Protein Aggregation after Transient Cerebral Ischemia (original) (raw)
Abstract
Protein aggregates containing ubiquitinated proteins are commonly present in neurodegenerative disorders and have been considered to cause neuronal degeneration. Here, we report that transient cerebral ischemia caused severe protein aggregation in hippocampal CA1 neurons. By using ethanolic phosphotungstic acid electron microscopy (EM) and ubiquitin immunogold EM, we found that protein aggregates were accumulated in CA1 neurons destined to die 72 hr after 15 min of cerebral ischemia. Protein aggregates appeared as clumps of electron-dense materials that stained heavily for ubiquitin and were associated with various intracellular membranous structures. The protein aggregates appeared at 4 hr and progressively accumulated at 24 and 48 hr of reperfusion in CA1 dying neurons. However, they were rarely observed in dentate gyrus neurons that were resistant to ischemia. At 4 hr of reperfusion, protein aggregates were mainly associated with intracellular vesicles in the soma and dendrites, and the nuclear membrane. By 24 hr of reperfusion, the aggregates were also associated with mitochondria, the Golgi apparatus, and the dendritic plasmalemma. High-resolution confocal microscopy further demonstrated that protein aggregates containing ubiquitin were persistently and progressively accumulated in all CA1 dying neurons but not in neuronal populations that survive in this model. We conclude that proteins are severely aggregated in hippocampal neurons vulnerable to transient brain ischemia. We hypothesize that the accumulation of protein aggregates cause ischemic neuronal death.
Keywords: brain ischemia, protein aggregation, ubiquitin, neuronal death, intracellular vesicles, mitochondrion, dendritic membranes, ethanolic phosphotungstic acid, electron microscopy, confocal microscopy
In rat cerebral ischemia models, a period of ischemia followed by reperfusion causes neuronal degeneration selectively in hippocampal CA1 pyramidal neurons after 48 hr of reperfusion but leaves dentate gyrus (DG), CA3, and most cortical neurons intact (Kirino, 1982; Pulsinelli et al., 1982; Smith et al., 1984). During the 48–72 hr delay period, the neurons destined to die look normal under the light microscope. At the ultrastructural level, however, disaggregation of polyribosomes, abnormalities of the Golgi apparatus, deposition of dark substances, and modification of postsynaptic densities have been reported (Kirino et al., 1984; Petito and Pusinelli, 1984; Rafols et al., 1995; Hu et al., 1998;Martone et al., 1999).
Protein polypeptide chains need to be folded into their native conformations to avoid aggregation. When newly synthesized polypeptide chains are in unfolded or misfolded states, their sticky hydrophobic segments are exposed on the surface. Without protection, these non-native proteins remain abnormal and are prone to aggregate. Abnormal proteins can be recognized and ubiquitinated by the ubiquitin system through a series of ATP-dependent reactions (Hershko and Ciechanover, 1998). Ubiquitination targets abnormal proteins to form ubiquitinated proteins (ubi-proteins) for degradation rather than chaperone-like protec- tion. Under pathological conditions, when abnormal proteins in cells are too numerous to be protected or quickly removed, they will aggregate through their hydrophobic segments. Abnormal protein aggregates have been observed consistently in almost all neurodegenerative diseases by ubiquitin immunogold electron microscopic (EM) analysis (Kakizuka, 1998). Thus, immunogold EM analysis has been used to identify protein aggregates associated with neurodegenerative disorders (Alves-Rodrigues et al., 1998).
During our previous study of synaptic structures using ethanolic phosphotungstic acid (EPTA) electron microscopy (Hu et al., 1998;Martone et al., 1999), we found that EPTA not only stained synapses and nuclei, but also additional dark aggregates throughout the soma and dendrites of postischemic dying neurons. These aggregates were not present in neurons destined to survive after ischemia or in sham-operated control neurons. This observation prompted us to conduct a series of experiments to investigate the nature of the aggregates and to study the mechanism of their formation. Because EPTA stains proteins rich in basic amino acids, we hypothesized that the aggregates may be composed of abnormal proteins. In the present study, this hypothesis is supported by the fact that the aggregates contain ubi-proteins, as demonstrated by ubiquitin-immunogold EM. These protein aggregates persistently accumulated on the membranes of mitochondria, vesicles, and dendrites in all CA1 dying neurons but not in the rest of surviving neurons after ischemia. We also provide evidence that ubi-proteins are persistently and progressively aggregated in CA1 dying neurons by high-resolution confocal microscopy. We hypothesize that persistent protein aggregation may cause neuronal death after ischemia.
MATERIALS AND METHODS
Ischemia model. Brain ischemia was produced using the two-vessel occlusion model in rats. All experimental procedures were approved by the committee on animal studies of the Queen's Medical Center (Honolulu, HI). Male Wistar rats (250–300 gm) were fasted overnight. Anesthesia was induced with 3% halothane followed by maintenance with 1–2% halothane in an oxygen/nitrous oxide (30/70%) gas mixture. Catheters were inserted into the external jugular vein, tail artery, and tail vein to allow blood sampling, arterial blood pressure recording, and drug infusion. Both common carotid arteries were encircled by loose ligatures. Fifteen minutes before ischemia induction and 15 min after ischemia, blood gases were measured and adjusted to PaO2 >90 mmHg and PaCO2 35–45 mmHg, pH 7.35–7.45, by adjusting the tide volume of respirator. Bipolar EEG was recorded before ischemia, continuously during the ischemic insult, and after ischemia until the rat recovered from the anesthesia. At the beginning of a 10 min steady-state period before induction of ischemia, the inspired halothane concentration was decreased to 0.5% and 150 IU/kg heparin was administered intravenously. Blood was withdrawn via the jugular catheter to produce a mean arterial blood pressure of 50 mmHg, and both carotid arteries were clamped. Blood pressure was maintained at 50 mmHg during the ischemic period by withdrawing or infusing blood through the jugular catheter. At the end of the ischemic period, the clamps were removed and the blood was reinfused through the jugular catheter, followed by 0.5 ml of 0.6 m sodium bicarbonate. In all experiments, brain temperature was maintained at 37°C before, during, and after ischemia (15 min of reperfusion). For biochemical studies of events occurring during 15 min of ischemia, tissue was obtained by freezing the brains in situ with liquid nitrogen at the end of 15 min ischemia. For rats subjected to reperfusion, halothane was discontinued at the end of ischemia and all wounds were sutured. The rats were reanesthetized, tracheotomized, and artificially ventilated at 2, 4, 24, and 48 hr of reperfusion. Rats were perfused with ice-cold 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 m cacodylate buffer for electron microscopy, with ice-cold 4% paraformaldehyde plus 0.1% glutaraldehyde in 0.1 m phosphate buffer for immunoelectron microscopy, and with ice-cold 4% paraformaldehyde in 0.1 m phosphate buffer for confocal microscopy. Sham-operated rats were subjected to the same surgical procedures but without induction of brain ischemia. Each experimental group consisted of at least three rats.
