The Brain Metabolite Kynurenic Acid Inhibits α7 Nicotinic Receptor Activity and Increases Non-α7 Nicotinic Receptor Expression: Physiopathological Implications (original) (raw)
Abstract
The tryptophan metabolite kynurenic acid (KYNA) has long been recognized as an NMDA receptor antagonist. Here, interactions between KYNA and the nicotinic system in the brain were investigated using the patch-clamp technique and HPLC. In the electrophysiological studies, agonists were delivered via a U-shaped tube, and KYNA was applied in admixture with agonists and via the background perfusion. Exposure (≥4 min) of cultured hippocampal neurons to KYNA (≥100 nm) inhibited activation of somatodendritic α7 nAChRs; the IC50 for KYNA was ∼7 μm. The inhibition of α7 nAChRs was noncompetitive with respect to the agonist and voltage independent. The slow onset of this effect could not be accounted for by an intracellular action because KYNA (1 mm) in the pipette solution had no effect on α7 nAChR activity. KYNA also blocked the activity of preterminal/presynaptic α7 nAChRs in hippocampal neurons in cultures and in slices. NMDA receptors were less sensitive than α7 nAChRs to KYNA. The IC50 values for KYNA-induced blockade of NMDA receptors in the absence and presence of glycine (10 μm) were ∼15 and 235 μm, respectively. Prolonged (3 d) exposure of cultured hippocampal neurons to KYNA increased their nicotinic sensitivity, apparently by enhancing α4β2 nAChR expression. Furthermore, as determined by HPLC with fluorescence detection, repeated systemic treatment of rats with nicotine caused a transient reduction followed by an increase in brain KYNA levels. These results demonstrate that nAChRs are targets for KYNA and suggest a functionally significant cross talk between the nicotinic cholinergic system and the kynurenine pathway in the brain.
Keywords: nicotinic ACh receptors, NMDA receptors, hippocampus, kynurenic acid, electrophysiology, brain slices
Kynurenic acid (KYNA), a product of tryptophan metabolism, has neuroprotective and neuroinhibitory properties that have been attributed to its action as a competitive antagonist at the glycine site on NMDA receptors (Stone, 1993;Parsons et al., 1997). However, high micromolar concentrations of KYNA are often needed to block NMDA receptor function (Stone, 1993). Considering that KYNA levels in brains of nonprimates and primates (including humans) range from low nanomolar to low micromolar (Moroni et al., 1988; Turski et al., 1988), it is uncertain whether endogenous KYNA levels are sufficiently high to block NMDA receptor activity (Scharfman et al., 1999; Urenjak and Obrenovitch, 2000).
Interest in the neurobiology of KYNA has gained momentum because of reports of changes in its brain content in several diseases. Cerebral KYNA levels are increased in patients with Alzheimer's disease (AD) (Baran et al., 1999), Down's syndrome (DS) (Baran et al., 1996), and schizophrenia (Schwarcz et al., 2001), and levels are decreased in patients with end-stage Parkinson's disease (PD) (Ogawa et al., 1992) and Huntington's disease (HD) (Beal et al., 1990). Manipulations of brain levels of KYNA in laboratory animals supported the concept that increased KYNA levels are neuroprotective and anticonvulsant, whereas decreased levels of the metabolite increase neuronal vulnerability (Pellicciari et al., 1994; Poeggeler et al., 1998; Cozzi et al., 1999).
The physiopathological consequences of relatively modest variations in brain levels of KYNA raised the possibility that KYNA might interact with high affinity with an as-of-yet unidentified target. Nicotinic receptors (nAChRs) were among the most likely candidates because (1) preliminary electrophysiological experiments revealed an exquisite sensitivity of α7 nAChRs to inhibition by KYNA (Hilmas et al., 2000), and (2) like NMDA receptors, nAChRs are involved in regulating neuronal plasticity (Albuquerque et al., 1997; Broide and Leslie, 1999;Mansvelder and McGehee, 2000) and survival in the brain (Zoli et al., 1999; Kihara et al., 2001).
Although numerous types of neuronal nAChRs have been identified (Lindstrom, 1996), those binding nicotine with high affinity (i.e., α4β2 nAChRs) and those binding α-bungarotoxin (α-BGT) (i.e., α7 nAChRs) are most prevalent in the brain (Lindstrom, 1996;Albuquerque et al., 1997). Notably, brain levels of nicotine-binding sites are reduced in patients with AD, PD, and DS (Perry et al., 1990;Hellstrom-Lindahl et al., 1999), and numbers of [125I]α-BGT-binding sites are decreased in selected brain regions of patients with schizophrenia and AD (Freedman et al., 2000; Court et al., 2001).
This study was designed to investigate the effects of KYNA on nAChRs in CNS neurons and to determine the effects of nicotine on KYNA levels_in vivo_. The results indicate that (1) α7 nAChRs are inhibited noncompetitively by KYNA (IC50 ∼7 μm), (2) NMDA receptors are less sensitive than nAChRs to inhibition by KYNA, (3) KYNA increases expression of non-α7 (most likely α4β2) nAChRs, and (4) repeated treatment of rats with nicotine causes a transient decrease followed by an increase in brain KYNA levels. Thus, KYNA may regulate neuronal excitability and plasticity by controlling function or expression, or both, of nAChRs in the CNS.
MATERIALS AND METHODS
Cultured hippocampal and cerebral cortical neurons. Primary cultures of cells dissociated from the hippocampi or cerebral cortex of 16- to 19-d-old rat fetuses (Sprague Dawley) were prepared according to the procedure described in Alkondon and Albuquerque (1993). Hippocampal cells were plated onto collagen-coated 35 mm Petri dishes (Nunc, Naperville, IL), whereas cerebral cortical cells were plated in 25 cm flasks (Falcon). Electrophysiological experiments were performed on hippocampal neurons cultured for 7–40 d, and binding assays were performed in 7-d-old primary cultures of cerebral cortex.
Rat hippocampal slices. Slices of 250 μm thickness were obtained using a Vibratome (Leica Microsystems, Wetzlar, Germany) from the hippocampi of 15- to 30-d-old Sprague Dawley rats and placed in a chamber containing artificial CSF (ACSF). The ACSF was bubbled continuously with a mixture of 95% O2 and 5% CO2 at room temperature (20–22°C) and had the following composition (in mm): NaCl 125, NaHCO3 25, KCl 2.5, NaH2PO4 1.25, CaCl2 · 2H2O 2, MgCl2 1, and glucose 25. Slices were allowed to equilibrate for at least 1 hr before they were transferred to the recording chamber (Alkondon et al., 1999).
Electrophysiological recordings. Transmembrane macroscopic currents were recorded according to the whole-cell mode of the patch-clamp technique (Hamill et al., 1981) using an LM-EPC-7 amplifier (List Electronics, Darmstadt, Germany). Signals were filtered at 1–2 kHz and either stored in a VCR for later analysis or directly sampled by a Pentium-III computer using the PCLAMP6 program (Axon Instruments, Foster City, CA). The composition of the external solution used to bathe the cultured neurons was (in mm): NaCl 165, KCl 5, CaCl2 · 2H2O 1, HEPES 5, dextrose 10 (pH was adjusted to 7.3 with NaOH; osmolarity ∼340 mOsm). Unless otherwise stated, atropine (1 μm) and tetrodotoxin (TTX; 200 nm) were added to the external solution to block muscarinic receptors and voltage-gated Na+channels, respectively. The internal solution for recordings from cultured neurons had the following composition (in mm): CsCl 80, CsF 80, EGTA 10, CsOH 22.5, and HEPES 10 (pH was adjusted to 7.3 with CsOH; osmolarity ∼340 mOsm). When whole-cell currents were evoked by activation of α7 nAChRs, rundown was minimized by adding the following ATP-regenerating compounds to the internal solution: Tris-phosphocreatine 20.0 mm, Tris-ATP 5.0 mm, and creatine-phosphokinase 50 U/ml. The concentrations of CsCl and CsF in the internal solution were decreased to 60 mm so that the osmolarity of the ATP-regenerating internal solution was maintained at ∼340 mOsm. The internal solution used for recordings from neurons in hippocampal slices had the following composition (in mm): EGTA 10, HEPES 10, Cs-methane sulfonate 130, CsCl 10, MgCl2 2, and QX-314 5 (pH was adjusted to 7.3 with CsOH; osmolarity ∼340 mOsm). When filled with the internal solutions described above, the recording pipettes had resistances of 2–5 MΩ (for cultured neurons) and 3–5 MΩ (for neurons in slices). No compensation was made for the access resistance, which was ≤15 MΩ. Results were not used when the access resistance changed significantly during the experiment.
Drug application. A standard U-shaped glass tube (U-tube) was used for drug delivery onto cultured neurons (Alkondon and Albuquerque, 1993). A modified U-tube was used for drug delivery onto neurons in hippocampal slices (Alkondon et al., 1999). In all experiments, antagonists were delivered to the neurons via the U-tube (as admixtures with agonists) and via the bath perfusion.
Hippocampal neurons cultured for 7–10 d were used in experiments designed to investigate the effects of 3 d exposure to KYNA on nicotinic sensitivity. In short, these experiments involved changing the culture medium with drug-containing medium on the first and last day before testing the nicotinic responsiveness of the neurons.
