities in brain morphology and behavior, allowed us to ask the primary question: whether neuronal absence of PS1 is sufficient to rescue the AD-related phenotypic defects of the APP[V717I] transgenic mice. These transgenic mice progressively and robustly recapitulate several of the most intrinsic features of AD pathology.
To experimentally address this question, PS1(n−/−) mice were crossed with APP[V717I] transgenic mice to obtain transgenic mice that were (1) homozygous for the targeted floxed PS1 gene and expressed (2) the thy1-Cre-recombinase transgene and (3) the thy1-APP[V717I] transgene. These mice, further denoted here as APPxPS1(n−/−), thereby expressed the human APP[V717I] mutant protein in precisely the same neurons that lack PS1 resulting from expression of Cre-recombinase, because both are controlled by the same mouse thy1-gene promoter (Fig.1) (Dewachter et al., 2000). Offspring with the APPxPS1(n−/−) genotype, identified with four independent PCR reactions, were born at the expected mendelian frequency, which again proved the absence of major embryonic and developmental defects.
Amyloid plaque formation is prevented in APPxPS1(n−/−) mice
The levels of the human amyloid peptides, derived from the human mutant APP[V717I] transgene, were dramatically lower in the brain of APPxPS1(n−/−) mice relative to the parent APP[V717I] transgenic mice (Fig. 5C), similar as noted for endogenous mouse Aβ (Fig. 2). Although the levels of full-length transgene APP remained unchanged (Fig. 5A), the human APP β-C-stubs also accumulated as expected (Fig.5B).
Fig. 5.

Inhibition of amyloid peptides and pathology in brain of APPxPS1(n−/−) mice. A, B, Western blotting of membrane proteins extracted from brain of three individual APP[V717I] transgenic mice (lanes marked_APP_) and from three individual APPxPS1(n−/−) transgenic mice. The Western blots were stained with monoclonal antibody 1G5 for full-length transgene membrane-bound APP (APPm) (∼110 kDa) (A) and with monoclonal antibody WO2 for human APP β-C-stubs (∼12 kDa) (B). C, Levels of soluble amyloid peptides (nanograms per gram brain tissue) in brain of APP and APPxPS1(n−/−) transgenic mice as measured with specific ELISA for human Aβ40 (red bars) and Aβ42 (blue bars) (mean with SEM, n = 6 for each genotype). D, E, Staining with thioflavinS (D) and immunostaining (E) of amyloid deposits in brain of APPxPS1(n−/−) (middle panels) and APP[V717I] transgenic mice (right panels). Note the absence of thioflavin-S staining and of immunoreaction in the subiculum of APPxPS1(n−/−) mice in contrast to the parent APP[V717I] transgenic mice. The left panels are a higher magnification of the areas selected in the middle panels. F,G, Amyloid load, expressed as relative surface area occupied by thioflavin-S-positive plaques (F) and of immune-positive plaques (G) in APP[V717I] and APPxPS1(n−/−) mice, all 16–18 months old (mean with SEM,n = 6).
In the brain of all APP[V717I] transgenic mice aged 10–12 months and older that we have examined to date, diffuse and senile amyloid plaques were invariably present and increased exponentially with age (Moechars et al., 1999; Dewachter et al., 2000; Van Dorpe et al., 2000). In contrast, however, no thioflavin-S-reactive amyloid plaques nor diffuse amyloid deposits could be detected in the brain of APPxPS1(n−/−) transgenic mice, even at the age of 18 months (Fig.5D_–_G).
The neuronal ablation of PS1 that decreased γ-secretase activity with concomitant inhibition of amyloid peptide production thereby also prevented development of the amyloid pathology, an important neuropathological hallmark of AD, in the APPxPS1(n−/−) mice.
Long-term potentiation in CA1 of PS1(n−/−), APP[V717I], and APPxPS1(n−/−) mice
Having demonstrated inhibition of the amyloid pathology, we addressed the question whether typical hippocampal defects, previously demonstrated in the parent APP[V717I] transgenic mice, were alleviated in the APPxPS1(n−/−) transgenic mice. Hippocampal LTP is a favored model of training-dependent enhancement of synaptic efficacy that can be induced experimentally by brief, high-frequency activation of specified synapses (for review, see Martin et al., 2000).
