Fibroblast Growth Factor-2 Activates a Latent Neurogenic Program in Neural Stem Cells from Diverse Regions of the Adult CNS (original) (raw)
Abstract
During development of the mammalian brain, both neurons and glia are generated from multipotent neural stem cells. Although neurogenesis ceases in most areas at birth, stem cells continue to generate neurons within the subventricular zone and hippocampal dentate gyrus throughout adult life. In this work, we provide the first demonstration that precursors native to regions of the adult brain that generate only glia can also generate neurons after exposure to FGF-2 in vitro. When progenitors isolated from hippocampal tissue were directly compared with cells isolated from the neocortex_,_ both populations were able to initiate a program of proliferative neurogenesis. Genetic marking and lineage analysis showed that a majority of the cells able to generate neurons were multipotent precursors; however, progeny from these precursors acquired the competence to differentiate into neurons only after exposure to FGF-2. The recruitment of similar FGF-2-responsive cells from the adult optic nerve, a structure well isolated from the neurogenic zones within the brain, confirmed that neuron-competent precursors naturally exist in widely divergent tissues of the adult brain.
Keywords: neural stem cells, adult, hippocampus, cortex, optic nerve, density gradient
Cell proliferation in the adult mammalian brain is ubiquitous but is primarily confined to the measured production of glia. Except for discrete regions in the hippocampus and the subventricular zone (SVZ), neurogenesis is conspicuously absent (Altman and Das, 1965, 1966; Bayer, 1982; Kaplan and Bell, 1984;Levison et al., 1993; Lois and Alvarez-Buylla, 1993; Luskin, 1993). The reasons why these areas continue to generate neurons are unknown, but primary cell cultures from the adult rodent brain are beginning to provide some insights. Cultures initiated from adult SVZ or hippocampal (HC) tissues contain proliferative neuronal and glial-restricted progenitors, as well as multipotent precursors with the characteristics of neural stem cells, i.e., the ability to self-renew and the ability to generate both neurons and glia (Gage et al., 1995b; Temple and Qian, 1996; Weiss et al., 1996; McKay, 1997). More recently, Johansson et al. (1999) have shown evidence that some of these stem-like cells may actually be ependyma. If ependymal cells are actually stem cells, it seems increasingly unlikely that neurogenesis is absent in other regions because of the lack of multipotent stem cells. In past work, we have suggested that stem cells may be more widely distributed because cells from non-neurogenic areas repeatedly passaged in the presence of high concentrations of basic fibroblast growth factor (FGF-2) do begin to generate neurons in vitro (Palmer et al., 1995; Shihabuddin et al., 1997). This observation is consistent with the isolation of neuronal progenitors from these areas, but the protracted times in culture suggest another explanation. It is known that stem cell cultures initiated from hippocampal tissues will spontaneously transform because of accumulated genetic abnormalities. Abnormalities in chromosome number can occur in as little as 30 population doublings (Palmer et al., 1997) and, as cells become increasingly aneuploid, it is possible that glial-restricted progenitors acquire capabilities beyond those available in vivo.
With current methodologies, it has been difficult to distinguish between the activation of a latent potential versus _in vitro_mutation. Unlike fetal tissues, which are easily dissociated and yield relatively abundant precursor populations, adult tissues yield few progenitors, and the progenitor preparations are contaminated with differentiated cells and tissue debris. The myelin-rich debris inhibits cell attachment and growth, whereas differentiated cells complicate the evaluation of lineage potential in acutely isolated cultures. Past studies have evaluated “progenitors” only after repeated passaging had eliminated the debris and differentiated cells (Gage et al., 1995a; Palmer et al., 1995, 1997). Even if these cells had remained diploid, they may have been altered dramatically in prolonged culture. A newly developed progenitor enrichment protocol has resolved many of these issues and has allowed us to evaluate the lineage potential of proliferative cells from cortex and hippocampus immediately after isolation from adult tissues. Surprisingly, both tissues yielded populations of multipotent precursors.