Electron and immunoelectron microscopy. Tissue sections from experimental and control animals were stained by either 1% EPTA (Fisher Scientific, Houston, TX) (Bloom and Aghajanian, 1968) or conventional osmium–uranium–lead staining. Briefly, coronal brain sections were cut at a thickness of 200 μm with a vibratome through the level of the dorsal hippocampus and post-fixed for 1 hr with 4% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4. For conventional osmium–uranium–lead staining, sections were post-fixed for 2 hr in 1% osmium tetroxide in 0.1m cacodylate buffer, rinsed in distilled water, and stained with 1% aqueous uranyl acetate overnight. Tissue sections were then dehydrated in an ascending series of ethanol to 100% followed by dry acetone, and embedded in Durcupan ACM resin. Thin sections were counterstained with lead citrate before examination in the electron microscope. For EPTA staining, sections were dehydrated in an ascending series of ethanol to 100% and stained for 50 min with 1% phosphotungstic acid (PTA) prepared by dissolving 0.1 gm of PTA in 10 ml of 100 ethanol and adding four drops of 95% ethanol. The EPTA solution was changed once after a 25 min interval during the staining. The sections were then further dehydrated in dry acetone and embedded in Durcupan ACM resin.
Immunoelectron microscopy was performed on postischemic and control brain tissues using a postembedding protocol. Brains were fixed in 4% paraformaldehyde containing 0.1% glutaraldehyde without secondary fixation with osmium tetroxide. Blocks of hippocampal tissue were dehydrated through 100% ethanol without osmication and then embedded in Durcupan ACM resin as above. Sections were cut at a thickness of 0.1 μm and collected on 300 mesh gold grids. Immunolabeling consisted of the following steps: (1) 5 min wash in 0.1 mPBS, pH 7.2; (2) 10 min in PBS containing 0.001% Triton X-100 (TX100); (3) 20 min blocking step in 1% BSA in PBS; (4) 2 hr incubation in anti-ubiquitin diluted 1:1000 in PBS [the monoclonal antibody against ubiquitin (MAB1510, Chemicon, Temecula, CA) has been extensively characterized by Western blot and immunocytochemistry and recognizes both free and bound ubiquitin (Morimoto et al., 1996)]; (5) three washes for 5 min each in PBS; (6) 5 min wash in 3% normal goat serum in PBS; (7) 1 hr incubation in 1:40 goat anti-mouse IgG conjugated to 10 nm gold (Stonybrook, NY) diluted in PBS; (8) three washes for 5 min wash each in PBS; and (9) three washes for 5 min each in double-distilled H2O. Grids were air-dried and counterstained with uranyl acetate and Satoh lead before examination in a JEOL (Peabody, MA) 100CX electron microscope. Some grids were also counterstained with ethanolic or aqueous phosphotungstic acid. Negative controls in which the primary antibody was omitted from the labeling sequence were also performed in both sham-operated and postischemic tissue.
Laser-scanning confocal microscopy. Double-labeled fluorescence immunocytochemistry was performed on coronal brain sections (50 μm) from sham-operated controls and animals subjected to 15 min of ischemia followed by 30 min, and 2, 4, 24, and 72 hr of reperfusion. The sections were transferred into a 24-well microtiter plate filled halfway with 0.01 m citric acid–sodium citrate buffer, pH 6.0, heated for 10 sec in a microwave set to 30% power. The sections were washed twice with 0.2% TX100–PBS for 10 min. Nonspecific binding sites were blocked with 3% BSA in PBS–0.2% TX100 for 30 min and incubated with the monoclonal anti-ubiquitin at a dilution of 1:400 in PBS containing 0.1% TX100 overnight at 4°C. The sections were washed three times for 10 min each in PBS containing 0.1% TX100 at room temperature and incubated in a mixture of fluorescein-labeled anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA) at a dilution of 1:200 and 15 μg/ml propidium iodide for 1 hr at room temperature. The sections were washed three times in PBS–0.1% TX100, mounted on glass slides, and coverslipped using Gelvatol. The slides were analyzed on a Bio-Rad (Hercules, CA) MRC 1024 laser-scanning confocal microscope.
RESULTS
EPTA stained protein aggregates in postischemic CA1 neurons
In our previous studies, we stained postsynaptic densities using EPTA in sham-operated control and postischemic brains (Hu et al., 1998;Martone et al., 1999). In this protocol, tissues are dehydrated through absolute ethanol without previous osmication, leading to extraction of much of the lipid content of membranes. Under these conditions, EPTA selectively stains synaptic structures and nuclei in normal brain tissue (Fig. 1, CA1, DG Sham). However, in the postischemic brains, EPTA strongly stained not only synapses and nuclei but also intracellular aggregates in the cell soma and dendrites in CA1 neurons (Fig. 1, CA1, 4h, 24h). As will be described below, these aggregates were associated with membranous structures, particularly vesicles and mitochondria (Figs. 1, 2). Although membranes were not directly visible in the EPTA-stained material because of lack of osmium and lipid extraction, they were often visible in negative contrast, allowing for easy identification of subcellular structures (Fig. 2). EPTA-stained aggregates were also present on the cytosolic face of the nuclear membrane at both 4 and 24 hr of reperfusion (Fig. 1, CA1, 4h,24h).
Fig. 1.