We measured LTP at the level of synapses between Schaffer's collaterals and CA1 pyramidal cells in brain slices from groups of transgenic mice with the four genotypes. Tetanic stimulation triggered a significantly impaired LTP in APP[V717I] transgenic mice (Fig.6A), confirming our previous findings (Moechars et al., 1999). The pattern of LTP induction in APPxPS1(n−/−) mice (Fig. 6C) was strikingly different as compared with single APP transgenic mice (Fig.6A). After an initial slight decrease, the slope of fEPSP approached the level measured in control mice, sharply contrasting with the progressively decreasing LTP measured in single APP[V717I] transgenic mice.
Fig. 6.
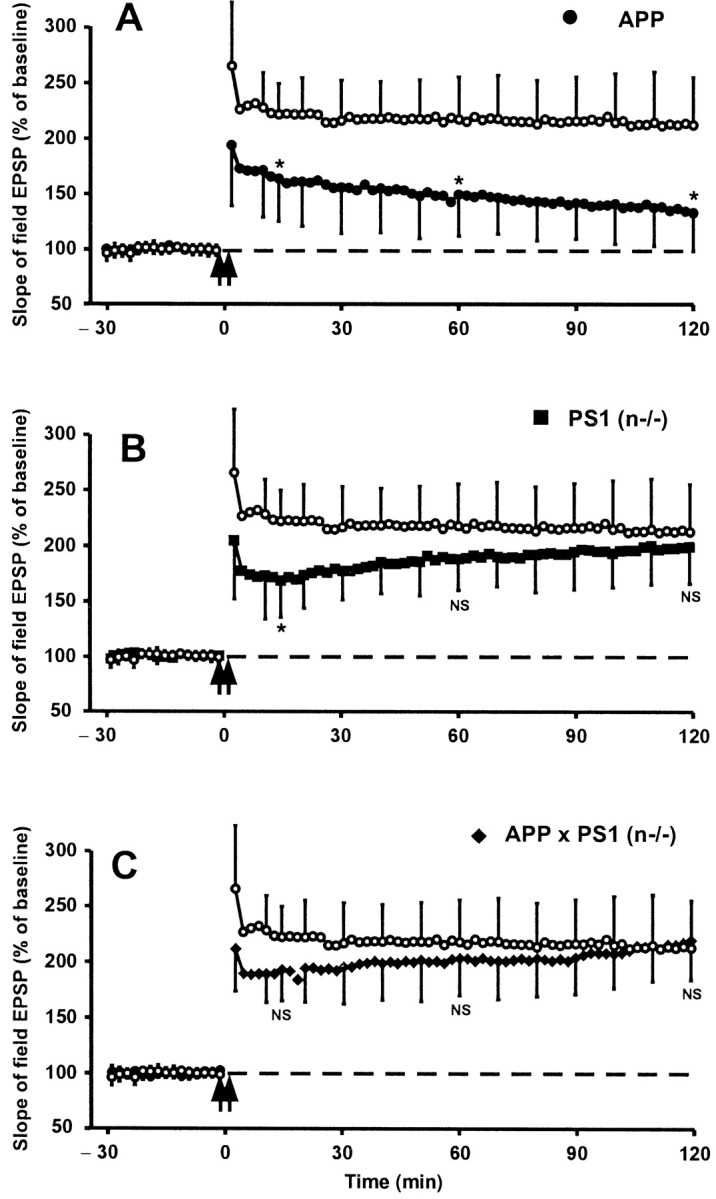
Long-term potentiation in hippocampal slices of APP[V717I], PS1(n−/−), and APPxPS1(n−/−) transgenic mice. The slope of field Schaffer's EPSP recorded before and after tetanic stimulation of Schaffer's collaterals in brain sections from APP[V717I] (A), PS1(n−/−) (B), and APPxPS1(n−/−) (C) mice in each panel compared with mice with floxed but active PS1 genes (○). Each data point shown is the mean ± SD of results from six individual mice of each genotype. A statistically significant decrease was evident for the fEPSP in APP[V717I] mice (p < 0.05; indicated by_asterisk_ in _A_), as described previously (Moechars et al., 1999; Schneider et al., 2001). The final LTP after 2 hr was not significantly different in PS1(n−/−) mice or in APPxPS1(n−/−) mice (_NS_; _p_ > 0.05) (B, C). Note the initial decrease 15 min after tetanic stimulation that is borderline statistically significant in the PS1(n−/−) mice (see Discussion).