MATERIALS AND METHODS
Tissue dissection. Three areas were dissected from adult rat brains as follows (Fig. 1). Rats (170–190 gm, Fisher 344 males or females; Harlan Sprague Dawley, Indianapolis, IN) were deeply anesthetized with a mixture of ketamine, xylazine, and acepromazine. Animals were decapitated, and whole brains were removed. First, ∼1.5 mm of each optic nerve were harvested rostral to, but not including, the optic chiasma. The brain was then bisected longitudinally, and each hippocampal lobe was separated from the overlaying cortical white matter using the natural separation line along the alveus hippocampus. The white matter of the fimbria and subiculum was removed as much as possible. Some white matter remained. Finally, a 1.5-mm-wide cortical ribbon containing parietal and frontal segments was dissected longitudinally, proximal to the central fissure. The pial and callosal surfaces were trimmed from each cortical ribbon to remove a majority of the meninges and white matter.
Fig. 1.

Isolation of endogenously proliferating cells from the adult hippocampus and cortex. A, Coronal section of the cortex and hippocampus of an adult rat immunostained for BrdU-labeled nuclei (white spots). Animals were injected with BrdU once each day for 6 d and then evaluated on the seventh day. Proliferative cells were abundant within the subgranule zone of the hippocampal dentate gyrus (b, B) and fimbrial ridge of the hippocampus (d, D). Closely juxtaposed pairs of daughter cells are clearly seen in white matter tracts, suggesting division in situ(D, E). Acid-stable vimentin immunoreactivity (blue) is clearly shown in the specialized ependymal cells lining the fimbrial surface. Although BrdU-positive proliferative cells were present in the cortical gray matter (c, C), they were much more abundant in subcortical white matter and adjacent ventricular zones (e, E). F, Tissues used for cultures included whole hippocampal lobes, cortical ribbons lateral to the midline (approximate lateral boundaries show by white bars in A), and sections of optic nerve rostral to the optic chiasma. G, Tissue was enzymatically dissociated and fractionated over a 50% Percoll gradient. Colored beads of known buoyancy were used to calibrate the gradient (left). The specific gravity of beads flanking the separation zone is shown (in grams per milliliter). Fractionated tissues generated two major bands of nucleated cells (right). The top band contained differentiated cells, myelin, and tissue debris. The bottom band contained undifferentiated progenitors and ependymal cells. A band of RBCs also formed immediately below the progenitor population (not visible here). H, The migration of progenitors in the gradient relative to that of calibrated beads demonstrates that adult-derived neural progenitors have a relatively high-buoyancy density of 1.065–1.075 gm/ml.
Tissue dissociation and fractionation. As described previously (Gage et al., 1995a; Palmer et al., 1995), tissues were finely minced and digested in a solution of papain (2.5 U/ml; Worthington, Freehold, NJ), DNase (250 U/ml, Worthington), and neutral protease (1 U/ml Dispase; Boehringer Mannheim, Indianapolis, IN) dissolved in HBSS. Cells and tissue fragments were washed three times with DMEM containing 10% fetal bovine serum (FBS) (Hyclone, Logan, UT). Whole digested tissue was then suspended in DMEM–10% FBS, filtered through a sterile 107 μm nylon mesh and thoroughly mixed with an equal volume of Percoll solution. The Percoll solution was made by mixing nine parts of Percoll (Amersham Pharmacia Biotech, Uppsala, Sweden) with one part 10× PBS (Irvine Scientific, Santa Ana, CA). The cell suspension was then fractionated by centrifugation for 30 min, 18°C, at 20,000 × g. Cell fractions were harvested and washed free of Percoll by three or more rinses in DMEM–10% FBS.
Cell culture. Percoll gradients were optimized using perpetualized hippocampal stem cell clones AP31 and PZ5a (Palmer et al., 1997). Cells were maintained on polyornathine/laminin (Porn/Lam)-coated dishes in growth medium consisting of DMEM/F-12 (1:1) supplemented with N2 supplement (Life Technologies, Gaithersburg, MD) and 20 ng/ml recombinant human FGF-2 prepared in_Escherichia coli_ (kindly provided by A. Baird, San Diego, CA). Cells fractionated on Percoll gradients were washed free of Percoll and plated onto Porn/Lam-coated multiwell slides (Fisher Scientific, Houston, TX) or Porn/Lam-coated tissue culture dishes (Fisher Scientific). For 36 hr acute cultures, cells were allowed to attach to glass slides for 36 hr in DMEM/F-12 (1:1) containing 10% FBS and subsequently fixed for 10 min using 4% paraformaldehyde in PBS. For long-term primary cultures, isolated cells were maintained for 24 hr in 10% FBS. The medium was then replaced with serum-free growth medium. Seventy-five percent of the medium was replaced with new growth medium every 48 hr. Cultures were passaged at confluence by exposing cells to trypsin–EDTA solution (Irvine Scientific). Detached cells were then rinsed once with DMEM/F-12 and replated at one-half of their original density in growth medium supplemented with 30% conditioned medium (medium exposed to cells for 24 hr before passaging). To promote differentiation, growth medium was replaced with DMEM/F-12 containing 1% FBS, 100 ng/ml all-trans retinoic acid, and 1 ng/ml FGF-2 (differentiation medium).
