A Functional Role for Intra-Axonal Protein Synthesis during Axonal Regeneration from Adult Sensory Neurons (original) (raw)
Abstract
Although intradendritic protein synthesis has been documented in adult neurons, the question of whether axons actively synthesize proteins remains controversial. Adult sensory neurons that are conditioned by axonal crush can rapidly extend processes in vitro by regulating the translation of existing mRNAs (Twiss et al., 2000). These regenerating processes contain axonal but not dendritic proteins. Here we show that these axonal processes of adult sensory neurons cultured after conditioning injury contain ribosomal proteins, translational initiation factors, and rRNA. Pure preparations of regenerating axons separated from the DRG cell bodies can actively synthesize proteins in vitro and contain ribosome-bound β-actin and neurofilament mRNAs. Blocking protein synthesis in these regenerating sensory axons causes a rapid retraction of their growth cones when communication with the cell body is blocked by axotomy or colchicine treatment. These findings indicate that axons of adult mammalian neurons can synthesize proteins and suggest that, under some circumstances, intra-axonal translation contributes to structural integrity of the growth cone in regenerating axons. By immunofluorescence, translation factors, ribosomal proteins, and rRNA were also detected in motor axons of ventral spinal roots analyzed after 7 d in vivo after a peripheral axonal crush injury. Thus, adult motor neurons are also likely capable of intra-axonal protein synthesis in vivo after axonal injury.
Keywords: mRNA localization, local protein synthesis, conditioned neuron, axonal regeneration, nerve regeneration
Localization of mRNAs to dendrites is now an accepted mechanism for targeting neuronal proteins to postsynaptic regions (Steward, 1997; Schuman, 1999; Martin et al., 2000). Although mRNAs are thought to be excluded from most adult vertebrate axons, intra-axonal translation has been demonstrated in neurons of several invertebrate species (Giuditta et al., 1991; Davis et al., 1992; van Minnen et al., 1997). There is evidence that some vertebrate axons also contain mRNAs. Axons of developing vertebrate neurons contain mRNAs and synthesize proteins (Knowles et al., 1996;Olink-Coux and Hollenbeck, 1996; Bassell et al., 1998; Eng et al., 1999). Axonal mRNAs have been detected in goldfish Mauthner cell (M-cell) and in mammalian hypothalamic and olfactory neurons (Mohr and Richter, 1992; Wensley et al., 1995; Koenig and Martin, 1996). Although M-cell axons contain rRNA and synthesize proteins (Koenig, 1991), there is surprisingly little evidence for local translation in vertebrate hypothalamic and olfactory neurons, the axons of which contain mRNAs but not other components needed for protein synthesis (Wensley et al., 1995; van Minnen et al., 1997). Thus, although protein synthesis has been shown to occur in axons of invertebrate and developing vertebrate neurons, it is unclear whether it occurs in axons of adult vertebrates. Even in neurons that are capable of intra-axonal protein synthesis, the biological relevance of this mechanism has remained unknown.
Very recently, Koenig and colleagues (2000) have shown evidence for RNA and ribosomes in adult mammalian axons, suggesting that we should reconsider the issue of whether mature axons can synthesize proteins. We showed previously that after a conditioning axonal crush lesion, cultured adult sensory neurons rapidly regenerate processes by translation of existing mRNAs (Twiss et al., 2000). Although this study pointed to the importance of translational control during regeneration, it did not address where the translation occurred. We now show that rat dorsal root ganglion (DRG) neurons can synthesize proteins directly within their regenerating processes. These processes show growth similar to regenerating axons in vivo (Smith and Skene, 1997) and contain axonal but not dendritic proteins by immunostaining. These regenerating sensory axons contain β-actin mRNA but not γ-actin mRNA, consistent with the differential localization of actin mRNAs in developing cortical neurons (Bassell et al., 1998). A previous study addressing the functional relevance of intra-axonal protein synthesis in embryonic sympathetic neurons saw no apparent effect on axonal growth when local protein synthesis was inhibited (Eng et al., 1999). In contrast, we show here that the axons of conditioned DRG neurons rapidly retract when intra-axonal protein synthesis is blocked but only after communication with the cell body is compromised by axotomy or inhibition of axonal transport. This indicates that intra-axonal protein synthesis is functionally relevant to the structure of the distal regenerating axon under some circumstances. Taken together, our data show that adult sensory neurons are not only capable of intra-axonal protein synthesis but also suggest that this mechanism facilitates axonal regeneration. The presence of similar translational machinery in motor axons suggests that this mechanism is not limited to sensory neurons.
MATERIALS AND METHODS
DRG culture. Cultures of conditioned primary sensory neurons from DRG were performed as described previously (Smith and Skene, 1997; Twiss et al., 2000). Briefly, 180–200 gm adult Sprague Dawley rats were subjected to sciatic nerve crush, and L4–5 DRGs were isolated for culturing 3–7 d later. Conditioned ganglia were dissociated with 500 U/ml collagenase (Sigma, St. Louis, MO) and 0.05% trypsin-EDTA (Life Technologies, Gaithersburg, MD). Dissociated DRGs were plated in DME/F12 medium containing N1 supplement and 10% horse serum. Cytosine arabinoside (10 μm; Sigma) was included to inhibit proliferation of non-neuronal cells, and 80 μm 5,6-dichlorobenzimidazole riboside (Sigma) was included to inhibit RNA synthesis (Smith and Skene, 1997).
Culture method for isolation of axons. DRGs were plated at two to five neurons per square millimeter into a tissue culture insert containing a polyethylene tetraphthalate (PET) membrane with 8 μm pores (Millipore, Bedford, MA) that had been coated with poly-l-lysine (Sigma) and laminin (Upstate Biotechnology, Lake Placid, NY). To evaluate axonal growth through the membrane, inserts were rinsed in PBS after 20 hr culturing and processed for immunofluorescence (see below). To isolate axons, the top membrane surface was scraped with a cotton-tipped applicator. Scraping was repeated three times with a fresh applicator altering the direction of scraping 90° each time. For isolation of cell bodies, the surface underneath the membrane was scraped in an identical manner. The scraped membrane was processed for immunofluorescence to evaluate the “axonal preparation” for complete removal of cell bodies and non-neuronal cells.
Electron microscopy. DRG cultures were grown on PET membranes for 18 hr and then fixed in PBS containing 3% formaldehyde and 1% glutaraldehyde for 20 min at room temperature. All subsequent steps were performed at room temperature unless indicated otherwise. Samples were transferred to fresh fixative for 3 hr at 4°C followed by 1% osmium tetroxide in PBS for 1 hr on ice. Samples were rinsed in water for 20 min and then treated with 2% aqueous uranyl acetate for 1 hr at 4°C. After rinsing in water for 20 min, samples were dehydrated in graded alcohol (30, 50, 70, and 95% ethanol for 20 min each, then 100% ethanol twice for 20 min). Samples were then treated with propylene oxide (PO) for 20 min, 2:1 PO/Spurr resin for 40 min, 1:2 PO/Spurr resin for overnight, and Spurr resin for 2 hr. Finally, samples were embedded in Spurr resin by polymerization in fresh resin at 60°C overnight. Sections 30–40 nm thick were cut perpendicular to the membrane surface using a Diatome diamond knife. Sections were stained with saturated aqueous uranyl acetate and lead citrate. Ultrastructural analysis was performed with a JEM1200-EX electron microscope (JEOL) at 80 kV, 50 μm objective aperture. Images were digitized by scanning film negatives at 1200 dpi resolution.
Immunofluorescence. All steps were performed at room temperature unless indicated otherwise. For most immunostaining experiments, dissociated DRGs were plated on coated glass coverslips at a density of five neurons per square millimeter. Cultures were rinsed 20–24 hr later with PBS and then fixed in buffered 4% paraformaldehyde for 20 min at room temperature. For tissue sections, ventral root and sciatic nerve were isolated from animals 7 d after sciatic nerve crush and fixed overnight in 4% paraformaldehyde. Tissues were equilibrated in 0.5 m sucrose for 4 hr, frozen, and then 6 μm sections were cut using a cryostat. In most experiments, cultures were incubated in 20 mm glycine for 10 min three times to quench autofluorescence; tissues were treated with 0.25 m NaBH4 for 30 min. Samples were rinsed in PBS, permeabilized in PBS containing 0.2% Triton X-100 for 15 min, and then incubated in 7.5% horse serum and 7.5% goat serum (HyClone) in PBS for 1 hr to block nonspecific binding. Cultures were then incubated overnight at 4°C with primary antibodies diluted in blocking buffer. The following primary antibodies were used: rabbit anti-L4 (1:1000) (Twiss et al., 2000), rabbit anti-L17 (1:100) (Laine et al., 1991), rabbit anti-L29 (1:100) (Hoke et al., 1998), human anti- ribosomal P protein (RPP) (1:1000) (Elkon et al., 1985, 1986); goat anti-eIF2α (1:50; Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-eIF4E (1:50; Santa Cruz Biotechnology), rabbit anti-eIF5 (1:1000; Santa Cruz Biotechnology), Y10B mouse anti-rRNA (1:500) (Lerner et al., 1981), mouse anti-GAP-43 (1:106) (Schreyer and Skene, 1991), mouse anti-neurofilament (NF) (1:1000; Zymed, San Francisco, CA), rabbit anti-NF (1:200; Chemicon, Temecula, CA), rabbit anti-S100 (1:1000; Sigma), and mouse anti-tubulin (Tub) βIII (1:400; Chemicon). Cultures were rinsed in PBS and incubated for 1 hr in secondary antibodies diluted in blocking buffer. The following secondary antibodies were used: FITC-conjugated goat anti-mouse, donkey anti-human, and donkey anti-goat antibodies (1:400; Jackson ImmunoResearch, West Grove, PA), and Texas Red-conjugated goat anti-rabbit antibody (1:400; ICN Biochemicals, Costa Mesa, CA). Cultures were rinsed in PBS and then mounted with Vectashield (Vector Laboratories, Burlingame, CA). Immunofluorescence was analyzed by standard fluorescent microscopy or laser scanning confocal microscopy. In all experiments, samples (coverslips, membranes, or tissue sections) were incubated without primary antibody to rule out nonspecific signals from secondary antibodies. Specificity of the anti-L4, -L17, -L29, -eIF2α, -eIF4E, and -eIF5 antibodies was evaluated by immunoblot of lysates from rat PC12 cells as described previously (Twiss et al., 2000).
Metabolic labeling. DRGs cultured on PET membrane were used for metabolic labeling. Scraped membranes (see above) were excised and incubated in Met/Cys-deficient medium containing 4 mCi/ml35S-Met/Cys (ICN) for 4 hr (membrane was always cut 2–3 mm central to the insert wall to exclude the most peripheral edge of the membrane from analyses). In some experiments, the axonal and cell body preparations were incubated in 10 μg/ml cycloheximide for 20 min before the addition of 1 mCi/ml35S-Met/Cys. Membranes were rinsed briefly in PBS, and cellular constituents were lysed in radioimmunoprecipitation assay buffer. Labeled protein lysates were analyzed by TCA precipitation followed by liquid scintillation counting (Twiss and Shooter, 1995). Lysates were also analyzed by 10% SDS-PAGE. Gels were treated with fluorographic reagent (NEN, Boston, MA), dried, and exposed to XAR5 or BioMax-MR film with appropriate intensifying screens (Kodak, Rochester, NY).