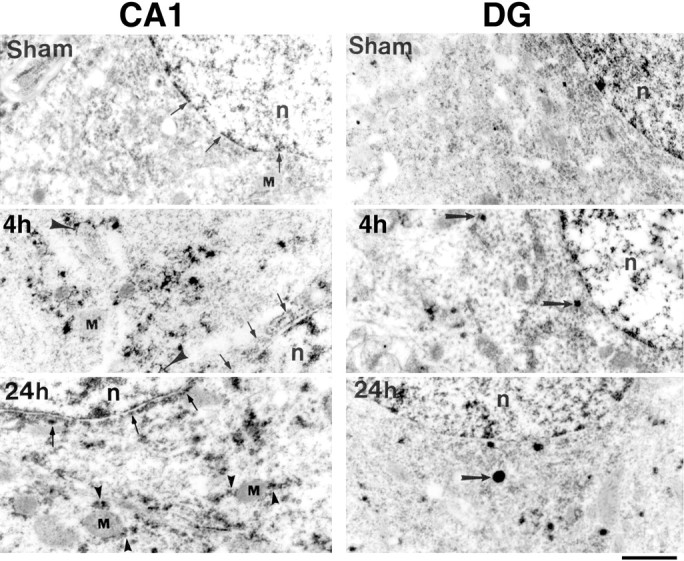
Electron micrographs of EPTA staining in the cell soma of CA1 pyramidal neurons and in DG granule cells in sham-operated controls (Sham) and rats subjected to 15 min ischemia followed by 4 and 24 hr of reperfusion. EPTA-stained materials were extensively distributed in the cytoplasm of postischemic CA1 neurons but not in control neurons. Many of the EPTA-stained proteins were attached to the membranes of intracellular vesicles, visible in negative contrast (arrowheads in_CA1_) at both 4 and 24 hr of reperfusion. At 24 hr of reperfusion, aggregates appeared on the membranes of mitochondria (M) as well (arrowheads in_CA1_). Dark aggregates were also attached to the cytoplasmic face of the nuclear membrane (small arrows) at both 4 and 24 hr in postischemic CA1 neurons but not in the control. The extensive EPTA-stained aggregates seen in postischemic CA1 neurons were not present in DG neurons, although intensely stained small, round structures were sometimes seen after ischemia (arrows in_DG_, 4h, 24h).n, Nucleus. Scale bar, 1 μm.
Fig. 2.
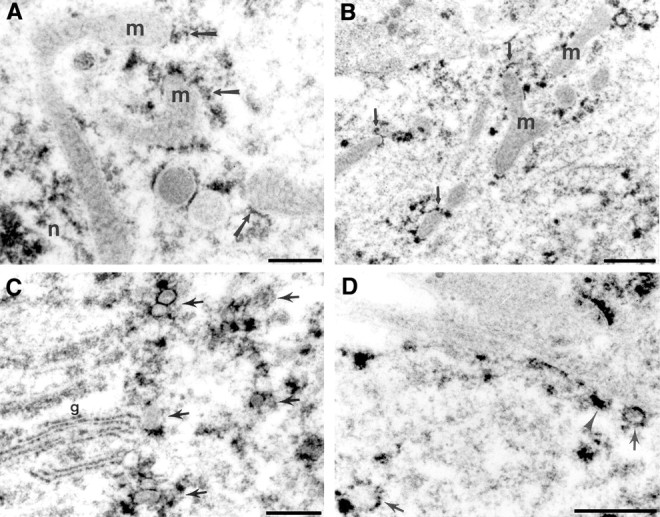
Electron micrographs of EPTA staining in the cell soma (A, C) and apical dendrite (B, D) of CA1 pyramidal neurons in the postischemic brain at 24 hr reperfusion. EPTA-stained protein aggregates (arrows in A,B) were associated with the outer membranes of mitochondria (m), recognized by their double membrane and cristae visible in negative contrast against the lightly stained matrix. In many cases, the stained deposits were localized at the poles of mitochondria (small arrows in_B_). EPTA-stained aggregates were also associated with the membranes of numerous round, clear vesicles, stained in negative contrast (arrows in C, D). In some cases, stained deposits on the membranes of the Golgi apparatus (g in C) were also observed. The vesicles surrounded the Golgi apparatus in the cell soma (arrows in C) and were close to the plasma membrane in dendrites (arrow in_D_). The arrowhead in _D_points to what appears to be a vesicle outlined by EPTA-stained aggregates partially fused with the plasmalemma, suggesting that vesicles are shuttling between the Golgi apparatus and plasma membrane.n, Nucleus. Scale bars: A,C, D, 0.5 μm; B, 1 μm.
EPTA-stained aggregates were observed in almost all CA1 pyramidal neurons, and the results were consistent in all three animals examined at 24 hr of reperfusion after 15 min of ischemia. We have not seen any similar EPTA-stained aggregates in CA1 neurons in any of the sham-operated control rats in this study or in our previous studies on synaptic modifications (Hu et al., 1998; Martone et al., 1999), nor have they been reported in numerous studies using the EPTA staining technique (Bloom and Aghajanian, 1966, 1968; Jones et al., 1974;Burry and Lasher, 1978). Compared with CA1 neurons, EPTA-stained aggregates were rarely found in DG granule neurons in all three animals in each experimental group after ischemia (Fig. 1, DG). Only small numbers of dark vesicles resembling lysosomes could be found surrounding the nuclei in DG neurons at 4 and 24 hr of reperfusion (Fig. 1, DG). Similarly, EPTA-stained aggregates were rarely found in CA3 and most neocortical neurons after ischemia (data not shown). Both of these populations are relatively resistant to ischemic cell death in this model.
By 24 hr of reperfusion, the aggregates were frequently attached to the cytoplasmic face of the outer mitochondrial membrane in both the soma and apical dendrites (Fig. 2A,B). Mitochondria were identified by their inner and outer membrane, clearly visible in negative relief (Fig.2A,B). EPTA also slightly stained the mitochondrial matrix, rendering the cristae visible as well. The aggregates were not distributed on all the mitochondrial membranes but were often located only on the poles (Fig.2A,B). Few aggregates were associated with mitochondria at 4 hr of reperfusion in either the cell soma or dendrites (Fig. 1, CA1, 4h).
EPTA aggregates were consistently observed attached to the cytoplasmic face of vesicles in the cell soma, ranging in size from 50 to 200 nm (Fig. 2C). Occasionally, EPTA also stained the membrane stacks of the Golgi apparatus (Fig. 2C), which were next to concentrations of stained vesicles. In dendrites, vesicles of similar size with associated aggregates were also found close to and attached to the cell membrane in CA1 dendrites (Fig. 2D), suggesting that they shuttled between the Golgi apparatus in the soma and the dendritic membrane, and were likely derived from the Golgi apparatus. In the region of CA1 stratum radiatum, EPTA-stained aggregates attached to the dendritic plasmalemma, as well as mitochondrial outer membranes, at 24 hr of reperfusion (Fig.3).
Fig. 3.
