A Prosurvival Function for the p75 Receptor Death Domain Mediated via the Caspase Recruitment Domain Receptor-Interacting Protein 2 (original) (raw)
Abstract
In addition to promoting cell survival, neurotrophins also can elicit apoptosis in restricted cell types. Recent results indicate that nerve growth factor (NGF) can induce Schwann cell death via engagement of the p75 neurotrophin receptor. Here we describe a novel interaction between the p75 receptor and receptor-interacting protein 2, RIP2 (RICK/CARDIAK), that accounts for the ability of neurotrophins to choose between a survival-versus-death pathway. RIP2, an adaptor protein with a serine threonine kinase and a caspase recruitment domain (CARD), is highly expressed in dissociated Schwann cells and displays an endogenous association with p75. RIP2 binds to the death domain of p75 via its CARD domain in an NGF-dependent manner. The introduction of RIP2 into Schwann cells deficient in RIP2 conferred NGF-dependent nuclear transcription factor-κB (NF-κB) activity and decreased the cell death induced by NGF. Conversely, the expression of a dominant-negative version of RIP2 protein resulted in a loss of NGF-induced NF-κB induction and increased NGF-mediated cell death. These results indicate that adaptor proteins like RIP2 can provide a bifunctional switch for cell survival or cell death decisions mediated by the p75 neurotrophin receptor.
Keywords: neurotrophin, apoptosis, Schwann cells, p75 receptor, death domain, receptor-interacting protein
Neurotrophins promote the differentiation, growth, and survival of diverse cell types in the nervous system (Lewin and Barde, 1996). Control of cell survival and death by the nerve growth factor (NGF) neurotrophin family is mediated by two transmembrane receptors, the Trk receptor tyrosine kinase and the p75 neurotrophin receptor (Chao, 1992). In the presence of TrkA the p75 receptor participates in the formation of high-affinity NGF binding sites and potentiates TrkA-mediated signal transduction to promote survival under low concentrations of neurotrophins. In the absence of Trk receptors, p75 is capable of initiating a cell death program in selected cells (Casaccia-Bonnefil et al., 1996; Frade et al., 1996;Bredesen and Rabizadeh, 1997; Bamji et al., 1998) and autonomous signaling that activates ceramide production, nuclear factor-κB (NF-κB), or c-Jun N-terminal kinase (JNK; Bothwell, 1996; Carter and Lewin, 1997). How these different activities are controlled by neurotrophins is not well understood.
In addition to neuronal functions, neurotrophins also regulate Schwann cell viability and development. Schwann cell migration is dependent on p75 signaling (Anton et al., 1994; Bentley and Lee, 2000). After nerve injury or in the absence of axons, Schwann cells express high levels of p75 (Lemke and Chao, 1988; Taniuchi et al., 1988). Schwann cells undergo apoptosis after axotomy or after trophic factor withdrawal. Apoptosis is relatively absent in Schwann cells cultured from p75−/− mice (Soilu-Hanninen et al., 1999; Syroid et al., 2000). Because the TrkA is not expressed in Schwann cells, these results indicate that NGF signaling via p75 is critical in regulating both survival and apoptotic events.
The p75 neurotrophin receptor is a member of the tumor necrosis factor (TNF) receptor superfamily (Smith et al., 1994; Wallach et al., 1999). The intracellular region of the p55 TNF receptor, Fas, and the p75 neurotrophin receptor contain a sequence that has been designated the death domain (Feinstein et al., 1995). Activation of the TNF receptor members leads to the recruitment of adapter proteins, including TNF receptor-associated factors (TRAFs), and TRADD (Hsu et al., 1995), FADD/MORT1 (Chinnaiyan et al., 1995), and receptor-interacting protein (RIP; Stanger et al., 1995; Hsu et al., 1996). Members of the TNF receptor family use these adaptor proteins to mediate downstream effector functions such as caspase activation, NF-κB, and JNK activation.
RIP2, also known as RICK or CARDIAK, is a protein kinase that contains a caspase recruitment domain (CARD) and associates with the TNF receptor complex (Inohara et al., 1998; McCarthy et al., 1998; Thome et al., 1998). In this study RIP2 was found to be expressed transiently in primary Schwann cells and to be capable of mediating NGF-dependent NF-κB activity. We find that the CARD domain of RIP2 binds to the death domain of p75 in a ligand-dependent manner. Surprisingly, the association of RIP2 with p75 results in enhanced NF-κB activity that blocks the apoptosis of Schwann cells induced by NGF. These results provide a new molecular explanation for how NGF plays a bifunctional role in determining cell survival and death decisions.
MATERIALS AND METHODS
Plasmids and constructs. HA-tagged p75 deletions were generated by using PCR. A common 5′-PCR primer (5′-GGATATGGTGACCACTGTGATG-3′) was used in conjunction with unique 3′-PCR primers for each particular deletion (Δ2, 5′-ATAAGGGCCCTCATGTGGCAGTGGACTCGCTG-3′; Δ3, 5′-ATAAGGGCCCTCAGCGTCGCAGGGCGGCTAAAAG-3′; and Δ4, 5′-ATAAGGGCCCTCAGGCACCCCAGCTGGCCAG-3′), and the pCDNA3 rat HA-tagged p75 cDNA was used as a template. All PCR products were cut with_Nar_I and _Apa_I and ligated into a _Nar_I- and _Apa_I-digested pCDNA rat HA-tagged p75 sequence. All constructs were verified by DNA sequencing (Rockefeller University, New York, NY).
The myc-tagged RIP2 cDNA and myc-tagged RIP2–ΔCARD cDNA constructs were in the vector pC1. The RIP2–CARD was generated by PCR by using a 5′-primer (5′-CGGGGTACCATGAAGCTGCATCACTGTCCTGG-3′) with a 3′-primer (5′-TTTTCCTTTTGCGGCCGCTTACATGCTTTTATTTTGAAGTAAATTTAAAGATGG-3′) and the pC1 RIP2 cDNA as a template. The PCR product was digested with_Kpn_I and _Not_I and ligated into a _Kpn_I- and _Not_I-digested RIP2 construct, with a _Kpn_I site generated between the myc epitope and RIP2 sequence (5′-CGGAATTCACCATGGAACAGAAATTGATCTCCGAAGAAGACTTGGGTACCATGAACGGGGAGGCCATCTGC-3′) and with a 3′-primer (5′-TTTTCCTTTTGCGGCCGCTTACATGCTTTTATTTTGAAGTAAATTTAAAGATGG-3′) and by using the pC1 RIP2 as a template.
For GST p75 constructs, rat p75 cDNA fragments encoding the entire intracellular domain (amino acids 245–396, p75IC) and the death domain-containing region (amino acids 302–396, p75DD) were amplified by PCR and subcloned into pGEX-4T1 vector (Amersham Pharmacia Biotech, Arlington Heights, IL).
_Human embryonic kidney (HEK) 293 cell culture and transfection._HEK 293 cells were cultured in DMEM plus 10% fetal calf serum (FCS) supplemented with penicillin–streptomycin (Life Technologies, Gaithersburg, MD) at 37°C in 5% CO2.
Isolation of Schwann cells. Sciatic nerves were dissected from P2 rats, minced, and incubated in HBSS containing 0.25% trypsin (Sigma, St. Louis, MO) and 0.25% collagenase (Sigma) for 30 min. The cells were triturated and plated on poly-d-lysine-coated six-well plates cultured in DMEM containing 10% fetal calf serum and 2 μm forskolin (Sigma).