RNA isolation. Total RNA was extracted from DRG cell body and axonal preparations, cultured Schwann cells, and adult rat tissues with 4 m guanidium isothiocyanate and phenol-chloroform (Chromozinski and Sacchi, 1987). Glycogen (Sigma) was used as a carrier for isopropanol precipitation in the DRG culture samples. RNAs were either used directly for Northern blotting or processed for RT-PCR as described in Results. “Polysomal” RNAs were isolated to determine whether the axonal mRNAs were actively translated. For this, scraped membranes were rinsed in PBS containing 0.1 mm cycloheximide and then lysed in 300 mmKCl, 2 mm MgCl2, 20 mm Tris · Cl, pH 7.4, 2 mm DTT, 0.05% deoxycholate, 0.1 mmcycloheximide, 50 U/ml RNase inhibitor (Panvera) for 20 min at 4°C (Baum and Wormington, 1985). The lysate was cleared by centrifugation at 13,500 rpm, 4°C, for 15 min. Ribosome-bound mRNAs were coimmunoprecipitated from the lysate by absorption with 5 μg Y10B antibody for 3.5 hr followed by anti-mouse IgG Agarose (Sigma) for 1 hr at 4°C. Immunocomplexed RNAs were collected by centrifugation, and pellets were washed twice in lysis buffer. After washing, RNA was extracted from the Agarose pellet as above. Where indicated RNAs were quantified by UV spectroscopy. Northern blotting was performed as described previously (Twiss et al., 2000). Lysates from PC12 cells were used for controls in the Y10B immunoprecipitation. PC12 cells were maintained in DMEM with 6% horse and 6% bovine calf sera (Hyclone) (Twiss and Shooter, 1995). To confirm that Y10B antibody coimmunoprecipitated ribosome-bound mRNAs, traditional polysomal RNA fractionation was performed with PC12 cell lysates using discontinuous 20% sucrose gradients as described previously (Baum and Wormington, 1985; Twiss et al., 2000).
RT-PCR. A commercial cDNA synthesis kit (Smart cDNA synthesis; Clontech, Cambridge, UK) was used for RT-PCR. This method provides an anchor at the 3′ end of first-strand cDNA by virtue of terminal transferase activity of Moloney murine leukemia virus-RT, allowing it to switch to a unique oligonucleotide as a second template on reaching the 5′ cap structure of the mRNA. This generates full-length, single-stranded cDNA with anchor sequence at 5′ and 3′ ends for subsequent PCR amplification (Matz et al., 1999). For standard RNA isolates from axons and cell bodies, 200 ng RNA was used as a template. For polysomal RNAs, one-third of the Y10B precipitate RNA was used as a template. RNA from human placenta (Clontech) or rat sciatic nerve served as positive control. First-strand reaction was diluted fivefold with 10 mm Tris · Cl, pH 7.6, 1 mmEDTA. Advantage 2 Taq polymerase (Clontech) was used for amplification with anchor primers. After a 1 min hot start at 95°C, reactions were cycled at 95°C × 5 sec, 65°C × 5 sec, and 68°C × 6 min in a PE2400 thermal cycler (Perkin-Elmer, Norwalk, CT). Fifteen microliter aliquots (100 μl reaction) were removed from every third cycle from 15–27 cycle (15–30 cycles for polysomal RNAs). Each aliquot was used for Southern blotting (Sambrook et al., 1989).
Hybridization. Northern and Southern blots were hybridized with 32P-labeled cDNA probes by standard methods as described previously (Twiss et al., 2000). cDNA probes for β-actin and γ-actin were provided by Peter Gunning, Children's Medical Research Institute (New South Wales, Australia) (Nudel et al., 1983; Erba et al., 1986). cDNA probe for NF-L was isolated by RT-PCR using PC12 RNA as a template. This cDNA was subcloned, and its sequence was verified by dideoxy sequencing (data not shown).
Microdissection and videomicroscopy. Time-lapse images were recorded using a Hamamatsu ORCA charge-coupled device camera mounted on a Zeiss inverted microscope and driven by Axiovision software. DRG cultures were plated onto coated coverslips at one to two neurons per square millimeter. Coverslips were transferred 14–20 hr after plating to a heated stage and equilibrated to 37°C with constant perfusion of fresh medium (maintained between 36.5 and 38°C). Axons were cut using a glass capillary microelectrode pulled to a sharp closed tip on a Sutter P-97 puller. In most cases, the remaining proximal portion of the axon as well as the cell body were moved ≥200 μm from the distal axon with the capillary needle. Images of anucleated axons were recorded once every minute for at least 20 min (up to 2 hr) and then treated with 10 μg/ml cycloheximide. In some instances, the anucleated axons were treated immediately with cycloheximide. To evaluate axonal protein synthesis in intact neurons, cultures were perfused with medium containing 10 μg/ml colchicine for 25 min followed by 10 μg/ml cycloheximide for 25 min. Images were captured every minute over the course of treatment.
Quantitative analysis of intra-axonal protein synthesis. Conditioned DRG neurons were cultured as above. After 16 hr in culture, neurons were treated with 10 μg/ml colchicine. Thirty minutes later, 10 μg/ml cycloheximide was added to the culture medium. After 30 min in cycloheximide (60 min in colchicine), cultures were fixed and processed for immunofluorescence. The length of the longest axon of individual neurons from control (untreated), colchicine, cycloheximide, and colchicine plus cycloheximide samples was measured under 125× magnification using an ocular micrometer (≥150 neurons per condition). To measure length of terminal axon branches, images of individual neurons from the control, colchicine, and colchicine plus cycloheximide samples were captured with a digital camera (200× magnification), and the length of the branches most distal from the cell body was measured (≥15 neurons per condition, ≥10 branches per neuron). Statistical significance of these values was analyzed using Student's _t_test.
RESULTS
Regenerating sensory axons contain components of the translational machinery
In previous studies, we showed that ribosomal protein L4 mRNA is translationally regulated during neurite regeneration from PC12 cells and that L4 translation is required for neurite regeneration from PC12 cells and for axonal regeneration from conditioned DRG neurons (Twiss et al., 2000). To initially address the role of L4 in nerve regeneration, we determined its subcellular localization. Adult rat DRG neurons were conditioned by axonal crush injury 3–7 d before culturing. By 18–24 hr in culture, these conditioned DRG neurons extend long processes in the absence of new gene transcription (Smith and Skene, 1997). Extension of such processes by DRG neurons in vitro represents regrowth or “regeneration” of axons that these mature neurons bore in vivo (Lindsay, 1988). In culture, these regenerating processes of conditioned DRG neurons show immunoreactivity for the axonal protein, Tau, but not for microtubule-associated protein 2 (MAP2) that is restricted to dendrites (Fig.1A,B). Thus, at least by morphologic parameters, these processes of cultured DRG neurons are axonal. Indirect immunofluorescence using a polyclonal antibody to rat L4 (Twiss et al., 2000) showed immunoreactivity in all cellular elements of these cultures (Fig. 1C). Surprisingly, the L4 antibody also labeled regenerating axons (Fig. 1C,arrowheads). Colabeling with monoclonal antibody to GAP-43 confirmed that these processes were derived from neurons and not closely apposed Schwann cells or other non-neuronal elements (Fig.1D). Although axonal L4 immunoreactivity was weak compared with the perikaryon, it was specific because preimmune serum did not stain any axonal processes (Fig.1E,F), and preincubating L4 antiserum with immunizing peptide eliminated specific fluorescence (data not shown). These immunocytochemical studies show that regenerating axons of conditioned sensory neurons contain ribosomal protein L4.
Fig. 1.

Rapidly growing axons of conditioned DRG neurons contain ribosomal protein L4. Four days after sciatic nerve crush, conditioned L4–5 DRG neurons were dissociated and cultured on coated coverslips for 22 hr in medium containing Ara-C and DRB. The long processes extended by these DRG neurons are strongly immunoreactive for Tau (A). Colabeling with monoclonal antibody, the dendritic protein MAP2 shows signals that are limited to the cell body and do not extend into the Tau-reactive processes (B). Polyclonal anti-ribosomal protein L4 antibody showed signals in both in the neuronal cell body and the axonal-like processes (C, arrowheads). Colabeling with a monoclonal antibody to GAP43 confirmed that the L4 immunoreactivity was within the neuronal processes (D,arrowheads). Preimmune serum for L4 showed a faint fluorescence in the cell body after long exposure times (2 min in_E_ vs 10 sec in C) and no signal in the axonal processes (E). Phase-contrast image of the same neuron in shown in E shows abundant axons extending from this sensory neuron cell body (F). Scale bars, 20 μm.
Although the presence of L4 in axons implies that regenerating sensory axons contain ribosomes, extra-ribosomal functions have been ascribed to some ribosomal proteins (Wool, 1996), and intra-axonal localization of L4 in sensory neurons thus could have merely represented an extra-ribosomal activity. To further test for the presence of ribosomes in axons, we asked whether regenerating axons contained other ribosomal proteins. The sensory axons also showed immunoreactivity for ribosomal protein L17, ribosomal protein L29, and RPPs (Fig.2A_–_C). It is possible that each of these ribosomal proteins could have some extra-ribosomal function in these regenerating axons. Indeed, L29 was isolated while cloning a cell surface heparin/heparin sulfate-binding protein that promotes cell adhesion (Liu et al., 1996; Hoke et al., 1998). However, we also found that regenerating axons contain other components of the translational machinery. First, antibodies to the translation initiation factors, eIF2α, eIF4e, and eIF5, showed signals within the axons (Fig.2D_–_F). Second, intra-axonal immunoreactivity was detected with an anti-28S rRNA antibody [Y10B (Lerner et al., 1981)] (Fig. 2G). These antibody signals appear valid because, by immunoblotting, anti-L17, -L29, -eIF2α, -eIF4E, and -eIF5 antibodies detected bands of appropriate molecular weight (Fig. 2H). Specificity of the anti-L4 and -RPP antibodies have been published previously (Elkon et al., 1985; Twiss et al., 2000), and as shown below, Y10B antibody preparation immunoprecipitates ribosomes (see Fig. 6) (Lerner et al., 1981; Garden et al., 1995).
Fig. 2.
