CD95 Ligand (Fas-L/APO-1L) and Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Mediate Ischemia-Induced Apoptosis in Neurons (original) (raw)
Abstract
Programmed cell death plays an important role in the neuronal degeneration after cerebral ischemia, but the underlying mechanisms are not fully understood. Here we examined, in vivo and_in vitro_, whether ischemia-induced neuronal death involves death-inducing ligand/receptor systems such as CD95 and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). After reversible middle cerebral artery occlusion in adult rats, both CD95 ligand and TRAIL were expressed in the apoptotic areas of the postischemic brain. Further recombinant CD95 ligand and TRAIL proteins induced apoptosis in primary neurons and neuron-like cells in vitro. The immunosuppressant FK506, which most effectively protects against ischemic neurodegeneration, prevented postischemic expression of these death-inducing ligands both in vivo and in vitro. FK506 also abolished phosphorylation, but not expression, of the c-Jun transcription factor involved in the transcriptional control of CD95 ligand. Most importantly, in lpr mice expressing dysfunctional CD95, reversible middle cerebral artery occlusion resulted in infarct volumes significantly smaller than those found in wild-type animals. These results suggest an involvement of CD95 ligand and TRAIL in the pathophysiology of postischemic neurodegeneration and offer alternative strategies for the treatment of cardiovascular brain disease.
Keywords: CD95 ligand, TRAIL, apoptosis, focal ischemia, neurons, lpr mouse
Focal ischemia in the mammalian brain provokes irreversible necrotic damage and generates areas “at risk” that may, with time, undergo apoptosis (Linnik et al., 1993;MacManus et al., 1993; Li et al., 1995; Vexler et al., 1997). In non-neuronal cells death-inducing ligands (DILs) such as CD95 ligand (CD95-L, also called APO-1L/Fas-L), tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL, also called APO-2L), and TNF-α are critically involved in the induction of apoptosis (Nagata, 1997). Few findings are reported about the involvement of DILs in apoptotic death in the adult nervous system, such as TNF-α production and expression of CD95 receptor (also called Fas or APO-1) in the postischemic brain (Matsuyama et al., 1995; Saito et al., 1996) or the cooperation of CD95-L with TNF-α for propagation of apoptosis in glioma cells (Mizuno and Yoshida, 1996). In the immature brain, CD95-L was found to be expressed in neurons, but the absence of a concomitant expression of its receptor questions the involvement of CD95-L at least in developmental neuronal death (French et al., 1996;Becher et al., 1998). However, in the adult brain the role of CD95-L and TRAIL under pathophysiological conditions remains to be defined.
Ischemic injury evokes a cellular stress response, which involves the activation of c-Jun N-terminal kinases/stress-activated protein kinases (JNK/SAPKs) (Knight and Buxton, 1996; Onishi et al., 1997; Herdegen et al., 1998). After translocation into the nucleus, JNK/SAPKs activate the c-Jun transcription factor by phosphorylation of the serine 63 and 73 residues in the transactivation domain (Hibi et al., 1993; Kyriakis et al., 1994; Kallunki et al., 1996) with subsequent induction of c-Jun/AP-1 target genes, including c-jun itself (Angel et al., 1988), TNF_-α (Kraemer et al., 1995), and_CD95-L (Faris et al., 1998; Kasibhatla et al., 1998). c-Jun is a potent inducer of apoptosis in several cell lines, including neuronal cells (Schlingensiepen et al., 1993; Bossy-Wetzel et al., 1997; Eilers et al., 1998; Watson et al., 1998). In hippocampal neurons, the disruption of the JNK3 locus prevents phosphorylation of c-Jun and protects against excitotoxic death (Yang et al., 1997). In the adult brain, the pattern of c-Jun phosphorylation and its link to apoptotic processes after neuronal injury are poorly understood.
In the present study, we have examined the participation of DILs and c-Jun in neuronal apoptosis induced by ischemia in vivo or other forms of cellular stress in vitro. We report that focal ischemia induces N-terminal phosphorylation of c-Jun and expression of CD95-L, TRAIL, and TNF-α. The capacity of DILs to induce apoptosis in neurons was proven in vitro, where treatment with DILs of primary neurons and neuron-like cells led to death of the majority of cells. FK506, an immunosuppressant shown to reduce ischemic damage in the brain (Sharkey and Butcher, 1994; Drake et al., 1996), completely prevented acute and delayed phosphorylation of c-Jun, expression of DILs, and concomitantly, the occurrence of apoptosis. lpr mice, deficient in a functionally active receptor for CD95-L, were significantly resistant against ischemia-induced neuronal damage. Our findings demonstrate that DILs such as CD95-L and TRAIL confer neuronal apoptosis after ischemia in the adult brain and that suppression of DILs, e.g., by FK506, may improve the neuronal survival after ischemic injury.
MATERIALS AND METHODS
Ischemic model. In male Sprague Dawley rats and female Bl/6 and lpr (Bl/6 background) mice, focal cerebral ischemia was induced by occlusion of the middle cerebral artery (MCA) as described previously (Zea Longa et al., 1989). A surgical nylon thread was advanced from the lumen of the common carotid artery up to the anterior cerebral artery to block the origin of the MCA for 90 min. MCA blood flow was restored by withdrawing the nylon thread. Deep anesthesia was reached by intraperitoneal injection of pentobarbital (100 mg/kg body weight) for rats or ketamine (150 mg/kg body weight) for mice. Animals were kept under anesthesia, and rectal temperature was controlled at or near 37°C with heating lamps throughout both the surgical procedure and the MCA occlusion period. After recirculation (12 and 24 hr and 3 and 5 d) for rats (each n = 5) and 24 hr for mice (each group n = 9), animals were deeply reanesthetized and killed by decapitation or intracardial perfusion. FK506 (1 mg/kg body weight) was applied intravenously between 5 and 15 min after the MCA occlusion. Sex differences between rats and mice are attributable to the availability in our animal facilities. In any case, we did not find sex differences in previous experiments, and the actual ones were exclusively performed within same-sex groups.