Data analysis. Peak amplitude, rise time, and decay-time constants (τdecay) of whole-cell currents and postsynaptic currents (PSCs) were analyzed using the PCLAMP6 program. Rundown of whole-cell currents triggered by activation of α7 nAChRs was corrected according to the following analysis: agonist pulses were applied to the neurons at 2 min intervals for as long as needed to stabilize the current amplitudes before exposure of the neurons to agonist–antagonist admixtures. After full reversibility of the effects of antagonists was achieved after washing of the neurons, agonist pulses were applied to the neurons at 2 min intervals for an additional 4–15 min. Plots of the amplitudes of agonist-evoked currents versus recording time were fitted by linear regression of the points obtained before exposure of the neurons to the antagonists and after wash. Then, the expected amplitudes of agonist-evoked currents at any given time were estimated. Concentration–response curves were fitted by the Hill equation: I = (_I_max × [_A_]nH)/([A_]nH+ EC50nH), where_I and _I_max are the measured and the maximum current amplitudes, respectively, [_A_] is the agonist concentration, nH is the Hill coefficient, and EC50 is the agonist concentration producing half-maximum response. Data are presented as mean ± SEM.
Binding assay. Primary cultures of rat cerebral cortex were used to assay nAChR binding. A solution of 0.02% EDTA was used to detach cells from the flasks, and mechanical scraping was performed to facilitate further detachment of cells. Four flasks of primary cerebral cortical cultures were combined for each control and treatment groups into separate culture tubes and centrifuged at 900 × _g_for 10 min at 8°C. The pellet was washed by resuspension in ice-cold Tris-HCl (20 mm, pH 7.4) and subsequently centrifuged again at 900 × g for 10 min at 8°C. The wash step was repeated twice. The final pellet was homogenized in 50 mm Tris-HCl buffer (pH 7.4) and kept on ice until assayed.
Binding of [3H] epibatidine (10 nm) to nAChRs was measured by vacuum filtration assay. Aliquots (25 μl) of the homogenized sample were added to the control and treatment tubes to begin the incubation. The preparation was then incubated for 60 min at 23°C with the radioactive ligand in a total volume of 250 μl buffer, in the absence and presence of its specific displacer. Bound radioactivity was separated from free ligand by vacuum filtration over Whatman GF/B polyethylenimine (0.05%)-treated filters and washed with 8 ml of ice-cold 0.9% saline solution. Radioactivity was determined by liquid scintillation spectroscopy. Nonspecific binding was determined using nonradioactive (+)epibatidine (100 μm). To determine the effect of prolonged exposure to KYNA on epibatidine binding, cultures of rat cerebral cortex were exposed for 3 d to 10 μm KYNA. The experiments consisted of changing the medium with drug-containing medium in the first and last day before the binding assay. KYNA was washed out during the preparation of the pellets. Epibatidine binding was assayed in untreated cultures and in age-matched, treated cultures. Protein measurements were done using the bicinchoninic acid assay (Pierce, Rockford, IL).
Nicotine treatment of rats. Adult male Sprague Dawley rats (180–200 gm) were used to evaluate the effect of repeated nicotine injections in vivo. Nicotine was dissolved in PBS and administered subcutaneously twice daily (every 12 hr) for 5 or 15 d. Control animals received identical PBS injections.
KYNA determination. One hour after the final nicotine or PBS injection, animals were decapitated, and the brain was rapidly removed from the skull and placed on ice. Brain regions of interest were dissected out, and the tissues were placed on dry ice and stored at −80°C. On the day of the assay, the tissue was thawed and homogenized (1:10, w/v) in ultra-pure water. A 300 μl aliquot of the homogenate was acidified with 75 μl of 6% perchloric acid. After centrifugation (10 min, 12,000 × g), an aliquot of the supernatant was diluted (1:1, v/v) with HPLC mobile phase containing 200 mm zinc acetate and 3.5% acetonitrile (pH 6.2). KYNA levels were then determined by HPLC with fluorescence detection (excitation wavelength 344 nm; emission wavelength 398 nm) as described by Wu et al. (1992).
Drugs used. Methyllycaconitine (MLA) citrate was a gift from Prof. M. H. Benn (Department of Chemistry, University of Calgary, Alberta, Canada). (+/−)Epibatidine[5,6-bicycloheptyl-3H] (specific activity = 33.8 Ci/mmol) was purchased from NEN Life Science Products (Boston, MA). DHβE hydrobromide was a gift from Merck, Sharp & Dohme (Rahway, NJ). KYNA was dissolved in DMSO and the appropriate concentration of DMSO was used in the control experiments. All other chemicals were purchased from Sigma (St. Louis, MO).
RESULTS
KYNA inhibits postsynaptic responses mediated by α7 nAChRs in cultured hippocampal neurons
In the presence of atropine (1 μm) and TTX (200 nm), application of ACh (1 mm) to ∼80% of the cultured hippocampal neurons evoked whole-cell currents that had the characteristics of responses mediated by α7 nAChRs. These currents, which are hereafter referred to as type IA currents (Alkondon and Albuquerque, 1993), were the result of activation of α7 nAChRs present in the somata and proximal dendrites of hippocampal neurons from which recordings were obtained. They decayed to the baseline before the end of the agonist pulse, were reversibly blocked by the α7 nAChR-selective antagonist MLA (1 nm) (Fig.1A) and could be evoked by the α7 nAChR-selective agonist choline.
Fig. 1.

KYNA-induced blockade of type IA currents in cultured hippocampal neurons. A, Fast desensitizing currents that were evoked by U-tube application of ACh to cultured hippocampal neurons were blocked after 10 min perfusion of the neurons with MLA (2 nm)-containing external solution. The effect of MLA was fully reversible after 15 min washing of the neurons.B, Sample recordings of ACh-evoked type IA currents obtained from hippocampal neurons before their exposure to KYNA (left traces), after 4–10 min perfusion with KYNA-containing external solution (middle traces), and after 10 min washing with KYNA-free external solution (right traces). Different neurons were exposed to each KYNA concentration. C, Concentration–response relationship for KYNA-induced blockade of ACh (1 mm)-evoked type IA currents. The rundown-corrected amplitude of type IA currents (see Materials and Methods) was taken as 1 and used to normalize the amplitude of type IA currents evoked by ACh in the presence of KYNA (0.1 μm to 1 mm). Each point and bar represent mean and SEM, respectively, of results obtained from 5–18 neurons. All experiments were performed in neurons perfused continuously with external solution containing atropine (1 μm) and TTX (200 nm). Membrane potential (A_–_C), −60 mV. Inset, Chemical structure of KYNA. D, KYNA-induced blockade of choline-evoked action potentials recorded from a cultured hippocampal neuron under cell-attached configuration. U-tube application of the α7 nAChR agonist choline (10 mm) to the neuron evoked action potentials (left trace). The response induced by choline was blocked by 4–6 min exposure of the neuron to KYNA (1 mm; middle trace). The effect of KYNA was reversed after 10 min washing of the neuron with external solution (right trace). Recordings were obtained in the presence of atropine (1 μm), picrotoxin (100 μm), CNQX (10 μm), and APV (50 μm). Pipette potential, −60 mV.
The amplitudes of ACh (1 mm)-evoked type IA currents were reduced after perfusion of cultured hippocampal neurons with external solution containing KYNA (0.1 μm to 1 mm) (Fig. 1B). At the lowest test concentration, i.e., 0.1 μm, KYNA reduced by ∼20% the amplitude of type IA currents. On average, it took 4 min to observe the onset of the inhibitory effect of KYNA on α7 nAChRs in cultured hippocampal neurons; this time varied with the concentration of KYNA, being shortest for the highest concentration of KYNA. The time needed for the effect to be maximal also decreased with increasing concentrations of KYNA; it ranged from 8–10 min for 0.1 μm KYNA to 4–6 min for 1 mm KYNA. In addition, inhibition of α7 nAChRs by KYNA was fully reversible after washing of the neurons with KYNA-free external solution for ≥10 min (Fig.1B).
The inhibitory effect of KYNA on α7 nAChRs was concentration dependent: the higher the concentration of KYNA, the larger the effect on the amplitude of type IA currents. Fitting the concentration–response relationship by the Hill equation revealed that KYNA blocks α7 nAChRs with an IC50 of 7.1 ± 0.9 μm and an nH of 0.34 ± 0.02 (Fig.1C). The lowest estimated effective concentration of KYNA was ∼1 nm. At 1 mm, KYNA reduced by 80–90% the amplitude of type IA currents. It also abolished fast current transients that represented action potentials resulting from activation of somatodendritic α7 nAChRs by choline (10 mm) applied to hippocampal neurons under the cell-attached condition (Fig. 1D).
The decay phase of type IA currents evoked by ACh in the absence and presence of KYNA (0.1 μm to 1 mm) was best fitted by a single exponential. The τdecay of type IA currents evoked by ACh (1 mm) was 49.1 ± 2.7 msec (n = 60 neurons). At concentrations ranging from 0.1 to 100 μm, KYNA had no effect on the decay phase of the currents. In the presence of KYNA (0.1–100 μm), τdecay of ACh-evoked type IA currents was 49.9 ± 2.7 msec (n = 60 neurons). At KYNA concentrations ≥300 μm, the reduction of the peak current amplitude was substantial, making it difficult to determine reliably the decay-time constant of the currents.
7-Chloro-KYNA, a derivative of KYNA that is more potent as an NMDA receptor antagonist than the parent compound (Leeson and Iversen, 1994), had very little effect on choline-evoked type IA currents. At 1 mm, 7-chloro-KYNA reduced by no more than 15% the amplitude of type IA currents (data not shown).