NF-κB luciferase assay. Schwann cells cultured for 4 d or for >30 d were plated at 20,000 cells per well of a 12-well plate. Cells were transfected with 75 ng of NF-κB luciferase reporter containing two κB sites in pBIIX–luc cDNA (Gu et al., 1998), 75 ng of LacZ cDNA, and 0.3 μg of cDNA, using the Effectene Transfection Reagent kit (Qiagen, Chatsworth, CA). After transfection the cells were re-fed with DMEM plus 0.5% serum and 100 ng/ml NGF (Harlan Bioproducts, Indianapolis, IN) for 24 hr. Cells were lysed and analyzed for luciferase activity by using the manufacturer's protocol (Promega, Madison, WI). HEK 293 cells were plated at 5 × 105 cells/well and transfected with a NF-κB reporter construct and RIP2, p75, or vector via the calcium phosphate method. After transfection the cells were re-fed with DMEM plus 1% FCS ± 100 ng/ml NGF for an additional 24 hr. Cells were lysed and analyzed for luciferase activity. All transfections included a LacZ construct to normalize for transfection efficiency.
Immunoprecipitation. For myc-RIP2 and p75 overexpression in HEK 293 cells, 1.5 × 106 cells in 10 cm tissue culture dishes were transfected with 10 μg of plasmid DNA (3 μg of p75, 5 μg of RIP2) by the calcium phosphate method. At 36 hr after transfection the cells were collected and aliquoted into microcentrifuge tubes at 2 × 106cells/ml. NGF (100 ng/ml) was added to the tubes for 5 min at room temperature, unless otherwise specified. Then the cells were spun down and lysed in 1 ml of Nonidet P-40 lysis buffer (1% Nonidet P-40, 20 mm Tris, pH 8.0, 200 mm NaCl, 1 mmEDTA, 2 μg/ml aprotinin, 1 μg/ml leupeptin, and 25 μg/ml phenylmethylsulfonyl fluoride). Lysates (800 μg/ml) were incubated with α-myc (2 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA) and protein A–Sepharose (Sigma) or with α-myc Affinity gel (4 μg/ml; Santa Cruz Biotechnology). The matrix was washed, and the immune complexes were resuspended in SDS-PAGE sample buffer and subjected to Western blot analysis (see below).
For the endogenous immunoprecipitation experiments the Schwann cells were isolated from the sciatic nerve taken from 120 pups [postnatal day 1 (P1)] and cultured as described above for 6 d. The cultures were washed and incubated in serum-free DMEM for 3 hr before 100 ng/ml NGF was added for 5 min. Then the cells were lysed in 1% Nonidet P-40 lysis buffer (see above). Lysates from Schwann cells (12 mg) were incubated with either anti-RIP2 (anti-RICK; StressGen, Sydney, Victoria, British Columbia) or normal rabbit IgG coupled to protein A–Sepharose overnight. Lysates (1.2 mg) also were subjected to immunoprecipitation with anti-RIP antibodies, followed by Western blotting with anti-p75. The matrix was washed, and the immune complexes were resuspended in SDS-PAGE sample buffer and subjected to Western blot analysis (see below).
Western blot analysis. Samples in SDS-PAGE sample buffer were resolved on a 10% SDS-polyacrylamide gel under reducing conditions. Proteins were transferred to polyvinylidene difluoride membrane, blocked with 5% milk, and incubated with a primary antibody, rabbit polyclonal α-p75 (α-9992), or rabbit polyclonal α-HA antibody (Immunotech, Westbrook, ME) at room temperature. The membrane was washed with TBST, incubated with a α-mouse or α-rabbit IgG horseradish peroxidase (Sigma), then processed by ECL (Amersham), and exposed to x-ray film. For endogenous protein expression in Schwann cells, sciatic nerves were taken from P1–P3 pups; primary cultures of Schwann cells were prepared on poly-d-lysine dishes. The cells were lysed in 1% Nonidet P-40 at 0, 2, 6, or 30 d of culture. Cell lysates (30 μg) were incubated in SDS-PAGE sample buffer and subjected to Western blot analysis with antibodies against RIP, RIP2, or p75.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) stain. After 3 d or >30 d in culture the Schwann cells were plated onto a poly-d-lysine-coated slides or chambers at 5000 cells per chamber. After two serum-free washes the cells were incubated in DMEM and low serum (0.5–1.0% FBS) with or without 100 ng/ml NGF for 24 hr. The cells were fixed with 4% paraformaldehyde for 1 hr at room temperature and were TUNEL labeled (Roche, Nutley, NJ). The cells also were stained with Hoechst for 20 min, and the coverslips were mounted with VectaShield (Vector Laboratories, Burlingame, CA). Cells were viewed at 40× on the Nikon scope, quantitated, and represented as the percentage of nuclei positive for TUNEL stain.
GFP cotransfection. After 3 d or >30 d in culture the Schwann cells were plated on poly-d-lysine-coated chambers at 5000 cells/chamber. Cells were transfected with 0.05 μg of GFP and 0.1 μg of dominant-negative RIP2 (ΔCARD), RIP2, or control vector. After 24 hr in DMEM and 10% FBS the cells were washed twice in serum-free media and incubated in low serum (0.5% FCS) with or without 100 ng/ml NGF. GFP-expressing cells were observed for apoptotic morphology 18–24 hr later. The data are represented as the percentage of GFP cells exhibiting apoptotic morphology.
GST p75 assay. p75 GST fusion proteins were prepared and immobilized on glutathione–Sepharose 4B beads (Amersham Pharmacia Biotech) by following the manufacturer's instructions. Full-length RIP2 was in vitro transcribed and translated from pCl vector, using the TNT-coupled reticulocyte system (Promega) with35S-methionine (Amersham Pharmacia Biotech). The generated 35S-labeled RIP2 protein was incubated with GST-p75IC or GST-p75DD BSA-coated beads at 4°C for several hours to overnight in TNE buffer [containing (in mm) 10 Tris, pH 8.0, 150 NaCl, and 1 EDTA plus 1% Nonidet P-40]. After the incubation the beads were washed with TNE buffer, and the bound proteins were separated by SDS-PAGE. The35S signal was enhanced by Amplify (Amersham Pharmacia Biotech) and exposed on x-ray film.
_Activating transcription factor-2 (ATF-2) luciferase assay._To assay for the activation of ATF-2, we cotransfected a Gal4–ATF2 (wild type) or a transactivation-incompetent mutant, Gal4–ATF-2 (T71A), with a Gal4-driven luciferase plasmid with the indicated constructs in HEK 293 cells. At 1 d after transfection the cells were maintained in DMEM plus 1% FBS and 100 ng/ml NGF. Then the cells were lysed and assessed for luciferase activity with a luminometer. All ATF-2 luciferase values were normalized to a mutant Gal4–ATF-2 (T71A) vector and presented as fold-over-vector transfection.
RESULTS
RIP2 binds to p75 in Schwann cells
NGF binding to p75 in Schwann cells results in the activation of the NF-κB transcription factor (Carter et al., 1996; Gentry et al., 2000). A class of adaptor proteins known to mediate TNF-induced NF-κB activity includes RIP and RIP2, both of which contain a serine/threonine kinase domain (Inohara et al., 1998; McCarthy et al., 1998; Thome et al., 1998). Because TNF receptor family members frequently bind to similar adaptor molecules to activate common signaling pathways, we examined the possibility that RIP and RIP2 are expressed in Schwann cells.
To investigate RIP and RIP2 expression in Schwann cells, we collected lysates from Schwann cells isolated from sciatic nerve and cultured them for different times. These lysates were analyzed by Western blot with antibodies directed against RIP, RIP2, or p75 (Fig.1A). The immunoblot shows that both RIP and RIP2 are highly expressed in primary cultures of Schwann cells (days 2 and 6), but RIP2 was no longer expressed in cultures of 30 d or more. As expected, p75 receptor expression also was induced and maintained in these Schwann cell cultures.
