Neural Cell Adhesion Molecule-Stimulated Neurite Outgrowth Depends on Activation of Protein Kinase C and the Ras–Mitogen-Activated Protein Kinase Pathway (original) (raw)
Abstract
The signal transduction pathways associated with neural cell adhesion molecule (NCAM)-induced neuritogenesis are only partially characterized. We here demonstrate that NCAM-induced neurite outgrowth depends on activation of p59_fyn_, focal adhesion kinase (FAK), phospholipase Cγ (PLCγ), protein kinase C (PKC), and the Ras–mitogen-activated protein (MAP) kinase pathway. This was done using a coculture system consisting of PC12-E2 cells grown on fibroblasts, with or without NCAM expression, allowing NCAM–NCAM interactions resulting in neurite outgrowth. PC12-E2 cells were transiently transfected with expression plasmids encoding constitutively active forms of Ras, Raf, MAP kinase kinases MEK1 and 2, dominant negative forms of Ras and Raf, and the FAK-related nonkinase. Alternatively, PC12-E2 cells were submitted to treatment with antibodies to the fibroblast growth factor (FGF) receptor, inhibitors of the nonreceptor tyrosine kinase p59_fyn_, PLC, PKC and MEK and an activator of PKC, phorbol-12-myristate-13-acetate (PMA). MEK2 transfection rescued cells treated with all inhibitors. The same was found for PMA treatment, except when cells concomitantly were treated with the MEK inhibitor. Arachidonic acid rescued cells treated with antibodies to the FGF receptor or the PLC inhibitor, but not cells in which the activity of PKC, p59_fyn_, FAK, Ras, or MEK was inhibited. Interaction of NCAM with a synthetic NCAM peptide ligand, known to induce neurite outgrowth, was shown to stimulate phosphorylation of the MAP kinases extracellular signal-regulated kinases ERK1 and ERK2. The MAP kinase activation was sustained, because ERK1 and ERK2 were phosphorylated in PC12-E2 cells and primary hippocampal neurons even after 24 hr of cultivation on NCAM-expressing fibroblasts. Based on these results, we propose a model of NCAM signaling involving two pathways: NCAM–Ras–MAP kinase and NCAM–FGF receptor–PLCγ–PKC, and we propose that PKC serves as the link between the two pathways activating Raf and thereby creating the sustained activity of the MAP kinases necessary for neuronal differentiation.
Keywords: NCAM, Ras–MAP kinase pathway, neurite outgrowth, PKC, MAP kinase activation, FGF receptor
The neural cell adhesion molecule NCAM induces neurite outgrowth via a homophilic binding mechanism by which NCAM on one cell binds to NCAM on an adjacent cell. NCAM is a member of the immunoglobulin superfamily and is expressed by almost all neural cells. Alternative splicing of the NCAM gene product results in translation of two transmembrane isoforms of 140 and 180 kDa and a 120 kDa glycosylphosphatidylinositol-linked isoform. NCAM plays a pivotal role in neuronal development, regeneration, and synaptic plasticity associated with learning and memory consolidation in the adult (Luthi et al., 1994; Rønn et al., 1995, 1998; Muller et al., 1996; Cremer et al., 1997; Uryu et al., 1999). NCAM null mice have a diminished olfactory bulb, presumably because of an impaired migration of olfactory neuronal precursor cells, as well as deficits in spatial learning and exploratory behavior (Cremer et al., 1994; Ono et al., 1994; Hu et al., 1996).
Homophilic NCAM binding thus modulates neuronal differentiation and plasticity. The underlying signal transduction mechanisms are currently being studied by many approaches. Clustering of NCAM on the cell surface by means of NCAM antibodies or cultivation of PC12 cells on monolayers of NCAM-expressing 3T3 cells result in changes in intracellular pH, Ca2+ concentration, and phosphoinositide turnover (Schuch et al., 1989; Doherty et al., 1991). By means of a coculture system allowing NCAM–NCAM interaction resulting in neurite outgrowth, it has been shown that NCAM stimulation leads to phosphorylation of the fibroblast growth factor (FGF) receptor (Williams et al., 1994a; Saffell et al., 1997), which in turn stimulates phospholipase Cγ (PLCγ). PLCγ converts phospholipids to diacylglycerol (DAG), which subsequently has been shown to be converted to arachidonic acid by diacylglycerol lipase, whereas protein kinase C (PKC) does not seem to be involved (Doherty et al., 1991). Arachidonic acid in turn has been suggested to induce an increased influx of extracellular calcium via calcium channels located in the plasma membrane resulting in neuronal cell differentiation (Williams et al., 1994b). On the other hand, NCAM-dependent neurite outgrowth is selectively abolished in fyn− neurons (Beggs et al., 1994), and the NCAM-140 isoform has been shown by means of immunoprecipitation to interact with two nonreceptor tyrosine kinases, p59_fyn_ and the focal adhesion kinase FAK (Beggs et al., 1994, 1997). A fraction of NCAM seems to associate constitutively with p59_fyn_, whereas cross-linking of NCAM at the neuronal cell surface by means of NCAM antibodies induces recruitment of FAK to the NCAM–p59_fyn_ complex, resulting in phosphorylation of both tyrosine kinases (Beggs et al., 1997), indicating the possibility that NCAM activates the Ras–mitogen-activated protein (MAP) kinase pathway through FAK (Schlaepfer et al., 1994; Marais and Marshall, 1996). This assumption is in accordance with the finding that antibody and NCAM-Fc fragment-induced NCAM clustering at the surface of differentiated mouse neuroblastoma cells transiently activates the MAP kinases extracellular signal-regulated kinases ERK1 and ERK2 (Schmid et al., 1999), again indicating a role of the Ras–MAP kinase pathway in NCAM-mediated signaling. On the other hand, it has been reported that NCAM added to astrocytes in culture inhibits cell proliferation by reducing MAP kinase activity (Krushel et al., 1998). Thus, the role of the Ras–MAP kinase pathway in NCAM-induced neuritogenesis needs clarification. Furthermore, it is important to assess whether cross talk between the above described different signaling pathways takes place.
NCAM has been shown to stimulate neurite outgrowth from various neurons in primary culture and from rat pheochromocytoma PC12 cells (for review, see Doherty and Walsh, 1994). In the present study, we used a subclone of PC12 cells to investigate the molecular mechanisms underlying NCAM-mediated neuronal differentiation and neurite outgrowth. PC12 cells, like most neurons, express the 140 and 180 kDa isoforms of NCAM, and when grown on monolayers of genetically modified fibroblasts expressing NCAM, PC12 cells extend significantly longer neurites than when grown on NCAM-negative fibroblasts (Doherty et al., 1991).
We here report that NCAM-mediated neurite outgrowth is dependent on activation of FAK and the Ras–MAP kinase signaling cascade. Furthermore, we show that activation of PKC also is necessary and provides a mechanism for modulation of the activity of the Ras–MAP kinase pathway, which presumably is necessary for NCAM-stimulated neuronal differentiation.
MATERIALS AND METHODS
Materials. Arachidonic acid was purchased from Sigma (St. Louis, MO). p59_fyn_ inhibitor PP2 [4-amino-5-(4-chlorophenyl)-7(_t_-butyl)pyrazolo[3,4-d]pyrimidine], PLC inhibitor U-73122 (1-[6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)a)mino)hexyl]-1H-pyrrole-2,5-dione), PKC inhibitor Calphostin C (UCN-1028c), and PKC activator phorbol-12-myristate-13-acetate (PMA) were from Calbiochem (La Jolla, CA). MAP kinase kinase (MEK) inhibitor PD98059 was from New England Biolabs (Beverly, MA). Polyclonal antibodies against the FGF receptor were purchased from Upstate Biotechnology (catalog #06–177; Lake Placid, NY). All reagents were added to the cultures immediately after seeding the PC12-E2 cells, in concentrations that have no nonspecific effects on the ability of monolayers to support neurite outgrowth. The C3 undeca peptide (ASKKPKRNIKA) synthesized as a dendrimer composed of four monomers coupled to a lysine backbone was a gift from Prof. Arne Holm (Royal Agricultural and Veterinary University, Copenhagen, Denmark). PhosphoPlus p42/44 MAP kinase (Thr202/Tyr204) antibody kit was from New England Biolabs.
cDNA constructs. The rat constitutively active MEK2 expression plasmid pRK5-MEK2-S222/226E was a gift from Dr. Klaus Seedorf (Hagedorn Research Institute, Gentofte, Denmark). An expression plasmid encoding a dominant negative form of the human Raf-1 protein (Bruder et al., 1992) was kindly provided by Dr. E. Lukanidin (Danish Cancer Society, Copenhagen, Denmark). Constitutively active and dominant negative Ras expression plasmids were generated by oligo-directed mutagenesis on c-Hras (Willumsen et al., 1991) by introducing G12V and G12V/S17N mutations, respectively. Both Ras-encoding plasmids and plasmids encoding rat constitutively active MEK1 (Bottorff et al., 1995) and vRaf (Rapp et al., 1983) were gifts from Dr. Berthe Willumsen (Institute of Molecular Biology, Copenhagen University, Copenhagen, Denmark). An expression vector encoding the enhanced variant of the Aequorea victoria green fluorescent protein (pEGFP-N1) was purchased from Clontech (Palo Alto, CA).