KYNA inhibits presynaptic responses mediated by α7 nAChRs in cultured hippocampal neurons
In the absence of TTX, and in the continuous presence of atropine (1 μm) and the glutamate receptor antagonists APV (50 μm) and CNQX (10 μm), bursts of postsynaptic currents could be recorded from cultured hippocampal neurons that were exposed for 5 sec to choline (10 mm) (Fig. 2). These currents were sensitive to blockade by picrotoxin (100 μm; data not shown) and are herein referred to as IPSCs. They were the result of GABA released by choline-induced activation of α7 nAChRs in GABAergic neurons synapsing onto the neurons from which recordings were obtained. In fact, previous studies have suggested that nAChRs are present in the somatodendritic and preterminal areas of GABAergic hippocampal neurons (Alkondon et al., 1999).
Fig. 2.
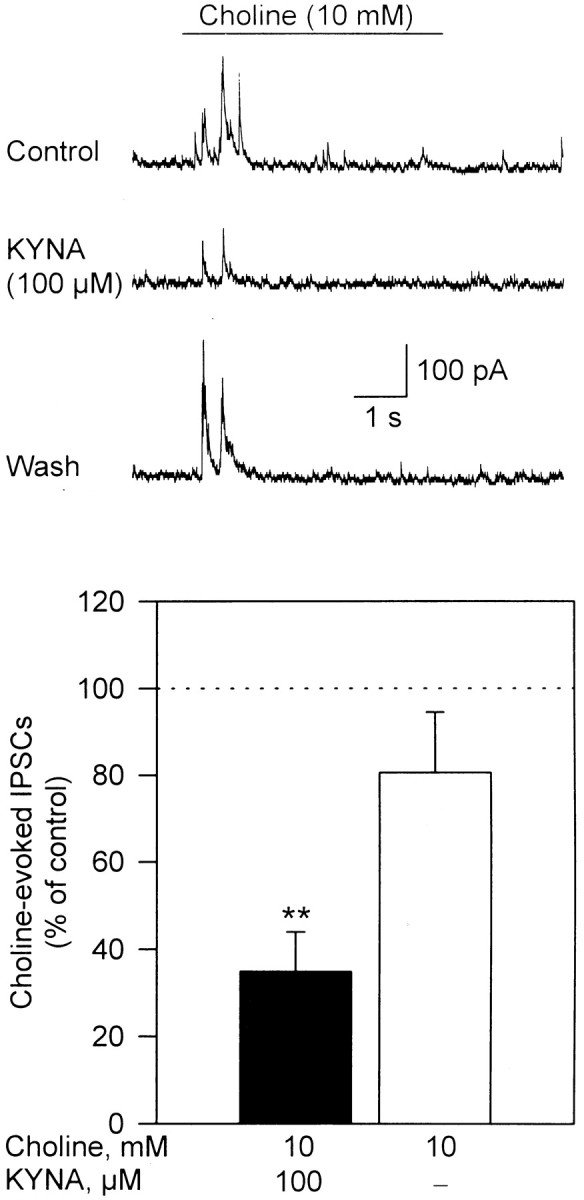
KYNA-induced blockade of IPSCs evoked by choline in cultured hippocampal neurons. Sample recordings of choline (10 mm)-evoked IPSCs obtained before (Control), after 10 min perfusion of the hippocampal neurons with external solution containing KYNA (100 μm), and after 20 min washing of the neuron with external solution (Wash) are shown. Recordings were obtained in the presence of atropine (1 μm), CNQX (10 μm), and APV (50 μm). Membrane potential, +40 mV. Graph shows quantification of the effect of KYNA on choline-triggered IPSCs. Total charge carried by IPSCs triggered by choline was estimated by the area under the curve during the 5 sec pulse application of choline. The total charge of choline-evoked IPSCs recorded before exposure of the neurons to KYNA was taken as 100% and used to normalize the responses recorded in the presence of KYNA and after washing of the neurons. Each graph bar and error bar represent mean and SEM, respectively, of results obtained from three neurons. **p < 0.01 (paired Student's_t_ test).
Choline-evoked IPSCs were blocked after 5 min perfusion of the hippocampal neurons with the external solution containing KYNA. At 100 μm, KYNA reduced by ∼60% the net charge carried by choline-evoked IPSCs (Fig. 2). The effect of KYNA was reversed after washing of the neurons with KYNA-free external solution. At 1 mm, a concentration sufficient to block >90% of the α7 nAChR activity, KYNA had no effect on whole-cell currents evoked by application of GABA (50 μm) to hippocampal neurons in culture; the peak amplitudes of GABA-evoked currents recorded in the presence of KYNA (1 mm) were ∼96% of those of control responses (n = 6). Taken together, these findings support the concept that postsynaptic GABAAreceptors are not sensitive to KYNA (Stone, 1993) and suggest that presynaptic and postsynaptic responses mediated by α7 nAChRs in cultured hippocampal neurons are equally sensitive to blockade by KYNA.
KYNA inhibits the activity of α7 nAChRs present in interneurons in hippocampal slices
Choline (10 mm, 6 sec) applied to CA1 interneurons of the stratum radiatum of rat hippocampal slices had effects that could be distinguished at different membrane potentials. At 0 mV, it triggered bursts of PSCs (Fig. 3). The events recorded at 0 mV were GABAergic in nature because the reversal potential for nicotinic and glutamatergic currents is close to 0 mV. As in the experiments performed in cultured neurons, choline-evoked GABAergic PSCs were the result of GABA released by activation of α7 nAChRs present in interneurons synapsing onto the neurons from which recordings were obtained (Alkondon et al., 1999).
Fig. 3.
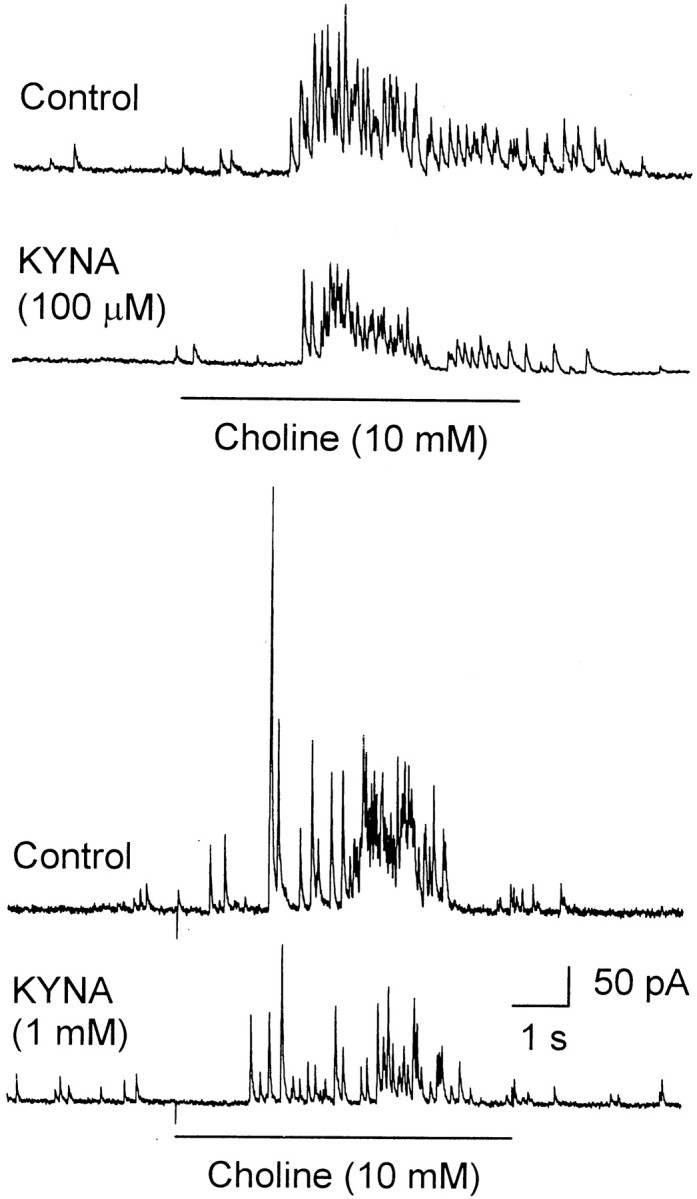
KYNA-induced blockade of choline-evoked IPSCs recorded from CA1 interneurons in rat hippocampal slices. Representative sample traces of choline (10 mm)-evoked GABAergic PSCs recorded before (Control) and after 25 min perfusion of the slices with ACSF containing KYNA (100 μm or 1 mm) are shown. Pairs of traces shown in the top and bottom panels were obtained from different neurons. Recordings were obtained in the presence of atropine (1 μm), CNQX (10 μm), and APV (50 μm). Methanesulfonate-based internal solution was used to fill the patch pipettes (see Materials and Methods). Membrane potential, 0 mV.
Perfusion of the slices with ACSF containing KYNA (100 μmor 1 mm) for 25 min resulted in the reduction of the total charge carried by choline-triggered GABAergic PSCs (Fig. 3). At 100 μm and 1 mm, KYNA reduced by 25–50 and 50–75%, respectively, the total charge carried by choline-triggered GABAergic PSCs (n = 3 for each concentration of KYNA). The magnitude of the effect of KYNA was time dependent. For instance, after 8 min perfusion of hippocampal slices with KYNA (1 mm), the total charge carried by choline-triggered GABAergic PSCs was reduced by only 30 ± 5% (n = 3 neurons). KYNA seemed less effective in inhibiting α7 nAChRs in hippocampal neurons in slices than in cultures. However, this apparent difference can be reconciled by considering that drug exchange in the slices is considerably slower than in the primary cultures and that KYNA is extremely hydrophobic.