Fig. 1.
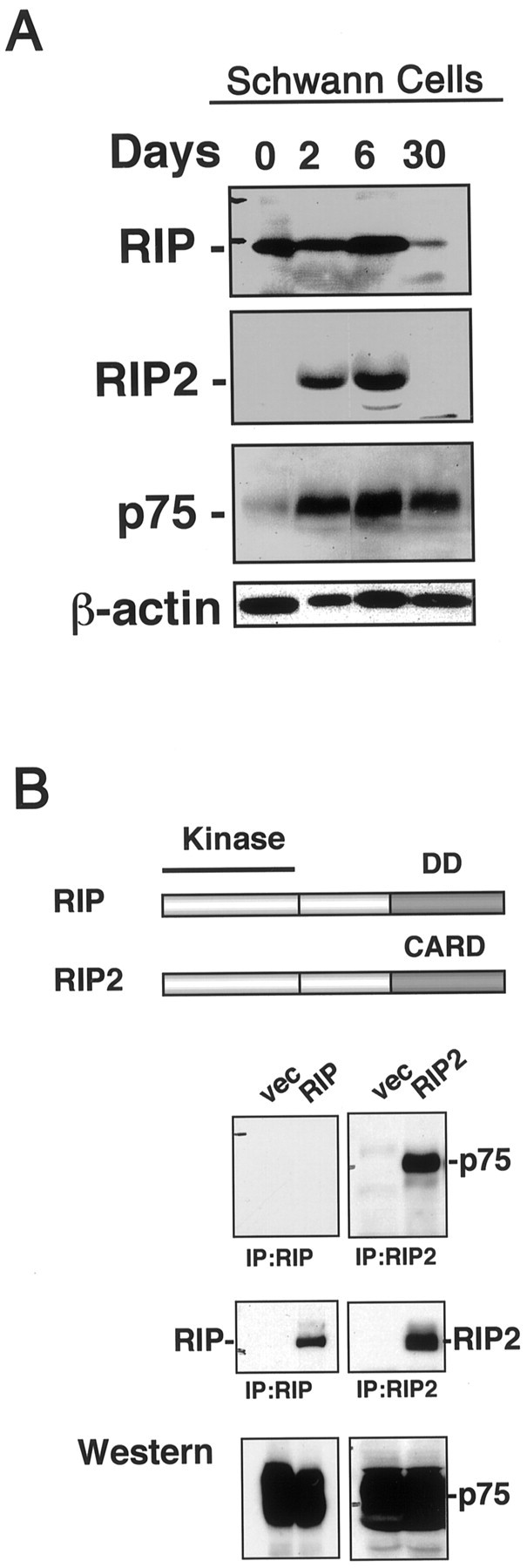
RIP2 binds to p75. A, Expression of RIP, RIP2, and p75 in Schwann cells. Schwann cells were isolated from P1 rat sciatic nerve and cultured for 0, 2, 6, and 30 d. Lysates were collected, and the expression of RIP, RIP2, and p75 was assessed by Western blot analysis. The blot was reprobed with α-actin as a loading control. B, Comparison of RIP and RIP2 proteins in p75 binding. HEK 293 cells were cotransfected with p75 and either Flag-tagged RIP or myc-tagged RIP2. Lysates were immunoprecipitated for RIP or RIP2 and were assessed for p75 association by immunoblotting (top). The blot was stripped and reprobed for Flag-RIP or myc-RIP2 (middle). Lysates were subjected to Western blot analysis with anti-p75 (bottom).
To identify whether RIP or RIP2 interacts with p75, we cotransfected cDNAs encoding p75 and epitope-tagged RIP or RIP2 into HEK 293 cells. Lysates were immunoprecipitated for either RIP or RIP2 and then immunoblotted for p75. Antibodies against RIP were unable to immunoprecipitate p75; however, a strong association between RIP2 and p75 was detected (Fig. 1B). This transfection experiment indicated that RIP2, and not RIP, associated with p75.
To determine whether this interaction is physiologically relevant, we assessed whether endogenous RIP2 protein interacts with p75 in Schwann cells. Lysates were collected from dissociated cultures and immunoprecipitated with anti-RIP2 and immunoblotted for p75. Incubation with anti-RIP2 antibody resulted in immunoprecipitation of p75 (Fig.2A). The association between p75 and RIP2 was observed after NGF treatment for 5 min. As a control, normal IgG did not produce a positive signal for p75. Coprecipitation of p75 was not observed with antibodies against RIP (Fig. 2B). These results indicate that an endogenous interaction could be observed specifically between p75 and RIP2 in Schwann cells from postnatal sciatic nerve. Moreover, only Schwann cell cultures treated with NGF produced an interaction between p75 and RIP2, indicating that this association was dependent on the NGF binding to p75.
Fig. 2.

An endogenous NGF-dependent association of RIP2 and p75 in Schwann cells. Schwann cells were isolated from P1–P3 sciatic nerve, cultured for 6 d, and then treated with NGF (100 ng/ml) for 5–10 min. The cells were lysed, immunoprecipitated with either anti-RIP2 (A) or anti-RIP (B), and subsequently immunoblotted for p75 (top panels). The same amount of lysate also was immunoprecipitated with normal IgG as a negative control. The blot was stripped and reprobed with anti-RIP2 (A) and anti-RIP (B) in the middle panels. p75 levels were confirmed by Western blotting in bottom panels. Two separate RIP2 immunoprecipitations are shown in A.
The RIP2 CARD binds to the p75 death domain
To determine whether RIP2 binds directly to p75, we translated the RIP2 protein in vitro with35S-methionine and then incubated it with GST fusion proteins containing the intracellular domain (IC) or the death domain (DD) of p75. Whereas RIP2 did not bind to GST protein alone, an interaction was observed with both the GST-p75 intracellular and death domain-containing proteins (Fig.3A), suggesting a direct association between RIP2 and the death domain of p75.
Fig. 3.

The CARD domain of RIP2 binds to the death domain of p75. A, In vitro binding.35S-labeled RIP2 was incubated with the following GST proteins: GST-p75 intracellular domain (IC), GST-p75 death domain (DD), or GST protein alone. The GST fusion proteins were isolated and subjected to electrophoresis on a 10% PAGE-SDS gel and exposed to film to detect radiolabeled RIP2 interaction (top). A Coomassie stain of the GST proteins is shown in the bottom panel. B, Mapping of the p75 domains responsible for RIP2 binding. HEK 293 cells were cotransfected with myc-tagged RIP2 and HA-tagged p75 serial deletions. The lysates were immunoprecipitated with anti-myc antibody and immunoblotted with anti-HA antibody to detect HA-p75 deletions (top). The blot was stripped and reprobed with α-myc antibody (middle). C, Mapping of the RIP2 domains responsible for p75 binding. HEK 293 cells were cotransfected with p75 and full-length myc-RIP2 (WT), myc-RIP2 construct with the CARD domain deleted (ΔCARD), or a myc-RIP2 CARD domain (CARD). The lysates were immunoprecipitated with anti-myc antibody and immunoblotted for p75 (top). The blot was stripped and reprobed with anti-myc for RIP2 (middle). Lysates were monitored for p75 levels (bottom).