For the cloning of the focal adhesion kinase-related nonkinase (FRNK) rat brain poly(A+)RNA (Clontech) was used as a template for reverse transcription using the antisense primer CAGACGGCCCA- GGTTTACTGATGAAC (position 2423–2445 of FAK; GenBank accession number AF 020777), followed by PCR amplification with the sense primer CTGTCATCAGTTGGAGCTGTGAGTG (position 3694–3718). The PCR product was reamplified with the nested primers 5′-GAGAAGGTACCGCAAGAAGAACGGATCA (position 2478–2505),introducing the underlined _Kpn_I restriction site, and 3′-CTGCTGGTGGAATTCTAGAHAAGATC (position 3638–3663), introducing the underlined _Xba_I restriction site. The upper primer also incorporated a mutation at position 2504 mutating Met 691 to Ile to prevent premature translation start. The resulting PCR product was ligated into pcDNA3.1(+) (Invitrogen, Groningen, Netherlands) and partially sequenced. Expression of full-length FRNK was verified using the Promega (Madison, WI) TnT T7 Quick Coupled Transcription/Translation System kit.
Cell culture and transfections. The PC12-E2 cell line (Wu and Bradshaw, 1995) was a gift from Dr. Klaus Seedorf. The cells were grown in DMEM supplemented with 5% fetal calf serum (FCS), 10% horse serum (HS), 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Life Technologies, Paisley, UK) at 37°C in a humidified atmosphere containing 5% CO2. The fibroblastoid mouse cell line L 929 (European Cell Culture Collection) was stably transfected with the eukaryotic expression vector pHβ-Apr-1-neo (Gunning et al., 1987) containing a full-length cDNA encoding human 140 kDa NCAM or the vector alone (Kasper et al., 1996). The NCAM cDNA did not contain exon VASE or exons a, b, c, AAG. The cells were routinely grown at 37°C, 5% CO2in DMEM supplemented with 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin. For transient transfection, PC12-E2 cells were seeded in 35 mm dishes at a density of 300,000 cells per dish and grown for 24 hr. Transfection into PC12-E2 cells was done by the Lipofectamine method with the PLUS Reagent according to the manufacturer's instructions (Life Technologies, Gaithersburg, MD) with the use of 3 μg of total DNA per 35 mm dish. Fifteen to 20 hr after transfection, PC12-E2 cells were seeded on top of confluent monolayers of fibroblasts in 35 mm dishes at a density of 60,000 cells per dish and grown for 24 hr in DMEM supplemented with 1% FCS and 1% HS, before image analysis. Transfected cells were identified by cotransfection with pEGFP-N1 (0.5 μg). The procedure was checked by immunostaining, showing a 85–95% concordance in expression of concomitantly transfected plasmids. Recording was done by computer-assisted microscopy using a Nikon (Tokyo, Japan) Diaphot inverted microscope and a Nikon Plan 20× objective. Images were grabbed with a video camera (Grundig Electronics) using the software package Prima created at the Protein Laboratory (Copenhagen, Denmark). The length of neuronal processes per cell was estimated using a stereological approach with the software package Process Length developed at the Protein Laboratory. Process Length superimposes an unbiased counting frame on images of cell cultures. This counting frame defines the fraction of the image to be evaluated and contains a number of horizontal test lines for determination of intersecting neurites. The absolute neurite length in micrometers per cell can subsequently be calculated from the ratio of the number of cell processes intersecting the test lines and the number of cell bodies within the counting frame (for details, see West and Gundersen, 1990; Ventimiglia et al., 1995; Geinisman et al., 1996).
Coculture of prenatal rat hippocampal neurons and mouse fibroblastoid L-cells with or without NCAM expression. Hippocampal cells were prepared from rat embryos at embryonic day 17 (E17) to E19 according to Trenkner and Sidman (1977). A suspension of dissociated cells was seeded on top of confluent monolayers of fibroblasts in 60 mm dishes at a density of 500,000 neurons per dish. Cells were maintained at 37°C, 5% CO2 in Neurobasal medium containing 20 mm HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.4% w/v BSA (Sigma), supplemented with B27 (Life Technologies). Cocultures were grown for 24 hr before analysis.
MAP kinase phosphorylation assay. PC12-E2 cells were plated in 35 mm dishes at a density of 200,000 cells per dish and grown for 24 hr, followed by further culturing in a low-serum medium (0.5% FCS) for 16 hr before stimulation. PC12-E2 cells were stimulated with the C3 peptide (0.54 μm) for 7 or 40 min. Alternatively, cocultures of PC12-E2 cells (100,000 cells per 35 mm dish) or hippocampal neurons cells (500,000 cells per 60 mm dish) grown on confluent fibroblasts without or with NCAM expression were analyzed after 24 hr of cultivation. For solubilization, cells were rinsed once in ice cold PBS containing 1 mm sodium orthovanadate, 0.5 mm phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 10 μg/ml aprotinin, and solubilized in lysis buffer (0.125 m Tris-HCl, 2% SDS, 10% glycerol, and 50 μm dithiothreitol, pH 7.5). Cell extracts were briefly sonicated (10–15 sec, 50 W) and clarified by centrifugation at 20,000 × g for 5 min at 4°C. Protein concentration was determined using the bicinchoninic acid assay (Pierce, Rockville, IL). Cell extracts were kept frozen at −80°C until use. Proteins in cell extracts (20 μg) were separated by SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Millipore, Bedford, MA). The membrane was blocked overnight at 4°C in Tris-buffered saline containing 0.05% (v/v) Tween 20 (TTBS) and 5% nonfat dry milk (w/v). Each sample was run in duplicate, and one was incubated for 1 hr at 25°C with anti-phosphoMAP kinase antibodies (diluted 1:1000) in 5% bovine serum albumin and TTBS. After washing in TTBS, the membrane was incubated with a goat anti-rabbit IgG horseradish peroxidase conjugate (diluted 1:2000) in 5% (w/v) nonfat dry milk and TTBS for 1 hr at room temperature. The membrane was again washed in TTBS, and immune complexes were visualized using enhanced chemiluminescence (Amersham, Buckinghamshire, UK). Another sample was probed in the same manner using an anti-MAP kinase antibody to detect total MAP kinase protein. For all assays, the exposed bands on x-ray film were quantified by scanning using the PrAverB image analysis program developed at the Protein Laboratory. The obtained values are normalized to the background brightness of the film, thus allowing the comparison between different films.
Statistics. For PC12-E2 cells and hippocampal neurons in coculture, the SE values indicate the variation between mean values obtained from at least four independent experiments. Statistical evaluation was performed by means of paired t test using the commercially available software package Fig-P, version 2.2 (Biosoft, Cambridge, UK).
RESULTS
NCAM-stimulated neurite outgrowth involves the FGF receptor, PLCγ, PKC, p59_fyn_, FAK, and the Ras–MAP kinase pathway
To investigate which signaling pathways are involved in NCAM-stimulated neurite outgrowth, we tested whether pharmacological compounds or transfection with cDNAs of signal transduction molecules alone or in combination could affect neuritogenesis promoted by NCAM. PC12-E2 cells were transiently transfected with either an empty expression plasmid or a plasmid coding for a signal transduction molecule together with pEGFP-N1 and cultured on a monolayer of control, NCAM-negative fibroblasts, or NCAM-positive fibroblasts (Fig.1). Figure 1 shows that PC12-E2 cells extend longer neurites when grown on NCAM-positive fibroblasts than on NCAM-negative fibroblasts.
Fig. 1.