Mechanism of action of KYNA on α7 nAChRs
Considering the slow onset of KYNA-induced inhibition of α7 nAChRs, experiments were designed to verify whether the effect of the drug on α7 nAChRs could be attributed to an intracellular mechanism of action. Type IA currents were recorded from hippocampal neurons using the ATP-regenerating internal solution containing either no KYNA or 1 mm KYNA. The amplitudes of type IA currents evoked by the first agonist pulse in each neuron tested were taken as 100% and used to normalize the current amplitudes recorded from that particular neuron at different time points. The absolute peak amplitudes of type IA currents evoked by the first agonist pulse in the absence and presence of intracellular KYNA were within the same range: 1229 ± 108 pA for control responses (n = 54 neurons) and 1664 ± 352 pA for responses recorded using the internal solution containing KYNA (n = 4 neurons). In addition, a plot of the normalized current amplitudes against recording time revealed that there were no significant differences between responses recorded in the absence and presence of intracellular KYNA (Fig. 4).
Fig. 4.

KYNA applied intracellularly has no effect on ACh-evoked type IA currents. Rundown of the peak amplitude of ACh (1 mm)-evoked type IA currents recorded from cultured hippocampal neurons using pipette solution without (Control) or with KYNA (1 mm) is shown. The amplitude of the currents evoked by the first ACh pulse in each neuron was taken as 1 and used to normalize the amplitude of currents recorded subsequently from that same neuron. Each symbol and bar represent mean and SEM, respectively, of results obtained from 54 neurons in the control group and 4 neurons in the test group. All recordings were obtained in the presence of TTX (200 nm) and atropine (1 μm). Membrane potential, −60 mV.
To investigate the nature of the blockade of α7 nAChRs by KYNA, ACh-evoked type IA currents were recorded before and after perfusion of the neurons with external solution containing KYNA (30 μm). Before exposure of the neurons to KYNA, increasing concentrations of ACh evoked type IA currents with progressively larger amplitudes. The maximal response occurred at 3–10 mm ACh, and the EC50 for ACh was 145.9 ± 11.9 μm (Fig. 5A). After perfusion with KYNA (30 μm), the maximal effect of ACh was reduced. Increasing concentrations of ACh did not surmount KYNA-induced inhibition of type IA currents (Fig.5A). In contrast, the EC50 for ACh was not changed significantly (Fig. 5B). Therefore, the interaction of KYNA with α7 nAChRs is noncompetitive with respect to the agonist. Also unchanged was the nH for ACh in evoking type IA currents.
Fig. 5.

KYNA-induced blockade of α7 nAChRs is noncompetitive and voltage independent. A, Semi-logarithmic plot of the concentration–response relationships for ACh in evoking type IA currents in cultured hippocampal neurons in the absence and presence of KYNA. Under the control condition, ACh (1 sec pulses; 30 μm to 10 mm) was applied to cultured hippocampal neurons. Rundown-corrected amplitudes of currents evoked by 10 mm ACh were taken as 1 and used to normalize the amplitudes of currents evoked by other ACh concentrations. In another set of experiments, after the control responses evoked by a given concentration of ACh were recorded, neurons were exposed for 4–8 min to KYNA (30 μm) and tested for their responsiveness to pulses of ACh plus KYNA. Rundown-corrected amplitudes of currents evoked by a given ACh concentration under control condition were then taken as 1 and used to normalize the amplitudes of currents evoked by ACh in the presence of KYNA. Membrane potential, −60 mV.B, Double-reciprocal plots of the concentration–response relationships for ACh in evoking type IA currents in the absence and presence of KYNA (30 μm). Each symbol and bar represent mean and SEM, respectively, of results obtained from 3–11 neurons. In some cases, error bars are not seen because they are smaller than the symbol size. C, Current–voltage relationships for responses evoked by ACh (1 mm) in the absence (●) or presence of KYNA (30 μm; ○). Under the control condition, rundown-corrected amplitudes of ACh (1 mm)-evoked currents recorded from neurons voltage clamped at −60 mV were taken as 1 and used to normalize the amplitudes of currents evoked by ACh at all membrane potentials. The plot of the normalized current amplitude versus membrane potential could be fitted by a straight line. The extrapolated reversal potential was ∼0 mV. Rundown-corrected amplitudes of type IA currents evoked by ACh (1 mm) at any membrane potential were taken as 1 and used to normalize the amplitudes of type IA currents evoked by pulses of ACh plus KYNA (30 μm) at that membrane potential. D, Plot of the ratio of the amplitudes of currents evoked by pulses of ACh plus KYNA and the amplitudes of currents evoked by ACh alone versus membrane potential. Each symbol and bar represent mean and SEM, respectively, of results obtained from three neurons. All experiments were performed in the presence of TTX (200 nm) and atropine (1 μm).
To determine the voltage dependence of KYNA-induced blockade of α7 nAChRs, type IA currents were evoked by ACh (1 mm) in the absence or presence of KYNA (30 μm) at several membrane potentials. At all membrane potentials, KYNA reduced the amplitude of type IA currents (Fig. 5C). Under control conditions, there was a linear relationship between amplitude of ACh-evoked type IA currents and membrane potentials ranging from −80 to 0 mV (Fig.5C). Inhibition of type IA currents by KYNA was voltage independent; the percentage reduction of the current amplitudes by KYNA was virtually the same at all membrane potentials (Fig. 5D). These data suggest that the binding site for KYNA on α7 nAChRs is not within the electrical field of the membrane.
KYNA on NMDA receptors in cultured hippocampal neurons: the effect of added glycine
A straightforward comparison of the sensitivity of nAChRs and NMDA receptors to blockade by KYNA would be made possible if the effects of the metabolite on both receptors were examined under the same experimental conditions. Thus, two sets of experiments were designed to determine the effects of KYNA on NMDA receptors. In one set, glycine was not added to the agonist solution. In the other set, the agonist solution consisted of NMDA (100 μm) plus glycine (10 μm).
In the absence of added glycine, 500 msec pulses of NMDA applied to the cultured hippocampal neurons elicited whole-cell currents that decayed much faster than currents evoked by the admixture of NMDA (100 μm) plus glycine (10 μm) (Figs.6A,7A). The τdecay of whole-cell currents evoked by NMDA was 305.1 ± 29.1 msec, whereas the τdecay of currents elicited by NMDA plus glycine was 645.7 ± 26.8 msec (n = 100 responses recorded from 12 neurons in each experimental group). In agreement with previous studies (Kleckner and Dingledine, 1988; Johnson and Ascher, 1992; Wang and MacDonald, 1995), glycine slowed down the rate of desensitization of the NMDA receptors.
Fig. 6.

KYNA-induced blockade of whole-cell currents evoked by NMDA in cultured hippocampal neurons. A,Traces are sample recordings of currents evoked by NMDA (100 μm) under control conditions (left traces), in the presence of KYNA (0.1 μm to 1 mm) after 4 min perfusion of the neurons with external solution containing KYNA (middle traces), and after 10 min washing of the neurons with KYNA-free external solution (right traces). B, Graph is the semi-logarithmic plot of the concentration-dependent inhibition of NMDA-evoked currents by KYNA. The amplitudes of currents evoked by NMDA under control condition were taken as 1 and used to normalize the amplitudes of currents recorded in the presence of KYNA. Each point and bar represent mean and SEM, respectively, of results obtained from 3–10 experiments. All recordings were obtained in the presence of TTX (200 nm). Membrane potential, −60 mV.
Fig. 7.

KYNA-induced blockade of whole-cell currents evoked by NMDA-plus-glycine in cultured hippocampal neurons.A, Traces are sample recordings of whole-cell currents evoked by NMDA (100 μm)-plus-glycine (10 μm) under control conditions (left traces), in the presence of KYNA (30 μm to 1 mm) after 4 min perfusion of the neurons with external solution containing KYNA (middle traces), and after 10 min washing of the neurons with KYNA-free external solution (right traces). B, Graph is semi-logarithmic plot of the concentration-dependent effect of KYNA on whole-cell current evoked by NMDA-plus-glycine. The amplitudes of currents evoked by NMDA-plus-glycine under control condition were taken as 1 and used to normalize the amplitudes of currents recorded in the presence of KYNA. Each point and bar represent mean and SEM, respectively, of results obtained from four to seven experiments. All recordings were obtained in the presence of TTX (200 nm). Membrane potential, −60 mV.
In several experiments, the effects of KYNA on whole-cell currents evoked by either NMDA or NMDA-plus-glycine were analyzed. Currents elicited by NMDA or NMDA-plus-glycine were recorded from neurons before and during their exposure to KYNA (0.1 μm to 1 mm). After 4 min perfusion of the neurons with external solution containing KYNA, the amplitudes of currents induced by NMDA (or NMDA-plus-glycine) in the continuous presence of the drug were smaller than those of currents recorded under control conditions. The effect of KYNA on the amplitude of NMDA-elicited currents was fully reversible and concentration dependent: the higher the concentration of the drug, the lower the amplitude of the currents. Fitting the concentration–response relationship by the Hill equation revealed that, in the absence of added glycine, KYNA blocks NMDA receptors with an IC50 of 15.4 ± 2.6 μm and an nH of 0.48 ± 0.04 (Fig.6B). In the presence of glycine (10 μm), the IC50 and the nH for KYNA in blocking NMDA receptors were 238.9 ± 5.8 μm and 0.93 ± 0.06, respectively (Fig.7B).