To map the binding domains between RIP2 and p75 further, we transfected HEK 293 cells with RIP2 and serial deletions of HA epitope-tagged p75 (Fig. 3B). The lysates were immunoprecipitated for myc-RIP2 and immunoblotted with a polyclonal anti-HA antibody to detect the p75 proteins. Each protein was monitored for expression after transfection. These binding experiments narrowed down the RIP2 binding region on p75 to the fifth α-helix in the death domain (Fig. 3B). Together, these experiments indicate that RIP2 binds directly to the p75 death domain.
RIP and RIP2 proteins display considerable homology in the protein kinase and intermediate domains but differ at the C-terminal region, with RIP containing a death domain and RIP2 containing a CARD domain (McCarthy et al., 1998) (Fig. 1B). Because p75 binds preferentially to RIP2 rather than to RIP, we hypothesized that the CARD domain of RIP2 may be important for the specificity of binding to p75. To identify the domain in RIP2 that is important for binding to p75, we transfected a full-length myc-RIP2 (WT), a myc-RIP2 missing the CARD domain (ΔCARD), and a myc-RIP2 construct expressing only the CARD domain (CARD) with p75 in HEK 293 cells and analyzed them for an association after immunoprecipitation of the RIP2 constructs (Fig.3C). Deletion of the CARD domain eliminated the ability of RIP2 to interact with p75, whereas a truncated RIP2 containing the CARD domain still associated with p75. These experiments indicate that the CARD domain of RIP2 is responsible for p75 binding.
NGF induces RIP2 and p75 association to mediate NF-κB activity
To verify the ligand dependency of this interaction, we cotransfected HEK 293 cells with p75 and RIP2 and treated them with NGF for different times. The lysates were immunoprecipitated for RIP2 and then immunoblotted for p75. The addition of NGF augmented the interaction between RIP2 and p75, peaking 5 min after NGF treatment (Fig. 4A). This experiment confirmed that RIP2 interacted with p75 in an NGF-dependent manner.
Fig. 4.

NGF binding to p75 induces NF-κB activity via RIP2. A, Time course of p75 and RIP2 association. HEK 293 cells were cotransfected with p75 and RIP2. The cells were collected and evenly aliquoted; 100 ng/ml NGF was added for different time points. Lysates were immunoprecipitated for RIP2 and immunoblotted for p75 (top). The blot subsequently was stripped and probed with anti-RIP2 antibody (bottom). A densitometric analysis of the ratio of p75 and RIP2 protein levels is displayed (bar chart).B, Reconstitution of NF-κB reporter gene activity by p75 and RIP2. HEK 293 cells were cotransfected with a NF-κB luciferase reporter and p75 or RIP2 alone or p75 plus RIP2. Cells were incubated for 24 hr with 100 ng/ml NGF, and NF-κB luciferase activity was measured. The average of three experiments is shown.
Neurotrophins activate the NF-κB transcription factor, which plays an important role in preventing apoptosis in many cell types (Beg and Baltimore, 1996; Lin et al., 1996). We assessed whether RIP2 was responsible for NGF-induced NF-κB activity. A NF-κB luciferase reporter construct was transfected in HEK 293 cells with p75, RIP2, or p75 plus RIP2. After transfection the cells were incubated with NGF for an additional 24 hr. NGF did not increase NF-κB activity in cells transfected with p75 or RIP2 alone (Fig. 4B). However, NGF increased NF-κB in HEK 293 cells cotransfected with p75 and RIP2. This suggests that RIP2 can mediate NGF-induced NF-κB activity in HEK 293 cells.
Regulation of NF-κB activity in Schwann cells
To determine whether RIP2 regulates NGF-induced NF-κB activity in Schwann cells, we transfected a NF-κB luciferase reporter construct into Schwann cells isolated from P1 sciatic nerve. The cells were cultured for 5 d and then transfected and maintained for 24 hr in the presence or absence of NGF. At this stage the Schwann cells expressed both p75 and RIP2 (RIP2+; Fig.1A). Consistent with previous results that used p65 nuclear translocation and electrophoretic mobility shift assays (Carter et al., 1996; Khursigara et al., 1999), NGF induced an increase in NF-κB activity (Fig. 5A). Induction of NF-κB activity by NGF was not detected in the long-term cultures of 30 d or more, a stage when these Schwann cells were deficient in RIP2 expression (RIP2−). These results indicated NGF-induced NF-κB activity, but only in Schwann cells expressing RIP2. The loss of RIP2 expression in long-term cultures correlated with the inability of NGF to activate NF-κB.
Fig. 5.

RIP2 mediates NGF-dependent NF-κB activity in Schwann cells. A, NGF induces NF-κB in 6 d, but not 30 d, Schwann cells. Schwann cells cultured for 6 or 30 d were transfected with an NF-κB luciferase reporter. After transfection the cells were incubated in low serum for 24 hr with or without NGF, and luciferase activity was measured.B, RIP2 expression confers NGF-dependent NF-κB activity in 30 d cultures. Schwann cells grown for 30 d do not express RIP2 (RIP2−). Cells grown for 30 d or more were transfected with a NF-κB luciferase reporter and either RIP2 or a vector control. After incubation with NGF (100 ng/ml) for 24 hr the NF-κB activity was measured. C, Dominant-negative RIP2 blocks NGF-dependent NF-κB activity. Schwann cells cultured for 6 d (RIP2+) were cotransfected with a NF-κB luciferase reporter and either DN RIP2 (ΔCARD) or vector control. Cells were incubated with 100 ng/ml NGF for 24 hr, and then NF-κB activity was measured. All experiments were an average of at least three separate experiments.
These findings establish two stages of Schwann cells, a short-term culture that is RIP2+ and a long-term culture that is RIP2−. To establish whether RIP2 is necessary for NF-κB signaling in Schwann cells, we expressed RIP2 in RIP2− Schwann cells. The full-length RIP2 cDNA was transfected along with an NF-κB luciferase reporter into these Schwann cells (RIP2−). As shown previously, NGF was unable to activate NF-κB activity in RIP2-deficient Schwann cells (Fig. 5B). Introduction of RIP2 in 30 d cell cultures increased NF-κB activity threefold above background. The addition of NGF to these transfected Schwann cells led to an increase in NF-κB activity (Fig. 5B). These findings indicate that the expression of RIP2 was sufficient for NGF-induced NF-κB activation in Schwann cells.
Dominant-negative RIP2
The loss of RIP2 was correlated with the inability of NGF to activate NF-κB activity in long-term cultures. To establish whether RIP2 is necessary for NF-κB activity in Schwann cells, we used a dominant-negative approach. To develop a dominant-negative RIP2 molecule capable of interfering with NF-κB activity, we noted that both the CARD and kinase domains of RIP2 are necessary for NF-κB activation (McCarthy et al., 1998). Therefore, we tested whether a RIP2 ΔCARD construct acted in a dominant-negative manner by coexpressing the full-length RIP2 cDNA with increasing amounts of RIP2 ΔCARD in HEK 293 cells. The full-length RIP2 protein was expressed equally in all conditions (data not shown). RIP2 ΔCARD (referred to as DN RIP2) reduced the ability of full-length RIP2 (WT) to activate NF-κB in a dose-dependent way, whereas DN RIP2 itself did not activate NF-κB (Fig. 6).
Fig. 6.

Dominant-negative RIP2 blocks RIP2-mediated, but not TRAF6-mediated, NF-κB activity. A, A NF-κB luciferase reporter plasmid (75 ng) was transfected in HEK 293 cells with 1 μg of full-length RIP2 (WT) and increasing doses of the dominant-negative DNRIP2 (ΔCARD) construct. The amount (in micrograms) of DN RIP2 used for each condition is indicated. Cells were transfected for 24 hr and harvested; then NF-κB activity was measured. B, Increasing amounts of DNRIP2 (in micrograms) were cotransfected with 1 μg of TRAF6 cDNA, a NF-κB luciferase plasmid (75 ng), in HEK 293 cells. After 24 hr the luciferase activity was assessed.
