Involvement of Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) and p53 in Neuronal Apoptosis: Evidence That GAPDH Is Upregulated by p53 (original) (raw)
Abstract
We recently reported that cytosine arabinoside (AraC)-induced apoptosis of cerebellar neurons involves the overexpression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The present study was undertaken to investigate whether p53 and/or Bax overexpression participates in the AraC-induced apoptosis of cerebellar granule cells and, if so, the relationship between p53 induction and GAPDH overexpression in these cells.
AraC-induced apoptosis of cerebellar granule cells was preceded by an increase in levels of p53 mRNA and protein detected between 1 and 8 hr after treatment. The mRNA level for a p53 target gene, Bax, was also increased. The increase in GAPDH mRNA lasted longer than that of either p53 or Bax, and the level of GAPDH protein in the particulate fraction increased after induction of GAPDH mRNA. The antisense oligonucleotide to p53 protected granule cells from AraC-induced chromatin condensation, internucleosomal cleavage, and apoptotic death. The inhibition of p53 expression by the p53 antisense oligonucleotide not only blocked the expression of Bax but also partially suppressed the increased GAPDH mRNA and protein levels. Conversely, the suppression of GAPDH expression and subsequent attenuation of apoptosis of granule cells by GAPDH antisense oligonucleotide did not influence the expression of p53 or Bax. Cerebellar granule cells prepared from p53 knock-out mice were resistant to AraC toxicity, and the p53 gene knock-out suppressed AraC-upregulated GAPDH expression. Moreover, infection of PC12 cells with an adenoviral vector containing p53 gene dramatically increased GAPDH expression and triggered cell apoptosis. These results suggest that AraC-induced apoptosis of cerebellar granule cells involves the expression of both GAPDH and p53 and that, similar to Bax, GAPDH is upregulated by p53 after exposure to the apoptotic insult.
Keywords: p53, GAPDH, cerebellar granule cell, PC12, cytosine arabinoside, apoptosis, adenovirus
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is an essential glycolytic enzyme that is expressed in all prokaryotic and eukaryotic organisms. Its role in cellular metabolism is to convert glyceraldehyde-3-phosphate to 1,3-diphosphoglycerate. Recent studies suggest that GAPDH is a multifunctional protein endowed with a number of diverse activities seemingly independent of its role in glycolysis. These activities include phosphorylating transverse-tubule proteins (Kawamoto et al., 1986), stimulating RNA transcription (Morgenegg et al., 1986), interacting with microtubules (Huitorel and Pantaloni, 1985), influencing RNA catalysis by binding to hammerhead ribozyme (Sioud and Jesperson, 1996), and acting as a diadenosine tetraphosphate binding protein to influence DNA replication and DNA repair (Baxi and Vishwanatha, 1995).
Cytosine arabinoside (1-β-d-arabinofuranosyl cytosine; AraC) is usually thought to kill proliferating cells through incorporation into DNA during replication, followed by chain termination (Hedley and McCulloch, 1996). However, we and others have reported that AraC induces apoptosis of postmitotic cerebellar granular cells cultured from postnatal rats (Dessi et al., 1995; Ishitani and Chuang, 1996; Saunders et al., 1997, 1999). AraC has been shown to induce apoptosis cerebellar granule cells in vitro in a concentration-dependent manner only when added within 24 hr after cell plating (Dessi et al., 1995). Previously, we found that AraC-induced apoptosis of granule cells can be blocked by inhibitors of transcription, translation, and deoxyribonuclease (Ishitani and Chuang, 1996). The AraC-induced apoptosis of granule cells is preceded by an increase in the levels of GAPDH mRNA and protein (Ishitani and Chuang, 1996), which is followed by nuclear translocation of the GAPDH protein (Saunders et al., 1997; Ishitani et al., 1998). Moreover, an antisense oligonucleotide against GAPDH attenuates GAPDH overexpression and nuclear translocation and robustly protects granule cells from apoptosis (Ishitani and Chuang, 1996; Saunders et al., 1997, 1999;Ishitani et al., 1998), suggesting that GAPDH plays a prominent role in AraC-induced apoptotic death. It has also been shown that GAPDH nuclear translocation is involved in apoptosis of non-neuronal and neuronal cells (Sawa et al., 1997).The role of GAPDH in the pathway and the events leading to its induction, however, are unknown.
Accumulating evidence supports the view that p53, a nuclear phosphoprotein, may be necessary for neuronal apoptosis. For example, cerebellar granule cells cultured from p53 null mice are resistant to treatments with excitotoxins (Xiang et al., 1996) or DNA-damaging agents, such as irradiation or AraC (Enokido et al., 1996a,b). Moreover, whole body γ-irradiation induces apoptosis of granule cells in wild-type, but not p53-deficient, neonatal mice (Wood and Youle, 1995). Other in vivo studies have shown that knocking out the p53 gene protects hippocampal neurons from seizure-induced cell death (Morrison et al., 1996), dopamine neurons from_N_-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced toxicity (Trimmer et al., 1996) and the brain from ischemia-induced infarction (Crumrine et al., 1994). Conversely, adenovirus-mediated transfection of the p53 gene in hippocampal or cortical neurons results in the apoptotic death of these neurons (Xiang et al., 1996; Jordan et al., 1997). In view of the emerging evidence suggesting a prominent role for p53 in neuronal apoptosis, this study examined whether p53 and its responsive gene, Bax, are overexpressed during AraC-induced apoptosis of granule cells. In addition, we explored whether GAPDH overexpression is regulated by p53 in granule cells treated with AraC and in other neurally related cell types.
MATERIALS AND METHODS
Primary cultures of cerebellar granule cells and treatment conditions. Cerebellar granule cells were prepared from 8-d-old Sprague Dawley rat pups or 7-d-old wild-type and p53 knock-out mouse pups (Donehower et al., 1992) supplied by Taconic Farms (Germantown, NY), as described previously (Nonaka et al., 1998). Briefly, cerebella were chopped into 400 μm cubes, and the cells were dissociated by trypsinization, followed by DNase treatment. The dissociated cells were resuspended in a basal modified Eagle's medium containing 10% fetal calf serum, 2 mm glutamate, 50 μg/ml gentamicin, and 25 mm KCl. Granule cells from rat were seeded at a density of 2.5 × 105 cells/cm2onto 24-well plates or 100 mm culture dishes and granule cells from mouse onto 96-well plates or 60 mm dishes precoated with poly-l-lysine. To induce cell apoptosis, AraC was added to culture medium to a final concentration of 300 μm 12 hr after cell plating. When used, oligonucleotides were added to the cultures 2 hr before the addition of AraC. After 24 hr of AraC treatment, viability of granule cells grown on plates was measured by MTT assays as described previously (Nonaka et al., 1998). The tetrazolium ring of MTT is reduced by active dehydrogenase in viable mitochondria, forming a blue-colored precipitate that was then dissolved in dimethyl sulfoxide and quantified spectrophotometrically at 540 nm.
Isolation of fragmented DNA. For DNA fragmentation analysis, cells grown on 100 mm dishes were also harvested after treatment essentially as described by Nonaka et al. (1998). Briefly, cells were washed with ice-cold PBS and then lysed in a buffer containing 10 mm Tris-HCl, pH 7.4, 10 mm EDTA, and 0.5% Triton X-100. Lysates were centrifuged at 10,000 × g for 10 min at room temperature. Fragmented DNA in the supernatant was precipitated by the addition of 400 μl of NaI (7.4 m) and 700 μl of isopropanol and then isolated by centrifugation at 12,000 ×g for 15 min. The DNA pellet was dissolved in 10 mm Tris-HCl, pH 7.4, containing 1 mm EDTA [Tris-EDTA (TE)]. The DNA samples were subjected to electrophoresis on 1.2% agarose gel, and the DNA bands were visualized by ethidium bromide staining.