Retroviral vectors and virus preparation. Steven Suhr (Salk Institute, La Jolla, CA) kindly provided the retrovirus-containing plasmid pNIT-GFP. pNIT-GFP contains replication-defective MoMLV-based retroviral elements designed to carry and express sequences encoding neomycin phosphotransferase (neo), tetracycline (tet) transactivator protein (Gossen and Bujard, 1992), and enhanced GFP. pNIT-GFP provides constitutive neo and tet-transactivator expression under the control of the MoMLV LTR. GFP is expressed under the control of the tetracycline-suppressible tet operator system. A stable NIT-GFP packaging cell line was generated by cotransfecting 293 cells with pLNIT-GFP and the packaging constructs pMD.G and pCMV-gp as described previously (Emi et al., 1991; Burns et al., 1993). The transiently produced virus was used to infect 293 cells carrying a stable integrant of pCMV-gp (293gp). Clones of G418-resistant 293gp/NIT-GFP cells were screened for single, unrearranged NIT-GFP integrants. Clone 293gp/NIT-GFPc4 was chosen for use in these studies. Virus containing supernatants was harvested from 293gp/NIT-GFPc4 after transfection with pMD.G. Viral stocks were then concentrated 100-fold by centrifugation at 50,000 × g for 90 min. Viral pellets were suspended in normal saline and again pelleted by centrifugation. Viral pellets were then resuspended in normal saline (∼0.001 × the volume of medium initially harvested). Final virus titers were ∼4 × 108neor colonies/ml of virus as measured by G418-resistant colony formation on National Institutes of Health 3T3 cells. No helper virus was detected (>1 colony-forming units per milliliter of unconcentrated supernatant) using a marker rescue assay. Concentrated virus was stored in small aliquots at −70°C before use.
Progenitor marking and clonal analysis. Acutely isolated progenitors were cultured for 7 d in growth medium, detached with trypsin–EDTA solution, washed one time with DMEM/F-12, and suspended to a final concentration of 106 cells/ml in growth medium supplemented with 1 μg/ml polybrene. Volumes of NIT-GFP virus sufficient to infect 5–50 cells were added to 0.5 ml of cells (500,000 cells) and then incubated for 30 min at 37°C. The cells were pelleted, resuspended in growth medium containing 30% conditioned medium, and plated into 10 cm Porn/Lam-coated tissue culture dishes. Twenty-four to 36 hr later, the locations of individual green cells were marked on each dish. The cells were then allowed to grow to confluence with monitoring of individual infected cells on each day (7 d). Adjacent clones closer than 1 cm apart were excluded from the study. The growth medium was replaced every other day and then switched on the seventh day to differentiation medium. Differentiation medium was replaced every day for 7 d, and then the confluent monolayers were fixed for 10 min with 4% paraformaldehyde and evaluated using immunofluorescence.
Immunofluorescent staining. Paraformaldehyde-fixed cells in suspension, cell monolayers, or 30 μm floating tissue sections were rinsed with PBS and then blocked for 30 min at room temperature in PBS containing 0.3% Triton X-100 and 5% preimmune donkey serum (PBS2+). Samples were then incubated in PBS2+ containing dilutions of up to four primary antibodies for 24–48 hr at 4°C. Samples were then washed twice with PBS for 10 min at room temperature and then a third time for 30 min at room temperature. Samples were then incubated at 4°C for an additional 24–48 hr with secondary antibodies conjugated to aminomethyl coumarin, fluorescein isothiocyanate, Texas Red, or cyanin 5. Secondary antibodies (donkey; Jackson ImmunoResearch, West Grove, PA) were used at a final dilution of 1:500 in PBS2+. The samples were then washed as above, treated with 10 mg/ml 4′, 6-diamidino-2-phenylindole (DAPI) (Sigma, St. Louis, MO) for 10 min and coverslipped in 20% polyvinylalcohol (20,000–30,000 MW; Air Products and Chemicals, Allentown, PA) in 50% glycerol (w/v) containing 2.5% w/v 1,4-diazobicyclo-[2.2.2]-octane (Sigma).