Cell culture and experimental treatment in vitro. Primary neuronal cultures were prepared from day 15 fetal rats as previously described (Dawson et al., 1991). In brief, cortical neurons were obtained after trituration in MEM with 20% horse serum, 25 mm glucose, and 2 mml-gutamine after a 30 min digestion in 0.025 trypsin/saline solution. Cells were plated in 24-well plates coated with polyornithine. After 4 d, cells were treated with 5-fluoro-2-deoxyuridine for another 4 d to inhibit proliferation of non-neuronal cells. Thereafter cell cultures were maintained in MEM, 10% horse serum, 25 mm glucose, and 2 mml-glutamine in an 8% CO2humidified incubator at 37°C. Neurons were allowed to mature for 12 d in culture before being used for experiments.
NT2 teratocarcinoma cells were grown and differentiated essentially as described previously (Pleasure et al., 1992), with the following modifications. NT2 cells were grown in serum-DMEM (DMEM supplemented with 10% fetal bovine serum, 5% horse serum, 100 U/ml penicillin, and 100 μg/ml streptomycin) at 37°C and 10% CO2. Differentiation was promoted with 10 μmretinoic acid for a total of 5 weeks. Cells were then enzymatically detached with 1 ml of trypsin/EDTA, mixed with 24 ml of serum-DMEM, and replated on a collagen-coated 15 cm dish. After 2 d, neuronal cells that were growing on top of a layer of non-neuronal cells were selectively removed by a short trypsination step with 1 ml of trypsin/EDTA, mixed with 10 ml of medium, and replated on a 10 cm dish. After 30 min, the supernatant that contained mostly neurons was transferred to a fresh collagen-coated 10 cm dish and treated with serum supplemented with 10 μm fluorodeoxyuridine, 10 μm uridine, and 1 μm cytosine arabinoside twice a week for a total of 2 weeks. The cells were enzymatically removed with 0.5 ml of trypsin/EDTA, mixed with 4.5 ml of serum-DMEM, and plated at a density of 10,000 cells/cm2 on collagen-coated tissue culture dishes or coverslips and used after 3 d. Collagen was prepared from rat tails by acetic acid extraction. Plate surfaces were coated with 5 μg/cm2 collagen in 20 mm acetic acid for 1 hr. Coverslips were pretreated with 100 μg/ml poly-l-lysine in borate buffer (1.24 gm of boric acid and 1.9 gm of Na-tetraborate in 400 ml of water).
The human neuroblastoma cell line SHEP and the human leukemic T cell line Jurkat were grown in RPMI-1640 medium supplemented with 10% heat-inactivated FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, 25 mm HEPES, and 2 mml-glutamine. Cycloheximide was used to block the synthesis of antiapoptotic molecules and to render SHEP cells sensitive to death-inducing, ligand-mediated apoptosis (von Reyher et al., 1998).
Cytostatic drugs were obtained from Sigma (Deisenhofen, Germany), all other cell culture products were obtained from Life Technologies (Paisley, Scotland). Doxorubicin was dissolved in sterile water, supplemented with 96% ethanol to a 90% stock solution, and stored in aliquots at −80°C. Cycloheximide stocks were dissolved in dimethylsulfoxide, and aliquots were stored at −20°C. Recombinant CD95-L (Alexis Corp., Grünberg, Germany) was freshly dissolved in medium. Recombinant TNF-α protein (Boehringer Mannheim, Mannheim, Germany) and recombinant TRAIL protein were prepared as described (Jeremias et al., 1998). FK506 stock solution (Fujisawa Pharmaceutical Co. Ltd., Osaka, Japan) was freshly dissolved in medium. Finally, the vehicles were diluted to 0.1%, a concentration that had no influence on apoptosis or expression of the genes examined. Cells were γ-irradiated in their flasks using a cesium radiator.
Assessment of cell death. Neuroblastoma cells were examined by fluorescence-activated cell sorting (FACS), and the percentage of cell death was determined by forward scatter/side scatter (FSC/SSC) analysis. Death of cortical neurons and NT2-N cells was assessed by trypan blue exclusion.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. Frontal cryostat sections (25 μm) were processed according to the terminal deoxynucleotidyl transferase (TdT)-mediated biotinylated UTP nick end-labeling (TUNEL) technique (Gavrieli et al., 1992). Nuclei were stripped from proteins by incubation in PBS with 1% Triton X-100 at 4°C overnight. Endogenous peroxidase was inactivated by covering the sections with 2% H2O2 for 5 min at room temperature. Sections were incubated with 0.3 μl pg Flu-dUTP (Amersham, Braunschweig, Germany), 1 μl of TdT, 10 μl of 5× TdT buffer, 2 μl of CoCl2 (Boehringer Mannheim), 0.3 μl of dATP (Perkin-Elmer, Norwalk, CT), and 36.4 μl of distilled H2O in a moist chamber at 37°C for 90 min. The reaction was terminated by transferring the slides to Tris-EDTA buffer at room temperature for 15 min. Normal nuclei, which contained only insignificant amounts of DNA 3′-OH ends, did not stain with this technique. Cells with necrotic morphology and detectable concentrations of DNA ends showed a more diffuse labeling compared with apoptotic nuclei. As a control, sections were incubated in the absence of either the enzyme or the nucleotide.
Measurement of infarct extension. lpr and wild-type (Bl/6) mice (each n = 9) were subjected to MCA filament occlusion for 90 min and reperfused for 24 hr as described. Forebrains were cut, and cryostat sections (20 μm thick), 400 μm apart, were silver-stained. In brief, sections were impregnated with a silver nitrate-lithium carbonate solution for 2 min and developed with a hydrochinone-formaldehyde solution for 3 min (Vogel et al., 1999). Stained sections were directly scanned (MCID-M4, 3.0; Imaging Research, St. Catherine’s, Ontario, Canada). The volume of infarction was determined by numeric integration of the scanned areas of marked pallor corrected for brain edema (Swanson et al., 1990;Lin et al., 1993). Significance was measured using a _t_test.