Northern blotting. Cerebellar granule cells were harvested after different time lengths of AraC treatment. Total RNA was extracted from cells with guanidine thiocyanate and isolated by cesium chloride gradient centrifugation as described previously (Fukamauchi et al., 1993), with modifications. After electrophoresis on 1% agarose gel containing formaldehyde, RNA was transferred to a Duralose membrane (Stratagene, La Jolla, CA) and hybridized with cDNA probes for Bax, Bcl-2, p53, and GAPDH, separately. The cDNA probes were labeled with [α-32P]dCTP by the random priming method. Hybridizations were performed at 42°C for 16 hr, followed by two washes at room temperature with 2 × SSC-0.1% SDS and then two additional washes at 50°C in 0.1 × SSC-0.1% SDS. Northern blots were quantified by Betascope Model 603 blot analyzer (Betagen, Waltham, MA).
Western blotting. To measure p53 and Bax protein levels, cells cultured in 60 or 100 mm dishes at indicated times were detached by scraping and sonicated 30 sec in Laemmli lysis buffer containing 6.25 mm Tris-HCl, pH 6.8, 2 mm EDTA, 15% sucrose, 10% glycerol, 3% SDS, and 0.7 m β-mercaptoethanol. For measurement of GAPDH immunoreactive protein, the samples were prepared as reported previously (Ishitani and Chuang, 1996), with slight modifications. Briefly, scraped cells were ruptured by sonication in 0.32m sucrose, and the homogenates were centrifuged at 2 × 105_g_ for 30 min. The particulate fraction (pellet) was resuspended in a small volume of lysis buffer. Protein concentration was determined by using the BCA protein assay kit (Pierce, Rockford, IL), and samples containing 10–30 μg of protein were loaded and separated by 12% SDS-PAGE. Proteins were subsequently transferred to an Immobilon-P membrane (Millipore, Bedford, MA). After blocking for 1 hr in PBST (1 × PBS and 0.1% Tween 20) containing 5% nonfat dry milk, blots were incubated for 1 hr at room temperature with antibodies against GAPDH (Advanced Immunochemical, Long Beach, CA), rat p53 (Pab240), human p53 (DO-1) (Santa Cruz Biotechnology, Santa Cruz, CA), or Bax (Ab-1; Oncogene Research, Cambridge, MA) in PBST containing 3% nonfat milk. Blots were washed four times in PBST during a 40 min period and were then incubated 1 hr with horseradish peroxidase-conjugated second antibodies in PBST containing 3% nonfat dry milk. Immunoreactivities of the protein bands were detected by enhanced chemiluminescent autoradiography (ECL kit; Amersham Pharmacia Biotech, Arlington Heights, IL) as instructed by the manufacturer. Western blots were quantitatively analyzed by capturing images on films using a CCD camera (Sierra Scientific, Sunnyvale, CA) and the Macintosh NIH Image 1.5 software (Wayne Rasband, National Institute of Mental Health, Bethesda, MD).
Morphological evaluation. Changes in nuclear morphology were examined by staining cerebellar granule cells with Hoechst 33258 (Nonaka et al., 1998). Cells growing on 35 mm dishes were fixed with 4% formaldehyde. After gentle washing with cold PBS, cells were stained with Hoechst 33258 (5 μg/ml in PBS) for 5 min at 4°C. Photomicrographs were taken using an inverted UV illumination microscope.
Oligonucleotide preparations. Antisense and sense oligonucleotides to GAPDH and p53 were synthesized by Cruachem (Dulles, VA). The first and last bases of each oligonucleotide were phosphorathioated. The GAPDH oligonucleotide sequence (GAPDH/AS) was 5′-GACCTTCACCATCTTGTCTA-3′, corresponding to a sequence of the rat GAPDH gene flanking the mRNA translation initiation region (GenBank GAPDH cDNA clone M17701). The p53 antisense oligonucleotide (p53/AS) sequence was 5′-GACCTCAGGTGGCTCATACGG-3′, corresponding to residues between 390 and 410 of rat p53 coding sequence (GenBank p53 cDNA clone X13058). The sequences of the GAPDH sense (GAPDH/S) and p53 sense (p53/S) oligonucleotides were the exact inverse of those of their corresponding antisense oligonucleotides. The stock concentration of oligonucleotides was 1 mm in TE buffer, pH 7.4.
Construction of adenoviral vectors and transfection into PC12 cells. The construction of adenovirus expressing human wild-type p53 cDNA (AdWTp53), β-galactosidase gene (Adβ-gal), and adenovirus containing no transgene (AdControl) have been described previously (Katayose et al., 1995). Briefly, the human wild-type p53 gene was cloned to the site downstream of cytomegalovirus promoter and upstream of SV40 polyadenylation sequence in the replication-defective recombinant adenovirus. Recombinant adenoviruses were propagated in human embryonic kidney 293 cells, purified by double cessium chloride gradient centrifugations, titrated, and stored at −70°C.
PC12 cells were maintained in 100 mm dishes containing RPMI medium 1640 plus 12% fetal bovine serum and 6% horse serum. Cells were infected with 20 pfu/cell of recombinant adenoviruses at 24 hr after plating. The infection efficiency of PC12 cells was determined by counting the β-galactosidase-positive cells under a light microscope at 24 hr after infection with Adβ-gal (Li et al., 1998).
RESULTS
Levels of GAPDH, p53, and Bax expression are increased during the AraC-induced apoptosis of cerebellar granule cells
Northern blot hybridization showed that the expression of GAPDH mRNA in freshly cultured granule cells increased from 1 to 24 hr after exposure to AraC (Fig. 1). p53 mRNA levels increased from 1 to 4 hr and then declined, whereas the Bax mRNA levels were elevated between 2 and 12 hr. Thus, the upregulation of GAPDH mRNA lasted longer than that of either p53 or Bax. The expression of Bcl-2 mRNA was very low and appeared to be decreased after 4 hr of AraC treatment.
Fig. 1.

Time course of AraC-induced changes in the mRNA levels of GAPDH, p53, Bax, and Bcl-2 in cerebellar granule cells. AraC was added to culture medium to a final concentration of 300 μm 12 hr after plating. At the indicated times after AraC exposure, cells were harvested for RNA isolation. An aliquot of110 of each sample was used for quantification of total cellular RNA. The RNA was allowed to migrate ∼5 mm from the sample well into 1% agarose gel, and the total RNA was quantified by image analysis (Fukamauchi et al., 1993). The same amount (10 μg) of total RNA from each sample was applied to each well of the agarose gel for Northern blot analysis. A, Northern blots;B, quantified results of blots shown in_A_.
Western blots and their quantified results showed that endogenous cellular p53 protein levels were increased at 1 hr, reached their peak (265% of the control) at 4 hr, and then decreased to less than the baseline value by 12 hr (Fig.2A,B). Levels of cellular Bax protein were increased at 2 hr and peaked at ∼8 hr (155% of the control). Compared with p53 and Bax protein, the GAPDH protein levels in the particulate fraction of AraC-treated granule cells increased rather late; the increase was detected at 8 hr, and reached its maximum (200% of the control) ∼24 hr after exposure.
Fig. 2.