Primary antibodies generated in mouse (mo), rat (rt), rabbit (rb), and guinea pig (gp) were used at the following concentrations: mo anti-type III β-tubulin (1:2000; Babco, Richmond, CA), mo anti-microtubule-associated protein 2abc (1:5000; Sigma), mo anti-neuronal nuclear antigen (1:20; hybridoma supernatant kindly provided by R. Mullen, University of Utah, Salt Lake City, UT), mo anti-O4 (1:4; hybridoma supernatant kindly provided by O. Boegler, University of California, San Diego, CA), mo anti-receptor-interacting protein (1:20; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), gp anti-glial fibrillary acidic protein (GFAP) (1:500; Advanced Immunochemical, Inc., Long Beach, CA), mo anti-A2B5 (1:100; Boehringer Mannheim), rt anti-bromodeoxyuridine (BrdU) (1:500; Accurate Chemicals, Westbury, NY), mo anti-Ox42 (1:1000; Chemicon, Temecula, CA), mo anti-Nestin (1:2000; Rat401; PharMingen, San Diego, CA), mo anti-vimentin (1:500; Amersham Pharmacia Biotech), rb anti-galactocerebroside (1:250; Advanced Immunochemicals),and rb anti-fibronectin (1:100; Telios Pharmaceuticals, San Diego, CA). Fluorescent samples were evaluated using a Bio-Rad (Hercules, CA) MRC1024UV confocal imaging system, which allows simultaneous evaluation of up to four separate fluorophores. When it was necessary to show nuclei in addition to four immunological markers, cells were first evaluated for immunological staining in the absence of DAPI and then counterstained with DAPI and reimaged.
RESULTS
A progenitor enrichment protocol based on buoyant density
Density gradient media have been used frequently to fractionate cells on the basis of buoyant density (Poduslo and Norton, 1975; Lisak et al., 1981; Pertoft and Laurent, 1982; Shank and Campbell, 1984). Recently, mitotic cells have been separated rapidly from postmitotic cells in the late embryo using density gradients generated with colloidal silica (Percoll; Pertoft and Laurent, 1982; Maric et al., 1997). In these studies, fetal postmitotic cells were found to have buoyant densities lower than 1.043 gm/ml, whereas mitotic cells and progenitors have densities higher than ∼1.056 gm/ml. To evaluate Percoll gradients for their use in fractionating multipotent precursors from the adult rat brain, we first determined the buoyant density of cells from perpetualized hippocampus-derived stem cell cultures (Palmer et al., 1997). These cultures are stem cell-derived and contain a mixture of lineages at various stages of differentiation. The most immature cells had remarkably high densities ranging from 1.065 to 1.075 gm/ml. On the assumption that the most immature cells within the adult brain would have similarly high densities, we optimized gradients using beads of known densities to generate steep density profiles spanning 1.060–1.075 gm/ml (Fig. 1). All cells with densities lower than 1.060 gm/ml were expected to form a band at the top of the gradient, whereas those with a buoyancy similar to the immature cultured progenitors would migrate into the gradient. Stem-like cells in perpetualized cultures typically formed a discrete band at the bottom of the gradient.
BrdU labeling was first used to mark endogenously proliferating cells for identification in situ. Adult rats were injected with BrdU once each day for 6 consecutive days, and then brains were collected for evaluation on day 7 (Fig. 1A). Two percent of all labeled nuclei within the hippocampus were found within the putative subventricular residuum, an area arbitrarily defined as a thin lamina extending inward 50 μm from the ependymal surface, including the hippocampus alveus but excluding ependymal cells. Ependymal cells proper accounted for 4% of the total labeled population, and a similarly small proportion was found within the neurogenic zone of the subgranular zone (SGZ) (8%) (Fig.1B). In contrast, 52% were present in the white matter of the fimbrial ridge (Fig. 1D), and the remainder were scattered throughout the parenchyma. A similar comparison of neocortical gray and white matter showed that 15% of the BrdU-labeled cells were present in the parenchyma of the cortex (Fig.1C). The remaining 85% were present in the subcortical white matter and associated SVZ (Fig. 1E). Although one might expect many of the BrdU-labeled cells to be derived from the local ventricular zone (Levison and Goldman, 1993; Luskin, 1993), ongoing studies indicate that most marked cells undergo DNA synthesis as resident populations within both gray and white matter (T. D. Palmer and F. H. Gage, unpublished observations).