Neuritogenic effect of NCAM on PC12-E2 cells. PC12-E2 cells were transiently transfected with pEGFP and grown for 24 hr on monolayers of fibroblasts without (a) or with (b) expression of human NCAM-140. Scale bar, 10 μm.
Because NCAM presumably activates the neuronal FGF receptor, resulting in a stimulation of PLCγ and DAG lipase to generate arachidonic acid (Williams et al., 1994a; Saffell et al., 1997), we tested whether perturbation of this intracellular pathway affected NCAM-dependent neurite outgrowth in the used test system. Initially, the effects of antibodies against the FGF receptor and the PLC inhibitor U-73122 on NCAM-stimulated neurite outgrowth were investigated. FGF receptor antibodies and the PLC inhibitor inhibited NCAM-stimulated neurite outgrowth but had no effect on neurite outgrowth on control cells (Fig.2A). By hydrolyzing inositol phospholipids, PLCγ generates DAG, a known stimulator of PKC, which in turn has been shown to activate the MAP kinase signal transduction pathway via the serine/threonine kinase Raf (Sozeri et al., 1992). To determine whether PKC plays a role in NCAM-stimulated neurite outgrowth, PC12-E2 cells were treated with the specific PKC inhibitor Calphostin C. This treatment led to an inhibition (Fig.2A) of NCAM-dependent neurite outgrowth. These results confirm that NCAM signaling involves the FGF receptor and PLCγ and indicate a thus far unrecognized role for PKC in NCAM-stimulated neurite outgrowth.
Fig. 2.
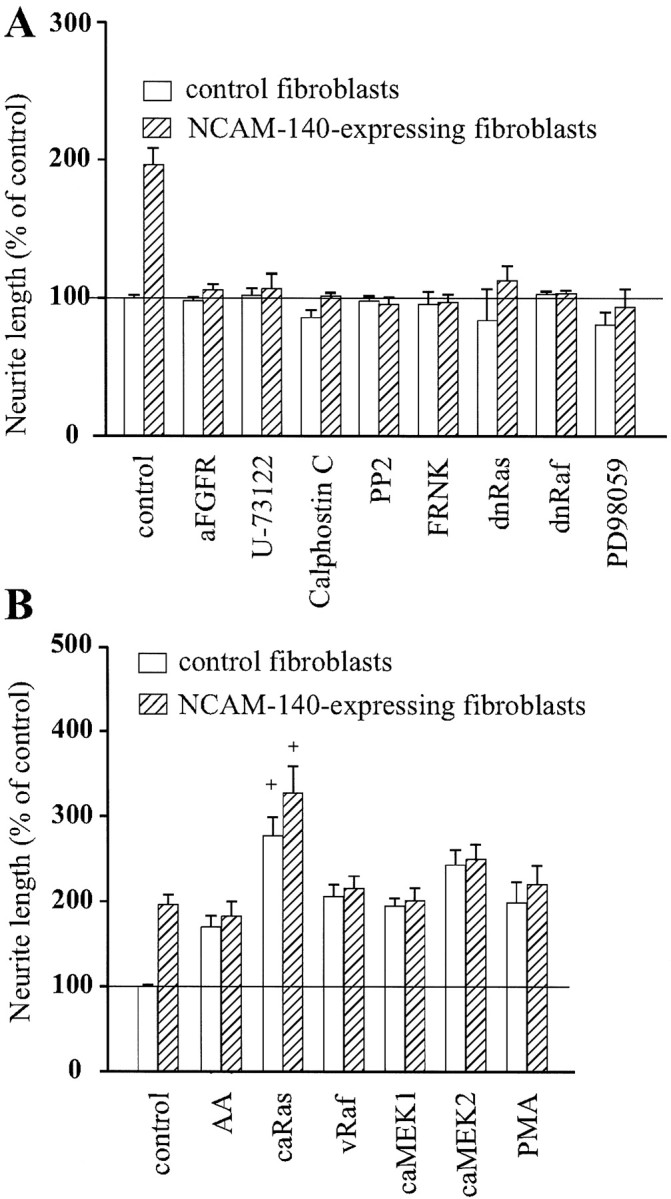
Effect of various compounds and signal transduction molecules on NCAM-induced neurite outgrowth. PC12-E2 cells were transiently transfected with pEGFP-N1 and an expression vector that was either empty (control) or encoding a signal transduction molecule. Alternatively, PC12-E2 cells were transfected with pEGFP-N1 alone and grown in the presence of a variety of compounds affecting signal transduction. The cells were grown for 24 hr on monolayers of fibroblasts with or without expression of human NCAM-140. Data are in all cases shown as means ± SE calculated from four or five independent experiments performed on different days. Between 200 and 300 cells were analyzed in each group in each individual experiment. The neurite length of vector-transfected cells on control fibroblasts was set to 100%, corresponding to an average value of 31 μm/cell. A, Inhibition of NCAM-stimulated neurite outgrowth from PC12-E2 cells by antibodies to FGF receptor (aFGFR) (1:1000), PLC inhibitor U-73122 (1 μm), PKC inhibitor Calphostin C (400 nm), p59_fyn_ inhibitor PP2 (24 μm), MEK inhibitor PD98059 (25 μm), and by expression of FRNK, dominant negative Ras (dnRas), or dominant negative Raf (dnRaf). The inhibition of NCAM-stimulated neurite outgrowth was in all cases statistically significant (p < 0.005). B, Effect of arachidonic acid (AA) (10 μm), PMA (10 ng/ml), and of constitutively active Ras (caRas), vRaf, or constitutively active MEK1 (caMEK1) and MEK2 (caMEK2). The stimulation of neurite outgrowth by the used activators was not significantly different from NCAM-induced stimulation, except for constitutively active Ras. +p < 0.05 when compared with control PC12-E2 grown on NCAM-expressing fibroblasts.
p59_fyn_ has been shown previously to be required for NCAM-stimulated neurite outgrowth, because cerebellar and dorsal root ganglion neurons from_fyn−_ mice displayed a complete inhibition of NCAM-dependent neurite outgrowth on NCAM-140-expressing fibroblast monolayers (Beggs et al., 1994). This finding was confirmed in our test system in which the p59_fyn_inhibitor PP2 was found to block growth of neurites from PC12-E2 cells over monolayers of NCAM-expressing fibroblasts (Fig.2A).
Although it has been shown that FAK is phosphorylated upon NCAM-140 clustering by NCAM antibodies (Beggs et al., 1997), a role for FAK in NCAM-dependent neurite outgrowth has so far not been established. We prepared a construct expressing FRNK, which represents the catalytically inert C-terminal domain of FAK harboring the focal adhesion targeting sequence. FRNK is not capable of autophosphorylation and therefore serves as a dominant negative form competing with FAK in focal adhesions. It was found that expression of FRNK inhibited NCAM-dependent neurite outgrowth (Fig. 2A), indicating that FAK is required for NCAM-stimulated neuritogenesis.
Because it has been proposed that FAK links integrin-mediated signals to the Ras–MAP kinase pathway (Schlaepfer et al., 1994; Chen et al., 1998), we surmised that FAK might also connect NCAM stimulation to this pathway. Therefore, we tested whether dominant negative forms of Ras and Raf affected NCAM-induced neurite outgrowth. Neurite outgrowth from PC12-E2 cells expressing these forms of Ras or Raf was inhibited when grown on NCAM-positive L-cells, whereas neurite outgrowth on control, NCAM-negative fibroblasts was unaffected (Fig. 2A). Moreover, treatment of PC12-E2 cells with the MEK inhibitor PD98059 also blocked neurite outgrowth on NCAM-expressing fibroblasts but had no effect on neurite outgrowth on control cells.
In contrast, arachidonic acid, which has been suggested to act as a second messenger for NCAM-dependent neurite outgrowth (Williams et al., 1994b), the PKC stimulator PMA, and constitutively active forms of Ras, Raf, MEK1, and MEK2, when expressed in PC12-E2 cells, all stimulated neurite outgrowth from the cells grown on control fibroblasts, without affecting NCAM-stimulated neurite outgrowth, with the exception of the constitutively active form of Ras, which not only strongly induced neurite outgrowth on control fibroblasts but also had an additive effect to the NCAM-induced stimulation (Fig. 2B). Therefore, these stimulators of neurite outgrowth, with the possible exception of Ras, may be used to rescue a pathway inhibited upstream of the activation step.
To conclude, NCAM-induced neurite outgrowth involves activation of p59_fyn_, FAK, PLCγ, and PKC and requires the Ras–MAP kinase signaling pathway.