Time course of AraC-induced increases in GAPDH, p53, and Bax protein levels in cerebellar granule cells. Freshly cultured cells were treated with AraC as described in Figure 1. At different time points, cells were collected in lysis buffer and sonicated for 30 sec. For measurement of GAPDH protein, an aliquot containing 10 μg of protein from the particulate fraction of the cells was loaded and separated by SDS-PAGE. For examination of p53 and Bax protein expression, an aliquot containing 30 μg of total protein was used for SDS-PAGE. A, Western blots;B, quantified results of blots shown in_A_. Note that protein levels of GAPDH, p53, and Bax were time dependently changed after AraC treatment.
p53 and GAPDH antisense oligonucleotides protect cerebellar granule cells against AraC-induced apoptosis
Cell viability, as measured by MTT assay at 24 hr after treatment with AraC, was reduced to 40.4 ± 3.2% (n = 4) (Fig. 3). Photomicrographs showed that AraC caused condensed and/or fragmented cell nuclei, which are characteristic of apoptosis (Fig. 4). Exposure to AraC for 24 hr also caused internucleosomal cleavage of the DNA (Fig. 5). To identify whether the increase in p53 expression was directly related to the cell death, granule cells were pretreated with 3 μmp53/AS before exposure to AraC. The cell viability for the p53/AS-pretreated group was 72 ± 4.0% of the control (n = 4), which was significantly greater than that of AraC alone (p < 0.001) (Fig. 3). Pretreatment with the corresponding p53/S was ineffective; cell viability was 44.6 ± 3.3% (_n_ = 4; _p_ > 0.05%). p53/AS, but not p53/S, markedly reduced chromatin condensation and nuclear fragmentation (Fig. 4) and essentially blocked internucleosomal DNA cleavage in AraC-treated cells (Fig. 5). These results suggest that AraC-induced apoptosis of granule cells is not only dependent on GAPDH, but on p53 as well. Moreover, similar to the GAPDH/AS (Ishitani and Chuang, 1996; Saunders et al., 1997), the p53/AS was effective in protecting granule cells from AraC-induced apoptosis.
Fig. 3.
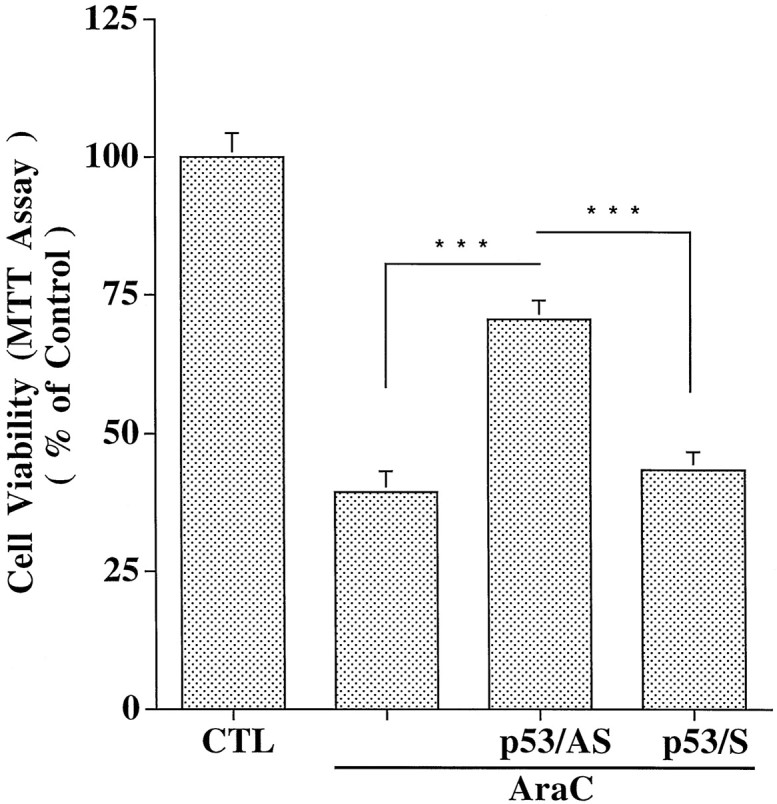
Antisense oligonucleotide to p53 protects against AraC-induced neuronal death. Cerebellar granule cells were pretreated with p53 oligonucleotides (3 μm) for 2 hr before the addition of AraC (300 μm). After 24 hr exposure to AraC, neuronal viability was measured using the MTT colorimetric assay. The results are expressed as percentage of mean ± SEM of control group from three independent experiments.CTL, Control. ***p < 0.001 using one-way ANOVA with Fisher's PLSD test.
Fig. 4.
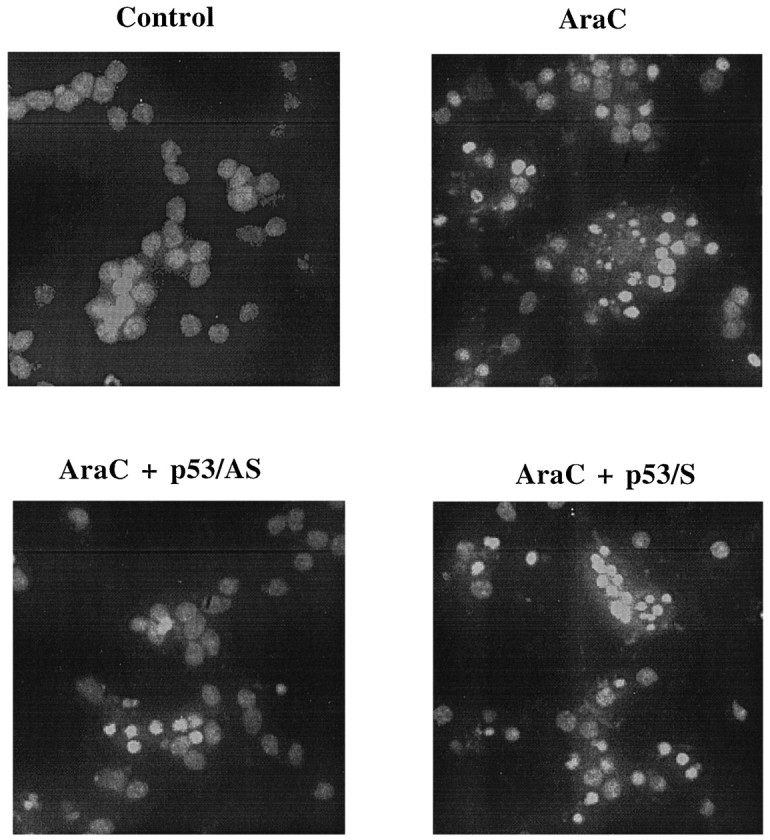
Antisense but not sense oligonucleotide to p53 protects against AraC-induced cell death: morphological studies. Cerebellar granule cells were treated as described in Figure 3. After exposure to AraC for 24 hr, cells were fixed with 4% formaldehyde and then stained with Hoechst dye 33258 (5 μg/ml in PBS) for 5 min at 4°C. Photomicrographs were taken using an inverted UV illumination microscope.
Fig. 5.
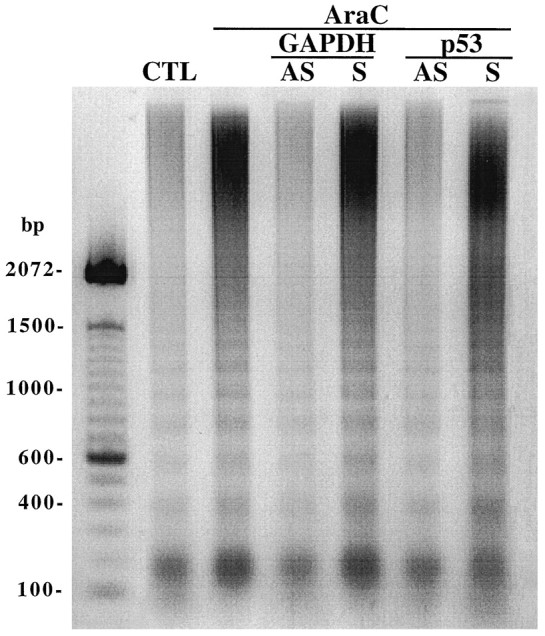
Antisense oligonucleotides to GAPDH and p53 reduce AraC-induced DNA fragmentation. Cerebellar granule cells were treated with AraC as described in Figure 3 and pretreated with GAPDH or p53 oligonucleotides (3 μm) for 2 hr. After exposure to AraC for 24 hr, soluble DNA was extracted from cells and subjected to electrophoresis as described in Materials and Methods. The DNA size markers are shown to the left. AS, Antisense oligonucleotide; S, sense oligonucleotide;CTL, control.