To determine whether endogenously proliferating progenitors could be isolated from adult tissue on the basis of buoyant density, adult rats were injected with BrdU four times over a 48 hr period. Whole hippocampal lobes or cortical ribbons (Fig. 1F) were dissociated and fractionated over Percoll gradients. Three visible bands of cells were formed (Fig. 1G). A layer of red blood cells (RBCs) formed near the bottom of the gradient. A band of nucleated cells with buoyant densities similar to those of cultured progenitors formed just above the RBC layer, and a majority of the remaining differentiated cells, as well as tissue fragments and myelinated neuropil, formed a large band at the top of the gradient. A small number of cells were diffusely distributed throughout the gradient. Hippocampal tissues yielded 1015 ± 31 cells/mg of tissue in the lower band, and cortical tissues yielded 732 ± 24 cells/mg of tissue (mean ± SEM; n = 5).
Cells from the upper and lower region of each gradient (Fig.1G) were collected. A fraction of each population was immediately fixed and evaluated for BrdU and lineage-specific markers (Table 1). The upper fractions were so contaminated with cell debris and myelin that it was very difficult to determine phenotype, with the exception of BrdU immunoreactivity in cell nuclei. For both HC and cortical tissues, there were very few BrdU-labeled cells in the high-buoyancy fraction (less than one cell detected in 5000 nuclei scored for both HC and cortex), and the few cells not trapped within aggregates of debris nearly all expressed neuronal or glial markers. BrdU-labeled cells were found predominantly in the lower band, with 0.7 or 0.1% of all hippocampal or cortical cells labeled. In both hippocampal and cortical preparations, <0.1% of the low-buoyancy cells were GFAP-immunoreactive astrocytes. Although a few immature neurons (β-tubulin) were present in the hippocampal population (0.1%), none were detected in the low-buoyancy cortical fraction. A significant fraction of cells were immunoreactive for vimentin (37 and 35% in hippocampus and cortex, respectively), and vimentin expression was rapidly upregulated with 87 and 89% of all cells expressing vimentin after 36 hr in culture. Vimentin, a marker attributed to ependymal cells, immature astrocytes, and radial glia_,_ is also known to be expressed by multipotent precursors in perpetualized neural precursor cultures (Levison and Goldman, 1993; Palmer et al., 1995; Luskin et al., 1997). Some cells in both fresh and 36 hr populations were weakly immunoreactive for O4, a marker first attributed to immature oligodendrocytes (Sommer and Schachner, 1981, 1982) and also expressed by FGF-2 stimulated multipotent precursors in vitro (Palmer et al., 1997). Nestin, a marker for immature precursors (Lendahl et al., 1990), was detected in 15 or 7% of the freshly isolated cells from HC or cortex, respectively, but was then transiently downregulated. At 36 hr in culture, very few cells expressed detectable nestin, yet 1 week later, virtually all cells in both hippocampal and cortical cultures were nestin-positive. Our paradigm involves an initial plating in 10% FBS. We suspect that exposure to serum followed by treatment with FGF-2 may be responsible for this modulation.
Table 1.