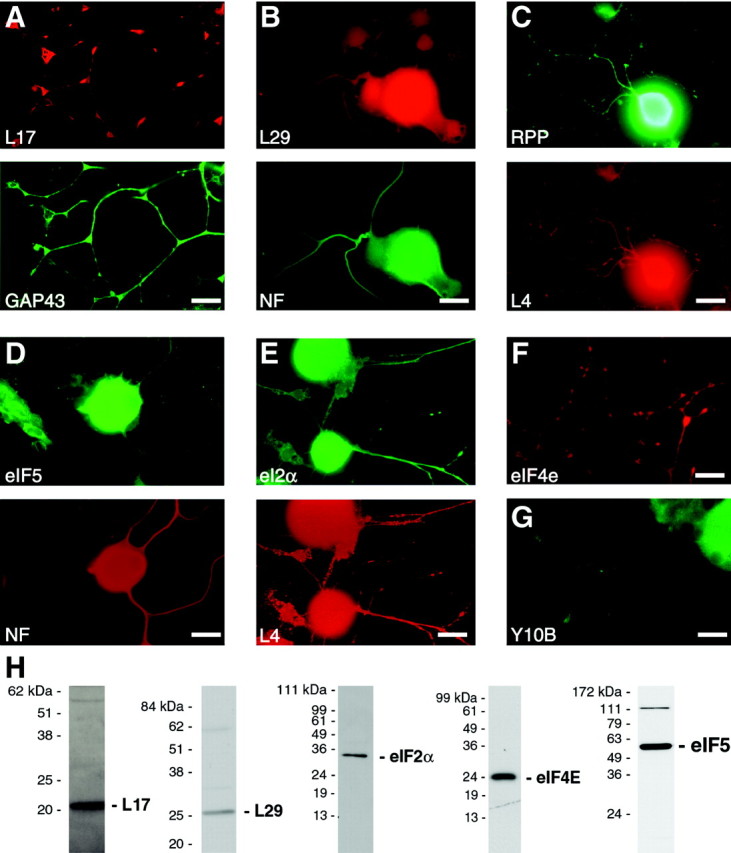
Regenerating sensory axons contain ribosomal proteins, rRNA, and translation factors. Conditioned DRG cultures were colabeled with antibodies to ribosomal proteins, translation factors, and rRNA. Processes that react with GAP43 and NF contain immunoreactivity for ribosomal proteins L17 and L29 (_A_and B, respectively). RPP shows a signal in the same neuronal processes that show immunoreactivity for L4 (C). NF-positive processes also contain eIF5 immunoreactivity (D), and eIF2α shows colocalization with immunoreactivity for L4 (E). Antibodies to eIF4e showed similar signals within the regenerating sensory axons (F). Finally, immunostaining with Y10B anti-28S rRNA antibody also shows a signal within the axons (G). Scale bars, 20 μm. Specificity of the anti-L17, -L29, -eIF2α, -eIF4E, and -eIF5 antibodies was evaluated by immunoblotting (H; 12% gel for L17, L29, eIF2α, and eIF4E, and 10% gel for eIF5). Each of these antibody preparations recognized one major band of the expected molecular weight. Similar data have been published for anti-L4 and -RPP antibodies (Elkon et al., 1985; Twiss et al., 2000), and Figure 6 shows immunoprecipitation using the Y10B antibody.
Fig. 6.

Intra-axonal mRNAs are translationally active in cultured DRG neurons. Coimmunoprecipitation of mRNAs with rRNA using Y10B antibody was used to determine whether intra-axonal mRNAs are translationally active. Lysates from PC12 cells were used to test the validity of this coimmunoprecipitation. A shows Northern blot analysis of polysomal RNAs and Y10B coimmunoprecipitated RNAs. Fractionation of β-actin mRNA in discontinuous 20% sucrose gradients is shown in lanes 1–4. Note that β-actin mRNA resides in the polysome fraction (P) rather than the subpolysome fraction (SP) (A,lanes 1–2). Addition of 50 mm EDTA before ultracentrifugation causes β-actin mRNA to shift from the_P_ to SP fraction (A,lanes 3–4). For Y10B immunoprecipitation, RNA was extracted from the Y10B immunocomplex (IC) and supernatant from immunoprecipitate (S) and equivalent proportions of these IC and S RNA fractions were used for Northern blotting. β-actin mRNA coimmunoprecipitated with Y10B (A, lanes 5–6). In lysates that were treated with EDTA to disrupt the 40S and 60S ribosome subunits, β-actin mRNA resided in the S fraction (A,lanes 7–8). Without addition of Y10B to the lysate, β-actin mRNA also resided in the S fraction (A,lanes 9–10). The Y10B coimmunoprecipitation is not limited to β-actin mRNA (data not shown). B and_C_ show virtual Northern blots of Y10B immunoprecipitates from the DRG axonal preparations. For this, axons were isolated from the DRG cultures (Fig. 3), and RNA from axonal Y10B immunocomplexes was used for RT-PCR. The Y10B immunoprecipitates contained β-actin and NF-L mRNAs (B and C, respectively). This suggests that the axonal mRNAs encoding β-actin and NF-L are actively translated in these regenerating sensory axons.
Confocal microscopy was used to obtain an estimate of the levels of translational machinery in these axons relative to the neuronal cell bodies. Figure 3, A and_B_, shows reconstructed three-dimensional images of an individual neuron that was colabeled with antisera to L4 and RPP. As expected, signals in the neuronal perikaryon are high compared with the overall signal intensity in the axons (Fig.3A,B, asterisk). However, there are foci of strong signal intensity in the axons, and the brightest signals for L4 and RPP colocalize (Fig.3A,B, arrows). Similar data have been obtained with the antibodies to translation factors and rRNA (data not shown). Confocal images of single optical planes through regions of high intra-axonal signal intensity show that L17 and rRNA assume a granular distribution in the axon (Fig.3C,D). Although our antibody preparations appeared specific, these data did not tell us whether these ribosomal constituents were assembled into ribosomes. To address this issue, we performed electron microscopy (EM) on the DRG cultures using uranyl acetate staining to enhance detection of nucleic acids. The proximal axonal segment contained electron-dense particles distributed along the rough endoplasmic reticulum (RER) and freely distributed within the axoplasm (Fig. 3F,G,insets). The distal segments of the axon also contained nonmembrane-bound electron-dense particles that measured 22–28 nm in diameter (Fig. 3G, arrows), which is the same size as those particles along the RER in the neuronal cell body (Fig.3F). These intra-axonal ribosome-like particles are also of the same diameter as those along the RER of mouse liver that was processed in an identical manner (data not shown) and are consistent with the rare EM reports of ribosomes in adult rodent axons (Zelená, 1970, 1972; Pannese and Ledda, 1991). Taken together, these data indicate that regenerating axons of cultured DRG neurons contain the necessary components for protein synthesis.
Fig. 3.
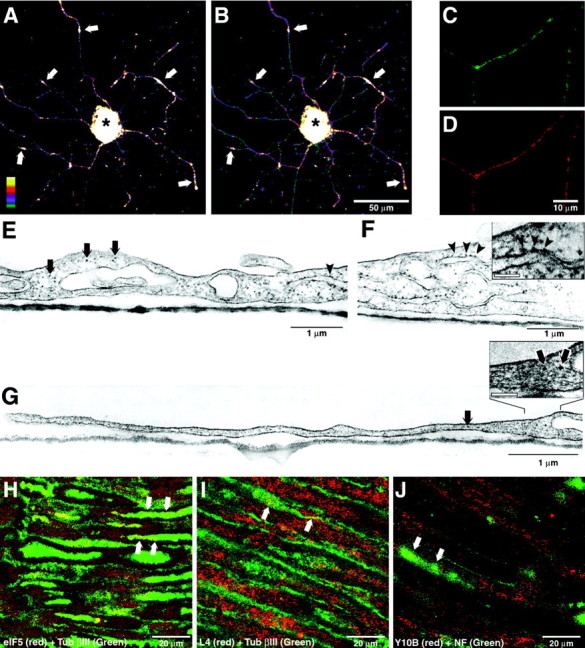
Abundance of translational machinery in regenerating axons. A, B, DRG cultures costained with antisera to ribosomal protein L4 and RPP were analyzed by laser scanning confocal microscopy. Neurons were scanned at 1 μm intervals over 24 optical (Z) planes. Digital three-dimensional images are displayed as a spectrum as indicated in the bottom left corner of A. The signal intensity for L4 and RPP in the cell body is saturated (asterisk); however, high signal intensity is also seen in the axons at >100 μm distance from the cell body, and the bright intra-axonal signals for L4 and RPP colocalize (arrows). Scale bar, 50 μm.C, D, A region of high intra-axonal fluorescence for ribosomal protein L17 and 28S rRNA (recognized with Y10b antibody) similar to those shown in A and_B_ is illustrated as a single Z-plane confocal image through the center of the axon. Note that the fluorescent signal for L17 and rRNA is granular rather than diffuse in these highly immunoreactive regions of the axon. Scale bar, 10 μm.E_–_G, Electron micrographs of DRG cultures stained with uranyl acetate show electron-dense particles in the proximal (E) and distal axonal segments (G) that are of the similar size to those seen along the RER in the cell body (F,arrowheads) and likely represent ribosomes. The proximal segment of the axon in E extends right to_left_ from the cell body in F and continues on right to left as the distal axonal segment shown in G. Ribosome-like particles are seen on the RER (arrowheads) and free within the axoplasm (arrows) in the proximal axonal segment (E). Ribosome-like particles are noted in the distal segment of the axon (G, arrows). At higher magnification, these electron-dense particles in the distal axon (G, inset) are approximately the same diam-eter as those along the RER in the cell body (F, inset). Scale bars:insets, 100 nm. H–J, Confocal images of ventral L4 spinal cord root (H_–_I) and sciatic nerve (J) illustrate signals for L4 (H), eIF5 (I), and 28S rRNA (J) that colocalize with intra-axonal signals for mouse anti-Tub βIII (H_–_I) and rabbit anti-NF (J). These confocal images represent a three-dimensional reconstruction of four Z-plane images taken at 0.4 μm intervals to provide optical sections through individual axons and exclude the myelin sheath and Schwann cell cytoplasm. The arrows in each panel indicate such optically isolated axons where non-neuronal components are excluded from consideration and show punctate intra-axonal signals for a ribosomal protein (H), translation factor (I), and rRNA (J). Scale bars, 20 μm.