Prolonged exposure of hippocampal and cerebral cortical neurons to KYNA increases expression of non-α7 (probably α4β2) nAChRs
It has been proposed that nAChR inhibition by some nicotinic antagonists leads to receptor upregulation (Dani and Heinemann, 1996). The nicotinic sensitivity of cultured hippocampal neurons that were exposed for 3 d to KYNA was therefore compared with that of untreated neurons. After the treatment and before the electrophysiological recordings, the neurons were perfused for 30 min with external solution to remove any remaining free metabolite and to reverse the blockade of α7 nAChRs by KYNA.
To examine changes in receptor function, nicotinic currents were evoked by ACh in 10 neurons of each treated culture dish and in 10 neurons of untreated dishes from the same batch. Three and four different cultures were treated with 1 and 10 μm KYNA, respectively. In this set of experiments, ACh was used as the agonist because, being a non-subtype selective nicotinic agonist, it could provide information regarding the function of different nAChR subtypes in the neurons. ACh-evoked currents were classified as type IA if they decayed to the baseline before the end of the agonist pulse and as non-type IA currents if they decayed to the baseline after the end of the agonist pulse. Most non-type IA currents had a rapidly decaying and a slowly decaying component, resembling type IB currents (Alkondon and Albuquerque, 1993); in these currents, the rapidly and slowly decaying components are generated by activation of α7 and α4β2 nAChRs, respectively (for review, see Albuquerque et al., 1997). In these composite responses, the amplitude of the slowly decaying component was taken as the amplitude of responses induced by non-type IA currents; the result of (total peak current amplitude) − (amplitude of the slowly decaying component) was taken as the amplitude of type IA current in that particular neuron. The other non-type IA currents that had a single slowly decaying component, resembling type II currents (Alkondon and Albuquerque, 1993), were most likely subserved by α4β2 nAChRs because this is the second prevalent nAChR subtype present in cultured hippocampal neurons (for review, see Albuquerque et al., 1997).
The average amplitude of ACh (1 mm)-evoked type IA currents recorded from neurons treated for 3 d with 1 or 10 μm KYNA was not statistically different from that of ACh-elicited currents recorded from batch-matched, untreated neurons (n = 30 or 40 neurons in each experimental group) (Table 1). However, after 3 d exposure of the hippocampal neurons to KYNA, there were changes in the pattern of nicotinic responsiveness. Of 40 neurons sampled from untreated cultures, 5 (12.5%) showed no response to ACh, 27 (67.5%) responded to ACh with fast-desensitizing, type IA currents, and 8 (20%) responded to ACh with non-type IA currents. In contrast, of 40 neurons that were sampled from cultures exposed to KYNA (10 μm), 2 (5%) showed no response to ACh, whereas 25 (62.5%) and 18 (45%) showed type IA and non-type IA currents in response to ACh, respectively (Table 1). Although the treatment with 10 μm KYNA increased significantly the non-α7 (most likely α4β2) nicotinic sensitivity of hippocampal neurons in culture, treatment with 1 μm KYNA did not have a significant effect. It should be noted that the absence of ACh-evoked whole-cell currents does not indicate that neurons have no nAChRs. A low number of somatodendritic nAChRs may not give rise to detectable macroscopic responses. Likewise, currents passing through nAChRs located in preterminal and terminal sites are unlikely to be detected in whole-cell recordings.
Table 1.
Effects of KYNA on nicotinic sensitivity of cultured hippocampal neurons
| Type IA currents | Non-type IA currents | |||
|---|---|---|---|---|
| Peak amplitude (pA) | % Neurons1-a | Peak amplitude (pA) | % Neurons1-a | |
| Control | 374 ± 114 | 53 (16/30) | 87 ± 20 | 43 (13/30) |
| KYNA (1 μm) | 189 ± 38 | 56 (17/30) | 52 ± 7 | 53 (16/30) |
| Control | 221 ± 50 | 67 (27/40) | 61 ± 9 | 20 (8/40) |
| KYNA (10 μm) | 180 ± 31 | 62 (25/40) | 47 ± 9 | 45 (18/40)1-b |
Increased nicotinic sensitivity of hippocampal neurons exposed for 3 d to KYNA could be a result of enhanced expression or sensitization of non-α7 (most likely α4β2) nAChRs to nicotinic agonists. In an attempt to determine whether prolonged exposure of CNS neurons to KYNA alters expression of α4β2 nAChRs, binding assays were performed on primary cultures of cerebral cortex using 10 nm [3H]epibatidine, a nicotinic agonist that binds with high affinity to α4β2 nAChRs (Houghtling et al., 1995). Nonspecific binding was determined using 100 μm unlabeled epibatidine. In three of four experiments, specific binding of [3H]epibatidine was 60 ± 12% higher in primary cerebral cortical cultures pretreated with KYNA than in age-matched cultures that had not been exposed to KYNA (untreated cultures: 31 ± 8 fmol/mg protein; treated cultures: 52 ± 16 fmol/mg protein; n = 3 cultures; p < 0.05 according to the paired Student's t test). In one experiment, no apparent change in epibatidine binding was detected. These initial binding results support the suggestion that prolonged exposure of CNS neurons to KYNA increases expression of α4β2 nAChRs.
Nicotine induces biphasic changes in KYNA levels in rat brain
Adult male rats received injections of PBS or nicotine (1 mg/kg, s.c.) twice daily for 5 or 15 d. Animals were killed 1 hr after the last injection, and the tissue content of KYNA was measured in three areas of the rat brain: striatum, hippocampus, and frontal cortex. The levels of KYNA were substantially lower in all three brain regions of rats treated for 5 d with nicotine than in the same brain areas of control animals (Table 2). In contrast, KYNA content in all three brain areas of rats treated with nicotine for 15 d was significantly higher than that in the same areas of control animals (Table 2). Thus, these results indicate that nicotine can dynamically control the levels of KYNA in the mammalian brain.
Table 2.
Effects of systemic nicotine treatment on KYNA levels in different areas of the rat brain
| KYNA (fmol/mg protein) | |||
|---|---|---|---|
| Striatum | Hippocampus | Frontal cortex | |
| 5 d | |||
| Control | 137 ± 9 | 180 ± 19 | 209 ± 14 |
| Nicotine | 103 ± 4* | 118 ± 7* | 141 ± 9* |
| Δ (%) | 75 | 66 | 67 |
| 15 d | |||
| Control | 154 ± 8 | 181 ± 7 | 199 ± 4 |
| Nicotine | 191 ± 10* | 213 ± 7* | 227 ± 10* |
| Δ (%) | 124 | 118 | 114 |
DISCUSSION
The present study demonstrates that KYNA inhibits noncompetitively α7 nAChRs and regulates α4β2 nAChR expression. The results also show that nicotine treatment affects brain levels of endogenous KYNA_in vivo_. This reciprocal functional relationship between KYNA and the nicotinic cholinergic system is likely to have a central role in modulating neuronal plasticity and viability in the CNS.
KYNA: an endogenous, noncompetitive antagonist of α7 nAChRs
Results obtained from the electrophysiological experiments performed in hippocampal neurons demonstrated that KYNA is a noncompetitive antagonist of α7 nAChRs and that somatodendritic and preterminal/presynaptic α7 nAChRs are equally sensitive to KYNA. Inhibition of α7 nAChRs by KYNA is voltage independent and therefore cannot be explained by open-channel blockade. Also, it does not involve changes in the rapid, agonist-induced desensitization of the receptors, because τdecays of type IA currents are unaffected by KYNA.
The slow onset of the effect of KYNA on α7 nAChRs is likely the result of slow association of the metabolite with the extracellular domain of the receptor, given that an intracellular action was ruled out. Alternatively, it could indicate that KYNA acts through another cell-surface receptor that influences α7 nAChR function via intracellular second messengers or protein–protein interactions, or both. An indirect action of KYNA seems unlikely, because such a pleiotropic action can potentially affect many other receptor systems. However, in addition to α7 nAChRs, only the glutamatergic ionotropic receptors are known to be inhibited by KYNA (Stone, 1993; Moroni, 1999).
The slope of the concentration–response relationship for KYNA was shallower than one would have expected for a simple mass interaction with a single site. The Hill coefficient below unity can be interpreted as negative cooperativity between the binding sites for KYNA on the nAChRs, whereby binding of KYNA to one site lowers the drug affinities to other receptor site(s). Negative cooperativity has been observed in other systems, including the ryanodine receptor (Carroll et al., 1991), ATP-sensitive K+ channels (Hehl and Neumcke, 1993), and muscle nAChRs (Chabala et al., 1986). The weak concentration dependence of KYNA-induced blockade of α7 nAChRs indicates that the receptor activity can be finely tuned within a wide range of KYNA concentrations and supports a modulatory role for KYNA on α7 nAChRs in vivo.
KYNA is more potent in inhibiting α7 nAChRs than NMDA receptors
It has been reported that KYNA binds with an apparent affinity of ∼8 μm to the glycine site of the NMDA receptor (Parsons et al., 1997). Here, in the absence of added glycine, KYNA blocked currents elicited by NMDA with an IC50 ∼15 μm. Glycine (10 μm) caused a parallel right shift of the concentration–response relationship for KYNA-induced block of NMDA receptors, increasing the IC50 for KYNA by ∼15-fold. This is in agreement with the concept that KYNA competes with glycine for the glycine site on NMDA receptors.