Markers expressed by freshly isolated cells or after 36 hr in culture (%)
| Lineage | Marker | Hippocampus | Cortex | Optic nerve | |||
|---|---|---|---|---|---|---|---|
| Lower 0 hr | Lower 36 hr | Upper 36 hr | Lower 0 hr | Lower 36 hr | Lower 36 hr | ||
| Progenitor | Nestin | 15 | 0.1 | <0.1 | 7 | <0.1 | <0.1 |
| A2B5 | 1 | 0.5 | nd | 0.8 | 0.7 | 1.2 | |
| O4 | 12 | 24 | 2 | 15 | 29** | 31 | |
| Vimentin* | 37 (34) | 87 (36) | 68 (15) | 35 (30) | 89 (15) | 76 (18) | |
| Glia | GFAP | <0.1 | 0.1 | 26 | <0.1 | <0.1 | <0.1 |
| RIP | <0.1 | <0.1 | 14 | <0.1 | <0.1 | <0.1 | |
| GalC | nd | <0.1 | 12 | <0.1 | <0.1 | <0.1 | |
| Neuron | βTubulin | 0.1 | 0.1 | 9 | <0.1 | <0.1 | <0.1 |
| Map2 | <0.1 | <0.1 | 18 | <0.1 | <0.1 | <0.1 | |
| NeuN | nd | <0.1 | 15 | <0.1 | <0.1 | <0.1 | |
| Other | Fibronectin | <0.1 | <0.1 | 2 | <0.1 | <0.1 | nd |
| Ox42 | <0.1 | <0.1 | 1 | <0.1 | <0.1 | nd | |
| ED-1 | <0.1 | <0.1 | <0.1 | <0.1 | nd | nd | |
| BrdU | 0.7 | 0.8 | <0.01 | 0.1 | 0.2 | nd |
The quality of the vimentin staining in the low-buoyancy fraction also proved to be informative. After 36 hr in culture, vimentin staining was intense in cells with a flattened, neurepithelial-like morphology and weak in phase-bright cells reminiscent of progenitors in long-term cultures. Upon acid pretreatment (required for the immunological detection of BrdU), only the intensely staining cells remained immunoreactive. This staining pattern was also seen _in vivo_in which the acid-stable vimentin immunoreactivity was restricted to the ependyma proper (Fig. 1D), whereas glia in the parenchyma exhibited a weaker, acid-labile staining. In addition, some of the cells in the low-buoyancy fraction were motile during the first few hours of culture. When stained with Coomassie blue, many of the vimentin-positive cells had easily detectable cilia typical of ependymal cells, and a small portion of cells were motile within the freshly isolated lower fraction (data not shown). On the basis of acid-stable vimentin staining, as many as 30–35% of the cells in the low-buoyancy fraction from both HC and cortex may have been ependymal cells.
Although BrdU-labeled cells fractionated to the low-buoyancy population, most of the isolated cells were unlabeled, consistent with the isolation of immature, yet relatively quiescent, precursors. To determine whether the unlabeled cells were competent to proliferate or simply lineage marker-negative terminally differentiated cells, hippocampus or cortex was fractionated, and cells from the low-buoyancy fraction were cultured in defined medium containing 20 ng/ml FGF-2 (DMEM/F-12 containing N2 supplement and 20 ng/ml FGF-2, growth medium). Cell division was monitored by counting cells and by treating replicate cultures with BrdU at different times after plating (Fig.2). After a delay of several days, cells began an exponential growth pattern that reached a steady state in 7–10 d. After 10 d, growth rates were similar to those of the perpetualized cultures, with ∼85% of the cells dividing in a given 24 hr period and >99% of the cells labeled after a 48 hr exposure to BrdU.
Fig. 2.

Proliferation of progenitors after exposure to FGF-2. Cells from the low-buoyancy fraction were plated at a density of 104/cm2 into defined medium containing 20 ng/ml FGF-2. To monitor proliferation, cells were pulsed with BrdU for 24 hr before being fixed after the indicated number of DIV, with the exception of day 21 in which each culture was passaged once and the cells labeled with BrdU for 72 hr before being fixed. At day 21, cultures initiated from optic nerve were also included, and >50,000 cells were scored in each culture. All cells were labeled, demonstrating that all cells were proliferative. The total number of cells per square centimeter was determined by counting nuclei (lines), whereas the percent of cells labeled with BrdU (mitotic index) was determined using immunofluorescent staining (bars). Values are mean ± SEM;n = 3 independent isolates. Ctx, Cortex; ON, optic nerve.
The freshly isolated cells also displayed a density-dependent growth that was similar to that seen in perpetualized stem cell cultures. Plating densities of ∼10,000 cells/cm2or higher were required for optimum proliferation, whereas cells plated at clonal densities (<1 cell/cm2) grew very slowly or not at all. By fractionating cells, not only was it possible to eliminate debris and differentiated cells, but those cells remaining could be plated immediately into culture at densities that promoted the recruitment of cells into cycle.