Arachidonic acid cannot rescue inhibition of p59_fyn_, FAK, or the Ras–MAP kinase pathway
PC12-E2 were grown in DMEM supplemented with 10 μmarachidonic acid in the presence of various inhibitors of NCAM-stimulated neurite outgrowth or transiently transfected with expression constructs encoding FRNK, dominant negative Ras, or dominant negative Raf. Figure 3A shows that arachidonic acid stimulated neurite outgrowth from PC12-E2 cells grown on NCAM-expressing and control fibroblasts in the presence of either antibodies to the FGF receptor or the PLC inhibitor U-73122 and, therefore, rescued the inhibition by these agents. However, arachidonic acid did not reverse the inhibitory effect of the p59_fyn_ inhibitor PP2, FRNK, dominant negative Ras, the MEK inhibitor PD98059, or the PKC inhibitor Calphostin C on NCAM-stimulated neurite outgrowth. Moreover, arachidonic acid was not able to stimulate growth of neurites over control, NCAM-negative fibroblasts, when any of these inhibitors was present. We therefore conclude that, although arachidonic acid may play a role in NCAM-stimulated neurite outgrowth, other factors such as PKC and the components of the Ras–MAP kinase pathway are also important in this process.
Fig. 3.
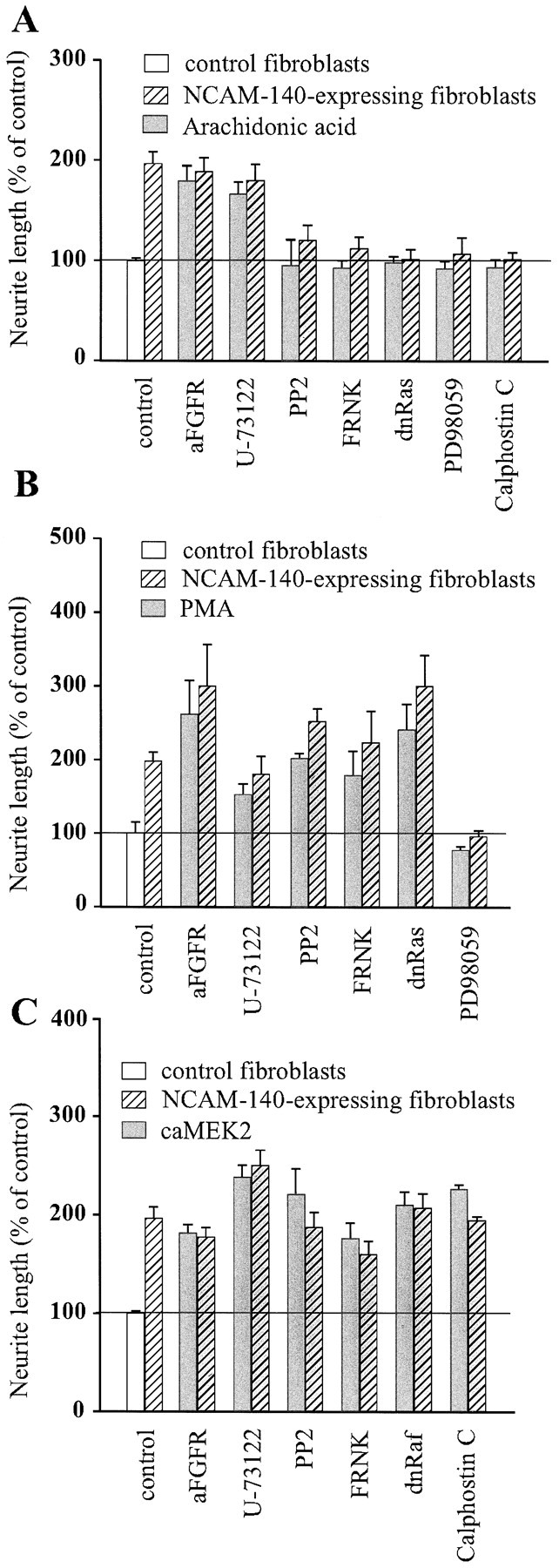
Effect of 10 μm arachidonic acid (A), 10 ng/ml PMA (B), and constitutively active MEK2 (C) on NCAM-specific neurite outgrowth affected by a series of inhibitors of signal transduction. PC12-E2 cells were transiently cotransfected with pEGFP-N1 and either an empty expression vector or one of the following expression plasmids: FRNK, dominant negative Ras, dominant negative Raf, or PC12-E2 cells transfected with pEGFP-N1 alone were grown in the presence of antibodies to the FGF receptor (diluted 1:1000), the PLCγ inhibitor U-73122 (1 μm), p59_fyn_ inhibitor PP2 (24 μm), the MEK inhibitor PD98059 (25 μm), or the PKC inhibitor Calphostin C (400 nm).
PMA cannot rescue inhibition of MEK
PKC is a serine/threonine kinase that is activated by DAG generated by PLC. PKC is a known activator of the Ras–MAP kinase signaling pathway (Sozeri et al., 1992; Morrison et al., 1996; Ueda et al., 1996), and the enzyme has been identified as a key mediator of basic FGF-induced process outgrowth in human oligodendrocytes (Oh et al., 1997). To study the role of PKC in NCAM-stimulated neurite outgrowth, we treated PC12-E2 cells with PMA to activate PKC in cells transfected with either FRNK or dominant negative Ras. Alternatively, the cells were treated with one of the following inhibitors of NCAM-specific neurite outgrowth: the p59_fyn_ inhibitor PP2, the MEK inhibitor PD98059, antibodies to the FGF receptor, or the PLC inhibitor U-73122. Figure 3B shows that, when PKC was stimulated, no agent significantly inhibited NCAM-specific neurite outgrowth, with the exception of the MEK inhibitor PD98059. Furthermore, stimulation of PKC by PMA also promoted neurite outgrowth on control, NCAM-negative fibroblasts in the presence of all inhibitors, with the exception of PD98059. These results indicate that PKC is a regulator of NCAM-stimulated neuritogenesis and that PKC probably connects NCAM signaling through the FGF receptor and PLCγ to the Ras–MAP kinase signaling pathway upstream of MEK.
Overexpression of MEK2 can rescue the effect of all tested inhibitors
To determine whether activation of the Ras–MAP kinase pathway could rescue NCAM-stimulated neurite outgrowth inhibited by the various inhibitors of signal transduction used in the present study, PC12-E2 cells were transiently transfected with constitutively active MEK2 and cotransfected with either FRNK or dominant negative Raf. PC12-E2 MEK2-transfected cells were also grown in the presence of the p59_fyn_ inhibitor PP2, antibodies against the FGF receptor, the PLC inhibitor U-73122, or the PKC inhibitor Calphostin C. Figure 3C shows that, when constitutively active MEK2 was expressed in PC12-E2 cells, none of the selected compounds had any inhibitory effect. These results indicate that the Ras–MAP kinase pathway is important in NCAM-stimulated neurite outgrowth.
Stimulation of NCAM with C3 peptide results in phosphorylation of ERK1 and ERK2
By screening a synthetic peptide library with the first immunoglobulin-like module of NCAM, a ligand, the C3 undeca peptide, was identified. The dendrimeric form of the C3 peptide composed of four monomers coupled to a lysine backbone has been shown to stimulate neurite outgrowth from both hippocampal neurons and PC12-E2 cells, apparently by activating a signaling pathway identical to that activated by NCAM–NCAM binding (Rønn et al., 1999). We used the C3 peptide as an NCAM stimulator and tested the effect of C3 treatment of PC12-E2 cells on phosphorylation of the terminal kinases in the Ras–MAP kinase cascade, ERK1 and ERK2. This was done by means of Western blotting using polyclonal antibodies, which specifically recognize the dually phosphorylated forms of ERK1 and ERK2. These two MAP kinases are activated by phosphorylation on Thr202 and Tyr204 by MEK1 and MEK2 (Crews et al., 1992). Phosphorylation of ERK1 and ERK2 in PC12-E2 cells was strongly increased as soon as after 7 min of treatment with the C3 peptide, and the activation lasted for at least 40 min (Fig.4A,B). The total amount of ERK1 and ERK2 was determined using antibodies that recognized both the active and inactive forms of the proteins, and no significant change in the levels of ERK1 and ERK2 proteins during C3 treatment was observed. These results support the conclusion that ERK1 and ERK2 are activated (phosphorylated) in response to stimulation of NCAM and that the activation is sustained for at least 40 min.
Fig. 4.