Immunohistochemistry. Coronal cryostat sections (40 μm) were processed for immunohistochemistry in 24-well microtiter plates. Sections were incubated for 48–72 hr with the primary antisera. Immunoreactivities were visualized by either a monoclonal or a polyclonal Cy3- and FITC-labeled secondary antibody (Dianova, Hamburg, Germany). To detect CD95-L we used a mouse monoclonal specific antibody (Transduction Laboratories, Lexington, KY), the monoclonal Nok-1 antibody (PharMingen, Hamburg, Germany), and the polyclonal P62 antibody (kindly provided by Dr. M. Hahne and Dr. J. Tschopp, University of Lausanne, Lausanne, Switzerland). These antibodies yielded similar immunohistochemical patterns for CD95-L (data not shown). For other proteins we used a polyclonal anti-c-Jun antiserum (Kovary and Bravo, 1991), a polyclonal antibody against the serine 73-phosphorylated c-Jun (kindly provided by Dr. M. Karin, University of San Diego, La Jolla, CA), a monoclonal MAP2 antibody (AP2ς, Boehringer Mannheim), and a monoclonal NeuN (antineuronal nuclei) antibody (MAB377; Chemicon, Temecula, CA).
Western blotting. Proteins were analyzed by Western blot analysis using a mouse monoclonal antibody specific for CD95-L (Transduction Laboratories) as described (Herr et al., 1997). Bound antibody was detected with anti-mouse horseradish peroxidase conjugate (Santa Cruz Biotechnology, Santa Cruz, CA) and enhanced chemoluminescence (Amersham).
RT-PCR. RNA preparation and RT-PCR were performed as described (Herr et al., 1996). Primer sequences were as follows: for human, CD95-L, 5′-ATG TTT CAG CTC TTC CAC CTA CAG A-3′ and 5′-CCA GAG AGA GCT CAG ATA CGT TGA C-3′; TRAIL, 5′-CAG GAT CAT GGC TAT GAT GGA GGT C-3′ and 5′-GCT GTT CAT ACT CTC TTC GTC ATT G-3′; _TNF_-α, 5′-ATG AGC ACT GAA AGC ATG ATC C-3′ and 5′-CTG GAG CTG CCC CTC AGC TTG AG-3′; and c-jun, 5′-CAG AGT TGC ACT GAG TGT GGC TG-3′ and 5′-ATG TGT CAA CAG CGC CTG GGC AGC A-3′; for rat, CD95-L, 5′-TTT TTC TTG TCC ATC CTC TG-3′ and 5′-CAG AGG GTG TGC TGG GGT TG-3′; and TRAIL, 5′-GGA CAC CAT TTC TAC AGT TC-3′ and 5′-AGT ATG TTT GGG AAT AGA TG-3′. β-Actin primers were obtained from Stratagene (Heidelberg, Germany). Twenty microliters of the PCR reaction were separated by electrophoresis on agarose gels and visualized after ethidium bromide staining.
RESULTS
Expression of DILs and phosphorylation of c-Jun after focal ischemia in the adult brain
Occlusion of the MCA for 90 min and consecutive recirculation in the adult rat produced a defined ischemic lesion with a necrotic area in the lateral striatum and adjacent neocortex and the penumbra, an area “at risk” where cells undergo apoptosis (Linnik et al., 1993) (Fig. 1a). We examined apoptosis by TUNEL in a subfield comprising the amygdala, piriform cortex, and entorhinal cortex. This region belongs to the penumbra area surrounding the lateral striatum, which even in cases with small infarcted lesions appeared ischemic. Apoptosis became apparent 12 hr after recirculation, reached a maximum at day 3, and was still visible in a substantial number of neurons after 5 d (Fig. 1b; data not shown). The intact nonischemic hemisphere served as control and did not exhibit TUNEL-positive cells (data not shown). In most cases, apoptotic cells were neurons, as detected by TUNEL and counterstaining with NeuN (Fig. 1b–d). Concomitant with the occurrence of apoptotic cells, CD95-L mRNA and protein were expressed between 12 hr and 5 d, with a maximum at day 3 as detected by RT-PCR, Western blotting, and immunohistochemistry (Figs.2a–c,3). In the ischemic penumbra, CD95-L was exclusively expressed in neurons (Fig. 3). In untreated or sham-operated rats the immunoreactivity for CD95-L was completely absent (data not shown). Similar to the kinetics of _CD95-L_expression, TRAIL mRNA levels increased in response to ischemia and recirculation with a maximum after 3 d (Fig.2c). In contrast to TRAIL and CD95-L,TNF-α mRNA was not consistently upregulated in the ischemic hemisphere. TNF-α mRNA exhibited a first peak after 24 hr, followed by a decline after 3 d and a second rise after 5 d (data not shown), consistent with previous findings (Saito et al., 1996).