At physiologically relevant concentrations (Moroni et al., 1988; Turski et al., 1988), KYNA inhibited α7 nAChRs more effectively than NMDA receptors. KYNA reduced α7 nAChR activity by 20 and 40%, respectively, at 0.1 and 1 μm. In the presence of a saturating concentration of glycine, 0.1 and 1 μm KYNA had no effect on NMDA receptors. Even when the glycine site was not saturated, KYNA at 0.1 and 1 μm reduced the NMDA receptor activity by no more than 10 and 20%, respectively.
The mechanism underlying the effects of endogenous KYNA on neuronal excitability and viability remains unclear (Schwarcz et al., 1992;Stone, 1993; Moroni, 1999; Scharfman et al., 1999; Urenjak and Obrenovitch, 2000; Erhardt et al., 2001). Some lines of evidence indicate that the glycine coagonist site on NMDA receptors may not be saturated (D'Angelo et al., 1990; Singh et al., 1990; Bergeron et al., 1998), so that low concentrations of KYNA could be sufficient to inhibit NMDA receptor activity in vivo. However, in light of the present data, it is likely that fluctuations in endogenous brain KYNA levels preferentially affect α7 nAChR function. A review of the neuroactive properties of KYNA in vivo and in vitro does not adequately resolve the question of whether the compound acts through α7 nAChRs or NMDA receptors. Qualitatively, the overall effects of α7 nAChR antagonists and NMDA receptor antagonists on neuronal plasticity and viability are similar and resemble those of KYNA. For instance, α7 nAChR antagonists and KYNA inhibit neurite outgrowth (Pearce et al., 1987; Pugh and Berg, 1994), decrease apoptotic neuronal death (Berger et al., 1998; Sun and Cheng, 1999), and are anticonvulsants (Damaj et al., 1999; Scharfman et al., 1999). Furthermore, α7 nAChR activation is normally associated with direct facilitation of glutamatergic transmission in the brain (Gray et al., 1996; Fu et al., 2000). Therefore, attenuation of glutamatergic function by KYNA in vivo (Pellicciari et al., 1994; Harris et al., 1998; Cozzi et al., 1999; Schwarcz et al., 1999) can be the result of inhibition of presynaptic α7 nAChRs in glutamatergic terminals.
KYNA enhances non-α7 nAChR sensitivity of CNS neurons
Prolonged exposure to KYNA increased α4β2 nAChR expression in CNS neurons as indicated by the binding and electrophysiological experiments performed in primary cultures of cerebral cortex and hippocampus, respectively. Increased α4β2 nAChR expression can be explained by enhanced de novo synthesis and decreased turnover of receptors (Peng et al., 1994). In general, “upregulation” of receptors is a compensatory mechanism for reduced receptor activity. However, non-α7 nAChRs are insensitive to inhibition by KYNA up to 1 mm (Bijak et al., 1991; Bertolino et al., 1997). Because α4β2 nAChR expression can be regulated by protein kinase A- and C-related mechanisms (Madhok et al., 1995; Gopalakrishnan et al., 1997), KYNA effects on α4β2 nAChR expression may be mediated by relatively slow changes in second messenger systems.
Reciprocal functional interactions between the nicotinic system and KYNA in the brain
In agreement with data showing that cigarette smoking reduces levels of endogenous KYNA (Milart et al., 2000), a 5 d treatment of rats with nicotine substantially reduced brain levels of KYNA. In contrast, treatment of rats for 15 d with nicotine slightly increased brain levels of KYNA. The initial reduction of KYNA levels by nicotine may be mediated indirectly by dopamine, because nAChR activation in dopaminergic neurons triggers release of dopamine (Marshall et al., 1997), a neurotransmitter that is known to reduce KYNA levels in the brain (Poeggeler et al., 1998). The transient nature of the nicotinic regulation of dopaminergic activity in the CNS (Kirch et al., 1987; Nisell et al., 1996) could explain why the reduction of KYNA levels subsides after prolonged nicotine treatment. Slowly developing adaptative changes in the tryptophan metabolic pathway in the brain can account for the slight enhancement of KYNA levels observed after 15 d treatment of the rats with nicotine. Nicotine-induced activation and desensitization of functional nAChRs present in astrocytes (Sharma and Vijayaraghavan, 2001), the major source of KYNA in the mammalian brain (Wu et al., 1992), may also control the levels of this metabolite in the CNS.
Physiopathological considerations
KYNA levels are altered in numerous multifactorial neurological disorders in which dysfunctions of various neurotransmitter systems are also evident. On the basis of results presented herein, it is tempting to speculate that α7 nAChR inhibition by elevated levels of KYNA is causally related to the hypoglutamatergic and hypocholinergic tones in schizophrenia and AD, respectively (Tamminga, 1998; Bartus, 2000), and to the cognitive deficits in patients with schizophrenia, AD, and DS (Court et al., 1999). Likewise, disinhibition of α7 nAChR activity consequent to decreased KYNA levels can explain the facilitation of excitotoxic damage in HD (Poeggeler et al., 1998). It is also conceivable that reduced levels of KYNA in the brain of patients with PD (Ogawa et al., 1992) contribute to decreased α4β2 nAChR expression, which correlates well with the severity of motor dysfunctions (Banerjee et al., 2000; Perry et al., 2000).
Fluctuations in brain KYNA levels can also underlie the involvement of cigarette smoking in the pathophysiology of these and other brain disorders. For example, cigarette smoking is more prevalent among individuals with schizophrenia than in normal subjects; it appears to be a means by which schizophrenics try to correct auditory gating deficits, which are caused by reduced α7 nAChR activity (Leonard et al., 2000). Nicotine at the concentrations found in the brain of cigarette smokers, although unable to activate α7 nAChRs (Alkondon et al., 2000; Almeida et al., 2000), can cause transient reduction in brain levels of KYNA. This should, in turn, disinhibit α7 nAChRs and thereby improve cognitive functions and auditory gating. Likewise, the lower incidence of PD among cigarette smokers compared with nonsmokers (Fratiglioni and Wang, 2000) could be explained at least in part by nicotine-induced increased levels of KYNA leading to higher expression of α4β2 nAChRs.
In summary, this is the first study to demonstrate reciprocal functional interactions between nAChRs and KYNA in the CNS. KYNA should be considered a neuromodulator in the nicotinic cholinergic system; it inhibits α7 nAChRs noncompetitively and voltage independently and increases α4β2 nAChR expression. Therefore, manipulations of the kynurenine pathway may represent a novel treatment strategy for conditions that are linked to abnormal function/expression of neuronal nAChRs.
Footnotes
This study was supported by United States Public Health Service Grants NS25296 (E.X.A.) and NS16102 (R.S.). We are indebted to Dr. Mohyee Eldefrawi for allowing the use of his laboratory and Dr. Ahmed Elnabawi for his expert assistance with the binding assays. The technical assistance of Bhagavathy Alkondon, Barbara Marrow, and Mabel Zelle is also gratefully acknowledged.
Correspondence should be addressed to Dr. Edson X. Albuquerque, Department of Pharmacology and Experimental Therapeutics, University of Maryland School of Medicine, 655 West Baltimore Street, Baltimore, MD 21201. E-mail: ealbuque@umaryland.edu.
A preliminary account of this study was presented at the 30th Annual Meeting of the Society for Neuroscience (2000).
