Properties and Plasticity of Paired-Pulse Depression at a Central Synapse (original) (raw)
Abstract
Synaptic depression was studied at the axo-axonic connection between the goldfish Mauthner axon and identified cranial relay interneurons using simultaneous presynaptic and postsynaptic recordings and a paired-pulse stimulus paradigm. We used interstimulus intervals (ISIs) ranging from 10 msec to 1 sec and a cycle time of ∼5 sec. Depression (Δ EPSP/EPSP1) was maximal at the shorter intervals (80%) and decreased exponentially with a τ ∼ 400 msec (360 ± 107 msec, mean ± SD). We found the amplitudes of the first and second EPSP were not correlated, indicating the magnitude of depression does not depend on the amount of transmitter released by the conditioning stimulus. At short ISIs, the latency of EPSP2 was 23% longer than that of EPSP1 and recovered to control with τ ∼ 400 msec, whereas rise time and decay time were not altered significantly. The latency distribution, which is determined by the timing of the first quantum released each trial, was used to derive α(t), the rate of evoked exocytosis after an action potential. α(t) was biphasic, and both components were consistently delayed during depression. Presynaptic manipulations of putative intracellular regulatory pathways, such as Ca2+ and GTPγS injections, preferentially affected the amplitude of EPSP1 or EPSP2. These results are not consistent with simple depletion of the available pool of synaptic vesicles as the major mechanism underlying depression. They rather suggest that it is attributable to a modification or refractoriness of the release process and that there may be multiple pathways subserving evoked exocytosis.
Keywords: paired-pulse depression, depletion, Mauthner cell, exocytosis, synaptic latency, rate of evoked transmitter release, cranial relay interneuron, secretory machinery, metaplasticity
Synaptic strength is a dynamic property such that a postsynaptic potential (PSP) exhibits short- and long-term changes in amplitude after different patterns of use. Long-term changes are believed to underlie learning and memory formation (Bear and Malenka, 1994), whereas those that are shorter lasting and that occur on the timescale of seconds or less play an important role in the dynamic function of neural networks. In some systems, one action potential leads to a decrease in the response to a second test stimulus. Historically, this depression has been related to depletion of the readily releasable pool of synaptic vesicles at junctions with a high probability of release (Liley and North, 1953;Elmqvist and Quastel, 1965; Thies, 1965; Rosenmund and Stevens, 1996;Dobrunz and Stevens, 1997; Goda and Stevens, 1998) Alternatively, depression may not be simply the consequence of release per se but may instead reflect a use-dependent modification of the functional state of the molecular machinery responsible for evoked exocytosis (Betz, 1970).
The depletion hypothesis has several implications and predictions that can be tested experimentally. Specifically, (1) when the paired-pulse paradigm is repeatedly applied with a fixed interstimulus interval, the degree of depression from trial to trial should be proportional to the amount of transmitter released by the preceding stimulus, (2) there should be no associated changes in the kinetics of release, (3) manipulations that selectively increase the probability of release should enhance the first response of a pair and reduce the second one, because the conditioning stimulus will evoke more release and a greater amount of depletion.
As part of the characterization of depression at the Mauthner (M-) axon output connections to cranial relay interneurons (CRNs) in the goldfish brainstem, we have tested these predictions. This connection mediates the cranial portion of the startle reflex (Hackett and Faber, 1983), and its general properties have been extensively characterized (Hackett and Faber, 1983; Hackett et al., 1989). Analogous to that studied in the hatchetfish (Auerbach and Bennett, 1969; Highstein and Bennett, 1975), it has a high safety factor associated with a high initial probability of release, and it exhibits a robust depression, as revealed by paired-pulse or repetitive stimulation (Hackett et al., 1989). Because of its accessibility for presynaptic manipulation of the release properties, along with the possibility to record simultaneously from both presynaptic and postsynaptic elements, it is an ideal preparation for investigating synaptic depression and its underlying mechanisms in vivo. Our results, including an increase in synaptic delay during depression, indicate the predictions of the depletion hypothesis are not satisfied and suggest instead that a change of the functional state of the exocytotic machinery is likely to underlie depression of this central synaptic contact. We also demonstrate that putative presynaptic intracellular regulatory pathways modify depression in a manner inconsistent with the depletion model, implying that the magnitude of depression itself can be modulated by metaplastic phenomena.
MATERIALS AND METHODS
Electrophysiology. For this study, 60 goldfish (Carassius auratus), 8- to 15-cm-long were perfused through the mouth with cold tap water and immobilized withd-tubocurarine injected intramuscularly (1–3 mg/gm, body weight). Surgical techniques were similar to those described previously (Faber and Korn, 1978). The cranium was opened dorsally to allow for simultaneous recordings near the midline of the brainstem between the vagal lobes and caudal to the facial lobe. The dura was removed above the vagal lobes, and the lobes were moved laterally, to expose the underlying surface of the brainstem and enable visual control of the microelectrode placement in the presynaptic Mauthner axon. At this level of the brainstem, this unbranched myelinated axon (60–100 μm diameter) is at most 100 μm below the dorsal surface of the brainstem and ∼150 μm from the midline (Fig.1A). Postsynaptic recordings were obtained from the axon of the CRN, which courses close to the M-axon in this region and receives axo-axonic synapses from it (Hackett and Faber 1983; Hackett et al., 1989). A stimulating electrode on the exposed caudal spinal column was used for antidromic activation of the M-axon.
Fig. 1.

Characteristics of the excitatory axo-axonal connection between the Mauthner axon and its postsynaptic target, the cranial relay neuron (CRN). _A,_Diagram showing the connections studied with paired intra-axonal recordings (V) and current injections (I) close to the site of the contact.P, Pressure injection of neurobiotin. B, Image of a 50 μm horizontal section of two neurobiotin-filled M-axons (M) and a CRN axon on the right side of the brainstem. Regions of two presumptive contact sites between the right M-axon and the CRN are enclosed in boxes. Scale bar, 250 μm. a, b, Higher magnification images of the contacts shown in top and _bottom_boxes of B, respectively. M-axon, _Red;_CRN, blue. Scale bar, 100 μm. C, Five superimposed records showing fluctuations in the amplitudes of EPSPs (top, Post) evoked by single presynaptic action potentials in the M-axon (bottom, Pre). Action potential of M-axon occasionally evoked a full-sized spike in the CRN (peak truncated, actual spike height = 80 mV).
The presynaptic and postsynaptic electrodes were filled with 5m KAc (resistance, ∼8 MΩ) and 2.5m KCl (resistance, ∼10 MΩ), respectively. In some experiments presynaptic calcium concentration was increased by pressure-injecting a 2 mm CaCl2solution into the M-axon, further identified by the characteristic waveform of its antidromic spike (Funch et al., 1984). The CRN was identified by a fast EPSP evoked by a presynaptic action potential, typically driven by a transmembrane current pulse of ∼15–20 nA. All recordings were in the current-clamp mode, and crosstalk between the microelectrodes was minimized electronically with an Axoprobe two electrode amplifier (Axon Instruments, Foster City, CA). Data were recorded on line, using a Macintosh Quadra 950 computer and acquisition software developed in the laboratory (sampling rate of 10–50 μsec) and on tape (PCM data recorder; Vetter Instruments), and analysis was with the same software and with procedures in Igor.