To demonstrate the specificity of the dominant-negative construct, we tested whether the DN RIP2 could reduce the ability of a unrelated protein, TRAF-6, to activate NF-κB. Overexpression of TRAF6 activates NF-κB in heterologous cells (Cao et al., 1996). Expression of DN RIP2 did not reduce the ability of TRAF6 to induce NF-κB in HEK 293 cells (Fig. 6). Expression of the dominant-negative RIP2 blocked NF-κB activity specifically through RIP2 and not through other adaptor molecules, such as TRAF6.
Blocking RIP2 function in Schwann cells
To establish the functional consequence of inhibiting RIP2 in Schwann cells, we introduced the DN RIP2 cDNA (ΔCARD) and a NF-κB luciferase reporter in primary Schwann cells expressing RIP2 (RIP2+). After transfection the cells were incubated with NGF for 24 hr, and NF-κB activity was measured. Expression of DN RIP2 blocked NGF-dependent NF-κB activation (Fig.5C). Hence, blocking RIP2 function specifically lowered the ligand-dependent induction of NF-κB in Schwann cells.
Influence of RIP2 in NGF-dependent cell death
Previous results using p65 nuclear translocation and electrophoretic mobility shift assays demonstrated that NGF induced an increase in NF-κB activity (Carter et al., 1996; Khursigara et al., 1999; Foehr et al., 2000; Gentry et al., 2000). The above results indicated that RIP2 was responsible for regulating NF-κB activity in Schwann cells. To determine whether RIP2 expression influenced Schwann cell viability, we evaluated the level of apoptosis of Schwann cells grown under very low serum conditions (0.5% FCS). Then the Schwann cells were assessed for TUNEL immunoreactivity. In RIP2+ cultures a baseline of 25% was observed to be TUNEL-positive. The addition of NGF did not produce a detectable change (Fig. 7). Quantitation of TUNEL-positive cells in RIP2-deficient cultures indicated a slightly higher background of cell death (35%). The addition of NGF increased the number of TUNEL-positive cells twofold. These findings suggested that NGF induced cell death preferentially in Schwann cells that do not express RIP2.
Fig. 7.

NGF induces apoptosis in Schwann cells in the absence of RIP2. Schwann cells were incubated in low serum with or without 100 ng/ml NGF for 18–24 hr. The cells were fixed and analyzed for TUNEL reactivity. RIP2 (RIP2 +) or RIP2-deficient (RIP2−) Schwann cells were quantified for the percentage of TUNEL-positive nuclei.
Because RIP2 association with p75 increased NF-κB activity, we hypothesized that RIP2 might protect Schwann cells from NGF-induced apoptosis. To assess the effects of endogenous RIP2, we cotransfected the DN RIP2 with a GFP marker plasmid and assessed the viability of RIP2+ cells after NGF treatment. GFP-expressing cells were identified and assessed for apoptotic morphology, including nuclear condensation and the presence of apoptotic bodies (Fig.8A).
Fig. 8.
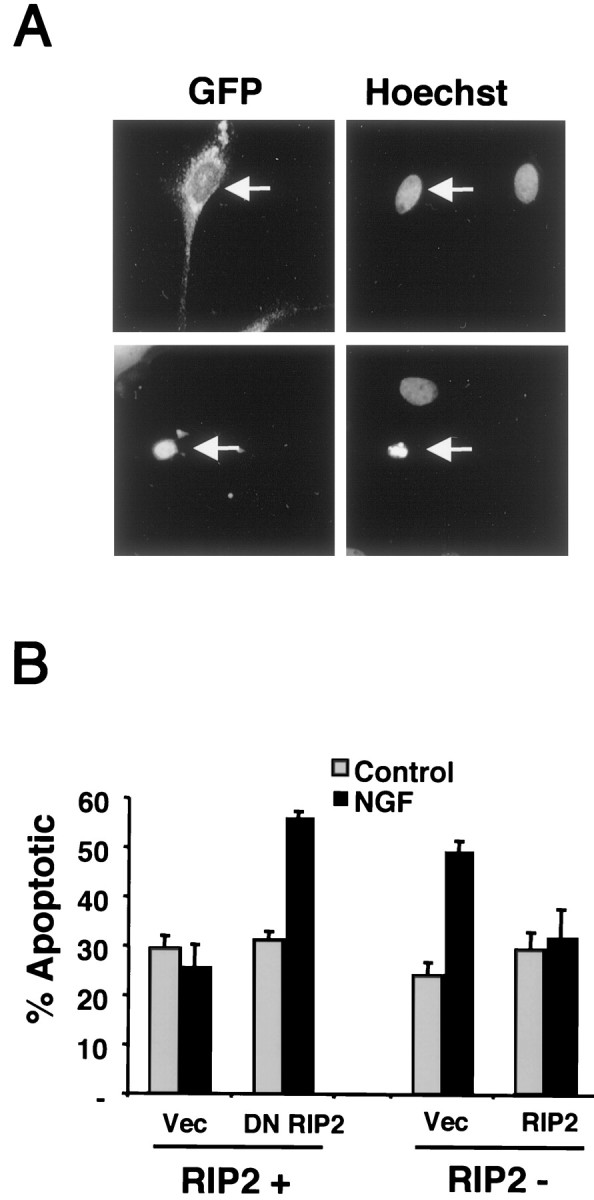
RIP2 blocks NGF-mediated apoptosis in Schwann cells. A, Detection of apoptotic nuclei in transfected Schwann cells. Schwann cells were cotransfected with GFP and a vector control. After transfection the cells were incubated in low serum with 100 ng/ml NGF for 18 hr. Cells were stained with Hoechst, and GFP-expressing apoptotic cells were identified and quantitated. The_top panels_ show two Hoechst-positive cells with normal nuclear morphology, a GFP-expressing cell (arrow), and an untransfected Schwann cell. The bottom panels show a GFP-expressing Schwann cell with apoptotic morphology (arrow) and a normal untransfected cell.B, Left, Dominant-negative RIP2 increases cell death. Schwann cells cultured for 6 d (RIP2 +) were cotransfected with GFP and either DN RIP2 or a vector control. Cells were incubated with 100 ng/ml NGF in low serum for 18 hr; apoptotic morphology was detected and quantitated. The cells were quantified for the percentage of GFP-expressing apoptotic cells. B, Right, RIP2 rescues NGF-dependent cell death. Schwann cells cultured for 30 d (RIP2−) were cotransfected with GFP and either full-length RIP2 or vector control and treated as described above. The number of GFP-expressing apoptotic cells was assessed by nuclear morphology after staining with Hoechst, as shown in_A_.
Cells transfected with the DN RIP2 construct displayed a marked increase in the number of apoptotic cells after NGF treatment (Fig. 8B). In contrast, transfection of a vector control did not produce increased cell death by NGF, similar to the TUNEL results (Fig. 7). These cell death measurements, together with the NF-κB response, suggest that interfering with endogenous RIP2 function inhibits NGF-mediated NF-κB activity (Fig. 5C), resulting in enhanced cell death by NGF (Fig. 8B).
RIP2 rescues Schwann cells from NGF-induced cell death
We next tested whether expressing RIP2 in long-term RIP2-deficient Schwann cells rescued the cells from NGF-induced cell death. We cotransfected RIP2 with a GFP plasmid into RIP2− Schwann cells and assessed apoptotic morphology in GFP-expressing cells. Expression of RIP2 abolished the ability of NGF to induce cell death under these conditions (Fig. 8B). Protection from NGF-induced cell death by RIP2 expression provides evidence that transcriptional events through NF-κB play an important role in regulating Schwann cell survival and that this response is directed by the p75 receptor.