Stimulation of NCAM in PC12-E2 cells by the C3 peptide induces phosphorylation of ERK1 and ERK2. A, PC12-E2 cells, serum-starved (0.5% FCS) for 16 hr, were incubated in PBS without or with the C3 peptide (0.54 μm) for 7 or 40 min. Cell extracts prepared in SDS-containing lysis buffer were subjected in duplicate to SDS-PAGE and immunoblotted using either polyclonal anti-phosphoMAP kinase antibodies or polyclonal anti-MAP kinase antibodies. B, Quantification of MAP kinase phosphorylation of experiments performed as shown in A. The activity is expressed relative to the control (0 min), which was set to 100%. Error bars indicate SEs based on five independent experiments. *p < 0.05; ** p< 0.01; paired t test.
NCAM-induced neurite outgrowth is the result of sustained phosphorylation of ERK1 and ERK2
To determine whether NCAM–NCAM interactions were able to induce long-term MAP kinase activation, lysates of PC12-E2 cells or primary hippocampal neurons in coculture with fibroblasts with or without NCAM expression grown for 24 hr were analyzed for ERK1 and ERK2 phosphorylation by means of Western blotting. As controls, lysates of confluent monolayers of NCAM-positive and -negative fibroblasts were analyzed. No activation of ERK1 and ERK2 was observed in monolayers of control or NCAM-expressing fibroblasts under the used conditions (Fig. 5A). At a longer exposure times, a very weak activation of the MAP kinases was, however, observed in the fibroblasts monolayers with a slightly higher level of phosphorylation in NCAM-positive fibroblasts than in NCAM-negative fibroblasts (data not shown). Consequently, any major phosphorylation of the MAP kinases ERK1 and ERK2 observed in the coculture systems could be attributed to phosphorylation of MAP kinases in PC12-E2 cells or neurons and not in the fibroblasts (Fig.5B,C). The MAP kinases ERK1 and ERK2 were strongly phosphorylated in PC12-E2 cells and neurons cultured on top of NCAM-positive fibroblasts compared with cells cultured on top of NCAM-negative fibroblasts in which phosphorylation clearly was weaker. The total amount of ERK1 and ERK2 was also determined, and the degree of phosphorylation of the MAP kinases in the PC12-E2 cells and the primary hippocampal neurons was quantified. A statistically significant increase in MAP kinase activation in the NCAM-stimulated cells could be demonstrated (Fig.5D,E). These results indicate that NCAM-induced neurite outgrowth may be the result of a sustained phosphorylation of MAP kinases ERK1 and ERK2.
Fig. 5.

NCAM-stimulated neurite outgrowth from PC12-E2 cells or neurons is the result of sustained MAP kinase phosphorylation. Fibroblast monolayers without (LVN) or with (LBN) expression of NCAM-140 (A), cocultures of fibroblasts with or without NCAM expression and PC12-E2 cells (B,D), or cocultures of fibroblasts with or without NCAM expression and primary hippocampal neurons (C,E) were grown for 24 hr. Cell lysates were submitted to SDS-PAGE in duplicate and immunoblotted using either polyclonal anti-phosphoMAP kinase antibodies or polyclonal anti-MAP kinase antibodies. Quantification of MAP kinase phosphorylation is shown in_D_ and E. ERK1 and ERK2 activation is expressed relative to the control (PC12-E2 or neurons cultured on monolayers of NCAM-negative fibroblasts) correcting for the total amount of ERK1 and ERK2 protein. Error bars indicate SEs based on four independent experiments. *p < 0.05; paired_t_ test.
DISCUSSION
The capacity of NCAM to induce neurite outgrowth has been attributed mainly to its activation of the FGF receptor with the subsequent activation of PLCγ to generate DAG, the conversion of DAG to arachidonic acid by DAG lipase, and the activation of neuronal calcium channels by arachidonic acid (Williams et al., 1994a,b; Saffell et al., 1997). On the other hand, Schmid et al. (1999) found that NCAM-Fc fragments or antibodies against NCAM elicited activation of ERK1 and ERK2 in neuroblastoma cells. Conversely, it has been reported recently that NCAM-mediated inhibition of astrocyte proliferation involves an inhibition of the Ras–MAP kinase pathway (Krushel et al., 1998). Based on these reports, we decided to test whether activation of the MAP kinases was required for NCAM-stimulated neurite outgrowth. To determine the roles of various signal transduction molecules in NCAM-induced neuritogenesis, we analyzed neurite outgrowth from PC12-E2 cells, which either transiently expressed a series of transduction molecules or were treated with selected pharmacological compounds followed by culturing on monolayers of NCAM-negative or NCAM-positive fibroblasts. NCAM, nerve growth factor (NGF), and FGF induce morphological and functional differentiation of PC12 cells into sympathetic-like neurons (Greene and Tischler, 1976; Togari et al., 1985; Fujita et al., 1989;Doherty et al., 1991) for which reason these cells were used in the present study. The obtained results identify two signaling cascades responsible for neurite outgrowth promoted by NCAM stimulation, as shown in Figure 6 in which it is indicated that NCAM-stimulated neurite outgrowth depends on the activation of both the Ras–MAP kinase pathway and the FGF receptor–PLCγ–PKC pathway and that both pathways are necessary.
Fig. 6.
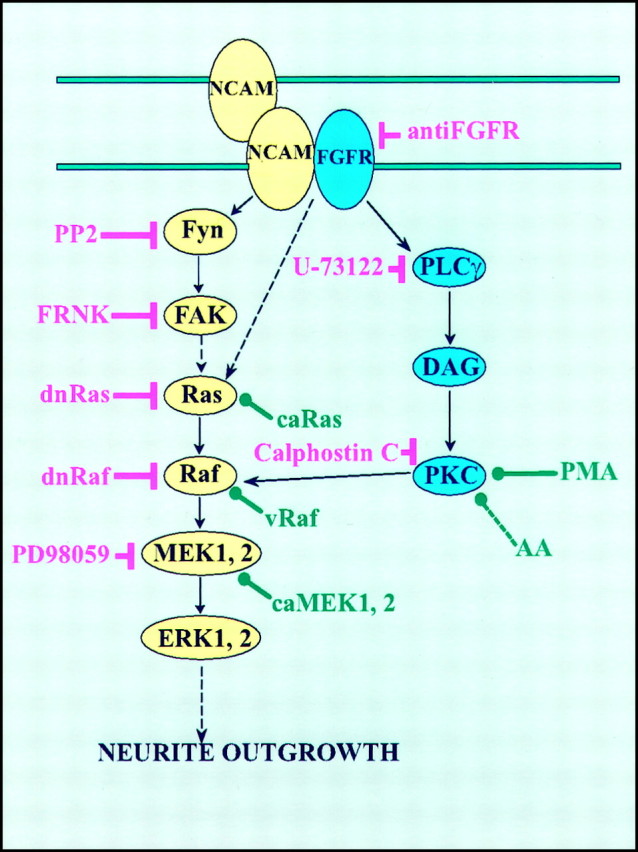
A model of signaling pathways involved in NCAM-induced neurite outgrowth. Inhibitors are shown in_red_, and activators are shown in_green_.
The signaling cascades are initiated by an interaction between NCAM at the surface of the PC12-E2 cells and NCAM expressed by supporting mouse fibroblasts. NCAM-dependent neurite extension by fyn−_neurons growing on monolayers of NCAM-expressing fibroblasts has been reported to be selectively abolished (Beggs et al., 1994), and the demonstration of inhibition of NCAM-specific neurite outgrowth by the p59_fyn inhibitor PP2 is consistent with this finding. Although it has been shown that FAK is transiently phosphorylated and recruited to the NCAM–p59_fyn_ complex (Beggs et al., 1997), the role of this interaction has so far not been determined. The inhibitory effect of expression of FRNK here clearly demonstrated the importance of FAK in NCAM-dependent neurite outgrowth. When FAK is stimulated, it becomes phosphorylated on Tyr397 and Tyr925, and this creates a binding site for the Grb2 adapter protein (Schlaepfer et al., 1994). Grb2 binding to FAK at Tyr925 may lead to the formation of a multiprotein signaling complex that promotes activation of Ras (Li et al., 1993; Schlaepfer et al., 1994). The ability of dominant negative Ras to block NCAM-stimulated neurite outgrowth further implicates Ras as a participant of this pathway. It was noted that neurite outgrowth from constitutively active Ras-expressing PC12-E2 cells on NCAM-positive fibroblasts was more prominent than neuritogenesis induced by NCAM alone. This probably reflects the multiplicity of the targets of Ras. A key downstream target of Ras is Raf, and an involvement of Raf in NCAM-stimulated neurite outgrowth was demonstrated by the ability of its dominant negative form to block the event and of vRaf to mimic NCAM-specific neurite extension. Activated Raf phosphorylates the MAP kinase kinases MEK1 and MEK2, which subsequently activate ERK1 and ERK2. An implication of MEK in NCAM signaling was confirmed by the fact that the MEK inhibitor PD98059 blocked NCAM-dependent neurite outgrowth and that overexpression of MEK2 could rescue all upstream inhibitions, which is also in accordance with the recent finding that the MEK inhibitor blocked NCAM-dependent neurite outgrowth from rat cerebellar neurons (Schmid et al., 1999).