Data analysis. The analysis program measured response amplitudes, their decay time constants (τ), time-to-peaks, and their onsets or latencies. The decay constant of individual EPSPs was measured assuming a monoexponential relationship, confirmed by curve fitting averaged responses. EPSP time-to-peak was defined as the rise time from 10 to 90% of peak value. Latency was defined as the time from the peak of the presynaptic action potential to the onset of the rising phase of the EPSP. The latter was measured with a computer-based algorithm that first determines the EPSP peak and then works backwards from the time at which the response is half-maximal toward that at which its first derivative approaches zero. Typically, the onset was 20 μsec later than that zero slope point. In some cases, latency could not be measured reliably because the EPSP onset was obscured by a coupling artifact caused by crosstalk from the presynaptic current pulse. A linear regression analysis was used to test for associations between variables. The coefficient of determination (_r_2), which indicated the proportion of the variance of the dependent variable attributable to covariation with the independent variable, was used to assess the quality of the regression model (Igor Wavemetrics).
Morphology. In some experiments electrodes were filled with a 4% solution of neurobiotin (_N_-(2-aminoethyl) biotinamide hydrocloride; Vector Laboratories, Burlingame, CA) in 2.5m KCl buffered with 10 mmHEPES, pH 7.2. Neurobiotin was injected iontophoretically in either or both axons, using +70 nA current pulses 400 msec in duration and repeated at 1 Hz. After those experiments, the animal was perfused intracardially with 4% paraformaldehyde in PBS, pH 7.4. The brain was post-fixed, sectioned at 50 μm sections horizontally (Vibratome), and reacted with Vectastain ABC reagent (Vector Laboratories) for 2 hr, rinsed, and then reacted with diaminobenzidine (DAB) to visualize the injected cells. Sections were mounted on gelatin-coated slides and coverslipped.
RESULTS
Paired-pulse depression at the Mauthner–CRN connection
Every 100–200 μm the M-axon issues a short (60 μm) collateral that terminates most often as a single ending (Fig.1B) (Celio et al., 1979; Funch et al., 1984) having multiple active zones (Model et al., 1975; Hackett et al., 1989). In the caudal brainstem, some of these outputs have connections with the axons of CRNs. The only sources of input to CRN axons are the two M-axons, an advantage for these studies for the analysis of spontaneous EPSPs. The junction is most likely cholinergic because we and others (Day et al., 1983) have observed that in goldfish and hatchetfish, superfusion with curare blocks the evoked and spontaneous EPSPs. Note that this preparation has several advantages for studying paired-pulse depression (PPD), including a high safety factor, which is presumably attributable in part to high probability release sites and underlies a pronounced depression with no obvious complications from superimposed facilitation. Finally the connection is axo-axonic, the presynaptic and postsynaptic recording sites can be within a few hundred micrometers of the contact zone, and both axons have large space constants (>2 mm; Auerbach and Bennett, 1969;Funch et al., 1984). Thus, postsynaptic filtering of the EPSPs is minimal (Faber et al., 1995, 1998).
At stimulating frequencies < 0.2 Hz, the evoked EPSP is sufficiently large that it triggers an action potential. For our experiments, a stimulus frequency (cycle time) of 0.5–5 Hz was used to reduce the underlying EPSP to a subthreshold level. That is, we used frequencies that produced some steady-state depression. At those frequencies there generally was trial to trial variability in the EPSP amplitude. These features are illustrated in Figure 1C, where examples of a spike-evoked EPSP are shown. In this case the M-axon was stimulated at 0.3 Hz, generally producing a subthreshold EPSP whose amplitude ranged from 5 to 10 mV, and in one trial the EPSP triggered a postsynaptic action potential. The size and duration of the presynaptic spike were constant during this series, indicating that, as expected, the EPSP fluctuations reflect variability in the release process per se. The illustrated EPSP had a 0.63 msec decay time constant and a latency of 0.27 msec, values typical for individual responses in all experiments.
Activity-dependent depression was quantified using a paired-pulse paradigm, with the first conditioning response being referred to as EPSP1, and the second or test response as EPSP2. These high output synapses depress rapidly, as revealed by the paired-pulse responses at an interval of 20 msec in Figure2A. In one group of connections (n = 7) the interstimulus interval (ISI) was progressively increased from 10 msec to 2 sec. The greatest level of depression was observed at intervals of <100 msec (Fig.2B), and for the illustrated experiment, recovery was best fit by a single exponential with a time constant (τdep) of 400 msec. Overall the mean τdep was 360 ± 107 msec, with a range from 200 to 450 msec (n = 6). Typically, recovery was slow with little change in the degree of depression in the first 30–50 msec, as shown in Figure 2B. To achieve a large number of paired responses per interval without concerns over interinterval differences, the majority of experiments described here used a fixed ISI, typically 150 msec.
Fig. 2.

Kinetic properties of paired-pulse depression.A, Averaged recordings (n = 10) of the EPSPs evoked in a CRN (top) by two successive M-axon spikes (bottom) separated by an interval of 20 msec, when depression is maximal. B, Plot of the decay of paired-pulse depression (ordinate) as a function of interspike interval (abscissa) in a different experiment. Each data point is an average from ≥50 trials, and in this experiment the stimulus paradigm was repeated at 2 Hz. Solid line, Best fit single exponential; τdep = 400 msec;_r_2 = 0.88.
The amplitude of EPSP2 is independent of the amplitude of EPSP1
The first test of the applicability of depletion model to depression at the M - axon to CRN connection was to ask if the amplitude of EPSP2 depended on that of EPSP1. As stated above, the model predicts that the magnitude of depression should vary from trial to trial, changing as a function of the amount of transmitter released by the conditioning stimulus. That is, on average the size of EPSP2 should decrease as that of EPSP1 increases. For these experiments we used a fixed ISI of either 40 or 150 msec (n = 23), and depression typically averaged ∼50%. The results in Figure3 are characteristic of the findings that, however, there was no relationship between the amplitudes of the two responses. The sample records were selected to demonstrate the same-sized depressed response could be observed for a threefold variation in the amplitude of the conditioning EPSP (2.86–8.57 mV). The scatterplot, from 1226 consecutive paired stimuli, demonstrates this independence and that there was sufficient variability to both responses to permit a fair test of the hypothesis (e.g., coefficient of variation of EPSP1, CV1 = 22%, CV2 = 32%). In addition, the slope of the regression line is 0.03 with a_r_2 value of 0.00, indicating the relationship is not different from one with a zero slope. Thus, the average amplitude of EPSP2 is the same when EPSP1 is large or small. In 23 experiments, the slope ranged from −0.08 to +0.28, and the _r_2 values ranged from 0.00 to 0.29. The relationship between the two EPSP amplitudes was deemed significant in only 8 of the 23 experiments (ANOVA, p< 0.01). However, even in those eight cases, the_r_2 values were low, suggesting other factors were more significant, and the slopes were positive, that is, the apparent relationship between the two variables was the opposite of that expected with the depletion model. Generally there was more variability in EPSP2 compared to EPSP1. The coefficient of variability (CV) calculated for EPSP1 ranged from 16–41%, compared to 25–50% for EPSP2. This is consistent with a lower quantal content for EPSP2 (Lin and Faber 1988). Overall, these findings indicate the major prediction of the depletion model is not met at the M-axon to CRN connection.
Fig. 3.

The magnitude of paired-pulse depression is independent of the amount of transmitter released by a conditioning stimulus. Left, Selected paired responses (top traces) where the magnitude of EPSP1 varies from 2.86 to 8.57 mV, but the size of EPSP2 remains relatively constant. Bottom trace, Typical presynaptic action potentials evoked in the M-axon by a transmembrane depolarization pulse. The interstimulus interval is indicated by the 150 msec bar. _Right,_Scatter plot of the amplitude of EPSP2 (ordinate) versus the size of the response to the corresponding conditioning stimulus (EPSP1). Note that EPSP2 varies independently of trial to trial fluctuations in the size of EPSP1. Line represents relationship generated by simple regression analysis, with slope of 0.03 and_r_2 value of 0.00. Same experiment as in left.