Translational machinery in spinal motor axons
Although we have detected protein synthesis machinery in sensory axons, it remained possible that we have illustrated a phenomenon unique to cultured DRG neurons. Although the peripheral branches of DRG neurons are described as “modified axons” (Peters et al., 1991), these sensory neurons are pseudounipolar in vivo, and their peripheral branch is functionally efferent rather than afferent. Also, the artificial ex vivo environment of our DRG culture may illicit a less mature state that allows mRNA and ribosomes to enter the axonal compartment. To determine whether injured axons of more polarized neurons contain ribosomes and translation factors in vivo, we have used immunostaining of ventral spinal nerve roots isolated 7 d after sciatic nerve crush. Confocal microscopy was used to optically section through individual axons that were identified by colabeling with antibodies to neurofilament or a neuronal-specific tubulin isoform (Tub βIII). eIF5 and L4 colocalized with Tub βIII in many axons and showed a punctate rather than diffuse intra-axonal signal (Fig. 3H,I). Similar results were obtained with anti-rRNA antibody, and sections stained without primary antibody showed no fluorescence (data not shown). Because spinal motor neurons are fully polarized compared with the pseudounipolar sensory neurons and the ventral root contains only motor axons, these localization data indicate that axons of injured motor neurons also contain components of the translational machinery. Furthermore, these data are consistent with a recent report that extruded axoplasm from ventral roots of normal adult rat shows immunoreactivity for rRNA and RPP and contains ribosomes (Koenig et al., 2000). The rRNA immunoreactivity that we have seen in the ventral root axons extends into the distal portion of the axons because the crushed sciatic nerve shows similar colocalization of 28S rRNA and neurofilament (Fig. 3J). Colabeling with antibodies to eIF5 or L4 and neuronal markers showed similar intra-axonal signals in the sciatic nerve (data not shown).
Regenerating sensory axons synthesize proteins
In contrast to other adult neurons, the DRG neurons are amenable to culture, and this provided us with a model system to address intra-axonal translation by biochemical and molecular techniques. We used an axonal preparation for incorporation of35S-labeled amino acids to determine whether the intra-axonal translational machinery actively synthesizes proteins. For this, we required a means to physically separate axons from their cell bodies. Torre and Steward (1992) developed a tissue culture system to isolate dendrites from cortical neurons, and we have modified this method to isolate regenerating axons (Torre and Steward, 1992). Conditioned DRG preparations were plated into tissue culture inserts that contained a translucent membrane with 8-μm-diameter pores. The sensory neurons readily extended axons through pores and then along the lower membrane surface (Fig.4A,B). Confocal microscope X–Y sections above (Fig. 4A) and below (Fig. 4B) membranes that had been labeled with anti-S100 antibodies to identify Schwann cells indicated that these cells do not traverse the membrane nor do they appear to enter the pores of the membrane.
Fig. 4.
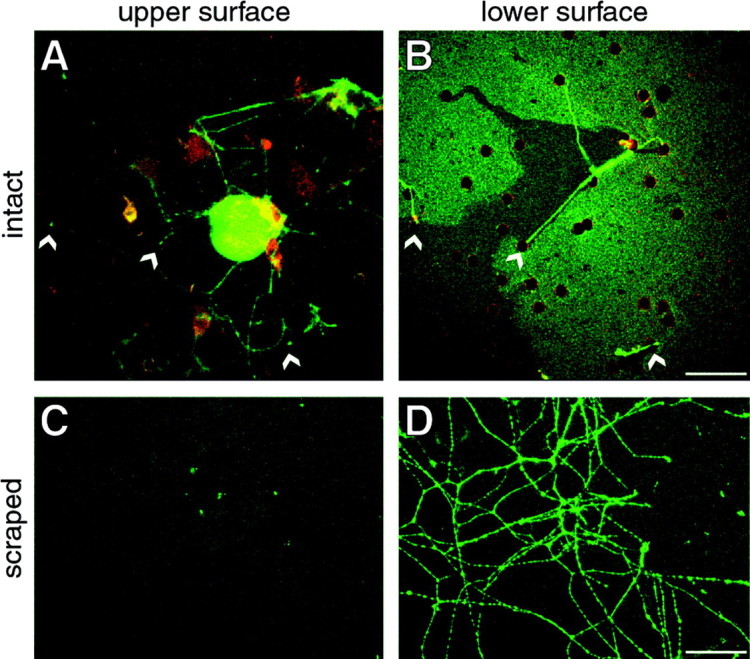
Culture system to isolate regenerating DRG axons. Dissociated cultures of conditioned DRG neurons were plated into a tissue culture insert containing a PET membrane with 8 μm pores. After 24 hr, the cultures were fixed, and indirect immunofluorescence was performed with antibodies to GAP-43 (shown in green) and S100 (shown in red). Confocal microscopy was used to image the membranes. A shows a three-dimensional reconstructed composite digital image of 19 × 1 μm Z planes along the top surface of the membrane. The _arrowheads_indicate axons that enter pores of the membrane. _B_displays a single X–Y plane image along the bottom surface of the membrane directly below that illustrated in A.Arrowheads indicate the pores where axons cross the membrane. Note that no S100-reactive cellular elements are seen along the bottom surface of the membrane. Non-neuronal elements were also not visible by phase-contrast images (data not shown). To isolate axons, the top surface of the membrane was scraped repetitively with a cotton-tipped applicator (see Materials and Methods). Scraped membranes were processed for immunostaining with antibodies to GAP43. Scraping removed all cellular elements from the top membrane surface (C; reconstructed composite digital image of 19 × 1 μm Z planes). Axons that had traversed the membrane remained adherent to the bottom surface of the membrane after scraping (D; single X–Y plane on bottom membrane surface). Scale bars, 50 μm.
To obtain an axonal preparation for biochemical analyses, we used a scraping method to remove cellular constituents from the top and bottom membrane surfaces (see Materials and Methods). Confocal imaging of such scraped membranes showed that all neuronal cell bodies and non-neuronal elements were effectively removed from the top surface of the membrane without stripping away axons that had extended through the membrane (Fig. 4C,D). Membranes were scraped along the top surface (axonal preparations) or bottom surface (cell body preparations) and then incubated in medium containing35S-Met/Cys. TCA precipitates from axonal preparations consistently showed ∼10% radioactivity of the cell body preparations. However, because axons traversed the membrane at random distances from the cell body and not all axons crossed the membrane, these TCA precipitates cannot be regarded as a quantitative estimate of intra-axonal protein synthesis. By SDS-PAGE analyses the axonal preparation clearly contains labeled proteins, and importantly, some newly synthesized proteins appeared to be enriched in the axonal preparation, because these were not represented in the cell body preparations (Fig. 5A). Specifically, labeled bands of ∼167, 160, 95, 68, 52, 40, 34, and 28 kDa were strongly enriched in the axonal preparations. To be certain that the axonal 35S signals represented newly synthesized protein, membranes scraped along the top or bottom surface were incubated in 35S-Met/Cys after pretreatment with a protein synthesis inhibitor. Cycloheximide effectively inhibited 35S-Met/Cys incorporation in both axonal and cell body preparations (Fig.5B). Thus, these regenerating sensory axons also actively synthesize proteins.
Fig. 5.

Regenerating sensory axons synthesize proteins and differentially localize β- and γ-actin mRNAs. A, DRG cultures were performed in tissue culture inserts as described above. After 18 hr in culture, the top or bottom surface of the membrane was scraped to yield an axonal or cell body preparation, respectively (note that the cell body preparation contains non-neuronal cells and neuronal processes that have not traversed the membrane pores). Membranes were then incubated in medium containing 4 mCi/ml 35S-Met/Cys for 4 hr. Axonal and cell body preparation lysates were fractionated on 10% SDS-PAGE gels and processed for fluorography. The axonal preparation required a much longer exposure time than the cell body preparation (1 vs 6 d). Autoradiograms show proteins of ∼167, 160, 95, 68, 52, 40, 34, and 28 kDa that appear enriched in the axonal preparations (dashes to right of autoradiogram). These data are representative of three independent metabolic labeling experiments. We cannot state that these proteins are uniquely synthesized in the axons because the high specific activity of the cell body lysates compared with the axonal lysates does not allow for matched exposure times. B, Axonal and cell body preparations were generated as above and incubated in 10 μg/ml cycloheximide for 20 min before metabolic labeling in 1 mCi/ml35S-Met/Cys for 4 hr and analyzed as in A. Note that cycloheximide completely inhibited incorporation of35S-Met/Cys into proteins in the cell body preparation (1 d exposure) and greatly diminished protein synthesis in the axonal preparation (6 d exposure). The labeled band at ∼70 kDa in the cycloheximide-treated axonal preparation may represent a protein derived from intra-axonal mitochondria. The lower isotope levels used for labeling compared with A account for different band intensities in the axonal preparation of this and A.C, RNA was extracted from axonal and cell body preparations from DRG neurons that had been plated for 18 hr and used for RT-PCR (see Materials and Methods). Aliquots were removed from the PCR at 21, 24, 27, and 30 cycles and used for virtual Northern blotting. Blots probed with β-actin cDNA showed a prominent band after a short exposure that corresponds to the full-length β-actin mRNA (3 hr exposure). In contrast, no signal for γ-actin could be detected in the axonal cDNA even with long exposure times (72 hr exposure), but γ-actin cDNA was readily detected in RT-PCR-amplified RNA of the DRG preparations that included the cell body (i.e., RNA from nonfractionated cultures) (3 hr exposure). Note that with matched exposure times, γ-actin cDNA appears to be even more abundant than β-actin cDNA in the cell body RT-PCR samples. D, To exclude this possibility of differential actin isoform expression by non-neuronal cells contaminating the axonal RNA preparation, total RNA was isolated from 7 d crushed [distal and proximal (ScNCrPr and ScN-CrD, respectively)] and naive rat sciatic nerve (ScN-N) and Schwann cell cultures (SC) and processed for standard Northern blotting. γ-actin mRNA was easily detected in all RNA preparations of sciatic nerve and in the RNA from purified Schwann cell cultures (24 hr exposure).
Regenerating sensory axons contain mRNAs
The above data indicated that regenerating sensory axons can synthesize at least some proteins. In developing cortical neurons, β-actin mRNA extends into axonal growth cones, whereas γ-actin mRNA is restricted to the cell body (Bassell et al., 1998). This occurs during a period of vigorous axonal growth similar to that of conditioned neurons. Thus, we asked whether regenerating sensory axon preparations contain actin mRNAs. RNA was extracted from axonal preparations and used for a coupled RT-PCR technique that amplifies full-length mRNA (Matz et al., 1999). Amplified cDNAs were then used for virtual Northern blots (see Materials and Methods). β-actin cDNA was detected in RT-PCR from axonal RNA preparations, but we could not detect γ-actin cDNA in axonal preparations even with at least 24-fold longer exposure duration (Fig. 5C). cDNAs amplified from whole DRG cultures showed a signal for γ-actin cDNA with short exposure time that appeared relatively even more abundant than the β-actin cDNA signal (Fig. 5C). Thus, these DRG cultures express γ-actin mRNA, but this transcript is excluded from the axonal RNA preparations. To rule out the possibility that we had detected distinct expression of actin genes by Schwann cells, we probed Northern blots of total RNA from purified Schwann cells (Notterpek et al., 1999) and naive and crushed sciatic nerve. γ-Actin mRNA was easily detected in these RNA preparations (Fig. 5D). Because these non-neuronal components of the DRG also express γ-actin mRNA, this indicates that β-actin mRNA in axonal RNA preparations was derived from axons rather than from non-neuronal elements. Moreover, this differential mRNA localization as well as the enrichment of proteins synthesized in the axonal preparation (Fig. 5A) point to the purity of our axonal preparations. Thus, conditioned DRG neurons differentially localize actin mRNAs, with γ-actin being restricted to the cell body and β-actin extending into axonal processes just as in developing cortical neurons (Bassell et al., 1998).