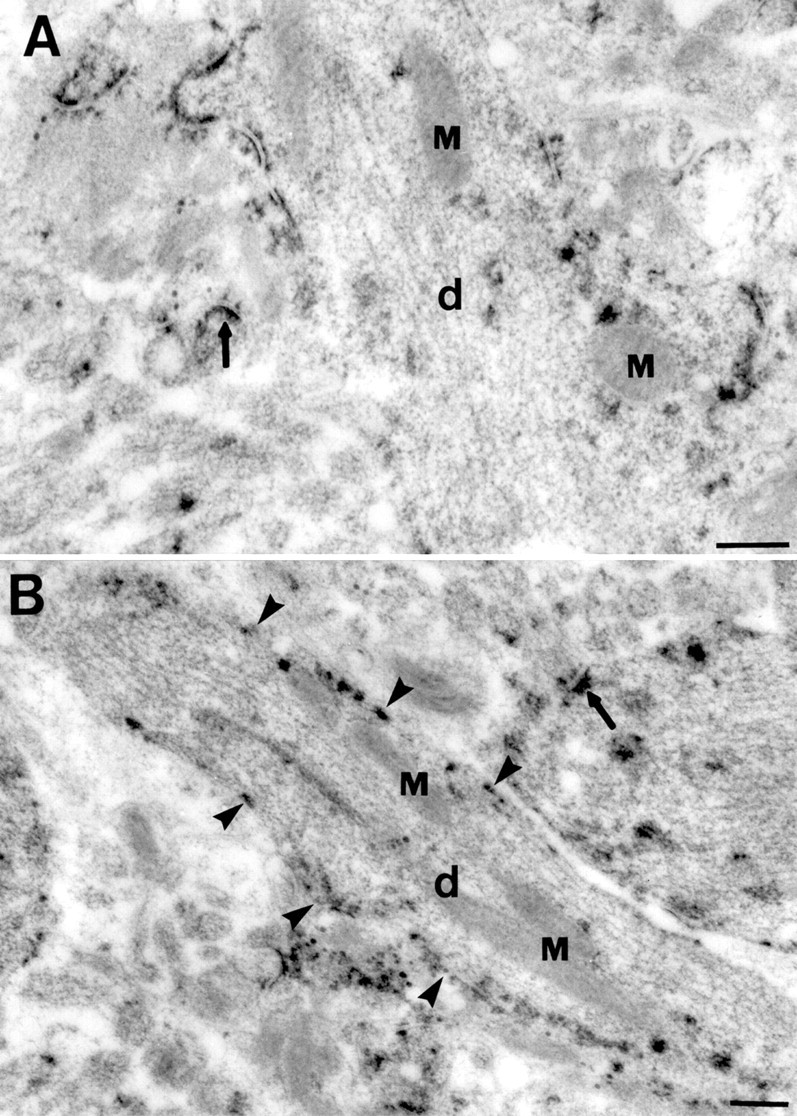
Electron micrographs of EPTA staining in dendrites (d) of CA1 pyramidal neurons in postischemic brains after 24 hr of reperfusion. EPTA preferentially stained postsynaptic densities in the neuropil of hippocampal stratum radiatum (arrows in A and B) The majority of EPTA-stained deposits in dendrites were associated with mitochondria (M) and the cytoplasmic face of the dendritic plasmalemma (arrowheads in_B_). Scale bars, 0.5 μm.
Comparison with osmium–uranium–lead staining
In material stained with conventional heavy metals (osmium–uranium–lead), electron-dense deposits were found in CA1 dying neurons (Fig. 4). Some of the osmium–uranium–lead-stained deposits seemed to colocalize with multivesicular bodies, whereas others appeared to distribute along with membranous structures, such as vesicles and the endoplasmic reticulum (Fig. 4, 24h). The osmium–uranium–lead-stained deposits were superimposed with other stained subcellular structures, and it was usually difficult to determine the relationship between them. The dark aggregates were more prominent and extensively distributed in the EPTA-stained material, and some notable differences in their appearance and distribution were present between the two methods. For example, the dark band surrounding the nuclear membrane observed in the EPTA sections at 4 and 24 hr of reperfusion (Fig. 1, CA1,4h, 24h) was barely visible in the osmium–uranium–lead-stained sections (Fig. 4). The osmium–uranium–lead-stained deposits were discrete and round in appearance with soft edges, whereas the EPTA-stained aggregates were directly apposed to membranous structures and more flocculent in appearance. However, the general distribution and the timing of appearance of the osmium–uranium–lead-stained deposits was similar to the aggregates in the EPTA sections, as was their selective presence in CA1 neurons but not DG neurons (data not shown).
Fig. 4.
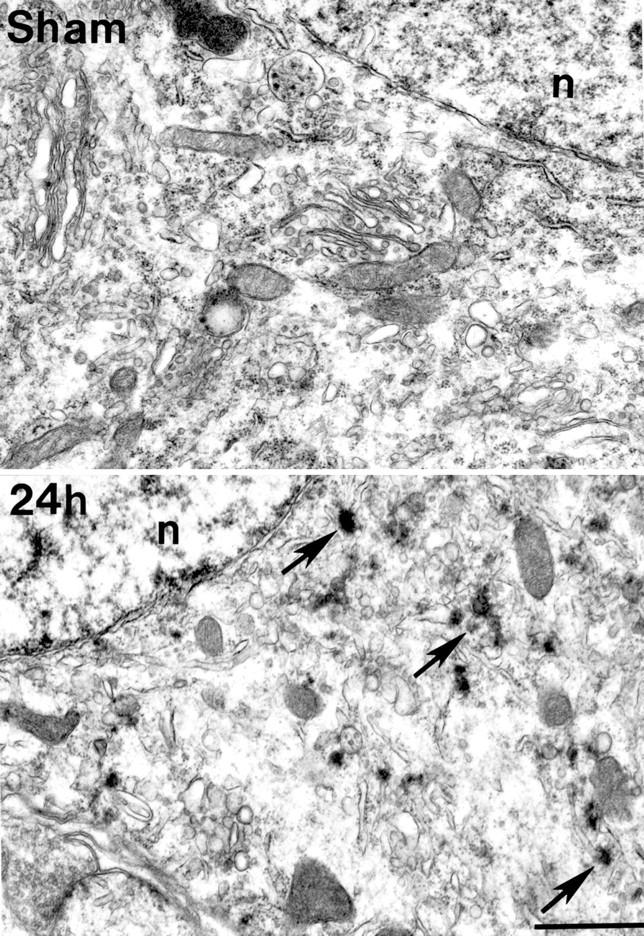
Electron micrographs of conventionally stained pyramidal cell somas in area CA1 from sham-operated controls (Sham) and in postischemic brains at 24 hr reperfusion (24h). Numerous round, electron-dense deposits were observed in the postischemic soma (arrows in_24h_) associated with membranous structures, including vesicles and the endoplasmic reticulum. Similar deposits were not visible in the control brain. n, Nucleus. Scale bar, 1 μm.