Change in kinetics of release after depression
The depletion model does not predict that depression would be associated with changes in the kinetics of release, for example, in its latency. However, in all cases with a fixed interstimulus interval, in which the EPSP onset was not obscured by a coupling artifact and could be measured reliably (n = 7), the latency of EPSP2 was greater than that of EPSP1. This effect is illustrated in Figure4A for two averaged sweeps, which are overlaid temporally such that the timing of the presynaptic spikes is the same, and the amplitude of EPSP2 is normalized to that of EPSP1. Scaling the response to the same amplitude is a method that avoids overestimating the latency of small-amplitude events. The latency of a compound EPSP is a measure of the earliest timing of exocytosis from a population of release sites (Stevens, 1968) because the different sites are not precisely synchronized. Therefore, histograms of measured latencies of EPSP1 or EPSP2 actually are the distribution functions for the first release events, and an increase in latency during paired-pulse depression could be attributable to a lower probability of occurrence of short-latency events (i.e., altered sampling from the same population of release times) or a shift to a new distribution. In the experiment of Figure 4B, the mean latency of EPSP2 is 29% longer than that of EPSP1, and the latency distribution shifts to the right with the longer latency values generally not being present in the distribution of EPSP1 latencies. This shift to the right in the latency histogram suggests there is a population of delayed release events during depression. Measurements of individual traces showed the average latency of EPSP2 was significantly longer than that of EPSP1 (lat1 = 0.35 ± 0.04 msec; lat2 = 0.43 ± 0.05 msec; p < 0.01;n = 6; Table 1), with the mean for EPSP2 being 23% longer than that of EPSP1. In addition, in six of the seven experiments in which ISI was varied systematically, latency 2 was significantly greater than latency 1, with the overall mean difference being 22% (n = 7; lat1 = 0.32 ± 0.03 msec; lat 2 = 0.39 ± 0.07 msec;p < 0.01).
Fig. 4.

Paired-pulse depression is associated with an increase in the latency of transmitter release. A, Top, Superimposed averaged records (n = 8) of EPSP1 (dashed line) and EPSP2 (solid trace), with the latter scaled in amplitude to match the size of EPSP1. Between solid and_dashed vertical lines,_ Latency of average EPSP1. Between two solid vertical lines, Latency of average EPSP2. The interval between the two stimuli was 150 msec. Bottom traces, Superimposed averaged M-axon action potentials that evoked the two responses. B, Histogram of the latency population of EPSP1 and EPSP2 evoked by stimulation of the M-axon at 1.4 Hz and with a 150 msec ISI. n = 564 pairs of responses from a different experiment than in A. Note shift to the right of latency values during depression (EPSP2, solid outline) indicating longer latency values, with a mean of 0.49 ± 0.09 msec. compared to the latency of EPSP1 (dashed outline), 0.38 ± 0.05 msec.
Table 1.
Summary of latency analysis of EPSP1 and EPSP2
| Experiment | EPSP 1 | EPSP 2 | Difference | Depression ratio (%) | |||
|---|---|---|---|---|---|---|---|
| Mean latency (msec) | Time of α Max (msec) | Mean latency (msec) | Time of α Max (msec) | Mean latency (msec) | Time of α Max (msec) | ||
| NB717 | 0.32 | 0.41 | 0.41 | 0.54 | 0.09 | 0.14 | 45 |
| 118 | 0.34 | 0.48 | 0.43 | 0.69 | 0.09 | 0.21 | 47 |
| 913 | 0.41 | 0.54 | 0.48 | 0.75 | 0.07 | 0.21 | 40 |
| MAP5 | 0.36 | 0.42 | 0.45 | 0.54 | 0.09 | 0.12 | 58 |
| Q127 | 0.29 | 0.39 | 0.35 | 0.51 | 0.06 | 0.12 | 57 |
| Q212 | 0.37 | 0.48 | 0.46 | 0.72 | 0.09 | 0.24 | 56 |
| AVE. (± SD) | 0.35 (0.04) | 0.45 (0.06) | 0.43 (0.05)* | 0.63 (0.11) | 0.08 (0.01) | 0.17 (0.05) | 51 (8) |
The magnitude of the increase in EPSP latency during PPD was directly related to the interstimulus interval, as illustrated by the averaged recordings in Figure5A for intervals from 10 to 350 msec. In this experiment, latency 2 was 0.52 msec at the shortest interval and decreased to 0.34 msec at the longest interval shown (the latencies of the corresponding conditioning responses were 0.35 and 0.29 msec, respectively). When the data from seven experiments were grouped on the basis of long (>100 msec) and short (10–100 msec) ISI, there was a clear decrease in the latency of EPSP2 at longer intervals, although the latency was still longer than that of EPSP1, as illustrated in Table 2. Because latency measurements from individual traces are subject to uncertainty associated with instrumental noise, we also determined the latency of the averages of all the responses at a given ISI, thereby reducing the signal-to-noise ratio, and the results were similar to those obtained with measurements from individual traces (data not shown). Finally, support for the conclusion that the increased latency of EPSP2 relative to that of EPSP1 is a function of the interval length was obtained by plotting the difference between the respective averages, or the latency differential, versus ISI. The results from the M-axon–CRN connection of Figure 5A are shown in 5_B1_, and they are characteristic of the finding that the latency differential decreased as an exponential (τLat = 125 msec) function of ISI. The data were not fit in this manner in other experiments, although it was clear that the latency differential decreased as the ISI increased.
Fig. 5.
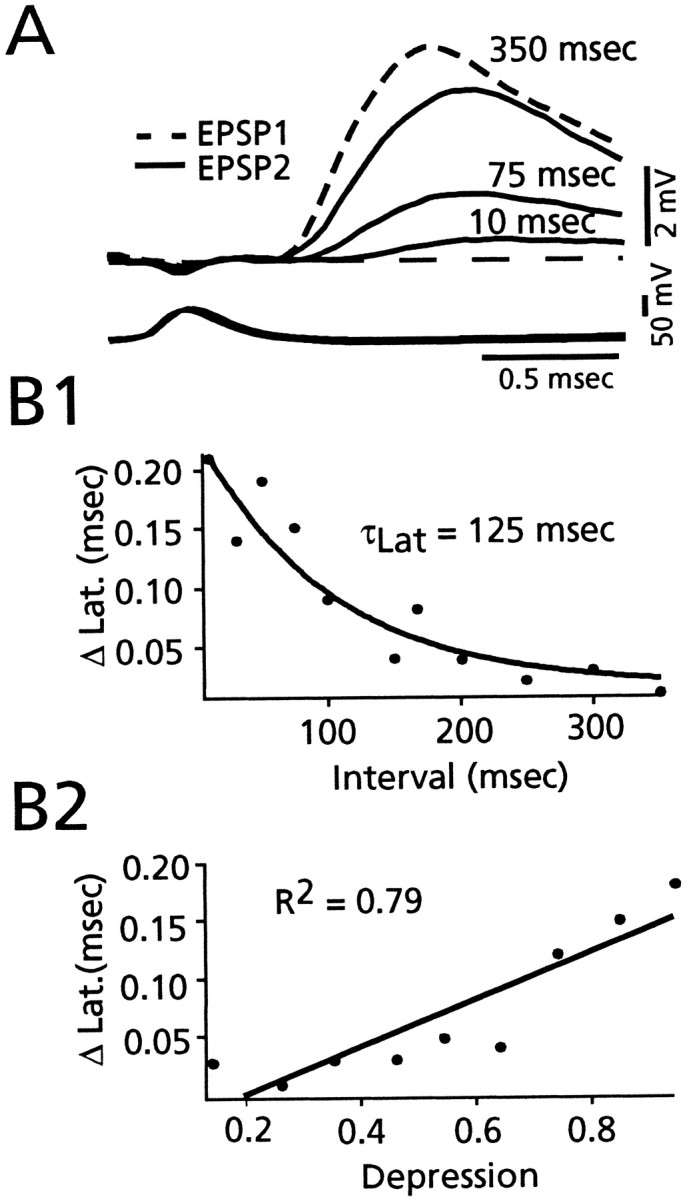
Latency differential between EPSP1 and EPSP2 is related to the interspike interval and to the magnitude of depression.A, Top, Superimposed averaged records (n > 20) of EPSP2 at the indicated intervals (10, 75, and 350 msec) and of EPSP1 at 350 msec (dashed response). Bottom traces, Superimposed averaged M-axon action potentials that evoked the CRN responses. Decay of EPSPs truncated. Dashed line, Baseline. _B1,_Plot of relationship between the ISI (abscissa) and the averaged latency difference (ordinate) between EPSP2 and EPSP1, for a different experiment. Solid line is exponential fit to decay phase, τLat = 125 msec. Data points represent averages of latency measurements (n > 20) for each interval. B2, Plot of relationship between the magnitude of depression (abscissa) and the averaged latency difference (ordinate). Linear regression (solid line) has a slope of 0.20, with r_2 = 0.79 (p < 0.01). Each data point represents an average of latency measurements (_n_ > 20) for averaged depression in 0.1 bins. Same experiment as_B1.