Abrogation of p53 expression suppresses GAPDH mRNA and protein levels
Northern blot results showed that p53/AS, but not p53/S, blocked AraC-induced upregulation of p53 and Bax mRNA, whereas neither GAPDH/AS nor GAPDH/S was effective (Figs. 6,7). AraC-induced GAPDH mRNA upregulation was blocked by GAPDH/AS and partially suppressed by p53/AS (Fig.8). p53/AS also concentration dependently suppressed AraC-increased levels of GAPDH protein in the particulate fraction of granule cells (Fig.9A,B). At concentrations of 1 and 3 μm, p53/AS suppressed upregulated GAPDH protein by 37.6 and 74.7%, respectively, whereas p53/S was ineffective at these two concentrations. At 5 μm, p53/AS completely blocked GAPDH protein upregulation, and p53/S was partially effective. Similar dose-dependent neuroprotective effects of p53/AS and p53/S on AraC-induced apoptosis were observed (data not shown). Therefore, oligonucleotide concentrations higher than 3 μm were not recommended. These results suggest that, during AraC-induced apoptosis of granule cells, GAPDH may function downstream of p53.
Fig. 6.
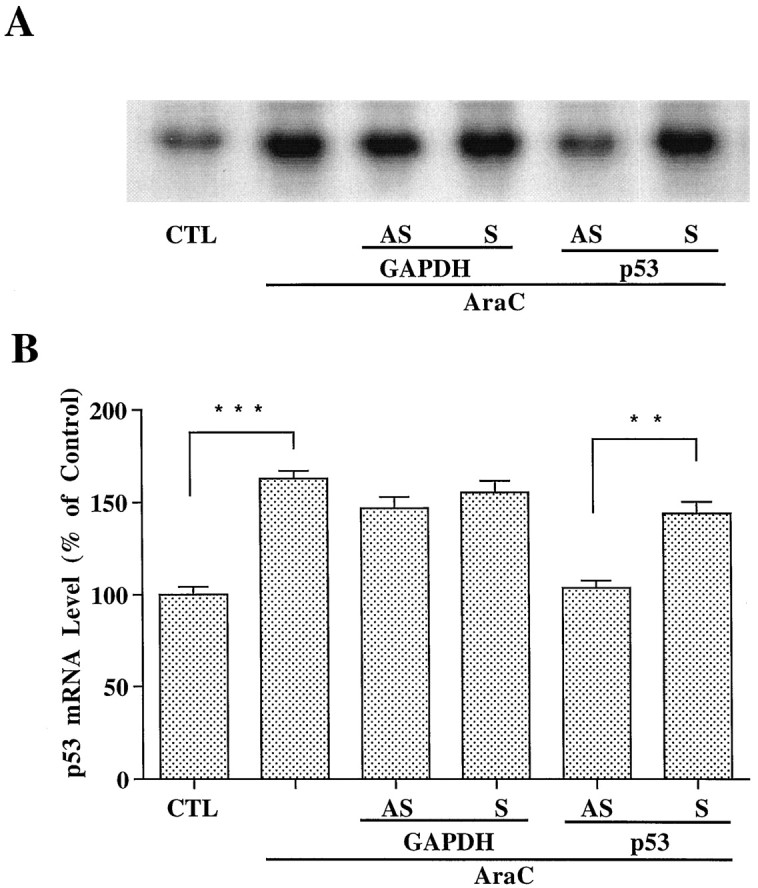
Antisense oligonucleotide to p53 but not GAPDH suppresses AraC-induced increases in p53 mRNA. Cerebellar granule cells were exposed to 300 μm AraC for 12 hr after plating. Cells were pretreated with GAPDH or p53 oligonucleotides for 2 hr before the addition of AraC. RNA was extracted from cells after exposure to AraC for 4 hr. A, Northern blot results of p53 mRNA levels from a typical experiment. B, Levels of p53 mRNA expressed as percent of the control quantitated with a Betascope Model 603 blot analyzer. Each _column_represents the mean ± SEM from three independent experiments.AS, Antisense oligonucleotide; S, sense oligonucleotide; CTL, control. **p< 0.01; ***p < 0.001 using one-way ANOVA with Fisher's PLSD test.
Fig. 7.
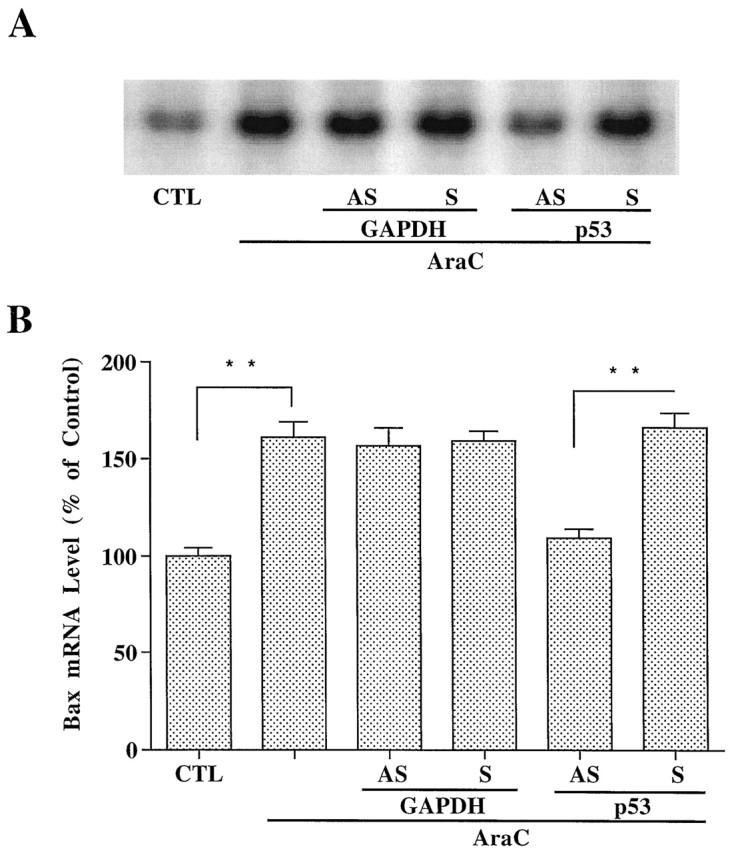
Antisense oligonucleotide to p53 inhibits AraC-induced Bax mRNA upregulation. Cerebellar granule cells were treated with AraC, and RNA was prepared as described in Figure 6.A, Northern blot results of Bax mRNA levels from a typical experiment. B, Levels of Bax mRNA expressed as percent of the control quantitated with a Betascope Model 603 blot analyzer. Each column represents the mean ± SEM from three independent experiments. AS, Antisense oligonucleotide; S, sense oligonucleotide;CTL, control. **p < 0.01 using one-way ANOVA with Fisher's PLSD test.
Fig. 8.