Analysis of lineage potential
The lineage potential of progenitors from cortex or hippocampus was determined by culturing low-buoyancy cells in growth medium for 14 d and then allowing cells to differentiate under conditions shown previously to stimulate both neuronal and glial differentiation (differentiation medium: 1 ng/ml FGF-2, 1% fetal bovine serum, and 100 nm all-trans retinoic acid) (Palmer et al., 1997; Takahashi et al., 1999). At 14 d, few of the cells expressed markers for neurons or glia. Of the total population in HC or cortical cultures, 0.8 or 0.2%, respectively, were immunoreactive for β-tubulin (neurons), 1.1 or 1.7% were immunoreactive for GFAP (astrocytes), and 24 or 25% were immunoreactive for O4, a marker often attributed to immature oligodendrocytes (Sommer and Schachner, 1981) but also expressed by multipotent progenitors in long-term FGF-2-stimulated cultures (Palmer et al., 1997). There were no galactocerebroside-positive oligodendrocytes (<0.01%) detected in either culture. After differentiation for 7 d, both cultures contained numerous cells from all three lineages. Cortical cultures tended to contain a higher proportion of astrocytes (36 vs 28% for the hippocampus). Both hippocampal and cortical cultures contained similar numbers of highly arborized oligodendrocytes (1 and 2%, respectively), and each contained significant numbers of neurons (8 and 3%, respectively). Figure3A shows a field of cells from cortical cultures in which all three lineages are present.
Fig. 3.
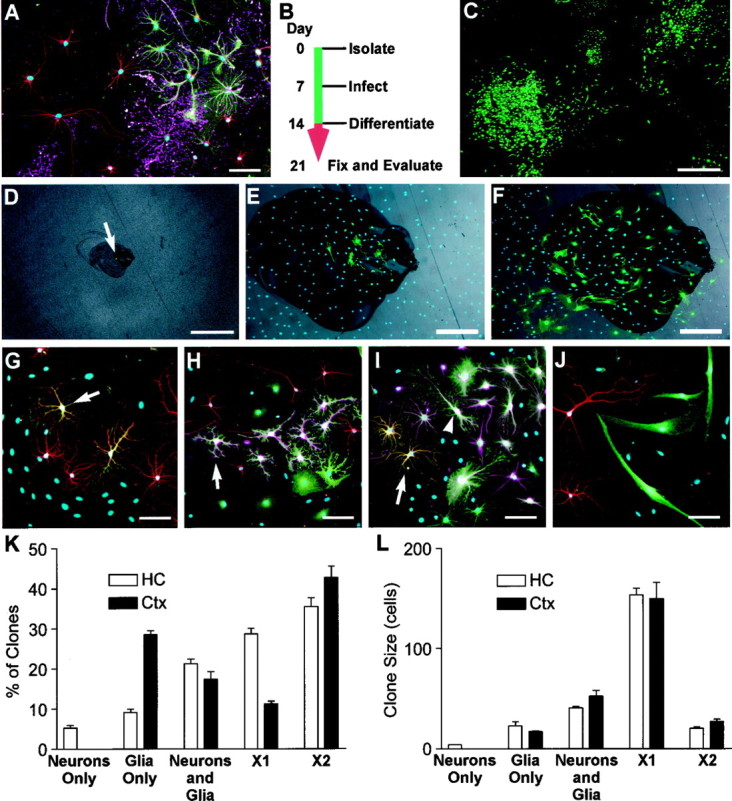
Lineage potential of freshly isolated progenitors. To determine the lineage potential of low-buoyancy cells from cortex or hippocampus, cells were cultured for 14 d and then allowed to differentiate (cortical cells shown in A–J).A, Bulk populations contained all three neural lineages. β-Tubulin-positive neurons are shown in red, GFAP-positive astrocytes in green, immature O4-positive oligodendrocytes in magenta, and nuclei in_blue_. B, To evaluate the lineage potential of single cells, cultures were treated with retroviral vectors on day 7, allowed to proliferate, and then induced to differentiate. Cell phenotypes were evaluated on day 21.C, A large variability in clone sizes and morphologies can be seen in the GFP-positive clones generated from an excess of virus. One hundred-fold less virus was used to generate well-separated clones scored in D–L.D–F, The locations of single green cells were marked 24–36 hr after infection (arrow in D). The same clone is shown at 5 d after infection and after fixation at day 21 (E, F).G–J, Lineage-specific markers were evaluated within each clone. Tubulin is shown in red, GFAP in_magenta_, and nuclei in blue.G, A two-cell clone containing only neurons. The neuronal cluster contains seven or more cells, but only two are marked with GFP (orange where red and_green_ overlap), suggesting that the virally marked cell was resident within a larger clone of neuroblasts.H, A clone containing only glia. Although neurons are present in this field (red), the GFP-marked clone (green) only contains GFAP-positive astrocytes (magenta within the green staining cell bodies, arrow). I, Both β-tubulin-positive (orange, arrow) and GFAP-positive cells (magenta and green_overlay, arrowhead) are present in this clone, demonstrating that the infected precursor was multipotent.J, Many small clones of large bipolar cells were negative for neuronal or glial markers. These cells grew slowly and were eventually outgrown by the neural progenitors in long-term cultures. K, L, Clones from at least three independent tissue preparations were scored for lineage and size (number of GFP-labeled cells per clone). Neurons Only, Only β-tubulin-positive cells; Glia Only, only GFAP- and/or O4-positive cells; Neurons and Glia, colonies with β-tubulin- and GFAP-positive cells; X1, large vimentin-positive clones negative for neuronal or glial markers similar to the large clone in the bottom left corner of_C; X2, small clones containing large bipolar cells as in J. Values are mean ± SEM. Scale bars: A, E–J, 50 μm;C, D, 400 μm.