Immunolocalization of ubiquitin
Because EPTA stains protein enriched with basic amino acid residues (Bloom and Aghajanian, 1966, 1968), the EPTA-stained aggregates in the postischemic neurons are likely composed of abnormal protein. Taking advantage of the observation that ubi-proteins are commonly present in protein aggregates in neurodegenerative diseases, we conducted ubiquitin immunogold electron microscopy (Fig.5). Immunolabeling was initially performed on material that had been stained using EPTA. However, the EPTA labeling was washed out of the tissue during the immunolabeling procedure. We then performed immunolabeling on sections that had been embedded without either osmication or EPTA staining and then counterstained the grids with a variety of methods, including PTA, osmium, and uranium–lead. In all cases, gold labeling for ubiquitin was clustered over heavy metal-stained aggregates in the postischemic brain. The best contrast was achieved with poststaining with uranium–lead so most analyses were performed on this tissue. Labeled aggregates were found close to the plasmalemma in dendrites (Fig.5A, arrowheads) and apposed to mitochondria and vesicles whose membranes stained in negative relief (Fig.5B), similar to the distribution of the EPTA-stained aggregates. In controls, ubiquitin immunogold labeling was randomly present in all neurons, as its name implies (Fig. 5C,arrowhead). Although several structures in the control tissue were stained with uranium and lead, these structures did not contain ubiquitin immunoreactivity (Fig. 5C). Negative controls in which the primary antibody was omitted showed very low amounts of nonspecific labeling (Fig. 5D). It should be pointed out that the density of the ubiquitin immunogold labeling in the brain sections is less than the densities of protein aggregates seen in EPTA staining and ubiquitin immunostaining of confocal microscopy (see below). This may be because of the limitations of the immunogold staining. It is known that secondary antibody-linked gold particles are often too big to penetrate into tissue efficiently, particularly for the dense protein aggregates. We believe that both EPTA and the secondary antibody for the confocal microscopy (see below) have much more chance to access the antigen relative to the immunogold-linked secondary antibody.
Fig. 5.

Ubiquitin immunogold labeling in the apical dendrites (d) of CA1 pyramidal neurons in the postischemic brain at 24 hr reperfusion (A,B) and in sham-operated control (C). Immunolabeling was performed on sections of brain that were not osmicated before embedding and were then counterstained with uranyl acetate and lead citrate. Heavy immunolabeling for ubiquitin was observed over the dark materials distributed along the dendritic plasmalemma (arrowheads in A) and associated with mitochondria (m in B) and vesicles (arrowhead in B). Immunolabeling in the control brain was usually present in the cytoplasm (arrowhead in C) and not with electron-dense structures. Immunostaining controls in which the primary antibody was omitted showed very little nonspecific labeling (D). Scale bar, 0.5 μm.
To further study the time course and distribution of protein aggregation after ischemia, we investigated ubi-protein aggregation in brain sections by high-resolution laser scanning confocal microscopy using an objective of PlanApo, 100×/1.40 oil. The brain sections were double-labeled with a monoclonal anti- ubiquitin antibody (green) and propidium iodide (red). Ubiquitin immunolabeling was evenly distributed in control neurons and 30 min of reperfusion in CA1, DG (Fig. 6,Sham, 30m), and CA3 and cortical neurons (Fig.7, Sham, 30min). At 4 hr of reperfusion, the immunolabeling pattern was clearly changed from an even distribution to a heterogeneous distribution, with anti-ubiquitin-positive aggregates scattered around the nuclei and in the dendrites, but disappeared in the nuclei (Fig. 6,4h). By 24 hr of reperfusion, the ubiquitin-positive aggregates were further enlarged to form patchy aggregates surrounding nuclei and close to the dendritic plasma membrane in CA1 neurons (Fig.6, 24h). However, in most DG, CA3, and cortical neurons, the ubiquitin immunolabeling had returned to a more even distribution but was induced to greater than control levels (Figs. 6, 7,24h). By 72 hr of reperfusion, the ubiquitin immunolabeling (green) was virtually absent in most CA1 pyramidal neurons (Fig. 6, 72h) but returned to control intensity in most DG, CA3, and cortical neurons (Figs. 6, 7, 72h). The nuclei of CA1 neurons stained with propidium iodide (red) were condensed, because the CA1 neurons were dead at this time point (Fig. 6, 72h).
Fig. 6.
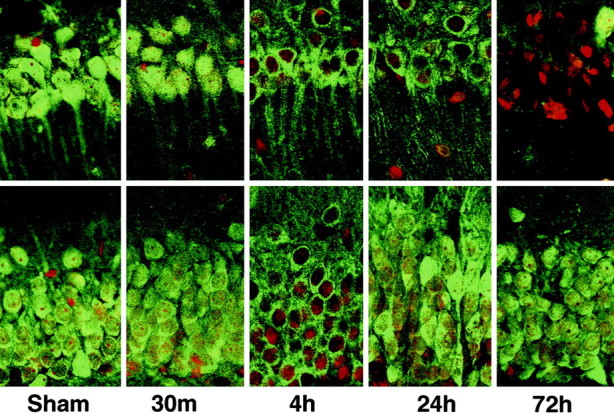
Confocal microscopic images of hippocampal neurons double labeled with anti-ubiquitin (green) and propidium iodide (red) in area CA1 (top panels) and the DG (bottom panels). Sections are shown from sham control (Sham) and from 30 min, and 4, 24, and 72 hr of reperfusion after 15 min of ischemia. The labeling pattern is clearly altered from an even to an aggregated distribution, persistently in CA1 neurons but transiently in DG neurons during reperfusion. Ubiquitin-labeled aggregates first appear as small dots at 4 hr reperfusion and progressively increase in size over time. By 24 hr of reperfusion, the aggregates form a patchy pattern surrounding the nuclei and close to the dendritic membrane. Ubiquitin immunostaining in the nuclei disappears after 4 hr of reperfusion in CA1 neurons.
Fig. 7.