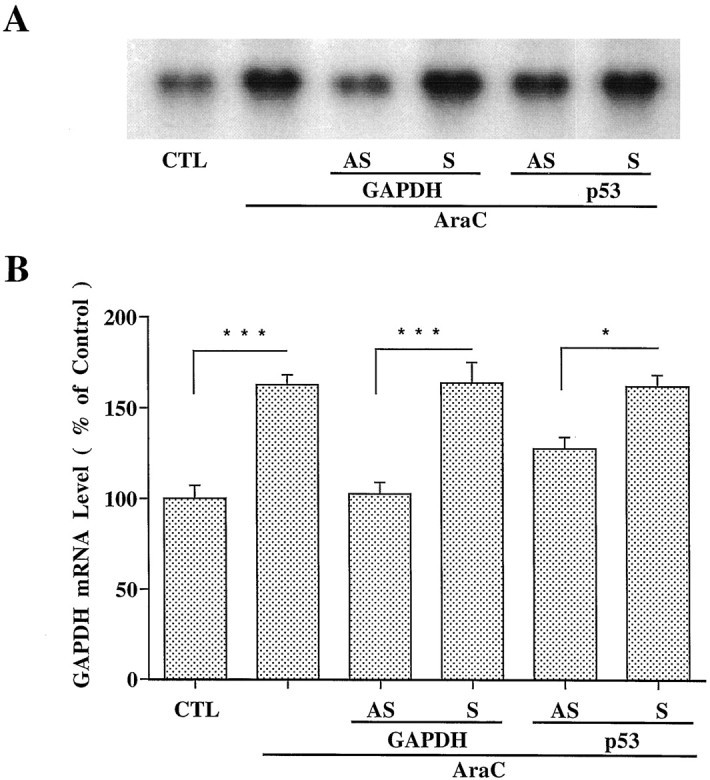
GAPDH and p53 antisense oligonucleotides inhibit AraC-induced GAPDH mRNA increase. Cerebellar granule cells were treated with AraC, and RNA was prepared as described in Figure 6.A, Northern blot results of GAPDH mRNA levels from a typical experiment. B, Levels of GAPDH mRNA expressed as percent of the control quantified with a Betascope Model 603 blot analyzer. Each column represents mean ± SEM from three independent experiments. AS, Antisense oligonucleotide; S, sense oligonucleotide;CTL, control. *p < 0.05; ***p < 0.001 using one-way ANOVA with Fisher's PLSD test.
Fig. 9.

Antisense oligonucleotide to p53 suppresses AraC-induced GAPDH protein increase in the particulate fraction of cerebellar granule cells. Cells were treated with AraC as described in Figure 6. After exposure to AraC for 24 hr, cells were harvested and the particulate (pellet) fraction was prepared as described in Materials and Methods. An aliquot of 10 μg of protein from each particulate fraction was used for Western blot analysis.A, Western blot results of GAPDH protein levels from a typical experiment. B, Quantified results of GAPDH protein levels expressed as percent of mean ± SEM of the untreated control from three independent experiments.CTL, Control. *p < 0.05 compared with the untreated control using one-way ANOVA with Fisher's PLSD test.
p53 gene knock-out in mice inhibits AraC-induced apoptosis and GAPDH expression in cerebellar granule cells
To further explore the role of p53 in mediating GAPDH induction, we used cerebellar granule cells from p53 knock-out mice. AraC treatment induced a concentration-dependent decrease in the viability of granule cells derived from wild type but had little or no effect in those cells derived from p53−/− mice (Fig.10A). Thus, after treatment with 1000 μm AraC for 24 hr, viabilities of granule cells derived from p53+/+ and p53−/− mice were 32.1 ± 1.5 and 81.4 ± 5.8%, respectively. These results confirm that p53 gene knock-out confers resistance to AraC-induced apoptotic cell death (Enokido et al., 1996b). Importantly, GAPDH protein levels were markedly elevated by AraC treatment in granule cells from p53+/+ mice but were only marginally increased in those cells from p53−/− mice (Fig. 10B).
Fig. 10.
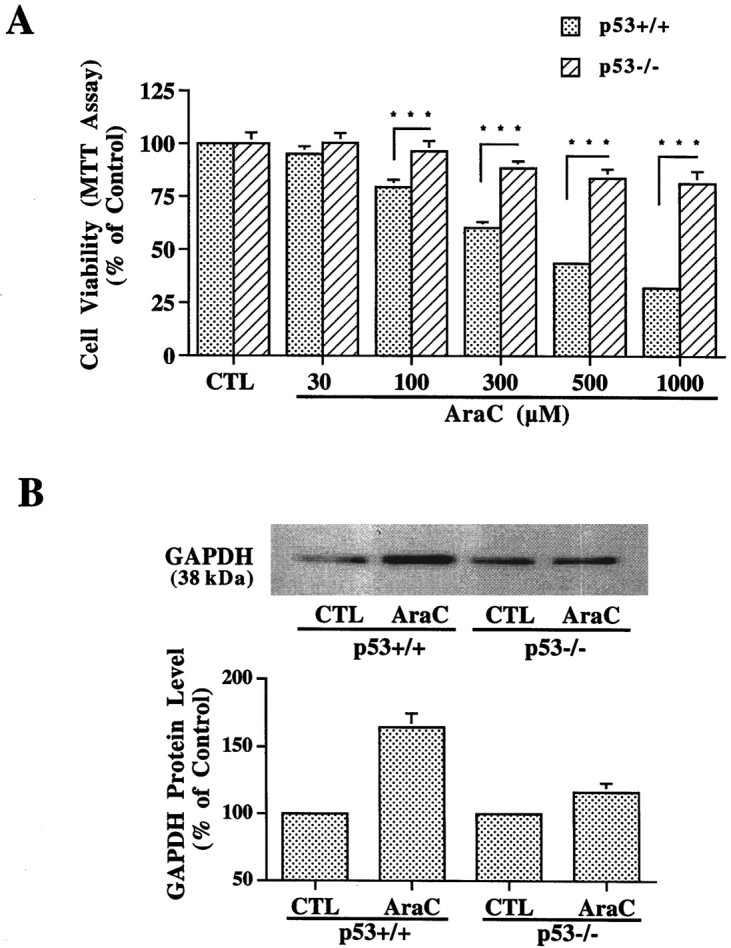
Knock-out of p53 gene suppresses AraC-induced neurotoxicity and GAPDH overexpression in mouse cerebellar granule cells. Granule cells were prepared from the cerebellum of 7-d-old wild-type (p53+/+) and p53 knock-out (p53−/−) mice (Donehower et al., 1992) according to the procedures described in Materials and Methods.A, At 12 hr after plating onto 96-well plates, various concentrations of AraC were added to the cultures, and the cell viability was determined 24 hr later by MTT assay. Results shown are mean ± SEM from six independent experiments. B, At 24 hr after plating onto 60 mm dishes, GAPDH protein levels in wild-type and p53-deficient mouse granule cells were determined by Western blot analysis. The top represents Western blot from a typical experiment. The bottom represents quantified results expressed as mean ± ranges from two independent experiments. Note that p53 gene knock-out resulted in not only inhibition of AraC toxicity but also suppression of AraC-induced GAPDH upregulation. CTL, Control. ***p < 0.001 compared with the corresponding results in p53-deficient mouse granule cells, using one-way ANOVA with Fisher's PLSD test.
p53 gene transfection into PC12 cells upregulates GAPDH protein and triggers apoptosis
To obtain additional evidence that GAPDH is regulated by p53, we infected PC12 pheochromocytoma cells with the recombinant adenovirus containing AdWTp53, Adβ-gal, or AdControl. The infection efficiency determined by counting β-galactosidase-positive cells was dependent on the dose of the recombinant adenovirus and found to be 82.9 ± 4.4% with 20 pfu/cell of Adβ-gal (data not shown). The p53 gene transfection of PC12 cells resulted in a time-dependent increase in the levels of exogenous p53 protein in the cell homogenate (Fig.11). Thus, the p53 protein levels were progressively increased from 12 to 48 hr after AdWTp53 infection. In contrast, infection with AdControl produced no detectable p53 protein. Interestingly, the levels of GAPDH protein in the particulate fractions were also markedly increased at 24 and 48 hr in cells infected with AdWTp53 but not AdControl. The p53 gene transfer into PC12 cells induced a time-dependent loss of cell viability with a detectable decrease at day 1, followed by a progressive decrease to <20% of the control at days 3 and 5 (Fig.12A). A dramatic increase in internucleosomal DNA cleavage was also observed at 2 d after AdWTp53 infection (Fig. 12B). In contrast, infection with AdControl induced neither cell death nor detectable DNA fragmentation throughout the time course studied.