REFERENCES
- 1.Albuquerque EX, Alkondon M, Pereira EFR, Castro NG, Schrattenholz A, Barbosa CTF, Bonfante-Cabarcas R, Aracava Y, Eisenberg HM, Maelicke A. Properties of neuronal nicotinic acetylcholine receptors: pharmacological characterization and modulation of synaptic function. J Pharmacol Exp Ther. 1997;280:1117–1136. [PubMed] [Google Scholar]
- 2.Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J Pharmacol Exp Ther. 1993;265:1455–1473. [PubMed] [Google Scholar]
- 3.Alkondon M, Pereira EFR, Eisenberg HM, Albuquerque EX. Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci. 1999;19:2693–2705. doi: 10.1523/JNEUROSCI.19-07-02693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alkondon M, Pereira EFR, Almeida LEF, Randall WR, Albuquerque EX. Nicotine at concentrations found in cigarette smokers activates and desensitizes nicotinic acetylcholine receptors in CA1 interneurons of rat hippocampus. Neuropharmacology. 2000;39:2726–2739. doi: 10.1016/s0028-3908(00)00156-8. [DOI] [PubMed] [Google Scholar]
- 5.Almeida LEF, Pereira EFR, Alkondon M, Fawcett WP, Randall WR, Albuquerque EX. The opioid antagonist naltrexone inhibits activity and alters expression of α7 and α4β2 nicotinic receptors: implications for smoking cessation programs. Neuropharmacology. 2000;39:2740–2755. doi: 10.1016/s0028-3908(00)00157-x. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee C, Nyengaard JR, Wevers A, de Vos RA, Jansen Steur EN, Lindstrom J, Pilz K, Nowacki S, Bloch W, Schroder H. Cellular expression of alpha7 nicotinic acetylcholine receptor protein in the temporal cortex in Alzheimer's and Parkinson's disease: a stereological approach. Neurobiol Dis. 2000;7:666–672. doi: 10.1006/nbdi.2000.0317. [DOI] [PubMed] [Google Scholar]
- 7.Baran H, Cairns N, Lubec B, Lubec G. Increased kynurenic acid levels and decreased brain kynurenine aminotransferase I in patients with Down syndrome. Life Sci. 1996;58:1891–1899. doi: 10.1016/0024-3205(96)00173-7. [DOI] [PubMed] [Google Scholar]
- 8.Baran H, Jellinger K, Deecke L. Kynurenine metabolism in Alzheimer's disease. J Neural Transm. 1999;106:165–181. doi: 10.1007/s007020050149. [DOI] [PubMed] [Google Scholar]
- 9.Bartus RT. On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp Neurol. 2000;163:495–529. doi: 10.1006/exnr.2000.7397. [DOI] [PubMed] [Google Scholar]
- 10.Beal MF, Matson WR, Swartz KJ, Gamache PH, Bird ED. Kynurenine pathway measurements in Huntington's disease striatum: evidence for reduced formation of kynurenic acid. J Neurochem. 1990;55:1327–1339. doi: 10.1111/j.1471-4159.1990.tb03143.x. [DOI] [PubMed] [Google Scholar]
- 11.Berger F, Gage FH, Vijayaraghavan S. Nicotinic receptor-induced apoptotic cell death of hippocampal progenitor cells. J Neurosci. 1998;18:6871–6881. doi: 10.1523/JNEUROSCI.18-17-06871.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergeron R, Meyer TM, Coyle JT, Greene RW. Modulation of N-methyl-d-aspartate receptor function by glycine transport. Proc Natl Acad Sci USA. 1998;22:15730–15734. doi: 10.1073/pnas.95.26.15730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertolino M, Kellar KJ, Vicini S, Gillis RA. Nicotinic receptor mediates spontaneous GABA release in the rat dorsal motor nucleus of the vagus. Neuroscience. 1997;79:671–681. doi: 10.1016/s0306-4522(97)00026-2. [DOI] [PubMed] [Google Scholar]
- 14.Bijak M, Jarolimek W, Misgeld U. Effects of antagonists on quisqualate and nicotinic receptor-mediated currents of midbrain neurones in culture. Br J Pharmacol. 1991;102:699–705. doi: 10.1111/j.1476-5381.1991.tb12236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broide RS, Leslie FM. The alpha7 nicotinic acetylcholine receptor in neuronal plasticity. Mol Neurobiol. 1999;20:1–16. doi: 10.1007/BF02741361. [DOI] [PubMed] [Google Scholar]
- 16.Carroll S, Skarmeta JG, Yu X, Collins KD, Inesi G. Interdependence of ryanodine binding, oligomeric receptor interactions, and Ca2+ release regulation in junctional sarcoplasmic reticulum. Arch Biochem Biophys. 1991;290:239–247. doi: 10.1016/0003-9861(91)90615-p. [DOI] [PubMed] [Google Scholar]
- 17.Chabala LD, Gurney AM, Lester HA (1986) Dose-response of acetylcholine receptor channels opened by a flash-activated agonist in voltage-clamped rat myoballs. 371:407–433. [DOI] [PMC free article] [PubMed]
- 18.Court J, Spurden D, Lloyd S, McKeith I, Ballard C, Cairns N, Kerwin R, Perry R, Perry E. Neuronal nicotinic receptors in dementia with Lewy bodies and schizophrenia: alpha-bungarotoxin and nicotine binding in the thalamus. J Neurochem. 1999;73:1590–1597. doi: 10.1046/j.1471-4159.1999.0731590.x. [DOI] [PubMed] [Google Scholar]
- 19.Court J, Martin-Ruiz C, Piggott M, Spurden D, Griffiths M, Perry E. Nicotinic receptor abnormalities in Alzheimer's disease. Biol Psychiatry. 2001;49:175–184. doi: 10.1016/s0006-3223(00)01116-1. [DOI] [PubMed] [Google Scholar]
- 20.Cozzi A, Carpenedo R, Moroni F. Kynurenine hydroxylase inhibitors reduce ischemic brain damage: studies with (m-nitrobenzoyl)-alanine and 3,4-dimethoxy-[-N-4-(nitrophenyl)thiazol-2yl]-benzenesulfonamide (Ro 61–8048) in models of focal or global brain ischemia. J Cereb Blood Flow Metab. 1999;19:771–777. doi: 10.1097/00004647-199907000-00007. [DOI] [PubMed] [Google Scholar]
- 21.Damaj MI, Glassco W, Dukat M, Martin BR. Pharmacological characterization of nicotine-induced seizures in mice. J Pharmacol Exp Ther. 1999;291:1284–1291. [PubMed] [Google Scholar]
- 22.D'Angelo E, Rossi P, Garthwaite J. Dual-component NMDA receptor currents at a single central synapse. Nature. 1990;346:467–470. doi: 10.1038/346467a0. [DOI] [PubMed] [Google Scholar]
- 23.Dani JA, Heinemann S. Molecular and cellular aspects of nicotine abuse. Neuron. 1996;16:905–908. doi: 10.1016/s0896-6273(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 24.Erhardt S, Oberg H, Engberg G. Pharmacologically elevated levels of endogenous kynurenic acid prevent nicotine-induced activation of nigral dopamine neurons. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:21–27. doi: 10.1007/s002100000325. [DOI] [PubMed] [Google Scholar]
- 25.Fratiglioni L, Wang HX. Smoking and Parkinson's and Alzheimer's disease: review of the epidemiological studies. Behav Brain Res. 2000;113:117–120. doi: 10.1016/s0166-4328(00)00206-0. [DOI] [PubMed] [Google Scholar]
- 26.Freedman R, Adams CE, Leonard S. The alpha7-nicotinic acetylcholine receptor and the pathology of hippocampal interneurons in schizophrenia. J Chem Neuroanat. 2000;20:299–306. doi: 10.1016/s0891-0618(00)00109-5. [DOI] [PubMed] [Google Scholar]
- 27.Fu Y, Matta SG, Gao W, Sharp BM. Local alpha-bungarotoxin-sensitive nicotinic receptors in the nucleus accumbens modulate nicotine-stimulated dopamine secretion in vivo. Neuroscience. 2000;101:369–375. doi: 10.1016/s0306-4522(00)00371-7. [DOI] [PubMed] [Google Scholar]
- 28.Gopalakrishnan M, Molinari EJ, Sullivan JP. Regulation of human alpha4beta2 neuronal nicotinic acetylcholine receptors by cholinergic channel ligands and second messenger pathways. Mol Pharmacol. 1997;52:524–534. [PubMed] [Google Scholar]
- 29.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 30.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 31.Harris CA, Miranda AF, Tanguay JJ, Boegman RJ, Beninger RJ, Jhamandas K. Modulation of striatal quinolinate neurotoxicity by elevation of endogenous brain kynurenic acid. Br J Pharmacol. 1998;124:391–399. doi: 10.1038/sj.bjp.0701834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hehl S, Neumcke B. Negative cooperativity may explain flat concentration-response curves of ATP-sensitive potassium channels. Eur Biophys J. 1993;22:1–4. doi: 10.1007/BF00205806. [DOI] [PubMed] [Google Scholar]
- 33.Hellstrom-Lindahl E, Mousavi M, Zhang X, Ravid R, Nordberg A. Regional distribution of nicotinic receptor subunit mRNAs in human brain: comparison between Alzheimer and normal brain. Brain Res Mol Brain Res. 1999;66:94–103. doi: 10.1016/s0169-328x(99)00030-3. [DOI] [PubMed] [Google Scholar]
- 34.Hilmas C, Rassoulpour A, Schwarcz R, Albuquerque EX. The brain metabolite kynurenic acid blocks α7 nicotinic acetylcholine receptors in rat hippocampal neurons in culture. Soc Neurosci Abstr. 2000;26:1403. [Google Scholar]
- 35.Houghtling RA, Davila-Garcia MI, Kellar KJ. Characterization of (±)-[3H]epibatidine binding to nicotinic cholinergic receptors in rat and human brain. Mol Pharmacol. 1995;48:280–287. [PubMed] [Google Scholar]