mRNAs within regenerating sensory axons are bound to ribosomes
The presence of translational machinery in regenerating sensory axons provides strong evidence that intra-axonal mRNAs are actively translated. To more specifically test whether the β-actin mRNA is used in the regenerating axons, we asked whether this transcript is bound to ribosomes in the axons. For this, we used Y10B anti-rRNA antibody to immunoprecipitate ribosomes (Lerner et al., 1981;Garden et al., 1995). By lysing cellular constituents under conditions classically used for polysome fractionation, mRNA/ribosome association is maintained, and mRNAs can be copurified with ribosomes (Baum and Wormington, 1985). We tested the validity of the Y10B immunoprecipitation using PC12 cell lysates. β-actin mRNA cofractionated with polysomal RNA by discontinuous 20% sucrose gradient ultracentrifugation (Fig.6A, _lane 1_-2). This is consistent with other cellular systems in which β-actin mRNA is always polysomal (Yenofsky et al., 1983; Biberman and Meyuhas, 1997). Addition of EDTA to the PC12 lysate before ultracentrifugation shifted the β-actin mRNA to the subpolysomal fraction (Fig. 6A, lanes 3–4). Because EDTA dissociates ribosomal subunits (Baum and Wormington, 1985), this shows that β-actin mRNA is also mostly polysomal in the PC12 cells. β-actin mRNA similarly coimmunoprecipitates with the Y10B antibody, but not if lysates were pretreated with EDTA to disrupt ribosome subunits (Fig. 6A, lanes 5–8). Control immunoprecipitates without Y10B primary antibody do not contain any β-actin mRNA (Fig. 6A, lanes 9–10). The lower RNA content of the axonal preparations required RT-PCR for detection. By virtual Northern blot, β-actin mRNA coimmunoprecipitated with axonal rRNAs, suggesting that intra-axonal β-actin mRNA was actively translated in regenerating sensory axons (Fig. 6B). mRNAs encoding NF proteins have been detected in squid giant axon and in goldfish M-cell axon (Giuditta et al., 1991; Weiner et al., 1996). Therefore, we asked whether the regenerating rat sensory axons also contained ribosome-bound NF-L mRNA. Reprobing the virtual Northern blots with a cDNA probe to rat NF-L mRNA showed that the axonal Y10B immunoprecipitates contained cDNAs with migration that corresponded to that of NF-L (Fig. 6C) (Dickson et al., 1986). Thus, these intra-axonal mRNAs that encode cytoskeletal proteins are bound to rRNA under biochemical conditions that maintain ribosome/mRNA interaction. Also, axonally synthesized proteins of ∼68 and 43 kDa, the molecular weights of NF-L and β-actin, respectively, were detected in the metabolic labeling experiments (Fig. 5A). This supports our contention that β-actin and NF-L mRNAs are translationally active within the axons. It is not clear how polymerization of NF triplet proteins into filaments in the growing axons would be affected by the local translation NF-L mRNA without the high and medium molecular weight forms (NF-H and NF-M, respectively). A band of the appropriate molecular weight for NF-H (160 kDa) is visible in the axonally synthesized protein preparations (Fig. 4A), but there is no clear representation of NF-M (130 kDa).
Intra-axonal mRNA translation contributes to growth cone maintenance
The above studies indicate that protein synthesis occurs directly within regenerating mammalian axons, but they do not provide any insight into what function intra-axonal protein synthesis may serve. Because actin polymerization is needed for growth cone motility (Okabe and Hirokawa, 1990, 1991), a local source of new β-actin protein should facilitate axonal growth. Such a mechanism has been suggested, but not proven, by Bassell and colleagues (Bassell et al., 1998; Zhang et al., 1999). Previous studies in compartmentalized cultures of neonatal sympathetic neurons failed to detect any morphological effect of inhibiting intra-axonal protein synthesis (Eng et al., 1999). However, our data point to a biological function for intra-axonal protein synthesis. We used time-lapse imaging to address the role of intra-axonal protein synthesis in regenerating axons using 14–20 hr cultures of conditioned DRG neurons. To completely exclude effects of cell body-derived proteins, axons were severed with a fine, closed tip glass capillary, and the DRG cell body was scraped well away from the anucleated axon. Distal portions of anucleated axons remained visually intact for at least 2 hr after the axon was severed (Fig.7A); occasionally, the proximal end of an anucleated axon would retract initially (Fig.7A, arrowheads), but the distal portion remained stable, neither elongating or retracting (Fig. 7B, top two panels). Treatment with the protein synthesis inhibitor cycloheximide caused growth cones of anucleated axons to retract (Fig.7B). Growth cone retraction was visible 10 min after addition of cycloheximide and also occurred in axons that were treated with cycloheximide immediately after removal of the cell body (Fig.7C). Axons that had not been severed from their cell body were not affected by cycloheximide treatment over this same time period (Fig. 7E). Similar results were obtained with anisomycin (data not shown). Although we cannot completely exclude a nonspecific effect on the axon structure by these translational inhibitors, both clearly blocked incorporation of35S-Met/Cys in the axonal preparation (Fig. 5). Furthermore, these inhibitors work by independent mechanisms: cycloheximide blocks A to P site translocation of peptidyl tRNA, and anisomycin inhibits peptidyl transferase activity (Gale, 1981).
Fig. 7.

Intra-axonal protein synthesis maintains the growth cone. To address the functional significance of intra-axonal protein synthesis, we used video microscopy to monitor changes in axons during inhibition of protein synthesis. Axons were anucleated using a glass capillary tube that had been pulled to a closed tip. Elapsed time is indicated in the top left corner of each image of a time-lapse sequence (A_–_E).A and B show the results of one experiment. A shows anucleated axons that were incubated for 20 min in complete medium. Scale bar, 20 μm. The_arrowhead_ indicates where the axon was severed with the axon coursing from left to right. Note that although the proximal portion of the axon retracts slightly (arrowheads), the distal axon remains intact. The_boxed region_ is shown at higher magnification in the time-lapse sequence in B. Scale bar, 10 μm. In the first two images the distal axon remains stable over 20 min. After addition of 10 μg/ml cycloheximide, the distal tip of the axon begins to retract over the 30–40 min of the time-lapse sequence (10–20 min after treatment). C shows high-magnification time-lapse sequence of a second experiment in which an axon was treated with cycloheximide immediately after severing or anucleation. Scale bar, 10 μm. Note that this anucleated axon is stable over the first 10 min but then begins to retract, similar to that seen in B. In a third series of experiments, the relevance of local protein synthesis in intact neurons was addressed by treating cultures with colchicine to decrease the influence of cell body-synthesized proteins by impeding axonal transport (D). A low-magnification time-lapse sequence of a culture preparation that was treated with 10 μg/ml colchicine for 25 min followed by 10 μg/ml cycloheximide for 25 min as indicated is shown in D. Most axonal branches were stable over the course of colchicine treatment (D, top two panels). However, during treatment with cycloheximide, many of the axonal branches retracted (D, bottom five panels). Scale bar, 60 μm. This axonal retraction required pretreatment with colchicine or axotomy because treatment of intact neurons with 10 μg/ml cycloheximide alone was without effect (E). Scale bar, 20 μm. The above series of time-lapse experiments have been repeated in three different DRG preparations in at least five neurons per preparation, using the protein synthesis inhibitor anisomycin, and yielded similar results (data not shown). To quantitate the axonal retraction in colchicine plus cycloheximide-treated neurons, 16 hr DRG cultures were treated with colchicine for 60 min (COL), cycloheximide for 30 min (CHX), or colchicine for 60 min plus cycloheximide for 30 min (COL + CHX). Control (Cntl) for these studies consisted of cultures that were allowed to grow in normal medium for 17 hr. The average length of most terminal axon branches was measured for each treatment paradigm. Axonal branch lengths in the CHX + COL were approximately half that of control, COL, or CHX paradigms (F) (average ± 2 × SEM). The differences between the COL + CHX, COL, and Cntl samples was statistically significant (p ≤ 0.0001 for COL + CHX vs Cntl, COL, and CHX samples).
Although there is evidence that the distal axon in invertebrates and some vertebrates remains viable for a period of time after axotomy (Rotshenker, 1981; Bittner, 1991; Chaudhry et al., 1992), the significance of protein synthesis in anucleated axons is limited. To address the functional relevance of intra-axonal protein synthesis in intact neurons, we have assessed the effect of translational inhibitors in neurons that were pretreated with colchicine to impede axonal transport. Time-lapse imaging showed that most axon branches remained intact over 25 min incubation in colchicine (Fig. 7D,top two panels). Distal portions of axon branches began to retract rapidly after subsequent treatment with cycloheximide (Fig.7D, bottom five panels). The axonal retraction was even more apparent in these intact, colchicine-treated neurons than in the anucleated axons, and curiously not all branches of an axon appeared equally affected by protein synthesis inhibition. Time-lapse studies performed with the protein synthesis inhibitor anisomycin produced identical results (data not shown). There was no axonal retraction when intact neurons were treated with cycloheximide (Fig.7E). To quantitate the axonal retraction, 16 hr DRG cultures were treated with colchicine for 60 min (COL), with cycloheximide for 30 min (CHX), or pretreated with colchicine for 30 min and then supplemented with cycloheximide for 30 min (COL + CHX). After a total of 17 hr in culture, the longest axon of neurons from each treatment paradigm was assessed. Average lengths of the longest axons were consistently shorter in the COL + CHX cultures, but the differences were only modest (data not shown). Because the time-lapse experiments in Figure 7D_suggested that not all branches of an axon were equally affected by protein synthesis inhibition, the length of individual axonal branches would likely provide a more valid quantitation of axonal retraction after inhibition of protein synthesis. Indeed, mean terminal axonal branch length in the control, COL, and CHX cultures (51, 51.75, and 49.5 μm, respectively) was almost double that of the COL + CHX cultures (26 μm) (Fig. 7F). By Student's_t test, the p values for COL + CHX compared with control, COL, and CHX data were clearly significant at_p_ < 0.0001. Taken together, these experiments indicate that, at the very least, intra-axonal protein synthesis contributes to structural maintenance of the distal regenerating axon when the supply of proteins from the cell body is compromised by axotomy or colchicine treatment.