Interestingly, the NCAM–Ras–MAP kinase signaling pathway itself appeared not to be sufficient for neurite extension, because antibodies to the FGF receptor and the inhibitors of PLC and PKC were able to block NCAM-induced neurite extension. When the FGF receptor is activated, PLCγ binds to the receptor and subsequently produces DAG. Arachidonic acid, generated from DAG by the action of DAG lipase, has been claimed to be a key second messenger of NCAM-induced neurite outgrowth. However, the role of arachidonic acid in the NCAM–Ras–MAP kinase signaling may need reinterpretation, because arachidonic acid did not modify the effect of the inhibitors of p59_fyn_, MEK, or PKC, or the effect of expression of FRNK, dominant negative Ras, or dominant negative Raf. Nevertheless, arachidonic acid was able to overcome the inhibition of antibodies to the FGF receptor and of the PLC inhibitor, the reason possibly being that the NCAM–Ras–MAP kinase pathway was only affected to a minor extent, if at all, in these experiments.
A role of PKC in NCAM signaling has been excluded previously based on experiments in which treatment of PC12 cells with high doses of PMA (thereby supposedly inhibiting PKC) or the PKC inhibitors H7 and staurosporine led to increased neurite outgrowth from PC12 cells on control fibroblasts without changing the neurite outgrowth on NCAM-expressing fibroblasts (Doherty et al., 1991). However, H7 and staurosporine can also inhibit other protein kinases, and the obtained results are therefore difficult to interpret. In contrast, we suggest a role for PKC in the NCAM–FGF receptor–PLCγ pathway based on experiments involving the specific PKC inhibitor Calphostin C and the PKC stimulator PMA, which is known to activate PKC when used in low concentrations (Roivainen et al., 1993). PKC is the major target of DAG (Inoue et al., 1977; Kishimoto et al., 1980), and PKC activation has been shown previously to enhance neurite outgrowth by its subsequent activation of the Ras–MAP kinase pathway (Hundle et al., 1995; Huang et al., 1995; Burry, 1998). When PKC was activated by PMA, inhibition of p59_fyn_, FAK, Ras, FGF receptor, or PLCγ did not block NCAM-dependent neurite outgrowth, indicating that not arachidonic acid but PKC may be the key molecule downstream of NCAM–FGF receptor–PLCγ-mediated signaling. Arachidonic acid has a potential of activating PKC, but probably only in the presence of DAG (Nishizuka, 1995), and according to our experiments a putative stimulation of PKC by arachidonic acid, unlike the stimulation by PMA, was not sufficient to rescue inhibition of the NCAM–Ras–MAP kinase pathway. Furthermore, arachidonic acid is generally not derived from DAG but from phospholipids by the action of phospholipase A2 (Nishizuka, 1992). Therefore, arachidonic acid probably neither participates in the NCAM–Ras–MAP kinase pathway nor in the NCAM–FGF receptor–PLCγ–PKC pathway but is a part of a third independent pathway involving the FGF receptor. The ability of the MEK inhibitor PD98059 to block NCAM-specific neurite outgrowth in the presence of the PKC activator PMA is consistent with the finding that PKC activates the MAP kinase pathway in a Raf-dependent manner (Marquardt et al., 1994;Ueda et al., 1996). PKC is also capable of phosphorylating growth-associated protein 43 (GAP-43), and a role for GAP-43 in NCAM-dependent neurite outgrowth has been proposed recently, although it was suggested that GAP-43 activation was mediated by arachidonic acid (Meiri et al., 1998). However, the ability of constitutively active MEK2 to reverse the inhibition of Calphostin C defines Raf, and not GAP-43, as the key target of PKC in NCAM signaling leading to induction of neurites.
NCAM did not stimulate neurite outgrowth when either the Ras–MAP kinase pathway or the FGF receptor–PKC pathway were blocked, and this indicates that these two pathways normally have to be simultaneously activated to induce neurite outgrowth via NCAM stimulation. The activation of Raf by both Ras and PKC probably provides a sustained activation of the MAP kinases. A sustained activation of the MAP kinases in PC12 cells has been shown to induce a differentiation response, in contrast to a brief, transient activation, which leads to proliferation (Heasley and Johnson, 1992; Nguyen et al., 1993;Traverse et al., 1994). It has been demonstrated that neuronal differentiation of PC12 cells induced by activation of receptor tyrosine kinases, such as the NGF receptor, the FGF receptor, and the platelet-derived growth factor receptor, depends on a sustained MAP kinase activity (Heasley and Johnson, 1992). In contrast, epidermal growth factor and insulin growth factor-I, which have been shown to only transiently activate MAP kinase activity, fail to differentiate PC12 cells (Heasley and Johnson, 1992). Phosphorylation of the MAP kinases, induced by the specific NCAM ligand, the C3 peptide, which has been shown to promote differentiation of hippocampal neurons and PC12-E2 cells, was lasting at least 40 min in contrast to the recently reported activation of ERK1 and ERK2 in response to antibody-induced NCAM clustering on the surface of already differentiated cells, which lasted only 10 min (Schmid et al., 1999). Therefore, we suggest that NCAM-stimulated neurite outgrowth is the result of a sustained phosphorylation of MAP kinases. This assumption is strongly supported by the fact that ERK1 and ERK2 were phosphorylated in PC12-E2 cells and neurons after 24 hr of growth on top of NCAM-expressing fibroblasts. A less pronounced phosphorylation of ERK1 and ERK2 was observed in PC12-E2 cells and neurons cultured on top of control fibroblasts, and this may be attributed to an integrin-dependent activation of the Ras–MAP kinase pathway (Schlapfer et al., 1998; Perron and Bixby, 1999).
Phosphorylated MAP kinases can directly activate gene expression by translocation into the nucleus and subsequent phosphorylation of transcription factors, such as Elk-1. Alternatively, phosphorylated MAP kinases are capable of activating a mitogen- and stress-activated protein kinase MSK1 (Deak et al., 1998), which in turn phosphorylates the cAMP-response element binding protein (CREB). It is worth noting that antibody clustering of NCAM recently has been reported to induce CREB phosphorylation (Schmid et al., 1999) and that both NCAM and CREB knock-out mice exhibit deficiencies in spatial learning (Bourtchuladze et al., 1994; Luthi et al., 1994). Furthermore, interference with the function of either NCAM or CREB has been shown to impair the establishment of long-term memory (Doyle et al., 1992; Bourtchuladze et al., 1994; Hu et al., 1996; Alexinsky et al., 1997). The MEK inhibitor PD98059 has been shown to markedly attenuate the induction of long-term potentiation in area CA1 (English and Sweatt,1997), implicating the Ras–MAP kinase cascade in this process. Moreover, recent studies also indicate that PKC stimulates CREB phosphorylation in the CA1 area of the hippocampus by activating ERK2 (Roberson et al., 1999). Thus, our results indicate that, by activating the Ras–MAP kinase signaling cascade, NCAM probably regulates transcription of genes responsible for long-term synaptic changes. To conclude, we have identified new mechanisms regulating NCAM-induced neuritogenesis of importance for brain development and neuroplasticity associated with regeneration and learning in the adult brain.
Footnotes
This work was supported by grants from the Lundbeck Foundation, the Danish Cancer Society, the Danish Medical Research Council, the Velux Foundation, Eva and Henry Frænkels Foundation, Agnes and Poul Friis' Foundation, and European Union Programme on Biotechnology BIO4-CT96–0450. We thank Dr. Berthe Willumsen for generously providing us with the vRaf, MEK1, and Ras-encoding plasmids, Dr. Klaus Seedorf for the PC12-E2 cell line and the MEK2-encoding plasmid, Dr. Eugene Lukanidin for the dominant negative Raf-1-encoding plasmid, and Dr. Arne Holm for the dendrimeric form of the C3-peptide.