Fig. 1.
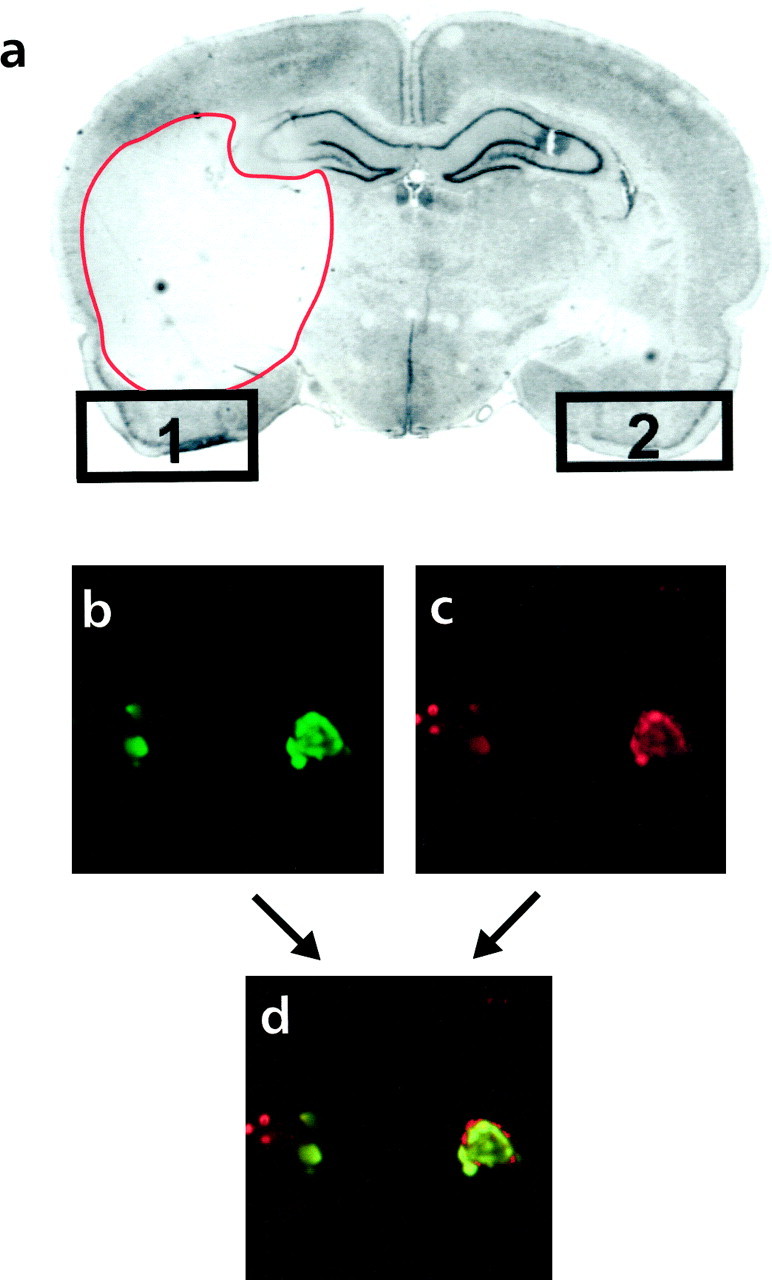
Neuronal cell death in the adult rat brain 3 d after ischemia and reperfusion (n = 5).a, Nissl staining. The ischemic area appears as a_white_ compartment limited with a red line_in the left hemisphere. Regions 1 and_2 mark the piriform cortex, ipsilateral and contralateral to the ischemic lesion, respectively. Similar results were obtained in five different animals. b–d, TUNEL-positive cells from region 1 (b, green) exhibit a neuronal phenotype, as detected by antineuronal nuclei (NeuN) antibody (c, red), as assessed by colocalization of NeuN and TUNEL labeling (d, yellow or red and_green_ in the same nucleus). Photographed with a 60× objective by confocal microscopy.
Fig. 2.
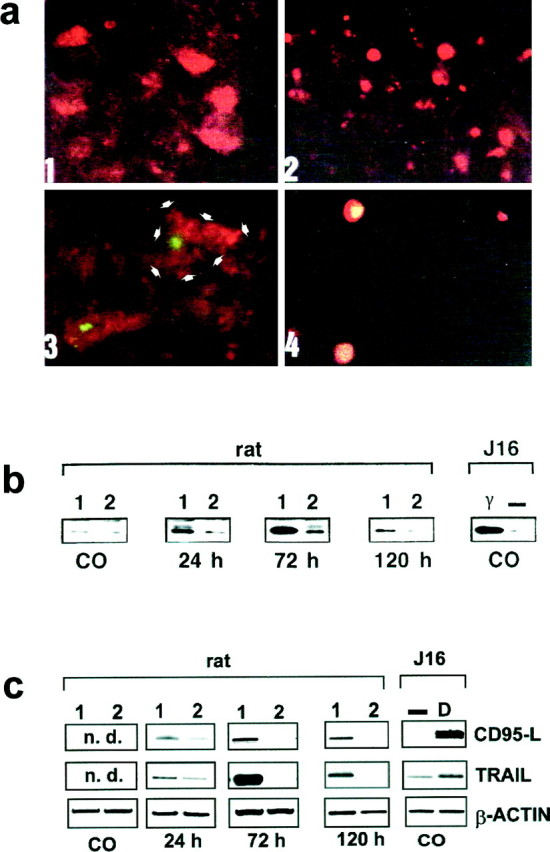
Enhanced phosphorylation of c-Jun and increased expression of CD95-L and TRAIL in the adult rat brain after ischemia (n = 5) and Jurkat-16 cells after γ radiation and doxorubicin (n = 5).a, Immunohistochemical signals from region 1 (see Fig.1a) 3 d after ischemia and recirculation. Photographed with a 40× objective. 1, Cytoplasmic neuronal CD95-L protein; 2, phosphorylation of serine 73 of c-Jun as detected by a Cy3-labeled secondary antibody (red); 3, TUNEL-positive neurons (green) and double labeling of TUNEL with CD95-L (red; the cell boundary is delineated with_arrowheads_); 4, colocalization of phosphorylated serine 73 of c-Jun with TUNEL-positive cells (orange–yellow). b, Expression of CD95-L protein after focal ischemia as detected by Western blotting. The specific band of the 39 kDa CD95-L protein was determined in regions_1_ and 2 (see Fig. 1a) at the indicated time points after ischemia and recirculation and in untreated controls (CO). CD95-L expression was also determined in leukemic Jurkat-16 (J16) T cells either untreated (−) or 3 d after γ irradiation with 10 Gy (γ). Equal protein loading was ensured by Ponceau red staining.c, Induction of CD95-L and_TRAIL_ mRNA after focal ischemia and recirculation at the indicated time points and in untreated controls (CO) as revealed by RT-PCR. n.d., Nondetectable. As a positive control, Jurkat cells (J16) were treated with 500 ng/ml doxorubicin, and RNA was isolated 4 hr later.
Fig. 3.

Expression of CD95-L in neurons 3 d after ischemia and reperfusion in the piriform cortex (corresponding to Fig.1a, region 1) as assessed by double labeling with MAP2 (red) and anti-CD95-L (yellow–green). Photographed with a 100× objective.
In sham-operated animals expression of the c-Jun transcription factor was similar to that under basal conditions, and phosphorylation of this factor was undetectable. In the postischemic brain, intense bilateral c-Jun expression was seen in a variety of forebrain areas, including the piriform and entorhinal cortex. By contrast, phosphorylation of c-Jun was virtually restricted to the penumbra area, and the distribution of the phospho-c-Jun labeled nuclei was congruent with the TUNEL-positive neurons (Fig. 2a). After 12 hr, ∼80% of the phospho-c-Jun-positive neurons were TUNEL-positive, and 23% of the TUNEL-positive neurons contained phospho-c-Jun. After 3 d, these values were ∼52 and 30%, respectively (data not shown).