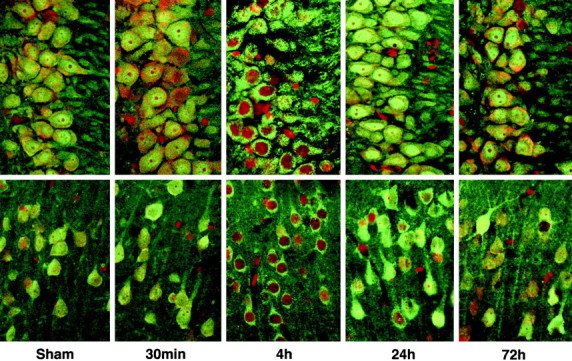
Confocal microscopic images of hippocampal area CA3 (top panels) and the neocortex (bottom panels) double labeled with anti-ubiquitin (green) and propidium iodide (red). Sections are shown from sham control (Sham) and from 30 min, and 4, 24, and 72 hr of reperfusion after 15 min of ischemia. The labeling pattern is transiently altered from an even to an aggregated distribution at 4 hr of reperfusion.
DISCUSSION
In this study, we demonstrated that intracellular membranous proteins were severely aggregated in dying CA1 neurons but not in neurons destined to survive after transient cerebral ischemia. These protein aggregates did not exist in control brains. The protein aggregates were clearly visible in postischemic CA1 neurons stained with EPTA and progressively accumulated during the postischemic phase until cell death. They were mainly located on membranes of intracellular vesicles and distributed in the cytoplasm of the cell body and close to dendritic membranes at 4 hr of reperfusion. By 24 hr of reperfusion, these aggregates also appeared on the membranes of mitochondria, the Golgi apparatus, and dendritic plasmalemma. The aggregates were strongly labeled with a ubiquitin antibody by immunogold electron microscopy. High-resolution confocal microscopy further confirmed that ubiquitin-immunoreactive aggregates were strongly and progressively accumulated around nuclei and close to the dendritic membrane in most CA1 neurons until their death. However, in neurons destined to survive, ubiquitin immunoreactivity showed only a transient redistribution at 4 hr of reperfusion. The pattern of ubiquitin immunostaining seen in the confocal images in the later period of reperfusion is similar to the distribution of EPTA-stained protein aggregates observed under the electron microscope.
Identity of the aggregates
At the electron microscopic level, the presence of abnormal protein aggregates in CA1 pyramidal neurons was most clearly demonstrated using the EPTA staining method of Bloom and Aghajanian (1966, 1968). It has been known for a long time that EPTA strongly stains proteins in synapses and nuclei but only slightly reacts with other subcellular structures as viewed by electron microscopy (Bloom and Aghajanian, 1968). Protein aggregates in cells consist of non-native polypeptide chains with very high density and are generally believed to aggregate through their hydrophobic segments. It is likely that the hydrophobic segments are concentrated inside, whereas segments rich in polar amino acids are exposed outside of the aggregates. Thus, staining of the aggregates with EPTA may be attributed to the very high density of the amino acid chains, as well as to the rearrangement of basic amino acids in the aggregates in the postischemic neurons. The identification of the stained deposits as aggregates of abnormal proteins is supported by the finding that the aggregates were strongly labeled with a ubiquitin antibody.
The present study is pertinent to two previous studies. It was first described in an early conventional (osmium–uranium–lead) EM study byKirino et al. (1984) that dark substances exist in postischemic neurons, which was later confirmed (Deshpande et al., 1992). However, the identity of these dark substances and their mode of formation were unknown. Also, the relationship of the dark substances with the subcellular structures was unclear. We also found the presence of osmium–uranium–lead-stained deposits in the CA1 neurons from the same brain tissues for the EPTA EM. Based on their overall distribution and their selective localization in CA1 neurons, we believe that the osmium–uranium–lead-stained deposits are a combination of normal membranous structures and some protein aggregates. This explanation is consistent with the finding that the ubiquitin immunogold-labeled aggregates can be stained with both PTA and uranium–lead. The protein aggregates were both more prominent and more extensively distributed in the EPTA-stained material and, as noted in Results, clearly differed somewhat in appearance. Part of the difference in stained materials between the two methods can be attributed to the lack of osmication before dehydration in the EPTA-stained sections. The lack of osmium leads to both extraction of many lipids from the tissue and also a lack of contrast of remaining membranous and other osmiophilic structures. Thus, EPTA-stained materials stand out from a relatively unstained background. Part of the difference between the two methods may also be attributable to differing affinities of heavy metal stains for certain subcellular structures. This may explain that why the EPTA EM shows some protein aggregates that cannot be seen with conventional EM.
Distribution of protein aggregates
ETPA-stained protein aggregates accumulate over time in almost all CA1 pyramidal neurons but rarely in DG, CA3, and most cortical neurons in the postischemic phase. They are not present in control neurons. The localization of the large ubiquitin-immunoreactive aggregates surrounding nuclei and along the dendritic membrane seen by confocal microscopy (Fig. 6, 24h) resembles the distribution of EPTA-stained aggregates under EM in the late period of reperfusion (Fig. 3), as is consistent with the labeling of the heavy metal-stained deposits with ubiquitin by immunoelectron microscopy (Fig. 5).
It is intriguing that ubiquitin immunostaining pattern transiently changes from relatively even to heterogeneous at 4 hr of reperfusion in most DG, CA3, and cortical neurons, but these populations do not show EPTA-stained aggregates under the electron microscope. On Western blots, increases in ubi-proteins in the membrane fraction peak as early as 30 min of reperfusion (earliest reperfusion point we studied) and then declines after 4 hr of reperfusion in CA1 neurons (B. R. Hu and B. K. Siesjö, unpublished data). The changes of ubi-proteins also occur, although to a less extent, in the surviving neuronal populations. However, even in CA1 neurons, EPTA-stained protein aggregates are not present at 30 min but appear at 4 hr and then progressively accumulate until neuronal death selectively in CA1 neurons. The evidence suggests that abnormal proteins become ubiquitinated on the membranous structures immediately after ischemia. These ubi-proteins may not completely aggregate into the visible EPTA-stained protein aggregates under the electron microscope. Meanwhile, the cell defense system for abnormal proteins may come into play to protect and/or to clear some of ubi-proteins immediately after ischemia. For unknown reasons, the cell defense system might be weaker or production of abnormal proteins might be greater in CA1 dying neurons than in surviving neurons after ischemia, which may result in severe and selective protein aggregation in CA1 dying neurons, eventually visible with ETPA staining by EM.