Role of TRAF6
Previous work suggested that TRAF6 recruitment to p75 was responsible for NF-κB activity (Khursigara et al., 1999; Ye et al., 1999; Foehr et al., 2000); however, efforts to reconstitute NGF-dependent NF-κB activation via p75 and TRAF6 in heterologous cells were unsuccessful (data not shown). In addition to the activation of NF-κB, TRAF proteins also regulate JNK activation (Song et al., 1997). Because neurotrophin binding to p75 can lead to JNK activation in oligodendrocytes (Casaccia-Bonnefil et al., 1996), sympathetic neurons (Bamji et al., 1998), and hippocampal neurons (Friedman, 2000), we tested the possibility that TRAF-6 was responsible for JNK activation.
To investigate whether TRAF-6 regulates JNK-mediated transcriptional events by NGF, we examined ATF-2, a transcription factor activated by JNK and p38 kinases. An ATF-2 reporter assay was used to measure ATF-2 activity in response to the NGF treatment of HEK 293 cells transfected with TRAF-6 and p75. A prominent NGF-dependent increase in ATF-2 activity was observed when both TRAF-6 and p75 were cotransfected, whereas expression of p75 or TRAF-6 alone did not lead to an increase in ATF-2 activity (Fig.9). This finding supports the involvement of TRAF-6 in JNK-dependent signaling by p75.
Fig. 9.

TRAF6 mediates NGF-induced ATF-2 activity. An ATF-2 luciferase reporter construct was cotransfected with p75 and TRAF6 in HEK 293 cells. After transfection the cells were treated with or without NGF for 24 hr and harvested; then ATF-2 activity was measured. Results reflect an average of three experiments.
DISCUSSION
In this study we have identified a Schwann cell protein, RIP2, that interacts with p75 and influences the ability of NGF to regulate survival decisions. The interaction between RIP2 and p75 was observed endogenously in Schwann cells. We found that NGF augmented the interaction between RIP2 and p75 and enhanced RIP2-mediated NF-κB activity in both HEK 293 and Schwann cells. Taking advantage of long-term cultures of Schwann cells, which become RIP2-deficient and more sensitive to apoptosis, we show that the expression of RIP2 promoted NGF-induced NF-κB activity and protected these cells from NGF-induced cell death. Interestingly, inhibiting RIP2 in freshly dissociated Schwann cells also led to NGF-induced cell death, indicating that p75 can initiate two separate pathways at the same time, NF-κB and cell death. Our data suggest that RIP2 plays a key role in NGF function in Schwann cells by leading to the activation of NF-κB and by negating death signals from p75.
Significantly, the interaction between RIP2 and p75 is ligand-dependent, in contrast to other TNF members, such as CD40 and TNF receptor. Previous overexpression studies with RIP or RIP2 in heterologous cells yielded enhanced apoptosis in addition to NF-κB (Inohara et al., 1998; McCarthy et al., 1998). Strikingly, in Schwann cells, RIP2 expression did not lead to an apoptotic outcome but, rather, to a prosurvival function. A unique finding of this study is that the CARD domain of RIP2 interacts with the death domain of p75. Most CARD-containing proteins are involved directly in the apoptotic pathway. These include caspases and Apaf-1 (Hofman et al., 1997). The death domain mediates protein–protein interactions and is essential for the transduction of cytotoxic signals (Feinstein et al., 1995). Unexpectedly, this study shows that the death domain of p75 may play a protective role in Schwann cells by binding to a CARD-containing protein. Previous analysis of the death domain of p75 indicated a different mode of action compared with the TNF and Fas receptors (Gu et al., 1999; Kong et al., 1999). We have found that recruitment of the RIP2 CARD domain to the p75 receptor death domain did not activate a cell death pathway but mediated anti-apoptotic signals.
This conclusion is consistent with recent microinjection experiments in sensory neurons in which p75-mediated apoptosis did not require the death domain (Coulson et al., 2000). Instead, a 29-amino-acid segment of the p75 juxtamembrane region was responsible for cell-killing properties. The effects of RIP2 in binding to the death domain of p75 and providing an anti-apoptotic response in Schwann cells are compatible with the results obtained in sensory neurons (Coulson et al., 2000). This outcome is also consistent with previous observations suggesting that p75 signaling can provide a prosurvival function (Rabizadeh et al., 1993; Cortazzo et al., 1996).
The ability of Schwann cells to receive survival signals is relevant to events that have been observed in vivo during development and axonal injury. One of the earliest responses is an elevated level of expression of NGF and the p75 neurotrophin receptor in Schwann cells (Heumann et al., 1987; Lemke and Chao, 1988). Expression of p75 is associated closely with Schwann cell development. Historically, presentation or sequestration of neurotrophins has been proposed for p75 receptors; however, there is little direct evidence for this function. On the other hand, migration of Schwann cells has been shown to be dependent on p75 receptors (Anton et al., 1994; Bentley and Lee, 2000). More recently, specific neurotrophins such as NGF stimulate the JNK and NF-κB pathways, leading to the possibility that the viability of Schwann cells may be regulated by neurotrophins. Indeed, Schwann cells undergo apoptotic cell death during development and after trophic factor withdrawal (Soilu-Hanninen et al., 1999; Syroid et al., 2000)
During the first week of injury Schwann cells are highly proliferative and express high levels of NGF and p75. This establishes an autocrine mechanism for NGF to signal via p75 in the absence of axons (Grinspan et al., 1996; Jessen and Mirsky, 1999). An NGF increase in NF-κB activity in the distal Schwann cells after a sciatic nerve injury may protect Schwann cells from early cell death. Indeed, an increase in NF-κB in distal Schwann cells after a sciatic nerve crush model has been observed (Frostick et al., 1998; Dobrowsky and Carter, 2000). If regeneration or axonal contact has not occurred after a nerve lesion, Schwann cell death increases significantly (Ferri and Bisby, 1999). One explanation to account for these apoptotic events involves p75 signaling. In fact, the absence of p75 in mice leads to less Schwann cell death in vivo (Ferri and Bisby, 1999; Soilu-Hanninen et al., 1999). Hence, a dual role for p75 signaling during nerve injury may depend on adaptor proteins such as RIP2.
Another set of adaptor proteins is the TRAF proteins, which are required for the activation of NF-κB and JNK by several members in the TNF receptor family (Wallach et al., 1999). Previous studies suggested that TRAF proteins associate with p75 and regulate NGF-mediated NF-κB activation (Khursigara et al., 1999; Ye et al., 1999; Foehr et al., 2000). Indeed, dominant-negative TRAF6 decreased NF-κB translocation in Schwann cells (Khursigara et al., 1999). However, this present study indicates that RIP2 is necessary for NGF-dependent NF-κB responses and that TRAF6 may be involved in mediating p75 JNK activity.
How are these observations reconciled? First, TRAF proteins may be acting downstream of RIP2 or in concert with RIP2. _In vitro_binding data indicate that TRAF proteins can bind to RIP2, and dominant-negative TRAF proteins block RIP2 activation of NF-κB (McCarthy et al., 1998). Second, TRAF6 may compete with RIP2 for binding to p75. As a result of overexpressing TRAF6, we have found that RIP2–p75 interactions are decreased (data not shown). This suggests there may be complexes of TRAF and RIP2 proteins with the p75 receptor. Third, dominant-negative forms of TRAF proteins, containing the TRAF domain, also can interact with NF-κB inducing kinase or IκB kinases (IKK) and inhibit the subsequent activation of NF-κB. IKK, which phosphorylates IκB and leads to the degradation of IκB and the translocation of NF-κB, also can be activated by either TRAF2 or RIP proteins (Devin et al., 2000).