The pattern of increased expression of _CD95-L, TRAIL,_TNF-α, and c-Jun in neurons after ischemia with recirculation resembles that seen in response to other forms of cellular stress. This is demonstrated in leukemic T cells (Jurkat), which have been γ-irradiated or treated with the cytotoxic drug doxorubicin, an anthracycline used in chemotherapy of human neurons (Fig.2b,c).
Induction of apoptosis by DILs in neurons_in vitro_
To directly support the possibility that these DILs mediate ischemia-induced apoptosis in the adult brain, we evaluated cell death after treatment of various neuronal populations with recombinant CD95-L, TRAIL, and TNF-α proteins (Fig.4). These ligands induced >60% cell death in primary and model (NT2-N) neurons. The latter cells, derived from a human teratocarcinoma cell line (NT2) by retinoic acid treatment, exhibit many of the features of fully polarized, postmitotic human CNS neurons, as shown by the presence of MAP2 (Fig.4d) (Pleasure et al., 1992). We found also that, like primary neurons, NT2N cells can express the receptors for DILs and be sensitive to DIL-induced apoptosis. In contrast, SHEP cells exposed to CD95-L and TNF-α exhibited a similar increase only after sensitization with cycloheximide, whereas TRAIL induced apoptosis in >60% of the cells both with or without cycloheximide (Fig. 4).
Fig. 4.

Induced specific cell death in neuroblastoma cells (SHEP; a), cortical neurons (b), and NT2-N cells (c) measured 24 hr after treatment with recombinant TNF-α, TRAIL, or CD95-L protein (each 100 ng/ml) alone (gray bars) or in the presence of 500 ng/ml cycloheximide (black bars). Cell death was assessed in SHEP cells by FACS analysis using FSC/SSC analysis and in cortical neurons and NT2-N cells by trypan blue exclusion. More than 90% of the NT2-N cells exhibited a neuronal phenotype, as assessed by immunostaining against MAP2 (d).Co, control.
FK506 suppresses c-Jun phosphorylation _in vivo_and DIL expression in vivo and in vitro
Recently it has been demonstrated that the immunosuppressant FK506 triggers neuroprotection after cerebral ischemia (Sharkey and Butcher, 1994; Drake et al., 1996) and prevents CD95-L-triggered apoptosis in non-neuronal cells (Brunner et al., 1996). Intravenous application of FK506 5–15 min after MCA occlusion reduced the area of infarction in the rat brain (Fig. 5b). Importantly, FK506 completely suppressed the expression of CD95-L (Fig.5c) and TRAIL (data not shown) and the occurrence of apoptotic nuclei. Moreover, FK506 prevented phosphorylation of c-Jun without affecting the distribution of c-Jun-immunoreactive neuronal nuclei (Fig. 5c). Finally, we investigated the effect of FK506 on DILs in neuronal cells in the absence of immune or glial cells. Therefore, neuroblastoma cells were incubated with doxorubicin, which triggers a cellular stress response (Herr et al., 1997) similar to γ irradiation (Chen et al., 1996) or ischemia. This drug induced upregulation of CD95-L, TRAIL, TNF-α, and c-jun_mRNA within 16 hr, as examined by RT-PCR (Fig. 5a). Coincubation with FK506 completely suppressed the doxorubicin-induced expression of_CD95-L, TRAIL, TNF-α and c-jun mRNA.
Fig. 5.
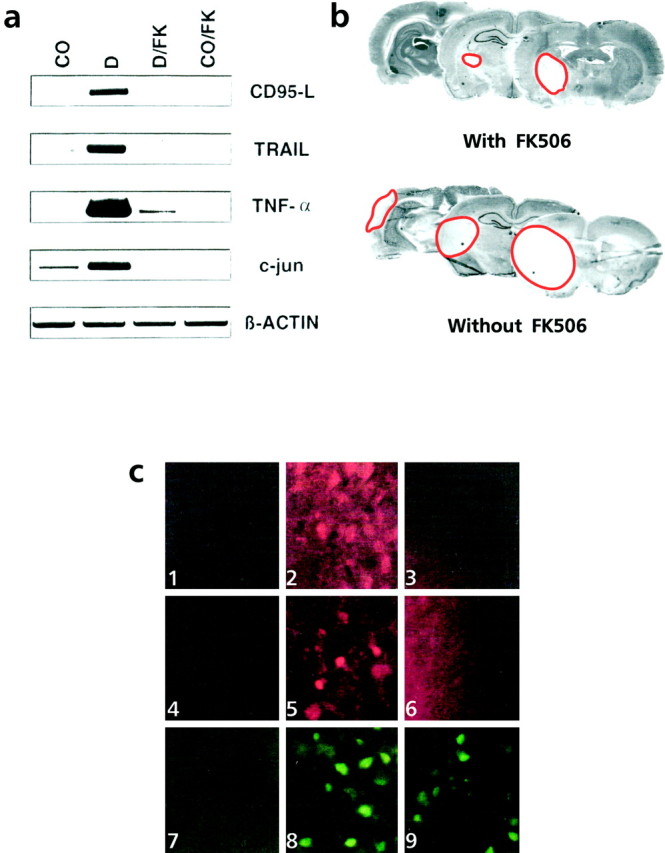
FK506 inhibits apoptosis and ischemia- or stress-induced upregulation of death-inducing ligands, c-jun, and c-Jun phosphorylation in vitro_and in vivo. a, Neuroblastoma cells (5 × 104 cells per well) were left either untreated (CO) or stimulated with 1 μg/ml doxorubicin alone (D), with doxorubicin in presence of 100 nm FK506 (D/FK), or with 100 nm FK506 alone (CO/FK) for 16 hr.CD95-L, TRAIL, TNF-α, and c-jun mRNA expression was assessed by RT-PCR. Expression of the_β-actin gene served as control for equal conditions.b, Reduction of the ischemic area 3 d after ischemia and reperfusion by FK506 as revealed by Nissl staining. The infarcted area is marked by a red line.c, Expression of CD95-L (1–3), phosphorylation of serine 73 of c-Jun (4–6), and c-Jun (7–9) was examined by immunohistochemistry in region 1 (see Fig.1) of untreated rats (1, 4, 7) 3 d after ischemia and recirculation (2, 5, 8) or 3 d after ischemia and recirculation with intravenous application of FK506 5–15 min after ischemia (3, 6, 9). FK506 antagonized the expression of CD95-L (3) and the phosphorylation of c-Jun (6) but did not affect the c-Jun expression (9). Similar results were obtained in five different experiments.