Protein aggregation as a cause of neuronal death
The cause and effect relationship between protein aggregation and subsequent neuronal death remains to be determined. This study has mainly shown that the distribution and time courses of protein aggregation correlate well with the distribution of the neuronal death. The studies of biochemical, genetic, or pharmacological interventions are essential for uncovering the roles of protein aggregation after brain ischemia. For this reason, we have recently been studying ischemic preconditioning on protein aggregation and the subsequent neuronal death and survival. Our results show that ischemic preconditioning can prevent both protein aggregation and neuronal death after ischemia (B. R. Hu and C. L. Liu, unpublished data). Taking into consideration that ischemic preconditioning induces heat shock proteins (HSPs) and that chaperone function of most HSPs is to prevent proteins from aggregation, ischemic preconditioning may protect neurons through preventing protein aggregation. The results of the present study suggest a new hypothesis for cell death occurring after ischemia. We propose that the progressive accumulation of protein aggregates in CA1 neurons may contribute to ischemic neuronal death in a number of ways. (1) The protein aggregates observed on the nuclear membrane after 4 hr of reperfusion may impair nuclear membrane function. (2) Accumulation of protein aggregates in the Golgi apparatus may damage post-translational protein modification, packing, and transportation. (3) Aggregation of abnormal proteins on mitochondrial membranes may result in overproduction of reactive oxygen species and severe secondary energy failure that initiates ischemic neuronal death. (4) Finally, aggregation of abnormal proteins on the neuronal cell membrane may signal microglia or other inflammatory cells to kill the neuron.
The hypothesis that aggregation of abnormal proteins leads to ischemic cell death is consistent with several other observations and mechanisms proposed for ischemic cell death. (1) Induction of HSPs by either preconditioning, viral infection, or transgenic overexpression before ischemia protects the neurons against ischemic insults (Kato et al., 1994; Hutter et al., 1996; Plumier et al., 1997; Yenari et al., 1998;Sharp et al., 1999). We have found that ischemic preconditioning inhibits formation of protein aggregates by confocal microscopy in the same ischemic model (Hu and Liu, unpublished data). (2) Persistent depression of protein synthesis may cause neuronal death (Hossmann, 1993). Abnormal proteins can shut off overall protein synthesis by phosphorylation of eIF-2α in cells of various origins (Matts et al., 1993). Protein synthesis initiation is severely depressed persistently in CA1 dying neurons but transiently in DG neurons after ischemia (Hu and Wieloch, 1993; Burda et al., 1994;DeGracia et al., 1997). (3) Ischemic acidosis and secondary energy failure cause neuronal death (Siesjö et al., 1996). Ischemic acidosis may contribute to protein aggregation because protein folding is pH-dependent (Kraig and Wagner, 1987). (4) Production of reactive oxygen species (Siesjö, 1988; Chan, 1996; Siesjöet al. 1999) after brain ischemia may lead to additional protein oxidation and aggregation. (5) Finally, protein aggregation on the dendritic membranes may activate microglia to induce an inflammatory reaction (Morioka et al., 1991; Gehrmann et al., 1992; Giulian, 1993).
Footnotes
This work was supported by National Institute of Health Grant NS36810 to B.R.H and by the Queen's Emma Foundation in Hawaii. The microscopy was performed, in part, at the National Center for Microscopy and Imaging Research, National Institutes of Health Grant RR04050.
Correspondence should be addressed to Dr. Bing-Ren Hu, Laboratory of Neurochemistry, Center for the Study of Neurological Disease, Queen's Medical Center, 1356 Lusitana Street, 8th Floor, Honolulu, HI 96813. E-mail: bhu@cns.queens.org.
REFERENCES
- 1.Alves-Rodrigues A, Gregori L, Figueiredo-Pereira ME. Ubiquitin, cellular inclusions and their role in neurodegeneration. Trends Neurosci. 1998;21:516–520. doi: 10.1016/s0166-2236(98)01276-4. [DOI] [PubMed] [Google Scholar]
- 2.Bloom FE, Aghajanian GK. Cytochemistry of synapses: selective staining for electron microscopy. Science. 1966;154:1575–1577. doi: 10.1126/science.154.3756.1575. [DOI] [PubMed] [Google Scholar]
- 3.Bloom FE, Aghajanian GK. Fine structural and cytochemical analysis of the staining of synaptic junctions with phosphotungstic acid. J Ultrastruct Res. 1968;22:361–375. doi: 10.1016/s0022-5320(68)90027-0. [DOI] [PubMed] [Google Scholar]
- 4.Burda J, Martin ME, Garcia A, Alcazar A, Fando JL, Salinas M. Phosphorylation of the alpha subunit of initiation factor 2 correlates with the inhibition of translation following transient cerebral ischaemia in the rat. Biochem J. 1994;302:335–338. doi: 10.1042/bj3020335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burry RW, Lasher RT. A quantitative electron microscopic study of synapse formation in dispersed cell cultures of the rat cerebellum stained either by Os-UL or by E-PTA. Brain Res. 1978;147:1–15. doi: 10.1016/0006-8993(78)90768-0. [DOI] [PubMed] [Google Scholar]
- 6.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 7.DeGracia DJ, Sullivan JM, Neumar RW, Alousi SS, Hikade KR, Pittman JE, White BC, Rafols JA, Krause GS. Effect of brain ischemia and reperfusion on the localization of phosphorylated eukaryotic initiation factor 2 alpha. J Cereb Blood Flow Metab. 1997;17:1291–1302. doi: 10.1097/00004647-199712000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Deshpande J, Bergstedt K, Linden T, Kalimo H, Wieloch T (1992) Ultrastructural changes in the hippocampal CA1 region following transient cerebral ischemia: evidence against programmed cell. [DOI] [PubMed]
- 9.Fink AL. Chaperone-mediated protein folding. Physiol Rev. 1999;79:425–449. doi: 10.1152/physrev.1999.79.2.425. [DOI] [PubMed] [Google Scholar]