Table 2.
Summary of paired-pulse depression at a variable interval
| Experiment | Freq (Hz) | ISI (n) | EPSP1 mV | EPSP2 mV | τ1 msec | τ2 msec | Lat 1 msec | Lat 2 msec |
|---|---|---|---|---|---|---|---|---|
| CRN 16 | 0.5 | short (134) | 4.04 (0.65) | 0.94 (0.28) | 1.58* (0.33) | 1.25 (0.41) | 0.39 (0.04) | 0.54* (0.06) |
| long (151) | 4.19 (1.1) | 1.58 (0.50) | 1.83* (0.48) | 1.52 (0.44) | 0.31 (0.09) | 0.40* (0.06) | ||
| CRN 17 | 0.3 | short (83) | 8.02 (1.1) | 1.44 (0.58) | 1.58* (0.17) | 1.41 (0.42) | 0.33 (0.07) | 0.47* (0.09) |
| long (122) | 6.53 (1.1) | 3.39 (0.73) | 1.81* (0.19) | 1.63 (0.32) | 0.33 (0.04) | 0.36* (0.04) | ||
| CRN 18 | 0.5 | short (53) | 8.29 (0.92) | 1.55 (0.70) | 1.05* (0.10) | 0.94 (0.30) | 0.30 (0.07) | 0.44* (0.11) |
| long (154) | 6.13 (1.1) | 3.62 (0.69) | 1.06* (0.10) | 0.89 (0.13) | 0.29 (0.03) | 0.32* (0.03) | ||
| CRN 19 | 0.5 | short (70) | 4.86 (0.95) | 1.24 (0.34) | 1.83* (0.34) | 1.73 (0.48) | 0.35 (0.06) | 0.43* (0.11) |
| long (150) | 3.86 (0.60) | 2.85 (0.27) | 1.69 (0.15) | 1.72 (0.21) | 0.31 (0.02) | 0.33* (0.03) | ||
| CRN 20 | 0.3 | short (66) | 7.26 (1.1) | 1.73 (0.48) | 0.94 (0.15) | 1.06* (0.33) | 0.27 (0.03) | 0.28 (0.03) |
| long (223) | 5.50 (1.3) | 2.99 (0.64) | 1.01 (0.13) | 1.14* (0.22) | 0.26 (0.04) | 0.27 (0.03) | ||
| CRN 21 | 0.5 | short (123) | 4.62 (1.4) | 1.46 (0.51) | 1.14* (0.34) | 0.67 (0.33) | 0.33 (0.04) | 0.46* (0.09) |
| long (75) | 4.96 (0.90) | 2.13 (0.55) | 1.01* (0.30) | 0.72 (0.25) | 0.30 (0.03) | 0.34* (0.04) | ||
| CRN 22 | 1 | short (399) | 5.75 (1.3) | 1.64 (0.48) | 1.10 (0.16) | 1.20* (0.36) | 0.31 (0.04) | 0.42* (0.09) |
| long (427) | 3.50 (1.1) | 1.91 (0.59) | 1.12 (0.22) | 1.22* (0.29) | 0.33 (0.05) | 0.38* (0.07) | ||
| Average | short | 6.12 (1.7) | 1.43 (0.27) | 1.32 (0.34) | 1.18 (0.34) | 0.33 (0.04) | 0.432-160 (0.09) | |
| long | 4.95 (1.2) | 2.64 (0.78) | 1.36 (0.39) | 1.26 (0.38) | 0.30 (0.02) | 0.342-160 (0.08) |
In addition to its inverse relationship to interval length, the latency differential was proportional to the magnitude of depression. When the average latency differential was plotted against the average magnitude of depression, the two grew in parallel (Fig.5B2). Specifically, these two parameters were linearly related (_r_2 value = 0.79;p < 0.01), and there was almost no difference between the latencies of conditioning and test EPSPs at the lowest level of depression (0.2 in Fig. 5B2). This relationship is not predicted by the depletion model and rather suggests that there is a modification of the release machinery after the conditioning response.
In contrast to the latency differential of EPSPs during PPD, two other kinetic parameters, the time constant of decay of EPSPs (τ) and time-to-peak (TTP), were not related to the magnitude of depression or the ISI. In the CRN, EPSPs are fast and not shaped by the membrane time constant (<50 μsec; Auerbach and Bennett, 1969; hatchetfish). These EPSPs decay with one or more exponentials, the dominant time constant being in the range of 1.32 to 1.41 msec for the first response and 1.18 to 1.60 msec for the second response. In neither the variable ISI groups nor the fixed ISI group was the overall mean of the decay time constant of EPSP1 found to be significantly different than that of EPSP2, although individual experiments did show significant differences in the mean values of the decay constants. Similarly, the overall mean time-to-peak of EPSP2 was not significantly different than that of EPSP1 (TTP1 = 0.74 ± 0.14 msec; TTP2 = 0.73 ± 0.16 msec; n = 7 fixed ISI group).
Estimating the release rate after an action potential
Stevens (1968) derived the rate of evoked exocytosis after an action potential, α(t), from the distribution of first latencies, dS/dt, at a connection that has a quantal content >1.0. Specifically α(t) = −(dP(0,t)/dt)/P(0,t), where P(0,t) is the probability that no quanta have been released at time t. Then_P_(0,t) = 1 −S(t), where S(t) is the cumulative latency distribution. Therefore α(t) = (dS/dt)/(1 − S(t)). We have calculated α(t) from the distribution of first latencies, and the corresponding cumulative histogram, in six experiments, to determine the waveform of the release rate. For this purpose, data were redigitized, using sampling rates of 5–10 μsec. The results confirm the shift in latency, and they reveal a biphasic release rate, as illustrated in Figure 6,A and B, for two different experiments. In both examples, the control waveform of α(t) starts at ∼0.21 msec after the peak of the presynaptic action potential, appears to rise slowly to an initial peak at ∼0.3–0.4 msec, and rises steeply to a second peak in another 100–200 μsec before falling sharply to zero. This waveform, which can be characterized by an early “foot” followed by a sharp peak, is typical of all experiments analyzed with this technique. Furthermore, the effect of depression on α(t) is striking, because both components are delayed (Fig. 6A,B). Table 1 compares the shift in mean EPSP latency in these six experiments during depression with the increase in the timing of the peak of α(t); whereas mean latency increased by 80 ± 10 μsec, the shift in the time at which α(t) was maximal was twice that, namely 170 ± 50 μsec. These results confirm the observed shift in latency and indicate the delay in the release rate is even greater then suggested from the latency distribution itself.