Resistance against focal ischemia in the mutant_lpr_ mouse
To further elucidate the involvement of DILs in postischemic neurodegeneration, we evaluated the extent of brain injury after MCA occlusion in lpr mice with mutant CD95. As control, we used Bl/6 mice to avoid known differences in infarct susceptibility depending on the genetic background of the mice used (Barone et al., 1993; Connolly et al., 1996). Wild-type and mutant mice were subjected to 90 min of MCA occlusion followed by 24 hr of reperfusion, after which the extent of infarct was determined by silver staining (Vogel et al., 1999) (Fig. 6b). The mean infarct volume in control animals was in good concordance with similar models from other groups (Eliason et al., 1997). The infarct volume was reduced by ∼66% (p < 0,004) in_lpr_ mice compared with wild-type mice [18.26 ± 5.78 (SE) vs 50.67 ± 8.73 mm3; each_n_ = 9; Fig. 6a_]. Interestingly, the surrounding cortex (penumbra area) was always spared in the_lpr mice (Fig. 6b). These results demonstrate an involvement of CD95 and other death systems such as TRAIL in ischemia-induced apoptosis.
Fig. 6.

Protection against focal ischemia in_lpr_ mice. a, Infarct volume after transient focal ischemia in wild-type (Control) and lpr mice. Data are presented as the means ± SEM (Control, n = 9;Lpr, n = 9). Significance was determined by t test. **p < 0.004. Data from individual animals are plotted as separate points overlaid on each histogram bar. Infarcted volume was determined by numeric integration of the scanned non-silver-impregnated areas corrected for brain edema. b, Infarct area (white area marked with red line) in the wild-type (Control) and lpr mice as revealed by silver staining. Note that in the lpr mouse despite an infarct volume (∼37 mm3) superior to the mean infarct volume (∼18 mm3), the surrounding cortex remains spared.
DISCUSSION
This study provides evidence that the neuronal suicide program after brain ischemia depends on the expression of the DILs CD95-L and TRAIL. We detected upregulation of these ligands in the postischemic brain and proved, in vitro, their potency to induce death in primary neurons, model neurons (NT2-N; Pleasure et al., 1992), and neuroblastoma (SHEP) cells. These findings imply that enhanced expression of DILs is involved in the neuronal apoptotic program in the adult nervous system. Alternatively, the presence of CD95-L in the developing brain might help in maintaining immune privilege rather than mediating neuronal death (French et al., 1996). The latter assumption relies on the findings that lpr mice, deficient in the CD95/CD95-L pathway, exhibit no abnormalities during embryonic development, and CD95 is undetectable in the immature brain. By contrast, expression of CD95 has been reported in neurons of the postischemic adult brain (Matsuyama et al., 1995). Indeed, we show that adult lpr mice are highly resistant to ischemia-induced neuronal damage. In these mice the cortex, an area where in wild-type animals most of the TUNEL-positive cells are found, was always spared. No significant differences in blood pressure or blood gases between wild-type and lpr mice were found, which could be responsible for nonspecific neuroprotection (data not shown). Thus, the reduction of infarcted volume in lpr mice does not result from hemodynamic alterations. Therefore, the CD95/CD95-L pathway might contribute to postischemic neurodegeneration in the adult CNS. For TRAIL, the cellular distribution remains to be defined, but induction of TRAIL in neuron-derived neuroblastoma cells and its ability to induce death in cortical and NT2-N neurons suggest the capacity of neurons to express TRAIL. The expression of DILs in the ischemic compartments of the brain and the propagation of death in neurons by DILs, together with the resistance of lpr mice to brain ischemia, allow the conclusion that CD95-L and, probably, TRAIL are executors of ischemia-mediated neuronal apoptosis.
Activation of the c-Jun transcription factor by N-terminal phosphorylation of its serine 73 residue in the ischemic hemisphere can be considered as a transcriptional effector for those apoptotic mechanisms depending on de novo synthesis. c-Jun-mediated neuronal death (Schlingensiepen et al., 1993; Ham et al., 1995) depends on N-terminal phosphorylation of c-jun by JNK/SAPKs (Virdee et al., 1997; Eilers et al., 1998; Watson et al., 1998). Recent data have provided evidence that c-Jun confers its apoptotic action by induction of CD95-L and TNF-α genes (Kraemer et al., 1995; Faris et al., 1998;Kasibhatla et al., 1998). Our data suggest that this transcriptional control might also be effective in the adult brain, because expression of the nonphosphorylated (i.e., inactive) form of c-Jun is not paralleled by either expression of CD95-L or appearance of TUNEL-positive cells. Moreover, activation of JNK/SAPK and subsequently c-Jun has been reported to lie downstream of the death domain of DIL receptors (Wilson et al., 1996; Goillot et al., 1997), resulting in a reinforcing apoptotic feedback.
Our observations on DIL expression and selective phosphorylation of c-Jun after ischemic injury in the adult brain opens a new dimension for understanding neurodegenerative disorders. Although the function of DILs is well characterized in mitotic cells such as lymphocytes, keratinocytes, and hepatocytes, the present data on DIL induction in postmitotic neurons suggest that cellular stress activates similar programs irrespective of the cellular phenotype. Therefore, therapeutic interventions that are successful in immunological disorders might also counteract the neuronal cell death program. The immunosuppressant FK506, which prevents activation of immune cells (Brunner et al., 1996), readily passes the blood–brain barrier and confers neuroprotection against ischemic injury (Sharkey and Butcher, 1994;Drake et al., 1996). Furthermore, we found that FK506 suppresses the induction of DILs in the adult brain and in cultures of neuron-derived neuroblastomas. Moreover, FK506 prevented the N-terminal phosphorylation of c-Jun in vivo. FK506 blocks the calcium-dependent activation of the phosphatase calcineurin (Snyder and Sabatini, 1995; Gold, 1997). Block of calcineurin could interfere with c-Jun-mediated transcription because the antiapoptotic protein Bcl-2 inhibits the activation of the calcineurin-mediated nuclear translocation of NF-AT (Shibasaki et al., 1997), which is a c-Jun-containing transcription factor. Most importantly, FK506 ameliorates mitochondrial dysfunction after transient focal cerebral ischemia (Nakai et al., 1997), a crucial event in the cellular commitment to the apoptotic program (Kroemer et al., 1997).