It is interesting to note that the pattern of LTP induction in PS1(n−/−) mice was different from that of wild-type mice (Fig.6B). In the initial phase the slope of the fEPSP was lower in sections from PS1(n−/−) mice relative to control mice, i.e., 168 ± 33% versus 221 ± 27%, 15 min after tetanic stimulation. Subsequently, and opposite to the monotonous decrease in APP[V717I] transgenic mice, the slope of fEPSP increased progressively in sections of PS1(n−/−) transgenic mice to attain a mean level that approached that of wild-type mice 2 hr after the tetanic stimulus (Fig. 6B).
These results indicated that the neuronal absence of PS1 might initially affect some mechanism(s) involved in the induction of hippocampal LTP. At the same time, the data demonstrate the restoration, at the end of the 2 hr observation window in this experimental model, of hippocampal LTP in the APPxPS1(n−/−) transgenic mice and hence strongly support the hypothesis that amyloid peptides are responsible for impaired LTP in single APP[V717I] transgenic mice.
Impaired object recognition of APP[V717I] and APPxPS1(n−/−) transgenic mice
The cognitive capacity of transgenic mice with the four genotypes was assessed in a paradigm of nonspatial visual recognition memory, by subjecting them to a “novel object” recognition task that is known to depend on hippocampal activity (Tang et al., 1999; Rampon et al., 2000). Basically, after training to familiarize all mice with a given object, they were tested for retention by confronting them with a novel object, next to and in addition to the familiar one.
The retention measured after 1 hr was similar for all four genotypes, displaying a similar increased preference for the novel object (results not shown). This proved that all the mice recognized and remembered the original object for at least 1 hr , thereby establishing that their motivation and their exploration capacity were intact. Testing after 3 hr demonstrated that the retention of the PS1(n−/−) mice was not significantly different from the control mice, whereas the APP[V717I] mice and the APPxPS1(n−/−) transgenic mice were both highly significantly impaired relative to the control group (p < 0.05 and p < 0.001, respectively) (Fig. 7). We therefore must conclude that the evident and documented impaired cognition of the APP[V717I] mice is not alleviated by the neuronal PS1 deficiency.
Fig. 7.

Analysis of cognition of wild-type, PS1(n−/−), APP[V717I], and APPxPS1(n−/−) transgenic mice in the object recognition task. In the novel-object recognition task, recognition memory is expressed as exploratory preference in the retention test. The recognition index =_t_B/(t_A +t_B) × 100, with_A and B, respectively, representing the familiar and the novel object. Exploratory preference for control mice, i.e., mice with floxed but active PS1 genes (bar marked_CO; n = 22), PS1(n−/−) mice (n = 17), APP[V717I] mice (_n_= 24), and APPxPS1(n−/−) (n = 18) transgenic mice. All mice were 3–6 months old. Retention was measured at 1 and 3 hr after training (mean ± SEM). At 3 hr after training, the APP[V717I] and APPxPS1(n−/−) mice were significantly impaired compared with control mice (respectively, p < 0.05, p < 0.001; determined by ANOVA analysis).
DISCUSSION
We report the generation and characterization of adult and aging mice with a neuron-specific deletion of PS1 in their brain. The successful generation of a “conditional” postnatal neuron-specific knock-out was evidenced by viable, fertile PS1(n−/−) mice that survived and were observed for up to 2 years. Thereby, we demonstrate that neuronal deficiency of PS1 is well tolerated in adult mouse brain, without any major effects on brain architecture and morphology. Evidently, the activity of PS2 might contribute to this apparent redundancy of PS1 in neurons of adult mouse brain. Although PS2 mRNA levels were unchanged, only a small increase in the level of the PS2 C-terminal fragment was observed, further supporting findings that demonstrated competition between PS1 and PS2 for (unknown) limiting factors (Thinakaran et al., 1997; Yu et al., 2000). Although it remains to be defined whether and how this minor increase in PS2 could account for the absent Notch-related phenotype in adult brain, it was clearly not able to compensate for the PS1 deficiency and the ensuing inhibited γ-secretase cleavage of APP.