DISCUSSION
The prevailing hypotheses of localized protein synthesis argue that ribosomes and mRNAs are actively excluded from axons of most vertebrate neurons (Steward, 1995). However, there are several lines of evidence indicating that intra-axonal translation occurs in some vertebrate neurons (van Minnen, 1994; Alvarez et al., 2000). Our studies point to a role for intra-axonal protein synthesis during nerve regeneration from adult neurons. DRG neurons from adult rats that are conditioned by axonal crush injury in vivo can rapidly regenerate processes in vitro independent of new gene expression (Smith and Skene, 1997). We reported recently that new protein synthesis is needed for this rapid growth (Twiss et al., 2000). Here we show that regenerating processes of conditioned sensory neurons contain ribosomal proteins, translation factors, rRNA, and mRNAs. By immunostaining, these DRG processes contain axonal and not dendritic markers. These axonal processes can synthesize proteins independent of the DRG cell body. The absence of any other cellular elements in our axonal preparations excludes any possible transfer of newly synthesized proteins from glia, as has been seen in invertebrates and recently suggested to occur in vertebrates (Lasek et al., 1974; Tytell and Lasek, 1984; Sotelo-Silveira et al., 2000).
mRNA localization in axons
Subcellular localization of mRNA to dendrites is well established in many different neuronal populations (Steward, 1997). Regulation of intra-dendritic translation by neurotransmitters and recent studies linking this local translation to synaptic plasticity indicate that dendritic protein synthesis fulfills a physiological function (Weiler and Greenough, 1993; Kang and Schuman, 1996; Steward and Halpain, 1999;Huber et al., 2000; Scheetz et al., 2000). In contrast to intra-dendritic protein synthesis, intra-axonal protein synthesis has received much less attention. Examples of mRNAs in the axonal compartment have been best characterized in rather unique neuronal populations of invertebrate, lower vertebrate, and (rarely) mammalian species (Mohr and Richter, 1992; van Minnen, 1994; Alvarez et al., 2000). Although early studies argued against the possibility that axons could synthesize proteins (Lasek et al., 1974; Tytell and Lasek, 1984), intra-axonal protein synthesis has now been proven in several invertebrate species (Capano et al., 1987; Giuditta et al., 1991; Davis et al., 1992; van Minnen et al., 1997). It has been suggested that these invertebrate axons are capable of protein synthesis because they share some morphological and physiological features with dendrites (Steward, 1995). Consistent with this, concentrations of rRNA have been detected in postsynaptic regions of the squid giant axon (Martin et al., 1989), and synaptic facilitation in _Aplysia_sensory/motor neuron cocultures is mediated by mRNA translation in sensory neuron processes (Martin et al., 1997).
Axons of mature vertebrate hypothalamic and olfactory neurons contain mRNAs, but these axons apparently do not contain any ribosomes (Jirikowski et al., 1990; Mohr and Richter, 1992; Tiedge et al., 1993;Wensley et al., 1995). Thus, it is not clear what biological significance mRNAs in such vertebrate neurons could provide, and this may represent incomplete axonal/dendritic polarity in these neuronal populations. One could similarly argue that processes of cultured DRGs are capable of protein synthesis because these neurons are never fully polarized, although these processes show axonal features in vitro. However, Koenig et al. (2000) recently demonstrated RNA and ribosomal P proteins in mammalian ventral spinal roots, and we have similarly detected rRNA, ribosomal protein, and translation factor in motor axons (Fig. 3). Thus, intra-axonal translation is likely not limited to the cultured sensory neurons. Developing vertebrate neurons are also capable of intra-axonal protein synthesis during periods of rapid axonal growth. As the axon is established during polarization of cultured embryonic cortical neurons, ribosomes and other RNAs migrate into this rapidly growing process (Kleiman et al., 1994; Bradke and Dotti, 1997). β-actin is encoded by one of the axonal mRNAs of embryonic cortical neurons (Bassell et al., 1998). Neurotrophin 3 increases neurite outgrowth in embryonic hippocampal neurons (Morfini et al., 1994), and it also enhances RNA localization in these developing axons, including localization of β-actin mRNA (Knowles and Kosik, 1997; Zhang et al., 1999). Axonal mRNA localization and translation may represent a developmental mechanism that neurons retain into adulthood and use for axonal regeneration.
Biological relevance of intra-axonal protein synthesis
The identity of the mRNAs that we and others have detected in axons provides some insight into the biological function of intra-axonal translation. Many of the mRNAs identified in invertebrate and developing vertebrate axons encode cytoskeletal proteins, including microtubule, microfilament, and intermediate filament proteins (Giuditta et al., 1991; Kaplan et al., 1992; Olink-Coux and Hollenbeck, 1996; Bassell et al., 1998; Eng et al., 1999). Neurite growth requires synthesis of new cytoskeleton. Local intra-dendritic mRNA translation may also contribute to growth because dendritic mRNA localization is first appreciable when dendrites begin to grow rapidly in developing hippocampal neurons (Kleiman et al., 1994). mRNAs encoding cytoskeletal proteins are included among those isolated from the dendritic growth cones of hippocampal neurons (Crino and Eberwine, 1996). The regenerating DRG axons contain mRNAs encoding the cytoskeletal proteins β-actin and NF-L, and tubulin (55 kDa) and NF-H (160 kDa) may also be among those proteins with synthesis that appears enriched in the axonal compartment. Determination of whether the proteins synthesized locally in axons are limited to cytoskeletal elements or also include transmembrane and secreted proteins will require further investigation.
Localization of β-actin in regenerating DRG processes specifically points to a growth-associated function for local translation. In other cellular systems, β-actin mRNA is enriched in regions of rapid cell movement, but γ-actin mRNA lies in non-motile regions of the cell (Kislauskis and Singer, 1992; Hill and Gunning, 1993; Kislauskis et al., 1993). Inhibition of β-actin mRNA localization in fibroblasts blocks migration, suggesting that its local translation contributes to cell motility (Kislauskis et al., 1994). Developing cortical neurons similarly localize β-actin mRNA to axonal growth cones and restrict γ-actin mRNA to the perikaryon (Bassell et al., 1998). Because the growth cone is a site of rapid actin polymerization and growth cone motility is required for axon growth (Okabe and Hirokawa, 1990, 1991),Bassell et al. (1998) hypothesized that local synthesis of β-actin protein is needed for axonal growth. A local source of β-actin should also prove advantageous to the regenerating axons.
Function of intra-axonal protein synthesis
As noted above, there are several examples of intra-axonal mRNA localization and even some biochemical examples of protein synthesis in developing axons. However, there is no concrete evidence for a biological function of intra-axonal protein synthesis. In earlier studies by Eng et al. (1999) using compartmentalized cultures of developing sympathetic neurons, local protein synthesis did not appear to contribute to axonal growth. Differences between the compartmentalized cultures and our DRG preparation may explain why we have detected a function for intra-axonal protein synthesis in the conditioned DRG neurons. First, Eng et al. (1999) studied a later stage of axonal extension than our DRG cultures. Processes must grow at least 1 mm in the compartmentalized cultures before they even reach the axonal chamber (Campenot, 1982), whereas our studies with DRGs during the first day in vitro were limited to axons of <1 mm length. Second, the etched growth surface of the compartmentalized culture preparation promotes directional axonal growth at the expense of branching (Campenot, 1982; Eng et al., 1999), whereas our data suggest that all branches of an axon may not be affected equally by local protein synthesis inhibition (Fig. 7D). Thus, local protein synthesis could play a role in axonal branching. On the other hand, our data are not completely inconsistent with those of Eng et al. (1999). Although we show that the distal axon rapidly retracts when local protein synthesis is inhibited in the DRG cultures (Fig. 7), this response was not apparent unless axonal transport was blocked or the cell body was removed from the axon. Inhibition of local protein synthesis in intact DRG neurons was without effect. Because anterograde axonal transport mechanisms active in the intact DRG neurons are maintained in the compartmentalized cultures of sympathetic neurons (Campenot et al., 1996), the functional significance of intra-axonal protein synthesis may be to complement the supply of proteins normally provided by axonal transport. Impeding axonal transport as we have done may force the axon to use an alternate source of proteins.
Local translation during axonal regeneration
Locally synthesized proteins of similar molecular weight to actin and tubulin have been identified by microelectrophoresis of lysates from regenerating axons of conditioned goldfish retinal explants (Koenig, 1989). However, studies in mammals addressing the role of local translation in nerve regeneration have not completely distinguished between proteins synthesized in the axon and those produced by non-neuronal cells (Tobias and Koenig, 1975; Edbladh et al., 1994; Gaete et al., 1998). Because we have eliminated non-neuronal cells from our axonal preparations, we show unequivocally that regenerating DRG axons synthesize proteins. The proteins encoded by the axonal mRNAs that we have identified are also synthesized in the cell body and transported down the axon by slow axonal transport (actin at 2–4 mm/d and NF at 0.25 mm/d) (Black and Lasek, 1979, 1980). Axonal transport increases after injury, and this could account for the more rapid axonal regeneration in conditioned retinal neurons (McQuarrie and Grafstein, 1982). However, axonal transport is not immediately increased, and growth cone formation and early regeneration in conditioned rat DRG neurons appear to be locally mediated rather than a response of the neuronal cell body (Sjoberg and Kanje, 1990).Koenig (1991) has argued that local synthesis of axonal proteins plays a role in initiating axonal regeneration. Onset of axonal regeneration_in vivo_ does occur earlier in conditioned than in naive goldfish retinal and rat DRG neurons (McQuarrie and Grafstein, 1981;Sjoberg and Kanje, 1990). In vitro, conditioned DRG neurons initiate process outgrowth faster than do naive DRG neurons(Smith and Skene, 1997; Lankford et al., 1998). However, such in vitro_studies, including our own, have used dissociated cultures consisting of DRG cell bodies without residual axon stumps, so axonal regrowth must be initiated by a cell body response. This does not exclude the possibility that the intra-axonal protein synthesis plays a role in the early stages of axonal regrowth in these dissociated cultures of injury-conditioned DRG neurons. The conditioned neuron may provide a unique situation in which intra-axonal translation facilitates rapid axonal regrowth. Future work will be needed to determine the role that local mRNA translation in axonal regeneration potentially plays_in vivo and what signals may regulate intra-axonal protein synthesis.
Footnotes
This work was supported by funds from the Christopher Reeve Paralysis Foundation (TB2-9903 to J.L.T.) and the National Science Foundation (IBN98–10803 to J.L.T.). We are grateful to Drs. Harley Kornblum, David Glanzman, Carolyn Schanen, and Michael Sofroniew for proofreading and insightful comments, and Dr. Matthew Schibler of the Carol Moss Spivak Imaging Center (University of California Los Angeles Brain Research Institute) for assistance with confocal microscopy. Drs. Lucia Notterpek and Brad Fletcher provided cultured Schwann cell RNA. Antibodies were kindly provided by Drs. D. Carson (L29), K. Elkon (ribosomal P proteins), M. Kilberg (L17), E. Rubin (Y10B), and P. Skene (GAP43). Graphical assistance for illustrations was provided by M. Blair Ligon (Meridith College, Raleigh, NC).
Correspondence should be addressed to Jeffery L. Twiss, Department of Pathology (Neuropathology), University of California Los Angeles School of Medicine, 10833 Le Conte Avenue, Los Angeles, CA 90095. E-mail:jtwiss@ucla.edu.