Our data shed new light on the mechanisms that propagate ongoing neuronal damage after ischemia in the adult mammalian brain and provide molecular targets for therapeutic intervention. Strategies aimed to repress either the death-inducing ligands TRAIL, CD95-L, and TNF-α or their transcriptional activators such as c-Jun open new perspectives for the treatment of stroke.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Research Grant He 1561 and University of Heidelberg Research Grant 72/96. We thank Dr. R. Bravo and Dr. M. Karin for providing anti-c-Jun antiserum and antiphosphorylated serine 73 c-Jun antibody, respectively. FK506 was generously provided by Dr. K. Muramoto (Fujisawa Pharmaceutical).
Drs. Martin-Villalba and Herr both contributed equally. Drs. Herdegen and Debatin share senior authorship.
Correspondence should be addressed to Dr. Klaus-Michael Debatin, University Children’s Hospital, Prittwitzstrasse 43, 89075 Ulm, Germany.
REFERENCES
- 1.Angel P, Hattori K, Smeal T, Karin M. The jun protooncogene is positively autoregualted by its product Jun/AP-1. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- 2.Barone FC, Knudsen DJ, Nelson AH, Feuerstein GZ, Willette RN. Mouse strain differences in susceptibility to cerebral ischemia are related to cerebral vascular anatomy. J Cereb Blood Flow Metab. 1993;13:683–692. doi: 10.1038/jcbfm.1993.87. [DOI] [PubMed] [Google Scholar]
- 3.Becher B, D’Souza SD, Troutt AB, Antel JP. Fas expression on human fetal astrocytes without susceptibility to Fas-mediated cytotoxicity. Neuroscience. 1998;84:627–634. doi: 10.1016/s0306-4522(97)00455-7. [DOI] [PubMed] [Google Scholar]
- 4.Bossy-Wetzel E, Bakiri L, Yaniv M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997;16:1695–1709. doi: 10.1093/emboj/16.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunner T, Yoo NJ, La Face D, Ware CF, Green DR. Activation-induced cell death in murine T cell hybridomas. Differential regulation of Fas (CD95) versus Fas ligand expression by cyclosporin A and FK506. Int Immunol. 1996;8:1017–1026. doi: 10.1093/intimm/8.7.1017. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y-R, Meyer CF, Tan T-H. Persistent activation of c-Jun N-terminal kinase 1 (JNK1) in γ-radiation-induced apoptosis. J Biol Chem. 1996;271:631–634. doi: 10.1074/jbc.271.2.631. [DOI] [PubMed] [Google Scholar]
- 7.Connolly ES, Winfree CJ, Stern DM, Solomon RA, Pinsky DJ. Procedural and strain-related variables significantly affect outcome in a murine model of focal cerebral ischemia. Neurosurgery. 1996;38:523–531. doi: 10.1097/00006123-199603000-00021. [DOI] [PubMed] [Google Scholar]
- 8.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drake M, Friberg H, Boris-Möller F, Sakata K, Wieloch T. The immunosuppressant FK506 ameliorates ischemic damage in the rat brain. Acta Physiol Scand. 1996;158:155–159. doi: 10.1046/j.1365-201X.1996.535298000.x. [DOI] [PubMed] [Google Scholar]
- 10.Eilers A, Whitfield J, Babij C, Rubin LL, Ham J. Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J Neurosci. 1998;18:1713–1724. doi: 10.1523/JNEUROSCI.18-05-01713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eliason MJL, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang Z-Q, Dawson TM, Snyder SH, Dawson VL. Poly (ADP-ribose)polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 12.Faris M, Kokot N, Latinis K, Kasibhatla S, Green DR, Koretzky GA, Nel A. The c-Jun N-terminal kinase cascade plays a role in stress-induced apoptosis in Jurkat cells by up-regulating Fas Ligand expression. J Immunol. 1998;160:134–144. [PubMed] [Google Scholar]
- 13.French LE, Hahne M, Viard I, Radlgruber G, Zanone R, Becker K, Müller C, Tschopp J. Fas and Fas ligand in embryos and adult mice: ligand expression in several immune-privileged tissues and coexpression in adult tissues characterized by apoptotic cell turnover. J Cell Biol. 1996;133:335–343. doi: 10.1083/jcb.133.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goillot E, Raingeaud J, Ranger A, Tepper RI, Davis RJ, Harlow E, Sanchez I. Mitogen-activated protein kinase-mediated Fas apoptotic signaling pathway. Proc Natl Acad Sci USA. 1997;94:3302–3307. doi: 10.1073/pnas.94.7.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gold B. FK506 and the role of immunophilins in nerve regeneration. Mol Neurobiol. 1997;15:285–306. doi: 10.1007/BF02740664. [DOI] [PubMed] [Google Scholar]
- 17.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Zaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 18.Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin M. Lasting N-terminal phosphorylation of c-Jun and activation of JNK/SAPK kinases following neuronal injury. J Neurosci. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herr I, Balemans L, Böhler T, Walczak H, Debatin K-M. Monitoring of CD95 (APO-1/Fas) ligand expression in human T cells by quantitative RT-PCR. Cell Death Differ. 1996;3:299–305. [PubMed] [Google Scholar]
- 20.Herr I, Wilhelm D, Böhler T, Angel P, Debatin K-M. Activation of CD95 (APO-1/FAS) signaling by ceramide mediates cancer therapy-induced apoptosis. EMBO J. 1997;16:6200–6208. doi: 10.1093/emboj/16.20.6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 22.Jeremias I, Herr I, Boehler T, Debatin K-M. TRAIL/APO-2-ligand-induced apoptosis in human T cells. Eur J Immunol. 1998;28:143–152. doi: 10.1002/(SICI)1521-4141(199801)28:01<143::AID-IMMU143>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 23.Kallunki T, Deng TL, Hibi M, Karin M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–939. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- 24.Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR. DNA damaging agents induce expression of Fas-ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kB and AP-1. Mol Cell. 1998;1:543–551. doi: 10.1016/s1097-2765(00)80054-4. [DOI] [PubMed] [Google Scholar]