We demonstrate a dramatic reduction of amyloid peptides, the primary index for the activity of PS1 as γ-secretase, in brain of adult PS1(n−/−) mice, with the concomitant expected accumulation of APP C-terminal fragments, extending and confirming in vivo, original observations in primary hippocampal neurons (De Strooper et al., 1998). The strongly decreased amyloid peptide production was evident in brain of both PS1(n−/−) and APPxPS1(n−/−) transgenic mice. The inhibition of amyloid peptide production completely prevented the late amyloid pathology, even at the age of 16–18 months. At this age, all parent APP[V717I] transgenic mice that we have analyzed to date contained, without exception, prominent brain amyloid plaque pathology (Moechars et al., 1999; Dewachter et al., 2000; Van Dorpe et al., 2000). Therefore, we confidently conclude that inhibition of PS1 is very efficient to prevent the typical amyloid plaque pathology that develops in aging APP transgenic mice. Given the demonstrated and documented neuronal specificity of the mouse thy-1 gene promoter in the selected transgenic mouse strains, this outcome also confirmed that the amyloid pathology is strictly neuronal in origin (Van Dorpe et al., 2000).
In our APP[V717I] transgenic mice, the pathological hallmark of AD, i.e., senile plaques, is preceded by earlier phenotypic changes that comprise impaired LTP and cognitive defects as early as age 4–6 months (Moechars et al., 1999; Schneider et al., 2001). Learning and memory are processes encoded in the brain by long-lasting modification of the efficacy of synaptic transmission, and hippocampal LTP is one model of this type of plasticity. The defect in LTP that was characteristic for the parent APP[V717] mice (Moechars et al., 1999; Schneider et al., 2001) was now observed to be practically corrected at the end of the 2 hr measuring window in brain slices from APPxPS1(n−/−) mice. These data support a critical role for amyloid peptides in the deficit in LTP in APP[V717] transgenic mice because it is almost completely rescued by the PS1 deficiency and the concomitant decreased γ-secretase activity. This is in agreement with the recent demonstration that intracerebroventricular injections of amyloid peptide fragments block LTP in the CA1 region of the rat hippocampus in vivo (Freir et al., 2001).
We and others have demonstrated that mutant PS1 affected calcium homeostasis and LTP (Guo et al., 1999; Leissring et al., 1999, 2000;Yoo et al., 2000; Schneider et al., 2001), whereas the current results demonstrated that the absence of PS1 also, directly or indirectly, affected the time course of LTP, especially the initial phase of the evoked fEPSP immediately after tetanic stimulation. This effect is likely to be linked to altered calcium ion homeostasis, although the precise nature of this effect or defect needs further study (Schneider et al., 2001). A possible explanation would be that the onset of LTP immediately after the short-lasting initial post-tetanic potentiation could be delayed, whereas alternatively unscheduled opening of Ca2+-activated channels could decrease the amplitude of the fEPSP (Nishiyama et al., 2000).
The failure of the PS1 deficiency to rescue or alleviate the impaired object recognition of the parent APP[V717I] transgenic mice was unexpected in light of the LTP results. Such a discrepancy between_in vitro_ hippocampal LTP and in vivo cognitive performance has been noted before, however, and in either direction: impaired LTP but normal behavior (Huang et al., 1995; Schurmans et al., 1997; Meiri et al., 1998; Okabe et al., 1998; Zamanillo et al., 1999;Ho et al., 2000) or impaired behavior with normal LTP (Rosahl et al., 1993; Silva et al., 1996; Matilla et al., 1998). This dissociation can be ascribed to contributions of other types of LTP or synaptic plasticity, or to the contribution of other parts of the hippocampus or the brain, to preserve particular forms of cognition. Alternatively, the mechanisms of memory consolidation could be affected, which would explain the persisting memory defect in APPxPS1(n−/−) mice despite the near rescue of LTP. In view of these results, it must be noted that besides the evident amyloid peptides, Aβ40 and Aβ42, our current data point to the C-terminal fragments of APP as additional contributors to neuropathological deficits. This being the case, the development and use of γ-secretase inhibitors as a therapy for AD needs to be considered carefully.
In conclusion, the neuronal deficiency of PS1 was well tolerated in PS1(n−/−) mice, yielding viable and fertile mice that behaved apparently normal. Most importantly, we demonstrated that the neuronal deficiency of PS1 inhibited amyloid peptide production, prevented plaque formation, and practically restored impaired LTP. Moreover, the data indicated a crucial role for the amyloid peptides in causing the deficit in LTP in the APP[V717I] transgenic mice. These results support the use of γ-secretase inhibitors as a therapy for AD. However, because the neuronal deficiency of PS1 and the consequent decrease of amyloid peptides failed to rescue the cognitive deficit in the APPxPS1(n−/−) transgenic mice, we believe that the use of γ-secretase inhibitors must be scrutinized further and also consider potential neurotoxic effects of accumulating C-terminal fragments of APP (Nalbantoglu et al., 1997; Suh, 1997; Chapman et al., 1999;Dewachter et al., 2000; Suh et al., 2000).