REFERENCES
- 1.Alvarez J, Giuditta A, Koenig E. Protein synthesis in axons and terminals: significance for maintenance, plasticity and regulation of phenotype. With a critique of slow transport theory. Prog Neurobiol. 2000;62:1–62. doi: 10.1016/s0301-0082(99)00062-3. [DOI] [PubMed] [Google Scholar]
- 2.Bassell GJ, Zhang H, Byrd AL, Femino AM, Singer RH, Taneja KL, Lifshitz LM, Herman IM, Kosik KS. Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J Neurosci. 1998;18:251–265. doi: 10.1523/JNEUROSCI.18-01-00251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baum E, Wormington W. Coordinate expression of ribosomal protein genes during Xenopus development. Dev Biol. 1985;111:488–498. doi: 10.1016/0012-1606(85)90500-7. [DOI] [PubMed] [Google Scholar]
- 4.Biberman Y, Meyuhas O. Substitution of just five nucleotides at and around the transcription start site of rat beta-actin promoter is sufficient to render the resulting transcript a subject for translational control. FEBS Lett. 1997;405:333–336. doi: 10.1016/s0014-5793(97)00234-2. [DOI] [PubMed] [Google Scholar]
- 5.Bittner GD. Long-term survival of anucleate axons and its implications for nerve regeneration. Trends Neurosci. 1991;14:188–193. doi: 10.1016/0166-2236(91)90104-3. [DOI] [PubMed] [Google Scholar]
- 6.Black M, Lasek R. Axonal transport of actin: slow component b is the principal source of actin for the axon. Brain Res. 1979;171:401–413. doi: 10.1016/0006-8993(79)91045-x. [DOI] [PubMed] [Google Scholar]
- 7.Black M, Lasek R. Slow components of axonal transport: two cytoskeletal networks. J Cell Biol. 1980;86:616–623. doi: 10.1083/jcb.86.2.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradke F, Dotti C. Neuronal polarity: vectorial cytoplasmic flow precedes axon formation. Neuron. 1997;19:1175–1186. doi: 10.1016/s0896-6273(00)80410-9. [DOI] [PubMed] [Google Scholar]
- 9.Campenot RB. Development of sympathetic neurons in compartmentalized cultures. II. Local control of neurite survival by nerve growth factor. Dev Biol. 1982;93:13–21. doi: 10.1016/0012-1606(82)90233-0. [DOI] [PubMed] [Google Scholar]
- 10.Campenot RB, Lund K, Senger DL. Delivery of newly synthesized tubulin to rapidly growing distal axons of sympathetic neurons in compartmented cultures. J Cell Biol. 1996;135:701–709. doi: 10.1083/jcb.135.3.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capano CP, Giuditta A, Castigli E, Kaplan B. Occurrence and complexity of polyadenylated RNA in squid axoplasm. J Neurochem. 1987;49:698–704. doi: 10.1111/j.1471-4159.1987.tb00950.x. [DOI] [PubMed] [Google Scholar]
- 12.Chaudhry V, Glass J, Griffin J. Wallerian degeneration in peripheral nerve disease. Neurol Clin. 1992;10:613–627. [PubMed] [Google Scholar]
- 13.Chromozinski P, Sacchi N. Single-step method of RNA isolation by guanidine isothiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 14.Crino PB, Eberwine J. Molecular characterization of the dendritic growth cone: regulated mRNA transport and local protein synthesis. Neuron. 1996;17:1173–1187. doi: 10.1016/s0896-6273(00)80248-2. [DOI] [PubMed] [Google Scholar]
- 15.Davis L, Dou P, DeWit M, Kater S. Protein synthesis within neuronal growth cones. J Neurosci. 1992;12:4867–4877. doi: 10.1523/JNEUROSCI.12-12-04867.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dickson G, Prentice H, Julien J-P, Ferrari G, Leon A, Walsh F. Nerve growth factor activates Thy-1 and neurofilament gene transcription in rat PC12 cells. EMBO J. 1986;5:3449–3453. doi: 10.1002/j.1460-2075.1986.tb04668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edbladh M, Tonge D, Golding J, Ekstrom PA, Edstrom A. Early regeneration in vitro of adult mouse sciatic axons is dependent on local protein synthesis but may not involve neurotrophins. Neurosci Lett. 1994;168:37–40. doi: 10.1016/0304-3940(94)90410-3. [DOI] [PubMed] [Google Scholar]
- 18.Elkon K, Parnassa A, Foster C. Lupus autoantibodies target ribosomal P proteins. J Exp Med. 1985;162:459–471. doi: 10.1084/jem.162.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elkon K, Skelly S, Parnassa A, Moller W, Danho W, Weissbach H, Brot N. Identification and chemical synthesis of a ribosomal protein antigenic determinant in systemic lupus erythematosus. Proc Natl Acad Sci USA. 1986;83:7419–7423. doi: 10.1073/pnas.83.19.7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eng H, Lund K, Campenot RB. Synthesis of β-tubulin, actin, and other proteins in axons of sympathetic neurons in compartmented cultures. J Neurosci. 1999;19:1–9. doi: 10.1523/JNEUROSCI.19-01-00001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erba HP, Gunning P, Kedes L. Nucleotide sequence of the human gamma cytoskeletal actin mRNA: anomalous evolution of vertebrate non-muscle actin genes. Nucleic Acids Res. 1986;14:5275–5294. doi: 10.1093/nar/14.13.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaete J, Kameid G, Alvarez J. Regenerating axons of the rat require a local source of proteins. Neurosci Lett. 1998;251:197–200. doi: 10.1016/s0304-3940(98)00538-2. [DOI] [PubMed] [Google Scholar]
- 23.Gale EF. The Molecular basis of antibiotic action, Ed 2. Wiley; New York: 1981. [Google Scholar]
- 24.Garden GA, Hartlage-Rubsamen M, Rubel EW, Bothwell MA. Protein masking of a ribosomal RNA epitope is an early event in afferent deprivation-induced neuronal death. Mol Cell Neurosci. 1995;6:293–310. doi: 10.1006/mcne.1995.1023. [DOI] [PubMed] [Google Scholar]
- 25.Giuditta A, Menichini E, Capano CP, Langella M, Martin R, Castigli E, Kaplan B. Active polysomes in the axoplasm of the squid giant axon. J Neurosci Res. 1991;28:21–28. doi: 10.1002/jnr.490280103. [DOI] [PubMed] [Google Scholar]
- 26.Hill MA, Gunning P. Beta and gamma actin mRNAs are differentially located within myoblasts. J Cell Biol. 1993;122:825–832. doi: 10.1083/jcb.122.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoke DE, Regisford EG, Julian J, Amin A, Begue-Kirn C, Carson DD. Murine HIP/L29 is a heparin-binding protein with a restricted pattern of expression in adult tissues. J Biol Chem. 1998;273:25148–25157. doi: 10.1074/jbc.273.39.25148. [DOI] [PubMed] [Google Scholar]
- 28.Huber K, Kayser M, Bear M. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1256. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- 29.Jirikowski GF, Sanna PP, Bloom FE. Messenger RNA coding for oxytocin is present in axons of the hypothalamoneurohypophysial tract. Proc Natl Acad Sci USA. 1990;87:7400–7404. doi: 10.1073/pnas.87.19.7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang H, Schuman EM. A requirement of local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1405. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 31.Kaplan B, Gioio A, Capano CP, Crispino M, Giuditta A. β-actin and β-tubulin are components of a heterogeneous mRNA population present in the squid giant axon. Mol Cell Neurosci. 1992;3:133–144. doi: 10.1016/1044-7431(92)90017-v. [DOI] [PubMed] [Google Scholar]
- 32.Kislauskis EH, Singer RH. Determinants of mRNA localization. Curr Opin Cell Biol. 1992;4:975–978. doi: 10.1016/0955-0674(92)90128-y. [DOI] [PubMed] [Google Scholar]
- 33.Kislauskis EH, Li Z, Singer RH, Taneja KL. Isoform-specific 3′-untranslated sequences sort alpha-cardiac and beta-cytoplasmic actin messenger RNAs to different cytoplasmic compartments. J Cell Biol. 1993;123:165–172. doi: 10.1083/jcb.123.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kislauskis EH, Zhu X, Singer RH. Sequences responsible for intracellular localization of beta-actin messenger RNA also affect cell phenotype. J Cell Biol. 1994;127:441–451. doi: 10.1083/jcb.127.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleiman R, Banker G, Steward O. Development of subcellular mRNA compartmentation in hippocampal neurons in culture. J Neurosci. 1994;14:1130–1140. doi: 10.1523/JNEUROSCI.14-03-01130.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knowles RB, Kosik KS. Neurotrophin-3 signals redistribute RNA in neurons. Proc Natl Acad Sci USA. 1997;94:14804–14808. doi: 10.1073/pnas.94.26.14804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knowles RB, Sabry JH, Martone ME, Deerinck TJ, Ellisman MH, Bassell GJ, Kosik KS. Translocation of RNA granules in living neurons. J Neurosci. 1996;16:7812–7820. doi: 10.1523/JNEUROSCI.16-24-07812.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koenig E. Cycloheximide-sensitive [35S]methionine labeling of proteins in goldfish retinal ganglion cell axons in vitro. Brain Res. 1989;481:119–123. doi: 10.1016/0006-8993(89)90491-5. [DOI] [PubMed] [Google Scholar]
- 39.Koenig E. Evaluation of local synthesis of axonal proteins in the goldfish Mauthner cell axon and axons of dorsal and ventral roots of the rat in vitro. Mol Cell Neurosci. 1991;2:384–394. doi: 10.1016/1044-7431(91)90025-j. [DOI] [PubMed] [Google Scholar]
- 40.Koenig E, Martin R. Cortical plaque-like structures identify ribosome-containing domains in the Mauthner cell axon. J Neurosci. 1996;16:1400–1411. doi: 10.1523/JNEUROSCI.16-04-01400.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koenig E, Martin R, Titmus M, Sotelo-Silveira JR. Cryptic peripheral ribosomal domains distributed intermittently along mammalian myelinated axons. J Neurosci. 2000;20:8390–8400. doi: 10.1523/JNEUROSCI.20-22-08390.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laine RO, Laipis PJ, Shay NF, Kilberg MS. Identification of an amino acid-regulated mRNA from rat liver as the mammalian equivalent of bacterial ribosomal protein L22. J Biol Chem. 1991;266:16969–16972. [PubMed] [Google Scholar]