Fig. 11.

p53 gene transfection into PC12 cells induces GAPDH overexpression. PC12 cells in cultures were incubated at 37°C for different lengths of time after infection with 20 pfu/cell of either AdWTp53 or AdControl. Western blotting for p53 was determined at indicated times as described in Materials and Methods using 30 μg of cell homogenate proteins. Western blotting for GAPDH was determined using 7 μg of 2 × 105_g_pellet proteins. Note that infection of cells with AdWTp53, but not AdControl, induced a dramatic time-dependent increase in p53 and GAPDH protein. The experiment has been repeated three times with similar results.
Fig. 12.
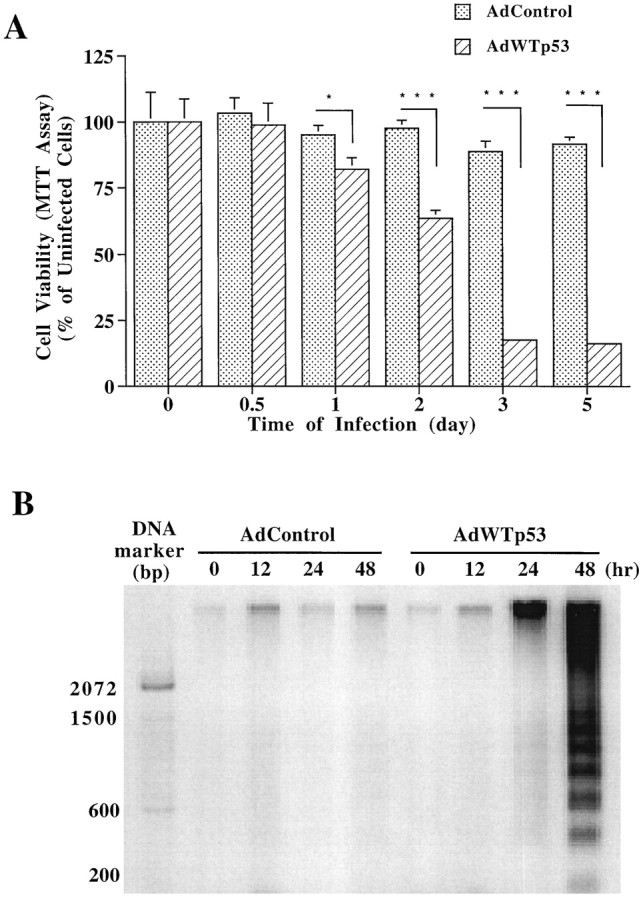
p53 gene transfection into PC12 cells induces delayed DNA fragmentation. Cells were infected with 20 pfu/cell of AdWTp53 or AdControl as described in the legend to Figure 11. At indicated times, cells were examined for viability using MTT assay (A) or for internucleosomal DNA cleavage using agarose gel electrophoresis (B). Results of MTT assay are expressed as percentage of mean ± SEM of the respective zero time control from eight independent determinations. The results of DNA agarose gel electrophoresis are from a typical experiment of three such experiments. Note that infection of cells with AdWTp53, but not AdControl, induced a time-dependent loss of cell viability and an increase in the level of internucleosomally cleaved DNA in the soluble DNA preparation. *p < 0.05; ***p < 0.001 using one-way ANOVA with Fisher's PLSD test.
DISCUSSION
The tumor suppressor p53 serves as a critical regulator in the cell cycle and in the apoptosis of normal cells after exposure to ionizing radiation or DNA-damaging agents. It has been suggested that p53 may act as a “guardian of the genome.” When DNA damage is mild, p53 halts cell cycle progression at the G1 phase through the induction of genes such as p21 and GADD45 (Pellegata et al., 1996) (for review, see Hughes et al., 1997), allowing DNA repair to occur before advancing through the cell cycle. However, when DNA damage is extensive and not repairable, p53 triggers apoptosis (Grasso and Mercer, 1997) (for review, see Yonish-Rouach, 1996; Hughes et al., 1997). These biochemical characteristics ensure that p53 plays a key role in controlling genomic stability (Lowe et al., 1993; Fukasawa et al., 1997). Elevated expression of p53 may initiate apoptosis by increasing the expression of the death gene Bax (Miyashita and Reed, 1995), decreasing the expression of the cytoprotective gene Bcl-2 (Selvakumaran et al., 1994) or inducing the transcription of redox-related genes that cause oxidative stress (Polyak et al., 1997).
When DNA is damaged in postmitotic neurons, p53 induction is associated with mechanisms underlying death rather than recovery (Sakhi et al., 1994; Wood and Youle, 1995; Enokido et al., 1996a; Jordan et al., 1997). Transfection studies have shown that overexpression of p53 per se is sufficient to induce neuronal apoptosis (Xiang et al., 1996;Jordan et al., 1997). In contrast, p53-deficient neurons are resistant to apoptosis induced by excitotoxins such as glutamate (Morrison et al., 1996). The precise mechanism by which p53 modulates neuronal viability has not yet been determined. However, it has been suggested that p53 may function by upregulating Bax and/or by downregulating Bcl-2, whereas generation of the reactive oxygen species and activation of caspases may be further downstream events of p53-dependent apoptosis (Johnson et al., 1996; Lotem and Sachs, 1996).
In this study, we extensively characterized AraC-induced apoptosis with a major emphasis on the role of p53 in the apoptotic process. We found that levels of p53 mRNA and protein in cerebellar granule cells were rapidly and markedly increased after treatment with AraC. Pre-exposure of these cells to p53/AS robustly protected these neurons from AraC-induced chromatin condensation, nuclear fragmentation, and internucleosomal DNA cleavage. Concomitant with these effects, the viability of AraC-treated cells was markedly enhanced. Additionally, AraC-induced apoptosis was shown to be suppressed in granule cells from p53 null mice (Enokido et al., 1996b; the present study). These results suggest that p53 plays an essential role in mediating AraC-induced apoptotic death of cerebellar granule cells. The overexpression of p53 induced by AraC also suggests that this apoptotic insult involves extensive DNA damage before p53 induction, as suggested previously (Tomkins et al., 1994; Enokido et al., 1996a,b). It is interesting to note that AraC-induced apoptosis of granule cells is preceded by an increase in the levels of both mRNA and protein of p53, suggesting an involvement of transcriptional regulation of p53.
p53 upregulation was immediately followed by increased expression of Bax; the latter was blocked by p53/AS but not p53/S, reinforcing the well established notion that Bax is a p53-responsive target gene and is involved in p53-mediated neuronal death (Hughes et al., 1997; Xiang et al., 1998). Importantly, GAPDH mRNA and protein levels were increased after p53 overexpression, and GAPDH upregulation was effectively suppressed by p53/AS. Conversely, neither p53 nor Bax overexpression was affected by GAPDH/AS, which is neuroprotective by blocking GAPDH upregulation and the subsequent nuclear translocation (Ishitani and Chuang, 1996; Saunders et al., 1997; Ishitani et al., 1998). Moreover, our results show that p53 gene knock-out suppressed AraC-induced GAPDH upregulation in mouse cerebellar granule cells compared with that in wild-type neurons. These observations suggest that GAPDH overexpression is an event downstream of p53 expression and may function independently of Bax in mediating AraC-induced apoptosis of granule cells. Moreover, our results raise the possibility that GAPDH is a novel target of p53. In further support of this notion, we found that high-efficiency transfection of p53 gene into PC12 cells by infection with AdWTp53 resulted in dramatic overexpression of not only p53 but also GAPDH protein, and induction of these proteins preceded internucleosomal DNA cleavage and extensive cell death. In contrast, infection with AdControl produced neither p53 and GAPDH increase nor apoptosis.