Correspondence should be addressed to Elisabeth Bock, Protein Laboratory, Institute of Molecular Pathology, University of Copenhagen, Panum Institute, 3C Blegdamsvej, Building 6.2, DK-2200 Copenhagen N, Denmark. E-mail: bock@plab.ku.dk.
REFERENCES
- 1.Alexinsky T, Przybyslawski J, Mileusnic R, Rose SP, Sara SJ. Antibody to day-old chick brain glycoprotein produces amnesia in adult rats. Neurobiol Learn Mem. 1997;67:14–20. doi: 10.1006/nlme.1996.3734. [DOI] [PubMed] [Google Scholar]
- 2.Beggs HE, Soriano P, Maness PF. NCAM-dependent neurite outgrowth is inhibited in neurons from Fyn-minus mice. J Cell Biol. 1994;127:825–833. doi: 10.1083/jcb.127.3.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beggs HE, Baragona SC, Hemperly JJ, Maness PF. NCAM140 interacts with the focal adhesion kinase p125 fak and the SRC-related tyrosine kinase p59fyn. J Biol Chem. 1997;272:8310–8319. doi: 10.1074/jbc.272.13.8310. [DOI] [PubMed] [Google Scholar]
- 4.Bottorff D, Stang S, Agellon S, Stone J. RAS signalling is abnormal in a c-raf1 MEK1 double mutant. Mol Cell Biol. 1995;15:5113–5122. doi: 10.1128/mcb.15.9.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 6.Bruder JT, Heidecker G, Rapp UR. Serum-, TPA-, and Ras-induced expression from Ap-1/Ets-driven promoters requires Raf-1 kinase. Genes Dev. 1992;6:545–556. doi: 10.1101/gad.6.4.545. [DOI] [PubMed] [Google Scholar]
- 7.Burry RW. PKC activators (phorbol ester or bryostatin) stimulate outgrowth of NGF-dependent neurites in a subline of PC12 cells. J Neurosci Res. 1998;53:214–222. doi: 10.1002/(SICI)1097-4547(19980715)53:2<214::AID-JNR10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 8.Chen HC, Chan PC, Tang MJ, Cheng CH, Chang TJ. Tyrosine phosphorylation of focal adhesion kinase stimulated by hepatocyte growth factor leads to mitogen-activated protein kinase activation. J Biol Chem. 1998;273:25777–25782. doi: 10.1074/jbc.273.40.25777. [DOI] [PubMed] [Google Scholar]
- 9.Cremer H, Lange R, Christoph A, Plomann M, Vopper G, Roes J, Brown R, Baldwin S, Kraemer P, Scheff S, Baernels D, Rajewsky K, Wille W. Inactivation of the N-CAM gene in mice results in size reduction of the olfactory bulb and deficits in spatial learning. Nature. 1994;367:455–459. doi: 10.1038/367455a0. [DOI] [PubMed] [Google Scholar]
- 10.Cremer H, Chazal G, Goridis C, Represa A. NCAM is essential for axonal growth and fasciculation in the hippocampus. Mol Cell Neurosci. 1997;8:323–335. doi: 10.1006/mcne.1996.0588. [DOI] [PubMed] [Google Scholar]
- 11.Crews CM, Alessandrini A, Erikson RL. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science. 1992;258:478–480. doi: 10.1126/science.1411546. [DOI] [PubMed] [Google Scholar]
- 12.Deak M, Clifton AD, Lucocq JM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doherty P, Walsh FS. Signal transduction events underlying neurite outgrowth stimulated by cell adhesion molecules. Curr Opin Neurobiol. 1994;4:49–55. doi: 10.1016/0959-4388(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 14.Doherty P, Ashton SV, Moore SE, Walsh FS. Morphoregulatory activities of NCAM and N-cadherin can be accounted for by G protein-dependent activation of L- and N-type neuronal Ca2+ channels. Cell. 1991;67:21–33. doi: 10.1016/0092-8674(91)90569-k. [DOI] [PubMed] [Google Scholar]
- 15.Doyle E, Nolan PM, Bell R, Regan CM. Intraventricular infusions of anti-neural cell adhesion molecules in a discrete posttraining period impair consolidation of a passive avoidance response in the rat. J Neurochem. 1992;59:1570–1573. doi: 10.1111/j.1471-4159.1992.tb08477.x. [DOI] [PubMed] [Google Scholar]
- 16.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 17.Fujita K, Lazarovici P, Guroff G. Regulation of the differentiation of PC12 pheochromocytoma cells. Environ Health Perspect. 1989;80:127–142. doi: 10.1289/ehp.8980127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geinisman Y, Gundersen HJ, van der Zee E, West MJ. Unbiased stereological estimation of the total number of synapses in a brain region. J Neurocytol. 1996;25:805–819. doi: 10.1007/BF02284843. [DOI] [PubMed] [Google Scholar]
- 19.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gunning P, Leavitt J, Muscat G, Ng SY, Kedes L. A human beta-actin expression vector system directs high-level accumulation of antisense transcripts. Proc Natl Acad Sci USA. 1987;84:4831–4835. doi: 10.1073/pnas.84.14.4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heasley LE, Johnson GL. The beta-PDGF receptor induces neuronal differentiation of PC12 cells. Mol Biol Cell. 1992;3:545–553. doi: 10.1091/mbc.3.5.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu H, Tomasiewicz H, Magnuson T, Rutishauser U. The role of polysialic acid in migration of olfactory bulb interneuron precursors in the subventricular zone. Neuron. 1996;16:735–743. doi: 10.1016/s0896-6273(00)80094-x. [DOI] [PubMed] [Google Scholar]
- 23.Huang J, Mohammadi M, Rodrigues GA, Schlessinger J. Reduced activation of RAF-1 and MAP kinase by a fibroblast growth factor receptor mutant deficient in stimulation of phosphatidylinositol hydrolysis. J Biol Chem. 1995;270:5065–5072. doi: 10.1074/jbc.270.10.5065. [DOI] [PubMed] [Google Scholar]
- 24.Hundle B, McMahon T, Dadgar J, Messing RO. Overexpression of epsilon-protein kinase C enhances nerve growth factor-induced phosphorylation of mitogen-activated protein kinases and neurite outgrowth. J Biol Chem. 1995;270:30134–30140. doi: 10.1074/jbc.270.50.30134. [DOI] [PubMed] [Google Scholar]
- 25.Inoue M, Kishimoto A, Takai Y, Nishizuka Y. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. II. Proenzyme and its activation by calcium-dependent protease from rat brain. J Biol Chem. 1977;252:7610–7616. [PubMed] [Google Scholar]
- 26.Kasper C, Stahlhut M, Berezin V, Maar TE, Edvardsen K, Kiselyov VV, Soroka V, Bock E. Functional characterization of NCAM fibronectin type III domains: demonstration of modulatory effects of the proline-rich sequence encoded by alternatively spliced exons a and AAG. J Neurosci Res. 1996;46:173–186. doi: 10.1002/(SICI)1097-4547(19961015)46:2<173::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 27.Kishimoto A, Takai Y, Mori T, Kikkawa U, Nishizuka Y. Activation of calcium and phospholipid-dependent protein kinase by diacylglycerol, its possible relation to phosphatidylinositol turnover. J Biol Chem. 1980;255:2273–2276. [PubMed] [Google Scholar]
- 28.Krushel LA, Tai MH, Cunningham BA, Edelman GM, Crossin KL. Neural cell adhesion molecule (N-CAM) domains and intracellular signaling pathways involved in the inhibition of astrocyte proliferation. Proc Natl Acad Sci USA. 1998;95:2592–2596. doi: 10.1073/pnas.95.5.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li N, Batzer A, Daly R, Yajnik V, Skolnik E, Chardin P, Bar-Sagi D, Margolis B, Schlessinger J. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature. 1993;363:85–88. doi: 10.1038/363085a0. [DOI] [PubMed] [Google Scholar]
- 30.Luthi A, Laurent JP, Figurov A, Muller D, Schachner M. Hippocampal long-term potentiation and neural cell adhesion molecules L1 and NCAM. Nature. 1994;372:777–779. doi: 10.1038/372777a0. [DOI] [PubMed] [Google Scholar]
- 31.Marais R, Marshall CJ. Control of the ERK MAP kinase cascade by Ras and Raf. Cancer Surv. 1996;27:101–125. [PubMed] [Google Scholar]