- 36.Johnson JW, Ascher P. Equilibrium and kinetic study of glycine action on the N-methyl-d-aspartate receptor in cultured mouse brain neurons. J Physiol (Lond) 1992;455:339–365. doi: 10.1113/jphysiol.1992.sp019305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T, Akaike A. Alpha7 nicotinic receptor transduces signals to PI3 kinase to block Aβ-induced neurotoxicity. J Biol Chem. 2001;276:13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- 38.Kirch DG, Gerhardt GA, Shelton RC, Freedman R, Wyatt RJ. Effect of chronic nicotine administration on monoamine and monoamine metabolite concentrations in rat brain. Clin Neuropharmacol. 1987;10:376–383. doi: 10.1097/00002826-198708000-00009. [DOI] [PubMed] [Google Scholar]
- 39.Kleckner NW, Dingledine R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science. 1988;241:835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- 40.Leeson PD, Iversen LL. The glycine site on the NMDA receptor: structure-activity relationships and therapeutic potential. J Med Chem. 1994;37:4053–4067. doi: 10.1021/jm00050a001. [DOI] [PubMed] [Google Scholar]
- 41.Leonard S, Breese C, Adams C, Benhammou K, Gault J, Stevens K, Lee M, Adler L, Olincy A, Ross R, Freedman R. Smoking and schizophrenia: abnormal nicotinic receptor expression. Eur J Pharmacol. 2000;393:237–242. doi: 10.1016/s0014-2999(00)00035-2. [DOI] [PubMed] [Google Scholar]
- 42.Lindstrom J. Neuronal nicotinic acetylcholine receptors. Ion channels. 1996;4:377–450. doi: 10.1007/978-1-4899-1775-1_10. [DOI] [PubMed] [Google Scholar]
- 43.Madhok TC, Matta SG, Sharp BM. Nicotine regulates nicotinic cholinergic receptors and subunit mRNAs in PC 12 cells through protein kinase A. Brain Res Mol Brain Res. 1995;32:143–150. doi: 10.1016/0169-328x(95)00073-2. [DOI] [PubMed] [Google Scholar]
- 44.Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- 45.Marshall DL, Redfern PH, Wonnacott S. Presynaptic nicotinic modulation of dopamine release in the three ascending pathways studied by in vivo microdialysis: comparison of naive and chronic nicotine-treated rats. J Neurochem. 1997;68:1511–1519. doi: 10.1046/j.1471-4159.1997.68041511.x. [DOI] [PubMed] [Google Scholar]
- 46.Milart P, Mlynarczyk M, Sikorski R. Influence of maternal smoking on kynurenic acid concentrations in umbilical cord blood. Ginekol Pol. 2000;71:843–847. [PubMed] [Google Scholar]
- 47.Moroni F. Tryptophan metabolism and brain function: focus on kynurenine and other indole metabolites. Eur J Pharmacol. 1999;375:87–100. doi: 10.1016/s0014-2999(99)00196-x. [DOI] [PubMed] [Google Scholar]
- 48.Moroni F, Russi P, Lombardi G, Beni M, Carlà V. Presence of kynurenic acid in the mammalian brain. J Neurochem. 1988;51:177–180. doi: 10.1111/j.1471-4159.1988.tb04852.x. [DOI] [PubMed] [Google Scholar]
- 49.Nisell M, Nomikos GG, Hertel P, Panagis G, Svensson TH. Condition-independent sensitization of locomotor stimulation and mesocortical dopamine release following chronic nicotine treatment in the rat. Synapse. 1996;22:369–381. doi: 10.1002/(SICI)1098-2396(199604)22:4<369::AID-SYN8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 50.Ogawa T, Matson WR, Beal MF, Myers RH, Bird ED, Milbury P, Saso S. Kynurenine pathway abnormalities in Parkinson's disease. Neurology. 1992;42:1702–1706. doi: 10.1212/wnl.42.9.1702. [DOI] [PubMed] [Google Scholar]
- 51.Parsons CG, Danysz W, Quack G, Hartmann S, Lorenz B, Wollenburg C, Baran L, Przegalinski E, Kostowski W, Krzascik P, Chizh B, Headley PM. Novel systemically active antagonists of the glycine site of the N-methyl-d-aspartate receptor: electrophysiological, biochemical and behavioral characterization. J Pharmacol Exp Ther. 1997;283:1264–1275. [PubMed] [Google Scholar]
- 52.Pearce IA, Cambray-Deakin MA, Burgoyne RD. Glutamate acting on NMDA receptors stimulates neurite outgrowth from cerebellar granule cells. FEBS Lett. 1987;223:143–147. doi: 10.1016/0014-5793(87)80525-2. [DOI] [PubMed] [Google Scholar]
- 53.Pellicciari R, Natalini B, Costantino G, Mahmoud MR, Mattoli L, Sadeghpour BM, Moroni F, Chiarugi A, Carpenedo R. Modulation of the kynurenine pathway in search for new neuroprotective agents. Synthesis and preliminary evaluation of (m-nitrobenzoyl)alanine, a potent inhibitor of kynurenine-3-hydroxylase. J Med Chem. 1994;37:647–655. doi: 10.1021/jm00031a015. [DOI] [PubMed] [Google Scholar]
- 54.Peng X, Anand R, Whiting P, Lindstrom J. Nicotine-induced upregulation of neuronal nicotinic receptors results from a decrease in the rate of turnover. Mol Pharmacol. 1994;46:523–530. [PubMed] [Google Scholar]
- 55.Perry E, Martin-Ruiz C, Lee M, Griffiths M, Johnson M, Piggott M, Haroutunian V, Buxbaum JD, Nasland J, Davis K, Gotti C, Clementi F, Tzartos S, Cohen O, Soreq H, Jaros E, Perry R, Ballard C, McKeith I, Court J. Nicotinic receptor subtypes in human brain aging, Alzheimer and Lewy body diseases. Eur J Pharmacol. 2000;393:215–222. doi: 10.1016/s0014-2999(00)00064-9. [DOI] [PubMed] [Google Scholar]
- 56.Perry EK, Smith CJ, Court JA, Perry RH. Cholinergic nicotinic and muscarinic receptors in dementia of Alzheimer, Parkinson and Lewy body types. J Neural Transm Park Dis Dement Sect. 1990;2:149–158. doi: 10.1007/BF02257646. [DOI] [PubMed] [Google Scholar]
- 57.Poeggeler B, Rassoulpour A, Guidetti P, Wu HQ, Schwarcz R. Dopaminergic control of kynurenate levels and N-methyl-d-aspartate toxicity in the developing rat striatum. Dev Neurosci. 1998;20:146–153. doi: 10.1159/000017309. [DOI] [PubMed] [Google Scholar]
- 58.Pugh PC, Berg DK. Neuronal acetylcholine receptors that bind alpha-bungarotoxin mediate neurite retraction in a calcium-dependent manner. J Neurosci. 1994;14:889–896. doi: 10.1523/JNEUROSCI.14-02-00889.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scharfman HE, Hodgkins PS, Lee SC, Schwarcz R. Quantitative differences in the effects of de novo produced and exogenous kynurenic acid in rat brain slices. Neurosci Lett. 1999;274:111–114. doi: 10.1016/s0304-3940(99)00690-4. [DOI] [PubMed] [Google Scholar]
- 60.Schwarcz R, Du F, Schmidt W, Turski WA, Gramsbergen JBP, Okuno E, Roberts RC. Kynurenic acid: a potential pathogen in brain disorders. Ann NY Acad Sci. 1992;648:140–153. doi: 10.1111/j.1749-6632.1992.tb24532.x. [DOI] [PubMed] [Google Scholar]
- 61.Schwarcz R, Ceresoli-Borroni G, Wu HQ, Rassoulpour A, Poeggeler B, Hodgkins PS, Guidetti P. Modulation and function of kynurenic acid in the immature rat brain. Adv Exp Med Biol. 1999;467:113–123. doi: 10.1007/978-1-4615-4709-9_17. [DOI] [PubMed] [Google Scholar]
- 62.Schwarcz R, Rassoulpour A, Wu H-Q, Medoff D, Tamminga CA, Roberts RC (2001) Increased cortical kynurenate content in schizophrenia. Biol Psychiatry, in press. [DOI] [PubMed]
- 63.Sharma G, Vijayaraghavan S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc Natl Acad Sci USA. 2001;98:4148–4153. doi: 10.1073/pnas.071540198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh L, Oles RJ, Tricklebank MD. Modulation of seizure susceptibility in the mouse by the strychnine-insensitive glycine recognition site of the NMDA receptor/ion channel complex. Br J Pharmacol. 1990;99:285–288. doi: 10.1111/j.1476-5381.1990.tb14695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stone TW. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev. 1993;45:309–379. [PubMed] [Google Scholar]
- 66.Sun A, Cheng J. Novel targets for therapeutic intervention against ischemic brain injury. Clin Neuropharmacol. 1999;22:164–171. [PubMed] [Google Scholar]
- 67.Tamminga CA. Schizophrenia and glutamatergic transmission. Crit Rev Neurobiol. 1998;12:21–36. doi: 10.1615/critrevneurobiol.v12.i1-2.20. [DOI] [PubMed] [Google Scholar]
- 68.Turski WA, Nakamura M, Todd WP, Carpenter BK, Whetsell WO, Jr, Schwarcz R. Identification and quantification of kynurenic acid in human brain tissue. Brain Res. 1988;454:164–169. doi: 10.1016/0006-8993(88)90815-3. [DOI] [PubMed] [Google Scholar]
- 69.Urenjak J, Obrenovitch TP. Kynurenine 3-hydroxylase inhibition in rats: effects on extracellular kynurenic acid concentration and N-methyl-d-aspartate-induced depolarization in the striatum. J Neurochem. 2000;75:2427–2433. doi: 10.1046/j.1471-4159.2000.0752427.x. [DOI] [PubMed] [Google Scholar]
- 70.Wang LY, MacDonald JF. Modulation by magnesium of the affinity of NMDA receptors for glycine in murine hippocampal neurones. J Physiol (Lond) 1995;486:83–95. doi: 10.1113/jphysiol.1995.sp020792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu HQ, Baran H, Ungerstedt U, Schwarcz R. Kynurenic acid in the quinolinate-lesioned rat hippocampus: studies in vitro and in vivo. Eur J Neurosci. 1992;4:1264–1270. doi: 10.1111/j.1460-9568.1992.tb00152.x. [DOI] [PubMed] [Google Scholar]
- 72.Zoli M, Picciotto MR, Ferrari R, Cocchi D, Changeux JP. Increased neurodegeneration during aging in mice lacking high-affinity nicotine receptors. EMBO J. 1999;18:1235–1244. doi: 10.1093/emboj/18.5.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]