Note added in proof. After submission of our manuscript, Yu et al. (2001) reached similar conclusions as far as the PS1(n−/−) are concerned. In this manuscript, we extend this research with the in-depth analysis of the cross of PS1(n−/−) mice with mutant APP transgenic mice.
Footnotes
This investigation was supported by the Fonds voor Wetenschappelijk Onderzoek (FWO)-Vlaanderen, by the Inter-University Attraction Pole program, by Vlaams Institut Biotechnologie, by European Economic Community (EEC)-Biotech, by EEC-5FP, by the Rooms-fund, by KULeuven Research Fund and by KULeuvenR&D, and by the Queen Elisabeth Fund for Medical Research. L.R. is a post-doctoral Researcher at the Belgian National Fund for Scientific Research, and I.D. is a post-doctoral fellow at Vlaams Institut Biotechnologie-FWO-Vlaanderen. The intellectual, technical, and material contributions of the following scientists are gratefully acknowledged: H. Van der Putten, P. Krimpenfort, A. Berns, U. Betz, K. Rajewsky, P. Saftig, K. Von Figura, E. Van Mechelen, H. Vanderstichele, G. Multhaup, and K. Beyreuther.
Correspondence should be addressed to Dr. Fred Van Leuven, Experimental Genetics Group (LEGT-EGG), Department of Human Genetics, K. U. Leuven, Campus Gasthuisberg, O&N 06, B-3000 Leuven, Belgium. E-mail:fredvl@med.kuleuven.ac.be.
REFERENCES
- 1.Akagi K, Sandig V, Vooijs M, Van der Valk M, Giovannini M, Strauss M, Berns A. Cre-mediated somatic site-specific recombination in mice. Nucleic Acids Res. 1997;25:1766–1773. doi: 10.1093/nar/25.9.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 4.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 5.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 6.Dewachter I, Van Dorpe J, Smeijers L, Gilis M, Kuiperi C, Laenen I, Caluwaerts N, Moechars D, Checler F, Vanderstichele H, Van Leuven F. Aging increased amyloid peptide and caused amyloid plaques in brain of old APP/V717I transgenic mice by a different mechanism than mutant presenilin1. J Neurosci. 2000;20:6452–6458. doi: 10.1523/JNEUROSCI.20-17-06452.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duff K, Rao MV. Progress in the modeling of neurodegenerative diseases in transgenic mice. Curr Opin Neurol. 2001;14:441–447. doi: 10.1097/00019052-200108000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Esler WP, Wolfe MS. A portrait of Alzheimer secretases: new features and familiar faces. Science. 2001;293:1449–1454. doi: 10.1126/science.1064638. [DOI] [PubMed] [Google Scholar]
- 9.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates (Franklin KBJ, ed). Academic; San Diego: 1997. [Google Scholar]
- 10.Freir DB, Holscher C, Herron CE. Blockade of long-term potentiation by beta-amyloid peptides in the CA1 region of the rat hippocampus in vivo. J Neurophysiol. 2001;85:708–713. doi: 10.1152/jn.2001.85.2.708. [DOI] [PubMed] [Google Scholar]
- 11.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 12.Golde TE, Younkin SG. Presenilins as therapeutic targets for the treatment of Alzheimer's disease. Trends Mol Med. 2001;7:264–269. doi: 10.1016/s1471-4914(01)02064-0. [DOI] [PubMed] [Google Scholar]
- 13.Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–106. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 14.Haass C, De Strooper B. The presenilins in Alzheimer's disease: proteolysis holds the key. Science. 1999;286:916–919. doi: 10.1126/science.286.5441.916. [DOI] [PubMed] [Google Scholar]
- 15.Handler M, Yang X, Shen J. Presenilin-1 regulates neuronal differentiation during neurogenesis. Development. 2000;127:2593–2606. doi: 10.1242/dev.127.12.2593. [DOI] [PubMed] [Google Scholar]
- 16.Hartmann D, De Strooper B, Saftig P. Presenilin-1 deficiency leads to loss of Cajal-Retzius neurons and cortical dysplasia similar to human type 2 lissencephaly. Curr Biol. 1999;9:719–727. doi: 10.1016/s0960-9822(99)80331-5. [DOI] [PubMed] [Google Scholar]