- 32.Marquardt B, Frith D, Stabel S. Signalling from TPA to MAP kinase requires protein kinase C, raf and MEK: reconstitution of the signalling pathway in vitro. Oncogene. 1994;9:3213–3218. [PubMed] [Google Scholar]
- 33.Meiri K, Saffell JL, Walsh FS, Doherty P. Neurite outgrowth stimulated by neural cell adhesion molecules requires growth-associated protein-43 (GAP-43) function and is associated with GAP-43 phosphorylation in growth cones. J Neurosci. 1998;18:10429–10437. doi: 10.1523/JNEUROSCI.18-24-10429.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morrison P, Saltiel AR, Rosner MR. Role of mitogen-activated protein kinase kinase in regulation of the epidermal growth factor receptor by protein kinase C. J Biol Chem. 1996;271:12891–12896. doi: 10.1074/jbc.271.22.12891. [DOI] [PubMed] [Google Scholar]
- 35.Muller D, Wang C, Skibo G, Toni N, Cremer H, Calaora V, Rougon G, Kiss JZ. PSA-NCAM is required for activity-induced synaptic plasticity. Neuron. 1996;17:413–422. doi: 10.1016/s0896-6273(00)80174-9. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen TT, Scimeca JC, Filloux C, Peraldi P, Carpentier JL, Van Obberghen E. Co-regulation of the mitogen-activated protein kinase, extracellular signal-regulated kinase 1, and the 90-kDa ribosomal S6 kinase in PC12 cells. Distinct effects of the neurotrophic factor, nerve growth factor, and the mitogenic factor, epidermal growth factor. J Biol Chem. 1993;268:9803–9810. [PubMed] [Google Scholar]
- 37.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 38.Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- 39.Oh LY, Goodyer CG, Olivier A, Yong VW. The promoting effects of bFGF and astrocyte extracellular matrix on process outgrowth by adult human oligodendrocytes are mediated by protein kinase C. Brain Res. 1997;757:236–244. doi: 10.1016/s0006-8993(97)00224-2. [DOI] [PubMed] [Google Scholar]
- 40.Ono K, Tomasiewicz H, Magnuson T, Rutishauser U. N-CAM mutation inhibits tangential neuronal migration and is phenocopied by enzymatic removal of polysialic acid. Neuron. 1994;13:595–609. doi: 10.1016/0896-6273(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 41.Perron JC, Bixby JL. Distinct neurite outgrowth signaling pathways converge on ERK activation. Mol Cell Neurosci. 1999;13:362–378. doi: 10.1006/mcne.1999.0753. [DOI] [PubMed] [Google Scholar]
- 42.Rapp UR, Goldsborough GE, Mark TI, Bonner J, Groffen FH, Reynolds JR, Stephenson JR. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc Natl Acad Sci USA. 1983;80:4218–4222. doi: 10.1073/pnas.80.14.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roivainen R, McMahon T, Messing RO. Protein kinase C isozymes that mediate enhancement of neurite outgrowth by ethanol and phorbol esters in PC12 cells. Brain Res. 1993;624:85–93. doi: 10.1016/0006-8993(93)90063-s. [DOI] [PubMed] [Google Scholar]
- 45.Rønn LC, Bock E, Linnemann D, Jahnsen H. NCAM-antibodies modulate induction of long-term potentiation in rat hippocampal CA1. Brain Res. 1995;677:145–151. doi: 10.1016/0006-8993(95)00147-i. [DOI] [PubMed] [Google Scholar]
- 46.Rønn LC, Hartz BP, Bock E. The neural cell adhesion molecule (NCAM) in development and plasticity of the nervous system. Exp Gerontol. 1998;33:853–864. doi: 10.1016/s0531-5565(98)00040-0. [DOI] [PubMed] [Google Scholar]
- 47.Rønn LC, Olsen M, Ostergaard S, Kiselyov V, Berezin V, Mortensen MT, Lerche MH, Jensen PH, Soroka V, Saffells JL, Doherty P, Poulsen FM, Bock E, Holm A. Identification of a neuritogenic ligand of the neural cell adhesion molecule using a combinatorial library of synthetic peptides. Nat Biotechnol. 1999;17:1000–1005. doi: 10.1038/13697. [DOI] [PubMed] [Google Scholar]
- 48.Saffell JL, Williams EJ, Mason IJ, Walsh FS, Doherty P. Expression of a dominant negative FGF receptor inhibits axonal growth and FGF receptor phosphorylation stimulated by CAMs. Neuron. 1997;18:231–242. doi: 10.1016/s0896-6273(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 49.Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- 50.Schmid RS, Graff RD, Schaller MD, Chen S, Schachner M, Hemperly JJ, Maness P. NCAM stimulates the Ras-MAPK pathway and CREB phosphorylation in neuronal cells. J Neurobiol. 1999;38:542–558. [PubMed] [Google Scholar]
- 51.Schuch U, Lohse MJ, Schachner M. Neural cell adhesion molecules influence second messenger systems. Neuron. 1989;3:13–20. doi: 10.1016/0896-6273(89)90111-6. [DOI] [PubMed] [Google Scholar]
- 52.Sozeri O, Vollmer K, Liyanage M, Frith D, Kour G, Mark GE, III, Stabel S. Activation of the c-Raf protein kinase by protein kinase C phosphorylation. Oncogene. 1992;7:2259–2262. [PubMed] [Google Scholar]
- 53.Traverse S, Seedorf K, Paterson H, Marshall CJ, Cohen P, Ullrich A. EGF triggers neuronal differentiation of PC12 cells that overexpress the EGF receptor. Curr Biol. 1994;4:694–701. doi: 10.1016/s0960-9822(00)00154-8. [DOI] [PubMed] [Google Scholar]
- 54.Trenkner E, Sidman RL. Histogenesis of mouse cerebellum in microwell cultures. Cell aggregation and migration, fiber and synapse formation. J Cell Biol. 1977;75:915–940. doi: 10.1083/jcb.75.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Togari A, Dickens G, Kuzuya H, Guroff G. The effect of fibroblast growth factor on PC12 cells. J Neurosci. 1985;5:307–316. doi: 10.1523/JNEUROSCI.05-02-00307.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ueda Y, Hirai S-i, Osada S-i, Suzuki A, Mizuno K, Ohno S. Protein kinase C activates the MEK–ERK pathway in a manner independent of Ras and dependent on Raf. J Biol Chem. 1996;271:23512–23519. doi: 10.1074/jbc.271.38.23512. [DOI] [PubMed] [Google Scholar]
- 57.Uryu K, Butler AK, Chesselet MF. Synaptogenesis and ultrastructural localization of the polysialylated neural cell adhesion molecule in the developing striatum. J Comp Neurol. 1999;405:216–232. doi: 10.1002/(sici)1096-9861(19990308)405:2<216::aid-cne6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 58.Ventimiglia R, Jones BE, Moller A. A quantitative method for morphometric analysis in neuronal cell culture: unbiased estimation of neuron area and number of branch points. J Neurosci Methods. 1995;57:63–66. doi: 10.1016/0165-0270(94)00126-2. [DOI] [PubMed] [Google Scholar]
- 59.West MJ, Gundersen HJ. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol. 1990;296:1–22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- 60.Williams EJ, Furness J, Walsh FS, Doherty P. Activation of the FGF receptor underlies neurite outgrowth stimulated by L1, N-CAM, and N-cadherin. Neuron. 1994a;13:583–594. doi: 10.1016/0896-6273(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 61.Williams EJ, Walsh FS, Doherty P. The production of arachidonic acid can account for calcium channel activation in the second messenger pathway underlying neurite outgrowth stimulated by NCAM, N-cadherin, and L1. J Neurochem. 1994b;62:1231–1234. doi: 10.1046/j.1471-4159.1994.62031231.x. [DOI] [PubMed] [Google Scholar]
- 62.Willumsen BM, Vass WC, Velu TJ, Papageorge AG, Schiller J, Lowy DR. The bovine papillomavirus E5 oncogene can cooperate with ras: identification of p21 amino acids critical for transformation by c-rasH but not v-rasH. Mol Cell Biol. 1991;11:6026–6033. doi: 10.1128/mcb.11.12.6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu YY, Bradshaw RA. PC12–E2 cells: a stable variant with altered responses to growth factor stimulation. J Cell Physiol. 1995;164:522–532. doi: 10.1002/jcp.1041640310. [DOI] [PubMed] [Google Scholar]