Our results show that levels of Bax mRNA and protein were rapidly and markedly increased by AraC in cerebellar granule cells. Other studies have suggested that Bax may produce cell death by induction of the mitochondrial permeability transition, which can result in disruption of plasma membrane integrity or release of cytochrome C from mitochondria (Pastorino et al., 1998; Chen and Chuang, 1999). The released cytochrome C may bind to Apaf-I, causing caspase cleavage of substrate proteins and, ultimately, cell death (Liu et al., 1996). The involvement of Bax in neuronal apoptosis seems to depend on the nature of stimuli. For example, Bax is required for apoptosis induced by deprivation of trophic factors and high potassium but is not involved in NMDA-induced excitotoxicity in some studies (Deckwerth et al., 1996;Miller et al., 1997).
The precise mechanism underlying the involvement of GAPDH in apoptosis is unclear. Because overexpressed GAPDH is translocated from the cytosol to the nucleus in neurons and non-neuronal cells (Saunders et al., 1997, 1999; Sawa et al., 1997; Ishitani et al., 1998; Shashidharan et al., 1999), one may surmise that GAPDH-dependent apoptosis is caused by a perturbation of nuclear functions. In this context, we found that AraC-induced GAPDH nuclear translocation occurs concurrently with a nuclear loss of dehydrogenase and uracil glycosylase activities (Saunders et al., 1999), suggesting that GAPDH undergoes structural and functional changes after transport to the nucleus. In the cytosol, GAPDH binds to glutamine repeats in huntingtin, a gene product of Huntington's chorea, and other polyglutamine-containing proteins associated with neurodegenerative diseases (e.g., ataxin for spinocerebellar ataxia) (Burke et al., 1996). Because N-terminal fragments of huntingtin and ataxin-3 are found in the intranuclear inclusions of cells in affected brain areas (Davies et al., 1997;DiFiglia et al., 1997; Paulson et al., 1997), it seems possible that GAPDH is a “carrier” mediating the transfer of these disease-related proteins to the nucleus, thus contributing to the progression of neuronal apoptosis.
Recently, it has been shown that the administration of_R_-deprenyl and its derivatives can protect cerebellar granule cells from AraC-induced apoptosis (Paterson et al., 1998). This neuroprotective effect could be attributable to the ability of these drugs to bind overexpressed GAPDH, thereby inhibiting the proapoptotic activity of GAPDH; such an effect was demonstrated in an independent study using neuroblastoma cells (Kragten et al., 1998). Clearly, molecular mechanisms underlying GAPDH-mediated apoptotic death in neurons and non-neuronal cells require future detailed investigation.
Footnotes
We thank Peter Leeds in our section for his kind assistance during the course of this study.
Correspondence should be addressed to Dr. De-Maw Chuang, Section on Molecular Neurobiology, Biological Psychiatry Branch, National Institute of Mental Health, National Institutes of Health, Building 10, Room 3N212, 10 Center Drive, MSC 1272, Bethesda, MD 20892-1272. E-mail: chuang@helix.nih.gov.
REFERENCES
- 1.Baxi MD, Vishwanatha JK. Uracil DNA glycosylase/glyceraldehyde-3-phosphate dehydrogenase is an Ap4A binding protein. Biochemistry. 1995;34:9700–9707. doi: 10.1021/bi00030a007. [DOI] [PubMed] [Google Scholar]
- 2.Burke JR, Enghild JJ, Martin ME, Jou YS, Myers RM, Roses AD, Vance JM, Strittmatter WJ. Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat Med. 1996;2:347–350. doi: 10.1038/nm0396-347. [DOI] [PubMed] [Google Scholar]
- 3.Chen R-W, Chuang D-M. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression: a prominent role in neuroprotection against excitotoxicity. J Biol Chem. 1999;272:6039–6042. doi: 10.1074/jbc.274.10.6039. [DOI] [PubMed] [Google Scholar]
- 4.Crumrine RC, Thomas AL, Morgan PF. Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. J Cereb Blood Flow Metab. 1994;14:887–891. doi: 10.1038/jcbfm.1994.119. [DOI] [PubMed] [Google Scholar]
- 5.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 6.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 7.Dessi F, Hoollard H, Moreau J, Ben-Ari Y, Charriaut-Marlangue C. Cytosine arabinoside induces apoptosis in cerebellar neurons in culture. J Neurochem. 1995;64:1980–1987. doi: 10.1046/j.1471-4159.1995.64051980.x. [DOI] [PubMed] [Google Scholar]
- 8.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 9.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 10.Enokido E, Araki T, Tanaka K, Aizawa S, Hatanaka H. Involvement of p53 in DNA strand break-induced apoptosis in postmitotic CNS neurons. Eur J Neurosci. 1996a;8:1812–1821. doi: 10.1111/j.1460-9568.1996.tb01325.x. [DOI] [PubMed] [Google Scholar]
- 11.Enokido E, Araki T, Tanaka K, Aizawa S, Hatanaka H. p53 involves cytosine arabinoside-induced apoptosis in cultured cerebellar granule neurons. Neurosci Lett. 1996b;203:1–4. doi: 10.1016/0304-3940(95)12247-8. [DOI] [PubMed] [Google Scholar]
- 12.Fukamauchi F, Saunders PA, Hough C, Chuang D-M. Agonist-induced down-regulation and antagonist-induced upregulation of m2- and m3-muscarinic acetylcholine receptor mRNA and protein in cultured cerebellar granule cells. Mol Pharmacol. 1993;44:940–949. [PubMed] [Google Scholar]
- 13.Fukasawa K, Wiener F, Vande Woude GF, Mai S. Genomic instability and apoptosis are frequent in p53 deficient young mice. Oncogene. 1997;15:1295–1302. doi: 10.1038/sj.onc.1201482. [DOI] [PubMed] [Google Scholar]
- 14.Grasso L, Mercer WE. Pathways of p53-dependent apoptosis. Vitam Horm. 1997;53:139–173. doi: 10.1016/s0083-6729(08)60706-2. [DOI] [PubMed] [Google Scholar]
- 15.Hedley DW, McCulloch EA. Generation of reactive oxygen intermediates after treatment of blasts of acute myeloblastic leukemia with cytosine arabinoside: role of bcl-2. Leukemia. 1996;10:1143–1149. [PubMed] [Google Scholar]
- 16.Hughes PE, Alexi T, Schreiber SS. A role of the tumor suppressor gene p53 in regulating neuronal apoptosis. NeuroReport. 1997;8:v–xii. [PubMed] [Google Scholar]
- 17.Huitorel P, Pantaloni D. Bundling of microtubules by glyceraldehyde-3-phosphate dehydrogenase and its modulation by ATP. Eur J Biochem. 1985;150:265–269. doi: 10.1111/j.1432-1033.1985.tb09016.x. [DOI] [PubMed] [Google Scholar]
- 18.Ishitani R, Chuang D-M. Glyceraldehyde-3-phosphate dehydrogenase antisense oligodeoxynucleotides protect against cytosine arabinonucleoside-induced apoptosis in cultured cerebellar neurons. Proc Natl Acad Sci USA. 1996;93:9937–9941. doi: 10.1073/pnas.93.18.9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishitani R, Tanaka M, Sunaga K, Katsube N, Chuang D-M. Nuclear localization of overexpressed glyceraldehyde-3-phosphate dehydrogenase in cultured cerebellar neurons undergoing apoptosis. Mol Pharmacol. 1998;53:701–707. doi: 10.1124/mol.53.4.701. [DOI] [PubMed] [Google Scholar]