- 17.Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Holtzman DM, Linden DJ, Zhuo M, Muglia LJ, Chatila TA. Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci. 2000;20:6459–6472. doi: 10.1523/JNEUROSCI.20-17-06459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 19.Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29:177–185. doi: 10.1023/a:1021691918517. [DOI] [PubMed] [Google Scholar]
- 20.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsiao K. Transgenic mice expressing Alzheimer amyloid precursor proteins. Exp Gerontol. 1998;33:883–889. doi: 10.1016/s0531-5565(98)00045-x. [DOI] [PubMed] [Google Scholar]
- 22.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 23.Huang YY, Kandel ER, Varshavsky L, Brandon EP, Qi M, Idzerda RL, McKnight GS, Bourtchouladze R. A genetic test of the effects of mutations in PKA on mossy fiber LTP and its relation to spatial and contextual learning. Cell. 1995;83:1211–1222. doi: 10.1016/0092-8674(95)90146-9. [DOI] [PubMed] [Google Scholar]
- 24.Ida N, Hartmann T, Pantel J, Schroder J, Zerfass R, Forstl H, Sandbrink R, Masters CL, Beyreuther K. Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- 25.Leissring MA, Paul BA, Parker I, Cotman CW, LaFerla FM. Alzheimer's presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J Neurochem. 1999;72:1061–1068. doi: 10.1046/j.1471-4159.1999.0721061.x. [DOI] [PubMed] [Google Scholar]
- 26.Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol. 2000;149:793–798. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 28.Matilla A, Roberson ED, Banfi S, Morales J, Armstrong DL, Burright EN, Orr HT, Sweatt JD, Zoghbi HY, Matzuk MM. Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J Neurosci. 1998;18:5508–5516. doi: 10.1523/JNEUROSCI.18-14-05508.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meiri N, Sun MK, Segal Z, Alkon DL. Memory and long-term potentiation (LTP) dissociated: normal spatial memory despite CA1 LTP elimination with Kv1.4 antisense. Proc Natl Acad Sci USA. 1998;95:15037–15042. doi: 10.1073/pnas.95.25.15037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moechars D, Lorent K, De Strooper B, Dewachter I, Van Leuven F. Expression in brain of amyloid precursor protein mutated in the alpha-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J. 1996;15:1265–1274. [PMC free article] [PubMed] [Google Scholar]
- 31.Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- 32.Nalbantoglu J, Tirado-Santiago G, Lahsaini A, Poirier J, Goncalves O, Verge G, Momoli F, Welner SA, Massicotte G, Julien JP, Shapiro ML. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature. 1997;387:500–505. doi: 10.1038/387500a0. [DOI] [PubMed] [Google Scholar]
- 33.Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- 34.Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- 35.Ohtsuka T, Ishibashi M, Gradwohl G, Nakanishi S, Guillemot F, Kageyama R. Hes1 and Hes5 as notch effectors in mammalian neuronal differentiation. EMBO J. 1999;18:2196–2207. doi: 10.1093/emboj/18.8.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okabe S, Collin C, Auerbach JM, Meiri N, Bengzon J, Kennedy MB, Segal M, McKay RD. Hippocampal synaptic plasticity in mice overexpressing an embryonic subunit of the NMDA receptor. J Neurosci. 1998;18:4177–4188. doi: 10.1523/JNEUROSCI.18-11-04177.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rampon C, Tang YP, Goodhouse J, Shimizu E, Kyin M, Tsien JZ. Enrichment induces structural changes and recovery from nonspatial memory deficits in CA1 NMDAR1-knockout mice. Nat Neurosci. 2000;3:238–244. doi: 10.1038/72945. [DOI] [PubMed] [Google Scholar]
- 38.Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC, Sudhof TC. Short-term synaptic plasticity is altered in mice lacking synapsin I. Cell. 1993;75:661–670. doi: 10.1016/0092-8674(93)90487-b. [DOI] [PubMed] [Google Scholar]
- 39.Schneider I, Reverse D, Dewachter I, Ris L, Caluwaerts N, Kuiperi C, Gilis M, Geerts H, Kretzschmar H, Godaux E, Moechars D, Van Leuven F, Herms J. Mutant presenilins disturb neuronal calcium homeostasis in the brain of transgenic mice, decreasing the threshold for excitotoxicity and facilitating long-term potentiation. J Biol Chem. 2001;276:11539–11544. doi: 10.1074/jbc.M010977200. [DOI] [PubMed] [Google Scholar]