Fig. 6.

The derived release rate is delayed during depression. A, B, Plots, from two experiments, of α(t) for both EPSP1 (solid line) and EPSP2 (dashed line), as calculated from the probability density for synaptic delays and the corresponding cumulative distribution. Open and filled arrows denote the early phase, or foot, and the peak of the second phase of α(t), respectively. Note that both phases are delayed during depression (EPSP2 vs EPSP1) and that the temporal separation of the two is increased. In A, bin size = 30 μsec; n = 1357. In_B_ bin size = 40 μsec; n = 400.
Plasticity of paired-pulse depression: further evidence for the independence of EPSP1 and EPSP2
If synaptic depression is primarily attributable to depletion, manipulations that alter the amount of transmitter released by a conditioning stimulus would have predictable effects on the size of EPSP2, and hence, on the depression. On the other hand, if the dominant factor were a functional change in the exocytotic machinery per se, it should be possible to modulate depression by differentially regulating the magnitudes of the successive responses in a pair. To test these two alternatives, we examined the effects of two common messengers or intermediates in signaling cascades, namely intracellular Ca2+, which usually signals activity-dependent changes (Nicoll et al., 1994), and heterotrimeric G-proteins, which often mediate the presynaptic effects of substances that modulate transmitter release (Hille, 1992). First, in seven experiments in which a fixed ISI (150 msec) was used, 2 mmCaCl2 was pressure-injected into the M axon. Elevating internal Ca2+ produced a marked increase in the amplitude of EPSP1, as expected. If this enhancement were attributable to a simple increase in release probability without modifying the size of the available pool, the depletion model would predict a corresponding reduction in the magnitude of EPSP2, with greater depression. However, both EPSPs increased in amplitude. This effect is illustrated in Figure7A for averaged recordings of EPSP1 (solid line) and EPSP2 (dashed line) before and after injection, and in both panels the conditioning and test traces are overlaid temporally so that the timing of the presynaptic spikes is the same. Note that the two responses did not increase by the same relative amounts. Rather, ∼15 min after calcium ion injection, the size of the illustrated EPSP2 had increased by 80%, compared to a 50% increase in EPSP1. Figure 7B1 illustrates the time course of the Ca2+ effect in this experiment and that the percentage of increase in EPSP2 was larger than that in EPSP1, a finding in all of the experiments (Table3). When the average size of EPSP2 at the conclusion of the recording session for each experiment (last 20 traces) was compared to the average control (20 traces before injection) there was a 67 ± 36% increase, significantly greater than the 42 ± 27% increase in the magnitude of EPSP1 (p < 0.05; n = 7; Table 3). As illustrated in Figure 7B1, the enhancement of amplitude typically began immediately after the injection, grew steadily during the next 10–15 min, and lasted for the remainder of the recording period (the longest duration being 30 min after injection). Such a large increase was never seen in control conditions, although great variability was observed in both responses. For comparison, the panel in Figure 7C shows the temporal history of EPSP1 and EPSP2 amplitudes in a control experiment with no Ca2+ in the presynaptic electrode. In this case, the response amplitudes measured after 30 min of recording with no injection were slightly less than at the beginning of the experiment. Overall, in the control experiments, EPSP1 and EPSP2, at comparable recording times, averaged 99.2 ± 12% and 93.2 ± 8% of the initial values, respectively (n = 5). In contrast to the change in amplitude after calcium injection, there was no change in latency of either response when compared to their control values.
Fig. 7.

Evidence that presynaptic Ca2+injection preferentially enhances the amplitude of EPSP2. A, Left and right, Superimposed averaged paired responses (n = 10), presynaptic spikes (below), and first (solid line) and second (dashed line) EPSPs (top) obtained before and after 2 mm Ca2+ injection, respectively. M-axon was stimulated at 1 Hz with an interval of 150 msec. B1, Amplitudes of EPSP1 and EPSP2 plotted as percentages of their control values versus time. Dashed line indicates 100% of control value, which was determined by averaging the traces during the preinjection period (∼2 min). Injection resulted in a greater percentage of increase in EPSP2 (open squares) than in EPSP1 (filled squares); 20 traces per square. B2, Plot, from experiment in B1, of depression versus time, before and after presynaptic injection of calcium. ISI was 150 msec. Each point is an average from 20 consecutive traces. Note that Ca2+ injection results in a persistent decrease in the magnitude of depression. C, Control. Similar plot as_B1_, however this is a different experiment with no Ca2+ in the electrode. M-axon was stimulated at 2 Hz with an interval of 150 msec. No increases are found in either amplitude, indeed both percentage amplitudes decrease.
Table 3.
Summary of changes after presynaptic Ca2+injection
| Experiment (Post-inj.n) | Increase (%) | Depression rate | Total time of postrecording | ||
|---|---|---|---|---|---|
| EPSP1 | EPSP2 | Control | Post-Inj. | ||
| RC1 (80) | 30 | 34 | 49 ± 20 | 50 ± 10 | 10 min |
| RC2 (170) | 29 | 33 | 62 ± 10 | 61 ± 10 | 10 min |
| RC4 (1000) | 20 | 85 | 64 ± 10 | 44 ± 20 | 30 min |
| RC5 (1500) | 46 | 73 | 47 ± 20 | 39 ± 10 | 30 min |
| RC7 (1600) | 86 | 131 | 66 ± 10 | 57 ± 10 | 30 min |
| RC10 (1200) | 13 | 36 | 44 ± 20 | 35 ± 20 | 10 min |
| RC11 (130) | 72 | 77 | 67 ± 10 | 64 ± 19 | 7 min |
| Average (± SD) | 42 ± 27 | 67 ± 363-150 | 57 ± 10 | 50 ± 113-150 |
Otherwise stated, injection of Ca2+ caused a decreased depression at the M-axon–CRN connection. An example of this is shown in Figure 7B2, where the magnitude of depression decreased by 9%, from a control level of 0.67 to 0.58 after injection. The figure also illustrates that depression did not return to control level (dashed line) at any time after the Ca2+ injection. The magnitude of depression decreased by 8–20% in four of the seven connections studied and was minimally affected in the remaining three (Table 3). To summarize, the results of the seven experiments listed in Table 3indicate that increasing the initial probability of release with Ca2+ injection does not result in an increased level of depression, as predicted by the depletion model. Rather, depression is actually decreased, a finding that is inconsistent with a simple increase in the available pool size and rather supports the notion that depression itself exhibits plasticity.
We also explored the possible role of heterotrimeric G-proteins in mediating a modulation of neurotransmitter release at the M-axon to CRN synapses. For this purpose, we injected GTPγS, a nonhydrolizable analog of GTP that irreversibly activates G-proteins (Bourne et al., 1990; Richmond and Haydon, 1993) into the M-axon. In a limited number of cases, GTPγS produced a distinctive effect characterized by a remarkable and selective enhancement of the first EPSP (Fig.8A,B1), along with a marked increase in the frequency of spontaneous EPSPs (Fig.8B2). Note that in the example of Figure8B1, spontaneous or asynchronous release followed the evoked response, a phenomenon not observed in control experiments. This combined effect was present in 3 of 10 experiments. For these three experiments, the injection of GTPγS resulted in an 83 ± 23% growth in the amplitude of EPSP1 versus only 17 ± 14% in that of EPSP2 (p < 0.05), and the frequency of spontaneous EPSPs increased twofold. However, kinetics of the first and second EPSPs were not altered after injection. The low efficacy of GTPγS injections is probably related to the distance between the injection site and the location of the terminals as well as difficulties in maintaining the activity of this compound during long recording sessions; however, when the injections were effective, the increase in the size of EPSP1 and the frequency of spontaneous EPSPs always occurred together. Interestingly, GTPγS modulation of PPD was different to that observed for Ca2+injections. Depression was enhanced, as in Figure 8A, but the increased size of EPSP1 was not paralleled by a reduction of EPSP2, which instead remained quite constant. Thus, the two presynaptic manipulations of intracellular regulatory mechanisms produced modifications of PPD inconsistent with those predicted by the depletion hypothesis.