- 10.Gehrmann J, Bonnekoh P, Miyazawa T, Hossmann KA, Kreutzberg GW. Immunocytochemical study of an early microglial activation in ischemia. J Cereb Blood Flow Metab. 1992;12:257–269. doi: 10.1038/jcbfm.1992.36. [DOI] [PubMed] [Google Scholar]
- 11.Giulian D. Reactive glia as rivals in regulating neuronal survival. Glia. 1993;7:102–110. doi: 10.1002/glia.440070116. [DOI] [PubMed] [Google Scholar]
- 12.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 13.Hossmann K-A. Disturbances of cerebral protein synthesis and ischemic cell death. Prog Brain Res. 1993;96:167–177. doi: 10.1016/s0079-6123(08)63265-3. [DOI] [PubMed] [Google Scholar]
- 14.Hu BR, Wieloch T. Stress-induced inhibition of protein synthesis initiation: modulation of initiation factor 2 and guanine nucleotide exchange factor activity following transient cerebral ischemia in the rat. J Neurosci. 1993;13:1830–1838. doi: 10.1523/JNEUROSCI.13-05-01830.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu BR, Park M, Martone ME, Fischer WH, Ellisman MH, Zivin JA. Assembly of proteins to postsynaptic densities after transient cerebral ischemia. Neuroscience. 1998;18:625–633. doi: 10.1523/JNEUROSCI.18-02-00625.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hutter JJ, Mestril R, Tam EK, Sievers RE, Dillmann WH, Wolfe CL. Overexpression of heat shock protein 72 in transgenic mice decreases infarct size in vivo. Circulation. 1996;94:1408–1411. doi: 10.1161/01.cir.94.6.1408. [DOI] [PubMed] [Google Scholar]
- 17.Jones DG, Dittmer MM, Reading LC. Synaptogenesis in guinea-pig cerebral cortex: a glutaraldehyde–PTA study. Brain Res. 1974;70:245–259. doi: 10.1016/0006-8993(74)90315-1. [DOI] [PubMed] [Google Scholar]
- 18.Kakizuka A. Protein precipitation: a common etiology in neurodegenerative disorders? Trends Genet 1998. 1998;14:396–402. doi: 10.1016/s0168-9525(98)01559-5. [DOI] [PubMed] [Google Scholar]
- 19.Kato H, Liu Y, Kogure K, Kato K. Induction of 27-kDa heat shock protein following cerebral ischemia in a rat model of ischemic tolerance. Brain Res. 1994;634:235–244. doi: 10.1016/0006-8993(94)91926-7. [DOI] [PubMed] [Google Scholar]
- 20.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 21.Kirino T, Tamura A, Sato K. Delayed neuronal death in the rat hippocampus following transient forebrain ischemia. Acta Neuropathol. 1984;64:139–147. doi: 10.1007/BF00695577. [DOI] [PubMed] [Google Scholar]
- 22.Kraig RP, Wagner RJ. Acid-induced changes of brain protein buffering. Brain Res. 1987;410:390–394. doi: 10.1016/0006-8993(87)90345-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martone ME, Jones YZ, Young SJ, Ellisman MH, Zivin JA, Hu BR. Modification of postsynaptic densities after transient cerebral ischemia: a quantitative and three-dimensional ultrastructural study. Neuroscience. 1999;19:1988–1997. doi: 10.1523/JNEUROSCI.19-06-01988.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matts RL, Hurst R, Xu Z. Denatured proteins inhibit translation in hemin-supplemented rabbit reticulocyte lysate by inducing the activation of the heme-regulated eIF-2 alpha kinase. Biochemistry. 1993;32:7323–7328. doi: 10.1021/bi00080a001. [DOI] [PubMed] [Google Scholar]
- 25.Morimoto T, Ide T, Ihara Y, Tamura A, Kirino T. Transient ischemia depletes free ubiquitin in the gerbil hippocampal CA1 neurons. Am J Pathol. 1996;148:249–257. [PMC free article] [PubMed] [Google Scholar]
- 26.Morioka T, Kalehua AN, Streit WJ. The microglial reaction in the rat dorsal hippocampus following transient forebrain ischemia. J Cereb Blood Flow Metab. 1991;11:966–973. doi: 10.1038/jcbfm.1991.162. [DOI] [PubMed] [Google Scholar]
- 27.Petito CK, Pusinelli WA. Delayed neuronal recovery and neuronal death in rat hippocampus following severe cerebral ischemia: possible relationship to abnormalities in neuronal processes. J Cereb Blood Flow Metab. 1984;4:194–205. doi: 10.1038/jcbfm.1984.28. [DOI] [PubMed] [Google Scholar]
- 28.Plumier JC, Krueger AM, Currie RW, Kontoyiannis D, Kollias G, Pagoulatos GN. Transgenic mice expressing the human inducible Hsp70 have hippocampal neurons resistant to ischemic injury. Cell Stress Chaperones. 1997;2:162–167. doi: 10.1379/1466-1268(1997)002<0162:tmethi>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 30.Rafols JA, Daya AM, O'Neil BJ, Krause GC, Neumar RW, White BC. Global brain ischemia and reperfusion: Golgi apparatus ultrastructure in neurons selectively vulnerable to death. Acta Neuropathol. 1995;90:17–30. doi: 10.1007/BF00294455. [DOI] [PubMed] [Google Scholar]
- 31.Sharp FR, Massa SM, Swanson RA. Heat-shock protein protection. Trends Neurosci. 1999;22:97–99. doi: 10.1016/s0166-2236(98)01392-7. [DOI] [PubMed] [Google Scholar]
- 32.Siesjö BK. Historical overview. Calcium, ischemia, and death of brain cells. Ann NY Acad Sci. 1988;522:638–661. doi: 10.1111/j.1749-6632.1988.tb33410.x. [DOI] [PubMed] [Google Scholar]
- 33.Siesjö BK, Katsura KI, Kristian T, Li PA, Siesjo P. Molecular mechanisms of acidosis-mediated damage. Acta Neurochir Suppl (Wien) 1996;66:8–14. doi: 10.1007/978-3-7091-9465-2_2. [DOI] [PubMed] [Google Scholar]
- 34.Siesjö BK, Hu BR, Kristian T. Is the cell death pathway triggered by the mitochondrion or the endoplasmic reticulum? J Cereb Blood Brain Metab. 1999;19:19–26. doi: 10.1097/00004647-199901000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Smith ML, Bendek G, Dahlgren N, Rosen I, Wieloch T, Siesjö BK. Models for studying long-term recovery following forebrain ischemia in the rat. A 2-vessel occlusion model. Acta Neurol Scand. 1984;69:385–401. doi: 10.1111/j.1600-0404.1984.tb07822.x. [DOI] [PubMed] [Google Scholar]
- 36.Yenari MA, Fink SL, Sun GH, Chang LK, Patel MK, Kunis DM, Onley D, Ho DY, Sapolsky RM, Steinberg GK. Gene therapy with HSP72 is neuroprotective in rat models of stroke and epilepsy. Ann Neurol. 1998;44:584–491. doi: 10.1002/ana.410440403. [DOI] [PubMed] [Google Scholar]