- 20.Johnson TM, Yu ZX, Ferrans VJ, Lowenstein RA, Finkel T. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc Natl Acad Sci USA. 1996;93:11848–11852. doi: 10.1073/pnas.93.21.11848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jordan J, Galindo MF, Prehn JH, Weichselbaum RR, Beckett M, Ghadge GD, Roos RP, Leiden JM, Miller RJ. p53 expression induces apoptosis in hippocampal pyramidal neuron cultures. J Neurosci. 1997;17:1397–1405. doi: 10.1523/JNEUROSCI.17-04-01397.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katayose D, Gudas J, Nguyen H, Srivastava S, Cowan KH, Seth P. Cytotoxic effects of adenovirus-mediated wild-type p53 protein expression in normal and tumor mammary epithelial cells. Clin Cancer Res. 1995;1:889–897. [PubMed] [Google Scholar]
- 23.Kawamoto RM, Caswell AH. Autophosphorylation of glyceraldehydephosphate dehydrogenase and phosphorylation of protein from skeletal muscle microsomes. Biochemistry. 1986;25:657–661. doi: 10.1021/bi00351a022. [DOI] [PubMed] [Google Scholar]
- 24.Kragten E, Lalande I, Zimmermann K, Roggo S, Schindler P, Muller D, van Oostrum J, Waldmeier P, Furst P. Glyceraldehyde-3-phosphate dehydrogenase, the putative target of the antiapoptotic compounds CGP 3466 and R-(−)-deprenyl. J Biol Chem. 1998;273:5821–5828. doi: 10.1074/jbc.273.10.5821. [DOI] [PubMed] [Google Scholar]
- 25.Li Z, Rakkar A, Katayose Y, Kim M, Shanmugam N, Srivastava S, Moul JW, McLeod DG, Cowan KH, Seth P. Efficacy of multiple administrations of a recombinant adenovirus expressing wild-type p53 in an immune-competent mouse tumor model. Gene Ther. 1998;5:605–613. doi: 10.1038/sj.gt.3300636. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome C. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 27.Lotem J, Sachs L. Differential suppression by protease inhibitors and cytokines of apoptosis induced by wild-type p53 and cytotoxic agents. Proc Natl Acad Sci USA. 1996;93:12507–12512. doi: 10.1073/pnas.93.22.12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 29.Miller TM, Moulder KL, Knudson CM, Creedon DJ, Deshmukh M, Korsmeyer SJ, Johnson EM., Jr Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 31.Morgenegg G, Winkler GC, Hubscher U, Heizmann CW, Mous J, Kuenzle CC. Glyceraldehyde-3-phosphate dehydrogenase is a nonhistone protein and a possible activator of transcription in neurons. J Neurochem. 1986;47:54–62. doi: 10.1111/j.1471-4159.1986.tb02830.x. [DOI] [PubMed] [Google Scholar]
- 32.Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nonaka S, Hough CJ, Chuang D-M. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-d-aspartate receptor-mediated calcium influx. Proc Natl Acad Sci USA. 1998;95:2642–2647. doi: 10.1073/pnas.95.5.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pastorino JG, Chen ST, Tafani M, Snyder JW, Farber JL. The overexpression of bax produces cell death upon induction of the mitochondrial permeability transition. J Biol Chem. 1998;273:7770–7775. doi: 10.1074/jbc.273.13.7770. [DOI] [PubMed] [Google Scholar]
- 35.Paterson IA, Zhang D, Warrington RC, Boulton AA. R-deprenyl and R-2-heptyl-N-methylpropargylamine prevent apoptosis in cerebellar granule neurons induced by cytosine arabinoside but not low extracellular potassium. Neurochemistry. 1998;70:515–523. doi: 10.1046/j.1471-4159.1998.70020515.x. [DOI] [PubMed] [Google Scholar]
- 36.Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 37.Pellegata NS, Antoniono RJ, Redpath JL, Stanbridge EJ. DNA damage and p53-mediated cell cycle arrest: a reevaluation. Proc Natl Acad Sci USA. 1996;93:15209–15214. doi: 10.1073/pnas.93.26.15209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 39.Sakhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber SS. p53 induction is associated with neuronal damage in the central nervous system. Proc Natl Acad Sci USA. 1994;91:7525–7529. doi: 10.1073/pnas.91.16.7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saunders PA, Chalecka-Franaszek E, Chuang D-M. Subcellular distribution of glyceraldehyde-3-phosphate dehydrogenase in cerebellar granule cells undergoing cytosine arabinoside-induced apoptosis. J Neurochem. 1997;69:1820–1828. doi: 10.1046/j.1471-4159.1997.69051820.x. [DOI] [PubMed] [Google Scholar]
- 41.Saunders PA, Chen R-W, Chuang D-W. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase isoforms during neuronal apoptosis. J Neurochem. 1999;72:925–932. doi: 10.1046/j.1471-4159.1999.0720925.x. [DOI] [PubMed] [Google Scholar]
- 42.Sawa A, Khan A, Hester LD, Snyder SH. Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death. Proc Natl Acad Sci USA. 1997;94:11669–11674. doi: 10.1073/pnas.94.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Selvakumaran M, Lin HK, Miyashita T, Wang HG, Krajewski S, Reed JC, Hoffman B, Liebermann D. Immediate early up-regulation of bax expression by p53 but not TGF beta 1: a paradigm for distinct apoptotic pathways. Oncogene. 1994;9:1791–1798. [PubMed] [Google Scholar]
- 44.Shashidharan P, Chalmers-Redman RM, Carlile GW, Rodic V, Gurvich N, Yuen T, Tatton WG, Sealfon SC. Nuclear translocation of GAPDH-GFP fusion protein during apoptosis. NeuroReport. 1999;10:1149–1153. doi: 10.1097/00001756-199904060-00045. [DOI] [PubMed] [Google Scholar]
- 45.Sioud M, Jesperson L. Enhancement of hammerhead ribozyme catalysis by glyceraldehyde-3-phosphate dehydrogenase. J Mol Biol. 1996;257:775–789. doi: 10.1006/jmbi.1996.0201. [DOI] [PubMed] [Google Scholar]
- 46.Tomkins CE, Edwards SN, Tolkovsky AM. Apoptosis is induced in post-mitotic rar sympathetic neurons by arabinoside and topoisomerase inhibitors in the presence of NGF. J Cell Sci. 1994;107:1499–1507. doi: 10.1242/jcs.107.6.1499. [DOI] [PubMed] [Google Scholar]
- 47.Trimmer PA, Smith TS, Jung AB, Bennett JP. Dopamine neurons from transgenic mice with a knockout of the p53 gene resist MPTP neurotoxicity. Neurodegeneration. 1996;5:233–239. doi: 10.1006/neur.1996.0031. [DOI] [PubMed] [Google Scholar]
- 48.Wood KA, Youle RJ. The role of free radicals and p53 in neuron apoptosis in vivo. J Neurosci. 1995;15:5851–5857. doi: 10.1523/JNEUROSCI.15-08-05851.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. Evidence for p53-mediated modulation of neuronal viability. J Neurosci. 1996;16:6753–6765. doi: 10.1523/JNEUROSCI.16-21-06753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yonish-Rouach E. The p53 tumor suppressor gene: a mediator of a G1 growth arrest and of apoptosis. Experientia. 1996;52:1001–1007. doi: 10.1007/BF01920109. [DOI] [PubMed] [Google Scholar]