Fig. 8.

Presynaptic injection of GTPγS selectively increases EPSP1. A, Left and _right,_Averaged paired responses (ISI = 20 msec; n = 20) obtained in control and 20 min after GTPγS injection.B1, B2, GTPγS injection also modifies spontaneous, or miniature EPSPs. B1, In another experiment EPSP1 increases in magnitude after injection, and the response is followed by a train of asynchronous EPSPs with slower than normal decay times (EPSP2 not shown; traces not averaged).B2, Recordings of synaptic noise obtained in absence of stimulation reveal an increased frequency of spontaneous EPSPs (*) after injection, with a prolonged decay time.
DISCUSSION
Our results address the questions of whether vesicle availability is the main determinant of the release probability and whether PPD can be modulated. One argument against depletion as the major determinant of depression is that the size of the test response (EPSP2) was independent of the magnitude of EPSP1, and hence of the amount of transmitter released by the first stimulus. This conclusion is based on the assumption that the test for a correlation is sensitive enough to detect the influence of depletion, which can be justified by observations that this connection has ∼25 release sites and a high initial probability of release (Hackett et al., 1989; Faber et al., 1998), such that on average >50% of the sites would be depleted by the test stimulus. Thus, it seems likely that the presynaptic stimulus triggers an additional process or processes that contribute to depression. The increased synaptic latency during depression is consistent with this concept, as is the plasticity of PPD. Similar findings have been found at inhibitory (Korn et al., 1984) and excitatory (Mori et al., 1994; Bellingham and Walmsley, 1999; Wu and Borst, 1999) synapses. In contrast, depression can be attributed to depletion at other junctions (Mori et al., 1994; Debanne et al., 1996;Thompson et al., 1998). Thus, different systems may use different mechanisms.
Modified kinetics of release during depression
The exocytotic rate is increased transiently by a presynaptic action potential, and this rate, α(t), is the product of the probability, p, of release of a quantum, and the number (n) of quantal release units (Stevens, 1968; Korn and Faber, 1987). Depression could be attributable to a decrease in either parameter, and the depletion model first became popular when it was assumed that n represented a releasable pool of vesicles (Liley and North, 1953; Elmqvist and Quastel, 1965; Thies, 1965). More recently, a different structural correlate has been identified for n, namely the number of active zones, each postulated to release the contents of at most one vesicle (Korn et al., 1982, 1994). This model would appear to preclude depletion as an explanation of depression. However it has been suggested that at each active zone p =_p_v_n_v, where_p_v is the release probability per vesicle, and _n_v is the number of docked vesicles (Rosenmund and Stevens, 1996; Dobrunz and Stevens, 1997; Goda and Stevens, 1998). This formulation might accommodate the depletion model if _n_v is small, although several considerations suggest that_p_v, or p itself, is the target for depression (Faber et al., 1998). They include the shifts in latency distributions and in α(t).
We find that α(t) is biphasic, in contrast to unimodal waveforms described for frog (Barrett and Stevens, 1972) and lobster (Parnas and Parnas, 1987) neuromuscular junctions. Whereas those connections were analyzed under conditions of relatively low release probability, p is quite high at the M-axon to CRN connection. When p is low, α(t) might be reduced to a single component. In addition, the synaptic delay at this junction is significantly shorter than that reported for mammalian synapses at the same temperature (18–20°C; see Sabatini and Regehr, 1996). While these and related issues require further study, our finding that α(t) is shifted to longer delays is again consistent with a change in the state of the release apparatus.
Alternative mechanisms of depression
As noted above, the depression described here could be attributable to a change in the functional state of the exocytotic machinery, a mechanism first suggested by Betz (1970). Our results also suggest this “refractoriness” is not restricted to sites that have undergone exocytosis, but is expressed at all release sites after a conditioning presynaptic stimulus. That is, if the mechanism underlying depression was operative only at sites that had contributed to the EPSP1, there would have been a negative correlation between the amplitudes of EPSP2 and EPSP1. Thus, we propose that the dominant mechanism in depression is triggered by the presynaptic action potential and occurs upstream of release itself. While the increase in synaptic delay may reflect such a process, these results do not rule out an additional contribution of depletion to depression.
The functional state of the secretory proteins (Calakos and Scheller, 1996) presumably plays a major role in shaping release, including its probability and synaptic delay, and this functional state is most likely affected by previous activity. One possible mechanism would be a conformational change of one or more secretory proteins, such as the putative Ca2+ sensor synaptotagmin (Martin et al., 1995; Hsu et al., 1996; Mochida et al., 1997; Geppert and Sudhof, 1998). Also, the calcium influx associated with the conditioning presynaptic potential might trigger two opposing Ca2+-dependent processes. A balance between secretory proteins, such as synaptotagmin and rab3, a release regulator implicated in the induction of long-term potentiation (Castillo et al., 1997), has been proposed to mediate a gain-setting mechanism (Geppert and Sudhof, 1998), and this balance might be modifiable by presynaptic activity. Finally, alternative models would involve a persistent inactivation of presynaptic Ca2+ channels, similar to that which contributes to posttetanic depression at the calyx of Held (Forsythe et al., 1998). Indeed, a cotransmitter, such as adenosine (Redman and Silinsky, 1994), might modulate channel activity.
The latency differential found during PPD may also be attributable to a modification in the functional state of the secretory proteins. In this context, it is important to note that facilitation at the crayfish neuromuscular junction is associated with an accelerated rate of transmitter release (Vyshedskiy and Lin, 1997). Thus, two forms of short-term plasticity are associated with latency shifts. One potential regulatory peptide would be SNAP-25 because cleavage of this peptide results in more variable and longer latencies (Trudeau et al., 1996, 1998). Alternatively, dilation of the fusion pore or desynchronization of fusion because of reduction in the number of fusion particles could affect release kinetics (Rahaminoff and Fernandez, 1997; Schweizer et al., 1998).
Other possible mechanisms for the increased synaptic delay include persistent inactivation of presynaptic Ca2+ channels, which however, does not alter release kinetics at the calyx of Held (Forsythe et al., 1998), and a slowing in action potential propagation at the branch points that give rise to the presynaptic collaterals. The latter is also unlikely: the collaterals are ∼50- to 100-μm-long, and to account for the observed shift in latency one would have to postulate a drop in conduction velocity to 0.6 to 1.2 m/sec for a neurite of ∼25 μm in diameter. If this delay were instead localized to the branch point itself, it would be detected with the axonal recording (Funch et al., 1984).
Plasticity of paired-pulse depression
We have described two examples of PPD modulated by a manipulation that selectively alters EPSP1 or EPSP2: (1) GTPγS only alters the size of EPSP1, and (2) an increased presynaptic Ca2+ concentration decreases depression by preferentially enhancing EPSP2. Furthermore, the Ca2+ effect persists for tens of minutes after brief injection of the cation. Therefore Ca2+ presumably initiates a cascade of events, leading to the long-lasting augmentation of evoked exocytosis. GTPγS would be expected to also initiate a cascade. These considerations suggest their effects mimic modifications that could be triggered physiologically. If so, they would represent a case of metaplasticity (Abraham and Bear, 1996), defined as a change or transformation in the way synaptic efficacy is modified.