Coexpression of TRAF6 with p75 led to an increase in ATF-2 activity in an NGF-dependent manner (Fig. 9). Together with the above considerations, the role of TRAF6 may be to mediate both p75-induced NF-κB and JNK activation. Indeed, NGF gives an increase in JNK phosphorylation in Schwann cells (data not shown).
Other adaptor proteins that bind to p75 have been reported recently. Three different proteins, NRIF (Casademunt et al., 1999), NADE (Mukai et al., 2000), and NRAGE (Salehi et al., 2000), bind sequences in the cytoplasmic domain of the p75 receptor; each contributes to apoptosis in immortalized cell lines or is correlated with neurotrophin-dependent cell death. It is not yet known whether RIP2 interferes with the binding of these proteins to p75 or exerts an effect on downstream apoptotic signaling via these proteins. Still other adaptor proteins, including RhoA GTPase (Yamashita et al., 1999) and the zinc finger protein SC-1 (Chittka and Chao, 1999), exert nonapoptotic activities such as neurite elongation and growth arrest.
In summary, our results provide evidence that p75 activates two independent pathways in Schwann cells, NF-κB and cell death. These dual roles of p75 are mediated by a receptor-interacting protein, RIP2. Therefore, prosurvival effects of NGF in cells may rely not only on TrkA signaling but also on p75-induced NF-κB activity. Because neurotrophins also have an ability to induce cell death in a number of cell types including sympathetic neurons, sensory neurons, oligodendrocytes, and Schwann cells via p75 signaling (Casaccia-Bonnefil et al., 1998; Majdan and Miller, 1999), the receptor-mediated mechanisms that dictate these survival and death decisions must be determined by uniquely defined cell-specific interactions.
Footnotes
This work was supported by National Institutes of Health Grants NS21072, CA56490, and HD23315 to M.V.C. We thank Ed Skolnik, Albert Kim, Ravi Tikoo, and Bruce Carter for advice and reagents. We also thank Simon Murray for assistance.
Correspondence should be addressed to M. V. Chao, Skirball Institute for Biomolecular Medicine, New York University School of Medicine, 540 First Avenue, New York, NY 10016. E-mail:chao@saturn.med.nyu.edu.
REFERENCES
- 1.Anton ES, Weskamp G, Reichardt LF, Matthew WD. Nerve growth factor and its low affinity receptor promote Schwann cell migration. Proc Natl Acad Sci USA. 1994;91:2795–2799. doi: 10.1073/pnas.91.7.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bamji S, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis, is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 4.Bentley C, Lee K-F. p75 is important for axon growth and Schwann cell migration during development. J Neurosci. 2000;20:7706–7715. doi: 10.1523/JNEUROSCI.20-20-07706.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bothwell M. p75NTR: a receptor after all. Science. 1996;272:506–507. doi: 10.1126/science.272.5261.506. [DOI] [PubMed] [Google Scholar]
- 6.Bredesen DE, Rabizadeh S. p75NTR and apoptosis: Trk-dependent and Trk-independent effects. Trends Neurosci. 1997;20:287–291. doi: 10.1016/s0166-2236(96)01049-1. [DOI] [PubMed] [Google Scholar]
- 7.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 8.Carter BD, Lewin GR. Neurotrophins live or let die: does p75NTR decide? Neuron. 1997;18:187–190. doi: 10.1016/s0896-6273(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 9.Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Bohm-Matthaei R, Baeuerle PA, Barde Y-A. Selective activation of NF-κB by nerve growth factor through the neurotrophin receptor p75. Science. 1996;272:542–545. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- 10.Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- 11.Casaccia-Bonnefil P, Kong H, Chao MV. Neurotrophins: the biological paradox of survival factors eliciting apoptosis. Cell Death Differ. 1998;5:357–364. doi: 10.1038/sj.cdd.4400377. [DOI] [PubMed] [Google Scholar]
- 12.Casademunt E, Carter BD, Benzel I, Frade JM, Dechant G, Barde YA. The zinc finger protein NRIF interacts with the neurotrophin receptor p75NTR and participates in programmed cell death. EMBO J. 1999;18:6050–6061. doi: 10.1093/emboj/18.21.6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao MV. Neurotrophin receptors: a window into neuronal differentiation. Neuron. 1992;9:583–593. doi: 10.1016/0896-6273(92)90023-7. [DOI] [PubMed] [Google Scholar]
- 14.Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 15.Chittka A, Chao MV. Identification of a zinc finger protein whose subcellular distribution is regulated by serum and nerve growth factor. Proc Natl Acad Sci USA. 1999;96:10705–10710. doi: 10.1073/pnas.96.19.10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cortazzo MH, Kassis ES, Sproul KA, Schor NF. Nerve growth factor (NGF)-mediated protection of neural crest cells from antimitotic agent-induced apoptosis: the role of the low-affinity NGF receptor. J Neurosci. 1996;16:3895–3899. doi: 10.1523/JNEUROSCI.16-12-03895.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coulson EJ, Reid K, Baca M, Shipham KA, Hulett SM, Kilpatrick TJ, Bartlett PF. Chopper, a new death domain of the p75 neurotrophin receptor that mediates rapid neuronal cell death. J Biol Chem. 2000;275:30537–30545. doi: 10.1074/jbc.M005214200. [DOI] [PubMed] [Google Scholar]
- 18.Devin A, Cook A, Lin Y, Rodriguez Y, Kelliher M, Liu Z-G. The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity. 2000;12:419–429. doi: 10.1016/s1074-7613(00)80194-6. [DOI] [PubMed] [Google Scholar]
- 19.Dobrowsky RT, Carter BD. p75 neurotrophin receptor signaling: mechanisms for neurotrophic modulation of cell stress? J Neurosci Res. 2000;61:237–243. doi: 10.1002/1097-4547(20000801)61:3<237::AID-JNR1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 20.Feinstein E, Kimchi A, Wallach D, Boldin M, Varfolomeev E. The death domain: a module shared by proteins with diverse cellular functions. Trends Biochem Sci. 1995;20:342–344. doi: 10.1016/s0968-0004(00)89070-2. [DOI] [PubMed] [Google Scholar]
- 21.Ferri CC, Bisby MA. Improved survival of injured sciatic nerve Schwann cells in mice lacking the p75 receptor. Neurosci Lett. 1999;272:191–194. doi: 10.1016/s0304-3940(99)00618-7. [DOI] [PubMed] [Google Scholar]
- 22.Foehr E, Lin X, O'Mahony A, Geleziunas R, Bradshaw R, Greene W. NF-κB signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J Neurosci. 2000;20:7556–7563. doi: 10.1523/JNEUROSCI.20-20-07556.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frade JM, Rodriguez-Tebar A, Barde Y-A. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature. 1996;383:166–168. doi: 10.1038/383166a0. [DOI] [PubMed] [Google Scholar]