- 25.Knight R, Buxton DB. Stimulation of c-Jun kinase and mitogen-activated protein kinase by ischemia and reperfusion in the perfused rat heart. Biochem Biophys Res Commun. 1996;218:83–88. doi: 10.1006/bbrc.1996.0016. [DOI] [PubMed] [Google Scholar]
- 26.Kovary K, Bravo R. Expression of different Jun and Fos proteins during the G0 to G1 transition in mouse fibroblasts: in vitro and in vivo associations. Mol Cell Biol. 1991;11:2451–2459. doi: 10.1128/mcb.11.5.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kraemer B, Wiegmann K, Kronke M. Regulation of the human TNF promoter by the transcription factor Ets. J Biol Chem. 1995;270:6577–6583. doi: 10.1074/jbc.270.12.6577. [DOI] [PubMed] [Google Scholar]
- 28.Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- 29.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad M, Avruch J, Woodgett RJ. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Chopp M, Jiang N, Zhang ZG, Zaloga C. Induction of DNA fragmentation after 10 to 120 minutes of focal cerebral ischemia in rats. Stroke. 1995;26:1252–1258. doi: 10.1161/01.str.26.7.1252. [DOI] [PubMed] [Google Scholar]
- 31.Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- 32.Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002–2008. doi: 10.1161/01.str.24.12.2002. [DOI] [PubMed] [Google Scholar]
- 33.MacManus JP, Buchan AM, Hill IE, Rasquinha I, Preston E. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neurosci Lett. 1993;164:89–92. doi: 10.1016/0304-3940(93)90864-h. [DOI] [PubMed] [Google Scholar]
- 34.Matsuyama T, Hata T, Yamamoto Y, Tagaya M, Akita H, Uno H, Wanaka A, Furujama J-I, Sugita M. Localization of Fas antigen mRNA induced in postischemic murine forebrain by in situ hybridization. Mol Brain Res. 1995;34:166–172. doi: 10.1016/0169-328x(95)00162-l. [DOI] [PubMed] [Google Scholar]
- 35.Mizuno M, Yoshida J. Tumor necrosis factor-α gene transfer augments anti-Fas antibody-mediated apoptosis in human glioma cells. Jpn J Cancer Res. 1996;87:543–547. doi: 10.1111/j.1349-7006.1996.tb00257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 37.Nakai A, Kuroda S, Kristian T, Siesjö BK. The immunosuppressant drug FK506 ameliorates secondary mitochondrial dysfunction following transient focal cerebral ischemia in the rat. Neurobiol Dis. 1997;4:288–300. doi: 10.1006/nbdi.1997.0146. [DOI] [PubMed] [Google Scholar]
- 38.Onishi I, Tani T, Hashimoto T, Shimizu K, Yagi M, Yamamoto K, Yoshioka K. Activation of c-Jun N-terminal kinase during ischemia and reperfusion in mouse liver. FEBS Lett. 1997;420:201–204. doi: 10.1016/s0014-5793(97)01517-2. [DOI] [PubMed] [Google Scholar]
- 39.Pleasure SJ, Page C, Lee VMY. Pure, postmitotic, polarized human neurons derived from NTera 2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J Neurosci. 1992;12:1802–1815. doi: 10.1523/JNEUROSCI.12-05-01802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saito K, Suyama K, Nishida K, Sei Y, Basile AS. Early increases in TNF-alpha, IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci Lett. 1996;206:149–152. doi: 10.1016/s0304-3940(96)12460-5. [DOI] [PubMed] [Google Scholar]
- 41.Schlingensiepen KH, Schlingensiepen R, Kunst M, Klinger I, Gerdes W, Seifert W, Brysch W. Opposite functions of Jun-B and c-Jun in growth regulation and neuronal differentiation. Dev Genet. 1993;14:305–312. doi: 10.1002/dvg.1020140408. [DOI] [PubMed] [Google Scholar]
- 42.Sharkey J, Butcher SP. Immunophilins mediate the neuroprotective effects of FK506 in focal cerebral ischaemia. Nature. 1994;371:336–339. doi: 10.1038/371336a0. [DOI] [PubMed] [Google Scholar]
- 43.Shibasaki F, Kondo E, Akagi T, McKeon F. Suppression of signalling through transcription factor NF-AT by interactions between calcineurin and bcl-2. Nature. 1997;386:728–731. doi: 10.1038/386728a0. [DOI] [PubMed] [Google Scholar]
- 44.Snyder SH, Sabatini DM. Immunophilins and the nervous system. Nat Med. 1995;1:32–37. doi: 10.1038/nm0195-32. [DOI] [PubMed] [Google Scholar]
- 45.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 46.Vexler ZS, Roberts TPL, Bolen AW, Derugin N, Arieff AI. Transient cerebral ischemia. J Clin Invest. 1997;99:1453–1459. doi: 10.1172/JCI119304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Virdee K, Bannister AJ, Hunt SP, Tolkovsky AM. Comparison between the timing of JNK activation, c-Jun phosphorylation, and onset of death commitment in sympathetic neurones. J Neurochem. 1997;69:550–561. doi: 10.1046/j.1471-4159.1997.69020550.x. [DOI] [PubMed] [Google Scholar]
- 48.Vogel J, Möbius C, Kuschinsky W (1999) Early delineation of ischemic tissue in rat brain cryosection by high contrast staining. Stroke, in press. [DOI] [PubMed]
- 49.von Reyher U, Strater J, Kittstein W, Gschwendt M, Krammer PH, Moller P. Colon carcinoma cells use different mechanisms to escape CD95-mediated apoptosis. Cancer Res. 1998;58:526–534. [PubMed] [Google Scholar]
- 50.Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J Neurosci. 1998;18:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson DJ, Fortner KA, Lynch DH, Mattingly RR, Macara IG, Posada JA, Budd RC. JNK, but not MAPK, activation is associated with Fas-mediated apoptosis in human T cells. Eur J Immunol. 1996;26:989–994. doi: 10.1002/eji.1830260505. [DOI] [PubMed] [Google Scholar]
- 52.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the JNK3 gene. Nature. 1997;289:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 53.Zea Longa E, Weinstein PR, Carlson S, Cummis R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]