Metaplasticity includes long- and short-term activity-dependent modifications (Huang et al., 1992; Wexler and Stanton, 1993; Fischer et al., 1997). The metaplasticities triggered by Ca2+ or GTPγS differ from the redistribution of synaptic efficacy in neocortex during repetitive stimulation, which apparently alters the content, not the gain, of the signal between neurons (Markram and Tsodyks, 1996). In contrast, both Ca2+ or GTPγS injections led to an increase in total synaptic efficacy, rather than a redistribution. Consideration of the properties of the M-cell network and the behavior it mediates may suggest a functional correlate of this metaplasticity, namely control of the frequency at which a fish can generate repetitive escape responses. A single action potential in the M-cell triggers an escape response (Zottoli, 1977), mediated supraspinally by suprathreshold excitation of CRNs, which in turn reliably activate cranial motoneurons (for review, see Faber et al., 1989; Korn and Faber, 1996; Zottoli and Faber, 2000). Activity-dependent depression of M-axon output synapses helps preclude rapid initiation of two sequential escapes that might be counterproductive. In fact, the time constant of recovery from depression is longer than the duration of the other predominant inhibitory process, feedback inhibition of the M-cell (Furshpan and Furukawa, 1963; 10–20 msec). Thus, altering the magnitude of spike-evoked depression can be expected to regulate the minimal interval at which a fish could execute double startle responses.
Footnotes
This work was supported by the National Institutes of Health (NS21838). We thank Carla Tyler-Polsz and Aaron Plante for technical support, and Maurice Volaski for software development and help in preparation of the figures.
Correspondence should be addressed to Dr. Donald S. Faber at his present address: Department of Neuroscience, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461.
Dr. Pereda's present address: Department of Neuroscience, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461.
REFERENCES
- 1.Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- 2.Auerbach AA, Bennett MVL. Chemically mediated transmission at a giant fiber synapse in the central nervous system of a vertebrate. J Gen Physiol. 1969;53:183–210. doi: 10.1085/jgp.53.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett EF, Stevens CF. Quantal independence and uniformity of presynaptic release kinetics at the frog neuromuscular junction. J Physiol (Lond) 1972;227:665–689. doi: 10.1113/jphysiol.1972.sp010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 5.Bellingham MC, Walmsley B. A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron. 1999;23:159–170. doi: 10.1016/s0896-6273(00)80762-x. [DOI] [PubMed] [Google Scholar]
- 6.Betz WJ. Depression of transmitter release at the neuromuscular junction of the frog. J Physiol (Lond) 1970;206:629–644. doi: 10.1113/jphysiol.1970.sp009034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature. 1990;348:125–132. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- 8.Calakos N, Scheller RH. Synaptic vesicle biogenesis, docking, and fusion: a molecular description. Physiol Rev. 1996;76:1–29. doi: 10.1152/physrev.1996.76.1.1. [DOI] [PubMed] [Google Scholar]
- 9.Castillo PE, Janz R, Sudhof TC, Malenka RC, Nicoll RA. The synaptic vesicle protein Rab3A is essential for mossy fiber long term potentiation in the hippocampus. Nature. 1997;388:590–593. doi: 10.1038/41574. [DOI] [PubMed] [Google Scholar]
- 10.Celio MR, Gray EG, Yasargil GM. Ultrastructure of the Mauthner axon collateral and its synapses in the goldfish spinal cord. J Neurocytol. 1979;8:19–29. doi: 10.1007/BF01206455. [DOI] [PubMed] [Google Scholar]
- 11.Day JW, Hall DH, Hall LM, Bennett MVL. α-Bungarotoxin labeling and acetylcholinesterase localization at the Mauthner fiber giant synapse in the hatchetfish. J Neurosci. 1983;3:272–279. doi: 10.1523/JNEUROSCI.03-02-00272.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Debanne D, Guerineau NC, Gahwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol (Lond) 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 14.Elmqvist D, Quastel DMJ. A quantitative study of end-plate potentials in isolated human muscle. J Physiol (Lond) 1965;178:505–529. doi: 10.1113/jphysiol.1965.sp007639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faber DS, Korn H. Electrophysiology of the Mauthner cell: basic properties, synaptic mechanisms, and associated networks. In: Faber DS, Korn H, editors. Neurobiology of the Mauthner cell. Raven; New York: 1978. pp. 47–131. [Google Scholar]
- 16.Faber DS, Fetcho JR, Korn H. Neuronal networks underlying the escape response in goldfish. General implications for motor control. In: Modulation of defined vertebrate neural circuits. Ann NY Acad Sci. 1989;563:11–33. doi: 10.1111/j.1749-6632.1989.tb42187.x. [DOI] [PubMed] [Google Scholar]
- 17.Faber DS, Waldeck RF, Pereda A. Multiple modes of transmitter release at a fast synapse. Soc Neurosci Abstr. 1995;21:336. [Google Scholar]
- 18.Faber DS, Waldeck RF, Pereda A. Synaptic depression at an identified central synapse: evidence for different constraints on evoked and spontaneous quanta. In: Faber DS, Redman SJ, Thompson SM, Altman JS, editors. Central synapses: quantal mechanisms and plasticity. HFSP; Strasbourg, France: 1998. pp. 158–168. [Google Scholar]
- 19.Fischer TM, Blazis DEJ, Priver NA, Carew TJ. Metaplasticity at identified inhibitory synapses in Aplysia. Nature. 1997;389:860–865. doi: 10.1038/39892. [DOI] [PubMed] [Google Scholar]
- 20.Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- 21.Funch PG, Wood MR, Faber DS. Localization of active sites along the myelinated goldfish Mauthner axon: morphological and pharmacological evidence for saltatory conduction. J Neurosci. 1984;4:2397–2409. doi: 10.1523/JNEUROSCI.04-09-02397.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furshpan EJ, Furukawa T. Two inhibitory mechanisms in the Mauthner neurons of the goldfish. J Neurophysiol. 1963;26:140–176. doi: 10.1152/jn.1963.26.1.140. [DOI] [PubMed] [Google Scholar]
- 23.Geppert M, Sudhof TC. Rab3 and synaptotagmin: The yin and yang of synaptic membrane fusion. Annu Rev Neurosci. 1998;21:75–95. doi: 10.1146/annurev.neuro.21.1.75. [DOI] [PubMed] [Google Scholar]
- 24.Goda Y, Stevens CF. Readily releasable pool size changes associated with long term depression. Proc Natl Acad Sci USA. 1998;95:1283–1288. doi: 10.1073/pnas.95.3.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hackett JT, Faber DS. Mauthner axon networks mediating supraspinal components of the startle response in the goldfish. Neuroscience. 1983;8:317–331. doi: 10.1016/0306-4522(83)90069-6. [DOI] [PubMed] [Google Scholar]
- 26.Hackett JT, Cochran SL, Greenfield LJ., Jr Quantal transmission at Mauthner axon target synapses in the goldfish brainstem. Neuroscience. 1989;32:49–64. doi: 10.1016/0306-4522(89)90107-3. [DOI] [PubMed] [Google Scholar]