- 24.Friedman W. Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci. 2000;20:6340–6346. doi: 10.1523/JNEUROSCI.20-17-06340.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frostick SP, Yin Q, Kemp GJ. Schwann cells, neurotrophic factors, and peripheral nerve regeneration. Microsurgery. 1998;18:397–405. doi: 10.1002/(sici)1098-2752(1998)18:7<397::aid-micr2>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 26.Gentry JJ, Casaccia-Bonnefil P, Carter BD. Nerve growth factor activation of nuclear factor κB through its p75 receptor is an anti-apoptotic signal in RN22 schwannoma cells. J Biol Chem. 2000;275:7558–7565. doi: 10.1074/jbc.275.11.7558. [DOI] [PubMed] [Google Scholar]
- 27.Grinspan JB, Marchionni MA, Reeves M, Coulaloglou M, Scherer SS. Axonal interactions regulate Schwann cell apoptosis in developing peripheral nerve: neuregulin receptors and the role of neuregulins. J Neurosci. 1996;16:6107–6118. doi: 10.1523/JNEUROSCI.16-19-06107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu C, Castellino A, Chan J-H, Chao MV. BRE: a modulator of TNF-α action. FASEB J. 1998;12:1101–1108. doi: 10.1096/fasebj.12.12.1101. [DOI] [PubMed] [Google Scholar]
- 29.Gu C, Casaccia-Bonnefil P, Srinivaran A, Chao M. Oligodendrocyte apoptosis mediated by caspase activation. J Neurosci. 1999;19:3043–3049. doi: 10.1523/JNEUROSCI.19-08-03043.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heumann R, Lindholm D, Bandtlow C, Meyer M, Radeke M, Shooter E, Thoenen H. Differential regulation of mRNA encoding nerve growth factor, its receptor in rat sciatic nerve during development, degeneration, regeneration: role of macrophages. Proc Natl Acad Sci USA. 1987;84:8735–8739. doi: 10.1073/pnas.84.23.8735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hofman K, Bucher P, Tschopp J. The CARD domain: a new apoptotic signaling motif. Trends Biochem Sci. 1997;22:155–156. doi: 10.1016/s0968-0004(97)01043-8. [DOI] [PubMed] [Google Scholar]
- 32.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 33.Hsu H, Huang J, Shu H-B, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 34.Inohara N, del Peso L, Koseki T, Chen S, Nu™ez G. RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J Biol Chem. 1998;273:12296–12300. doi: 10.1074/jbc.273.20.12296. [DOI] [PubMed] [Google Scholar]
- 35.Jessen KR, Mirsky R. Schwann cells and their precursors emerge as major regulators of nerve development. Trends Neurosci. 1999;22:402–410. doi: 10.1016/s0166-2236(98)01391-5. [DOI] [PubMed] [Google Scholar]
- 36.Khursigara G, Orlinick J, Chao M. Association of the p75 neurotrophin receptor with TRAF6. J Biol Chem. 1999;274:2597–2600. doi: 10.1074/jbc.274.5.2597. [DOI] [PubMed] [Google Scholar]
- 37.Kong H, Kim AH, Orlinick JR, Chao MV. A comparison of the cytoplasmic domains of the Fas receptor and the p75 neurotrophin receptor. Cell Death Differ. 1999;6:1133–1142. doi: 10.1038/sj.cdd.4400587. [DOI] [PubMed] [Google Scholar]
- 38.Lemke G, Chao MV. Axons regulate Schwann cell expression of major myelin and NGF receptor genes. Development. 1988;102:499–504. doi: 10.1242/dev.102.3.499. [DOI] [PubMed] [Google Scholar]
- 39.Lewin GR, Barde Y-A. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- 40.Lin Z, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 41.Majdan M, Miller F. Neuronal life and death decisions: functional antagonism between the Trk and p75 neurotrophin receptors. Int J Dev Neurosci. 1999;17:153–161. doi: 10.1016/s0736-5748(99)00016-7. [DOI] [PubMed] [Google Scholar]
- 42.McCarthy JV, Ni J, Dixit VM. RIP2 is a novel NF-κB-activating and cell death-inducing kinase. J Biol Chem. 1998;273:16968–16975. doi: 10.1074/jbc.273.27.16968. [DOI] [PubMed] [Google Scholar]
- 43.Mukai J, Hachiya T, Shoji-Hoshino S, Kimura M, Nadano D, Suvanto P, Hanaoka T, Li Y, Ire S, Greene L, Sato T. NADE, a p75NTR-associated cell death executor, is involved in signal transduction mediated by the common neurotrophin receptor p75NTR. J Biol Chem. 2000;275:17566–17570. doi: 10.1074/jbc.C000140200. [DOI] [PubMed] [Google Scholar]
- 44.Rabizadeh S, Oh J, Zhong LT, Yang J, Bitler CM, Butcher LL, Bredesen DE. Induction of apoptosis by the low affinity NGF receptor. Science. 1993;261:345–348. doi: 10.1126/science.8332899. [DOI] [PubMed] [Google Scholar]
- 45.Salehi A, Roux P, Kubu C, Zeindler C, Bhakar A, Tannis L, Verdi J, Barker P. NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron. 2000;27:279–288. doi: 10.1016/s0896-6273(00)00036-2. [DOI] [PubMed] [Google Scholar]
- 46.Smith CA, Farrah T, Goodwin RG. The TNF receptor superfamily of cellular and viral proteins: activation, costimulation, and death. Cell. 1994;76:959–962. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 47.Soilu-Hanninen M, Ekert P, Bucci T, Syroid D, Bartlett P, Kilpatrick T. Nerve growth factor signaling through p75 induces apoptosis in Schwann cells via a Bcl-2-independent pathway. J Neurosci. 1999;19:4828–4838. doi: 10.1523/JNEUROSCI.19-12-04828.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song HY, Regnier CH, Kirschning CJ, Goeddel DV, Rothe M. Tumor necrosis factor (TNF)-mediated kinase cascades: bifurcation of nuclear factor-κB and c-Jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proc Natl Acad Sci USA. 1997;94:9792–9796. doi: 10.1073/pnas.94.18.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stanger BZ, Leder P, Lee T-H, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1995;81:513–524. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 50.Syroid DE, Maycox PJ, Soilu-Hanninen M, Petratos S, Bucci T, Burrola P, Murray S, Cheema S, Lee KF, Lemke G, Kilpatrick TJ. Induction of postnatal Schwann cell death by the low-affinity neurotrophin receptor in vitro and after axotomy. J Neurosci. 2000;20:5741–5747. doi: 10.1523/JNEUROSCI.20-15-05741.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taniuchi M, Clark H, Schweitzer J, Johnson E. Expression of nerve growth factor receptors by Schwann cells of axotomized peripheral nerves: ultrastructural location, suppression by axonal contact, and binding properties. J Neurosci. 1988;8:664–681. doi: 10.1523/JNEUROSCI.08-02-00664.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thome M, Hofmann K, Burns K, Martinon F, Bodmer J-L, Mattmann C, Tschopp J. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr Biol. 1998;8:885–888. doi: 10.1016/s0960-9822(07)00352-1. [DOI] [PubMed] [Google Scholar]
- 53.Wallach D, Varfolomeev E, Malinin N, Goltsev Y, Kovalenko A, Boldin M. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. 1999;17:331–367. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- 54.Yamashita T, Tucker K, Barde Y. Neurotrophin binding to the p75 receptor modulates Rho activity, axonal outgrowth. Neuron. 1999;24:585–593. doi: 10.1016/s0896-6273(00)81114-9. [DOI] [PubMed] [Google Scholar]
- 55.Ye X, Mehlen P, Rabizadeh S, VanArsdale T, Zhang H, Shin H, Wang JJL, Leo E, Zapata J, Hauser CA, Reed JC, Bredesen DE. TRAF family proteins interact with the common neurotrophin receptor and modulate apoptosis induction. J Biol Chem. 1999;274:30202–30208. doi: 10.1074/jbc.274.42.30202. [DOI] [PubMed] [Google Scholar]