- 43.Lankford K, Waxman S, Kocsis J. Mechanisms of enhancement of neurite regeneration in vitro following a conditioning sciatic nerve lesion. J Comp Neurol. 1998;391:11–29. doi: 10.1002/(sici)1096-9861(19980202)391:1<11::aid-cne2>3.0.co;2-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lasek R, Gainer H, Przybylski R. Transfer of newly synthesized proteins from Schwann cells to the squid giant axon. Proc Natl Acad Sci USA. 1974;71:1118–1192. doi: 10.1073/pnas.71.4.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lerner E, Lerner M, Janeway C, Steitz J. Monoclonal antibodies to nucleic acid-containing cellular constituents: probes for molecular biology and autoimmune disease. Proc Natl Acad Sci USA. 1981;78:2737–2741. doi: 10.1073/pnas.78.5.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindsay R. Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J Neurosci. 1988;8:2394–2405. doi: 10.1523/JNEUROSCI.08-07-02394.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu S, Smith SE, Julian J, Rohde LH, Karin NJ, Carson DD. cDNA cloning and expression of HIP, a novel cell surface heparin sulfate/heparin-binding protein of human uterine epithelial cells and cell lines. J Biol Chem. 1996;271:11817–11823. doi: 10.1074/jbc.271.20.11817. [DOI] [PubMed] [Google Scholar]
- 48.Martin KC, Casadio A, Zhu H, E Y, Rose JC, Chen M, Bailey CH, Kandel ER. Synapse-specific, long-term facilitation of Aplysia sensory to motor synapses: a function for local protein synthesis in memory storage. Cell. 1997;91:927–938. doi: 10.1016/s0092-8674(00)80484-5. [DOI] [PubMed] [Google Scholar]
- 49.Martin KC, Barad M, Kandel ER. Local protein synthesis and its role in synapse-specific plasticity. Curr Opin Neurobiol. 2000;10:587–592. doi: 10.1016/s0959-4388(00)00128-8. [DOI] [PubMed] [Google Scholar]
- 50.Martin R, Fritz W, Guiditta A. Visualization of polysomes in the postsynaptic area of the squid giant synapse by electron spectroscopic imaging. J Neurocytol. 1989;18:11–18. doi: 10.1007/BF01188419. [DOI] [PubMed] [Google Scholar]
- 51.Matz M, Shagin D, Bogdanova E, Branova O, Lukyanov S, Diatchenko L, Chenchik A. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res. 1999;27:1558–1560. doi: 10.1093/nar/27.6.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McQuarrie IG, Grafstein B. Effect of condition lesion on optic nerve regeneration in goldfish. Brain Res. 1981;218:253–264. doi: 10.1016/0006-8993(81)90128-1. [DOI] [PubMed] [Google Scholar]
- 53.McQuarrie I, Grafstein B. Protein synthesis and axonal transport in goldfish retinal ganglion cells during regeneration accelerated by a conditioning lesion. Brain Res. 1982;251:25–37. doi: 10.1016/0006-8993(82)91270-7. [DOI] [PubMed] [Google Scholar]
- 54.Mohr E, Richter D. Diversity of messenger RNAs in the axonal compartment of peptidergic neurons in the rat. Eur J Neurosci. 1992;4:870–876. doi: 10.1111/j.1460-9568.1992.tb00197.x. [DOI] [PubMed] [Google Scholar]
- 55.Morfini G, DiTella M, Feiguin F, Carri N, Caceres A. Neurotrophin-3 enhances neurite outgrowth in cultured hippocampal pyramidal neurons. J Neurosci Res. 1994;39:219–232. doi: 10.1002/jnr.490390212. [DOI] [PubMed] [Google Scholar]
- 56.Notterpek L, Snipes GJ, Shooter EM. Temporal expression pattern of peripheral myelin protein 22 during in vivo and in vitro myelination. Glia. 1999;25:358–369. [PubMed] [Google Scholar]
- 57.Nudel U, Zakut R, Shani M, Neuman S, Levy Z, Yaffe D. The nucleotide sequence of the rat cytoplasmic beta-actin gene. Nucleic Acids Res. 1983;11:1759–1771. doi: 10.1093/nar/11.6.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Okabe S, Hirokawa N. Turnover of fluorescently labeled tubulin and actin in the axon. Nature. 1990;343:479–482. doi: 10.1038/343479a0. [DOI] [PubMed] [Google Scholar]
- 59.Okabe S, Hirokawa N. Actin dynamics in growth cones. J Neurosci. 1991;11:1918–1929. doi: 10.1523/JNEUROSCI.11-07-01918.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Olink-Coux M, Hollenbeck PJ. Localization and active transport of mRNA in axons of sympathetic neurons in culture. J Neurosci. 1996;16:1346–1358. doi: 10.1523/JNEUROSCI.16-04-01346.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pannese E, Ledda M. Ribosomes in myelinated axons of the rabbit spinal ganglion neurons. J Submicrosc Cytol Pathol. 1991;23:33–38. [PubMed] [Google Scholar]
- 62.Peters A, Palay S, Webster H. The fine structure of the nervous system, Ed 3. Oxford UP; New York: 1991. [Google Scholar]
- 63.Rotshenker S. Sprouting and synapse formation by motor axons separated from their cell bodies. Brain Res. 1981;223:141–145. doi: 10.1016/0006-8993(81)90813-1. [DOI] [PubMed] [Google Scholar]
- 64.Sambrook J, Fritsch E, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 65.Scheetz A, Nairn A, Constantine-Paton M. NMDA receptor-mediated control of protein synthesis at developing synapses. Nat Neurosci. 2000;3:211–216. doi: 10.1038/72915. [DOI] [PubMed] [Google Scholar]
- 66.Schreyer DJ, Skene JH. Fate of GAP-43 in ascending spinal axons of DRG neurons after peripheral nerve injury: delayed accumulation and correlation with regenerative potential. J Neurosci. 1991;11:3738–3751. doi: 10.1523/JNEUROSCI.11-12-03738.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schuman EM. mRNA trafficking and local protein synthesis at the synapse. Neuron. 1999;23:645–648. doi: 10.1016/s0896-6273(01)80023-4. [DOI] [PubMed] [Google Scholar]
- 68.Sjoberg J, Kanje M. The initial period of peripheral nerve regeneration and the importance of the local environment for the conditioning lesion effect. Brain Res. 1990;529:79–84. doi: 10.1016/0006-8993(90)90812-p. [DOI] [PubMed] [Google Scholar]
- 69.Smith DS, Skene P. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci. 1997;17:646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sotelo-Silveira J, Calliari A, Kun A, Benech J, Sanguinetti C, Chalar C, Sotelo J. Neurofilament mRNAs are present and translated in normal and severed sciatic nerve. J Neurosci Res. 2000;62:65–74. doi: 10.1002/1097-4547(20001001)62:1<65::AID-JNR7>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 71.Steward O. Targeting of mRNAs to subsynaptic microdomains in dendrites. Curr Opin Neurobiol. 1995;5:55–61. doi: 10.1016/0959-4388(95)80087-5. [DOI] [PubMed] [Google Scholar]
- 72.Steward O. mRNA localization in neurons: a multipurpose mechanism? Neuron. 1997;18:9–12. doi: 10.1016/s0896-6273(01)80041-6. [DOI] [PubMed] [Google Scholar]
- 73.Steward O, Halpain S. Lamina-specific synaptic activation causes domain-specific alterations in dendritic immunostaining for MAP2 and CAM kinase II. J Neurosci. 1999;19:7834–7845. doi: 10.1523/JNEUROSCI.19-18-07834.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tiedge H, Zhou A, Thorn N, Brosius J. Transport of BC1 RNA in hypothalamo-neurohypophyseal axons. J Neurosci. 1993;13:4214–4219. doi: 10.1523/JNEUROSCI.13-10-04214.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tobias G, Koenig E. Influence of nerve cell body and neurolemma cell on local axonal protein synthesis following neurotomy. Exp Neurol. 1975;49:235–245. doi: 10.1016/0014-4886(75)90207-1. [DOI] [PubMed] [Google Scholar]
- 76.Torre ER, Steward O. Demonstration of local protein synthesis within dendrites using a new cell culture system that permits the isolation of living axons and dendrites from their cell bodies. J Neurosci. 1992;12:762–772. doi: 10.1523/JNEUROSCI.12-03-00762.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Twiss JL, Shooter EM. Nerve growth factor promotes neurite regeneration in PC12 cells by translational control. J Neurochem. 1995;64:550–557. doi: 10.1046/j.1471-4159.1995.64020550.x. [DOI] [PubMed] [Google Scholar]
- 78.Twiss J, Smith D, Chang B, Shooter E. Translational control of ribosomal protein L4 is required for rapid neurite extension. Neurobiol Dis. 2000;7:416–428. doi: 10.1006/nbdi.2000.0293. [DOI] [PubMed] [Google Scholar]
- 79.Tytell M, Lasek R. Glial polypeptides transferred into the squid giant axon. Brain Res. 1984;324:223–232. doi: 10.1016/0006-8993(84)90032-5. [DOI] [PubMed] [Google Scholar]
- 80.van Minnen J. RNA in the axonal domain: a new dimension in neuronal functioning? Histochem J. 1994;26:377–391. doi: 10.1007/BF00160050. [DOI] [PubMed] [Google Scholar]
- 81.van Minnen J, Bergman J, Kesteren EV, Smit A, Geraerts W, Lukowiak K, Hasan S, Syed N. De novo protein synthesis in isolated axons of identified neurons. Neuroscience. 1997;80:1–7. doi: 10.1016/s0306-4522(97)00137-1. [DOI] [PubMed] [Google Scholar]
- 82.Weiler IJ, Greenough WT. Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proc Natl Acad Sci USA. 1993;90:7168–7171. doi: 10.1073/pnas.90.15.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weiner O, Zorn A, Krieg P, Bittner G. Medium weight neurofilament mRNA in goldfish Mauthner axoplasm. Neurosci Lett. 1996;213:83–86. doi: 10.1016/0304-3940(96)12860-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wensley CH, Stone DM, Baker H, Kauer JS, Margolis FL, Chikaraishi DM. Olfactory marker protein mRNA is found in axons of olfactory receptor neurons. J Neurosci. 1995;15:4827–4837. doi: 10.1523/JNEUROSCI.15-07-04827.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wool IG. Extraribosomal functions of ribosomal proteins. Trends Biochem Sci. 1996;21:164–165. [PubMed] [Google Scholar]
- 86.Yenofsky R, Cereghini S, Krowczynska A, Brawerman G. Regulation of mRNA utilization in mouse erythroleukemia cells induced to differentiate by exposure to dimethyl sulfoxide. Mol Cell Biol. 1983;3:1197–1203. doi: 10.1128/mcb.3.7.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zelená J. Ribosome-like particles in myelinated axons of the rat. Brain Res. 1970;24:359–363. doi: 10.1016/0006-8993(70)90120-4. [DOI] [PubMed] [Google Scholar]
- 88.Zelená J. Ribosomes in myelinated axons of dorsal root ganglia. Z Zellforsch Mikrosk Anat. 1972;124:217–229. doi: 10.1007/BF00335680. [DOI] [PubMed] [Google Scholar]
- 89.Zhang HL, Singer RH, Bassell GJ.Neurotrophin regulation of beta-actin mRNA and protein localization within growth cones. J Cell Biol 147199959–70.0 [DOI] [PMC free article] [PubMed] [Google Scholar]