- 27.Highstein SM, Bennett MV. Fatigue and recovery of transmission at the Mauthner fiber-giant fiber synapse of the hatchetfish. Brain Res. 1975;98:229–242. doi: 10.1016/0006-8993(75)90003-7. [DOI] [PubMed] [Google Scholar]
- 28.Hille B. G protein-coupled mechanisms and nervous signalling. Neuron. 1992;9:187–195. doi: 10.1016/0896-6273(92)90158-a. [DOI] [PubMed] [Google Scholar]
- 29.Hsu SF, Augustine GJ, Jackson MB. Adaptation of Ca2+ - triggered exocytosis in presynaptic terminals. Neuron. 1996;17:501–512. doi: 10.1016/s0896-6273(00)80182-8. [DOI] [PubMed] [Google Scholar]
- 30.Huang Y-Y, Colino A, Selig DK, Malenka RC. The influence of prior synaptic activity on the induction of long-term potentiation. Science. 1992;255:730–733. doi: 10.1126/science.1346729. [DOI] [PubMed] [Google Scholar]
- 31.Korn H, Faber DS. Regulation and significance of probabilistic release mechanisms at central synapses. In: Edelman GM, Gall WE, Cowan WM, editors. Synaptic function. Wiley; New York: 1987. pp. 57–108. [Google Scholar]
- 32.Korn H, Faber DS. Escape behavior—brainstem and spinal cord circuitry and function. Curr Opin Neurobiol. 1996;6:826–832. doi: 10.1016/s0959-4388(96)80034-1. [DOI] [PubMed] [Google Scholar]
- 33.Korn H, Mallet A, Triller A, Faber DS. Transmission at a central synapse. II. Quantal description of release with a physical correlate for binomial n. J Neurophysiol. 1982;48:679–707. doi: 10.1152/jn.1982.48.3.679. [DOI] [PubMed] [Google Scholar]
- 34.Korn H, Faber DS, Burnod Y, Triller A. Regulation of efficacy at central synapses. J Neurosci. 1984;4:125–130. doi: 10.1523/JNEUROSCI.04-01-00125.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korn H, Sur C, Charpier S, Legendre P, Faber DS. The one-vesicle hypothesis and multivesicular release. In: Stjarne L, Greengard P, Grillner S, Hokfelt T, Ottoson D, editors. Molecular and cellular mechanisms of neurotransmitter release. Raven; New York: 1994. pp. 301–322. [DOI] [PubMed] [Google Scholar]
- 36.Liley AW, North KAK. An electrical investigation of effects of repetitive stimulation on mammalian neuromuscular junction. J Neurophysiol. 1953;16:509–527. doi: 10.1152/jn.1953.16.5.509. [DOI] [PubMed] [Google Scholar]
- 37.Lin J, Faber DS. Synaptic transmission mediated by single club endings on the goldfish Mauthner cell. II. Plasticity of excitatory postsynaptic potentials. J Neurosci. 1988;8:1313–1325. doi: 10.1523/JNEUROSCI.08-04-01313.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Markram H, Tsodyks M. Redistribution of synaptic efficacy between neocortical pyramidal neurons. Nature. 1996;382:807–810. doi: 10.1038/382807a0. [DOI] [PubMed] [Google Scholar]
- 39.Martin KC, Hu Y, Armitage BA, Siegelbaum SA, Kandell ER, Kaang B-K. Evidence for synaptotagmin as an inhibitory clamp on synaptic vesicle release in Aplysia neurons. Proc Natl Acad Sci. 1995;92:11307–11311. doi: 10.1073/pnas.92.24.11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mochida S, Fukada M, Niinobe N, Kobayashi H, Mikoshiba K. Roles of synaptotagmin C2 domains in neurotransmitter secretion and inositol high-polyphosphate binding at mammalian cholinergic synapses. Neuroscience. 1997;77:937–943. doi: 10.1016/s0306-4522(96)00572-6. [DOI] [PubMed] [Google Scholar]
- 41.Model PG, Highstein SM, Bennett MVL. Depletion of vesicles and fatigue of transmission at a vertebrate central synapse. Brain Res. 1975;98:209–228. doi: 10.1016/0006-8993(75)90002-5. [DOI] [PubMed] [Google Scholar]
- 42.Mori A, Takahashi T, Miyashita Y, Kasai H. Two distinct glutamatergic synaptic inputs to striatal medium spiny neurones of neonatal rats and paired-pulse depression. J Physiol (Lond) 1994;476:217–228. doi: 10.1113/jphysiol.1994.sp020125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nicoll RA, Castillo PE, Weisskopf MG. The role of Ca2+ in transmitter release and long-term potentiation at hippocampal mossy fiber synapses. Adv Second Messenger Phosphoprotein Res. 1994;29:497–505. doi: 10.1016/s1040-7952(06)80031-5. [DOI] [PubMed] [Google Scholar]
- 44.Parnas J, Parnas I. Influence of depolarizing pulse duration on the time course of transmitter release in lobster. J Physiol (Lond) 1987;388:487–494. doi: 10.1113/jphysiol.1987.sp016626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rahaminoff R, Fernandez JM. Pre- and postfusion regulation of transmitter release. Neuron. 1997;18:17–27. doi: 10.1016/s0896-6273(01)80043-x. [DOI] [PubMed] [Google Scholar]
- 46.Redman RS, Silinsky EM. ATP released together with acetylcholine as the mediator of neuromuscular depression at frog motor nerve endings. J Physiol (Lond) 1994;477:117–127. doi: 10.1113/jphysiol.1994.sp020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richmond J, Haydon PG. Rab effector domain peptides stimulate the release of neurotransmitter from cell cultured synapses. FEBS Lett. 1993;326:124–130. doi: 10.1016/0014-5793(93)81775-u. [DOI] [PubMed] [Google Scholar]
- 48.Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 49.Sabatini BL, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature. 1996;384:170–172. doi: 10.1038/384170a0. [DOI] [PubMed] [Google Scholar]
- 50.Schweizer FE, Dresbach T, DeBello WM, O'Connor V, Augustine GJ, Betz H. Regulation of neurotransmitter release kinetics by NSF. Science. 1998;279:1203–1206. doi: 10.1126/science.279.5354.1203. [DOI] [PubMed] [Google Scholar]
- 51.Stevens CF. Synaptic physiology. Proc IEEE. 1968;56:916–930. [Google Scholar]
- 52.Thies RE. Neuromuscular depression and the apparent depletion of transmitter in mammalian muscle. J Neurophysiol. 1965;28:427–442. doi: 10.1152/jn.1965.28.3.427. [DOI] [PubMed] [Google Scholar]
- 53.Thompson SM, Debanne D, Capogna M. Presynaptic determinants of synaptic efficacy in hippocampal pyramidal neurons. In: Faber DS, Redman SJ, Thompson SM, Altman JS, editors. Central synapses: quantal mechanisms and plasticity. HFSP; Strasbourg, France: 1998. pp. 247–254. [Google Scholar]
- 54.Trudeau L-E, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- 55.Trudeau L-E, Fang Y, Haydon PG. Modulation of an early step in the secretory machinery in hippocampal nerve terminals. Proc Natl Acad Sci USA. 1998;95:7163–7168. doi: 10.1073/pnas.95.12.7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vyshedskiy A, Lin J-W. Change of transmitter release kinetics during facilitation revealed by prolonged test pulses at the inhibitor of the crayfish opener muscle. J Neurophysiol. 1997;78:1791–1799. doi: 10.1152/jn.1997.78.4.1791. [DOI] [PubMed] [Google Scholar]
- 57.Wexler EM, Stanton PK. Priming of homosynaptic long-term depression in hippocampus by previous synaptic activity. NeuroReport. 1993;4:591–594. doi: 10.1097/00001756-199305000-00034. [DOI] [PubMed] [Google Scholar]
- 58.Wu LG, Borst JG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/s0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- 59.Zottoli SJ. Correlation of the startle reflex and Mauthner cell auditory responses in unrestrained goldfish. J Exp Biol. 1977;66:243–254. doi: 10.1242/jeb.66.1.243. [DOI] [PubMed] [Google Scholar]
- 60.Zottoli SJ, Faber DS. Mauthner cell: what has it taught us? The Neuroscientist. 2000;6:26–38. [Google Scholar]