- 40.Schurmans S, Schiffmann SN, Gurden H, Lemaire M, Lipp HP, Schwam V, Pochet R, Imperato A, Bohme GA, Parmentier M. Impaired long-term potentiation induction in dentate gyrus of calretinin-deficient mice. Proc Natl Acad Sci USA. 1997;94:10415–10420. doi: 10.1073/pnas.94.19.10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 42.Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 43.Silva AJ, Rosahl TW, Chapman PF, Marowitz Z, Friedman E, Frankland PW, Cestari V, Cioffi D, Sudhof TC, Bourtchuladze R. Impaired learning in mice with abnormal short-lived plasticity. Curr Biol. 1996;6:1509–1518. doi: 10.1016/s0960-9822(96)00756-7. [DOI] [PubMed] [Google Scholar]
- 44.Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci USA. 1999;96:6959–6963. doi: 10.1073/pnas.96.12.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spittaels K, Van den Haute C, Van Dorpe J, Geerts H, Mercken M, Bruynseels K, Lasrado R, Vandezande K, Laenen I, Boon T, Van Lint J, Vandenheede J, Moechars D, Loos R, Van Leuven F. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. 2000;275:41340–41349. doi: 10.1074/jbc.M006219200. [DOI] [PubMed] [Google Scholar]
- 46.Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- 47.Suh YH. An etiological role of amyloidogenic carboxyl-terminal fragments of the beta-amyloid precursor protein in Alzheimer's Disease. J Neurochem. 1997;68:1781–1791. doi: 10.1046/j.1471-4159.1997.68051781.x. [DOI] [PubMed] [Google Scholar]
- 48.Suh YH, Kim HS, Lee JP, Park CH, Jeong SJ, Kim SS, Rah JC, Seo JH, Kim SS. Roles of A beta and carboxyl terminal peptide fragments of amyloid precursor protein in Alzheimer disease. J Neural Transm [Suppl] 2000;58:65–82. doi: 10.1007/978-3-7091-6284-2_6. [DOI] [PubMed] [Google Scholar]
- 49.Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- 50.Tesseur I, Van Dorpe J, Spittaels K, Van den Haute C, Moechars D, Van Leuven F. Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am J Pathol. 2000;156:951–964. doi: 10.1016/S0002-9440(10)64963-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, Sisodia SS. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J Biol Chem. 1997;272:28415–28422. doi: 10.1074/jbc.272.45.28415. [DOI] [PubMed] [Google Scholar]
- 52.Vanderstichele H, Van Kerschaver E, Hesse C, Davidsson P, Buyse MA, Andreasen N, Minthon L, Wallin A, Blennow K, Vanmechelen E. Standardization of measurement of beta-amyloid(1–42) in cerebrospinal fluid and plasma. Amyloid. 2000;7:245–258. doi: 10.3109/13506120009146438. [DOI] [PubMed] [Google Scholar]
- 53.Van Dorpe J, Smeijers L, Dewachter I, Nuyens D, Spittaels K, Van Den Haute C, Mercken M, Moechars D, Laenen I, Kuiperi C, Bruynseels K, Tesseur I, Loos R, Vanderstichele H, Checler F, Sciot R, Van Leuven F. Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the london mutant of human APP in neurons. Am J Pathol. 2000;157:1283–1298. doi: 10.1016/S0002-9440(10)64644-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vassar R, Citron M. Abeta-generating enzymes: recent advances in beta- and gamma-secretase research. Neuron. 2000;27:419–422. doi: 10.1016/s0896-6273(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 55.Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- 56.Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim TW. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 57.Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, St. George-Hyslop P. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000;407:48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- 58.Yu H, Saura CA, Choi SY, Sun LD, Yang X, Handler M, Kawarabayashi T, Younkin L, Fedeles B, Wilson MA, Younkin S, Kandel ER, Kirkwood A, Shen J. APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron. 2001;31:713–726. doi: 10.1016/s0896-6273(01)00417-2. [DOI] [PubMed] [Google Scholar]
- 59.Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Koster HJ, Borchardt T, Worley P, Lubke J, Frotscher M, Kelly PH, Sommer B, Andersen P, Seeburg PH, Sakmann B. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]