To determine whether the neurons in the cortical populations were derived from proliferative multipotent precursors, retroviral marking was used to evaluate the lineage potential of cells within the low-buoyancy fractions. Cells were first stimulated with FGF-2 for 7 d to induce proliferation (a prerequisite for retroviral infection), and then retroviruses carrying a GFP transgene were used to infect the proliferating population. Individual infected cells were marked and allowed to proliferate within the noninfected bulk population for an additional 7 d. The resulting colonies were then induced to differentiate in differentiation medium (marking scheme shown in Fig. 3B). Figure 3C shows a typical population of colonies generated using an excess of virus [multiplicity of infection (m.o.i.), ∼ 0.01]. To avoid overlapping colonies, ∼50 infectious units were used in each assay below (m.o.i. of ∼10−4). The number of colonies generated per 10 cm assay dish ranged from 4 to 21. To determine whether individual colonies were indeed clonal, the positions of individual marked cells were documented 24–36 hr after infection, and then colony growth was monitored daily (Fig.3D–F). The incidence of closely juxtaposed cells giving rise to overlapping colonies with such low virus concentrations was insignificant (two clones were excluded in 543 clones scored). The remaining quantitation was performed as a “colony assay” on the assumption that all colonies separated by a centimeter or more were derived from single cells.
Clones were grouped into five categories based on the expression of lineage-specific markers and morphology (Fig.3G–J,K–L). Neuron-only clones were infrequent (5.3%) in the hippocampal preparations and rare (<0.1%) in cortex-derived cultures (Fig. 3G). Some clones were glial-restricted and contained only GFAP-positive astrocytes and/or O4-positive oligodendrocytes (Fig. 3H). A small but significant proportion of the marked cells (21 and 17% from hippocampus and cortex, respectively) produced a mixture of glia and neurons (Fig. 3I). The remaining clones were negative for all three lineage markers. The marker-negative cells were further divided into two clone types; one type was very large and consisted of flattened phase-dark cells strongly immunoreactive for the acid-stable vimentin epitope (vimentin staining not shown). One such colony can be seen in Figure 3C (bottom left). The remaining marker-negative clones were small and contained large bipolar cells with simple, large-caliber processes (Fig. 3J).
When scored by size (Fig. 3L), neuron-only clones contained few cells, whereas glial-restricted progenitors and multipotent progenitors generated colonies of intermediate size. The largest clones were the lineage marker-negative, vimentin-positive clones. Although the large size may suggest a faster growth rate in the 7 d after infection, we found this large size to be an artifact caused by continued growth in differentiation medium. When observed during the first 7 d after viral infection, the clones with the flattened phase-dark morphology typical of these large lineage-negative clones actually grew more slowly than the other clones being monitored. Continued growth in differentiation medium was confirmed by repeating these experiments in the presence of BrdU during differentiation. We found that the large marker-negative clones were uniformly labeled with BrdU, whereas cells from the smaller neuron-only, glia-only, or mixed clones were unlabeled (data not shown).
The clonal analysis demonstrated that approximately half of the lower fraction of cells from both cortex and hippocampus was made up of cells that formed large colonies but did not differentiate into neurons or glia. Although the acid-stable vimentin staining in these cells is consistent with an ependymal origin, the lineage of these cells has not been determined. The remaining cells consisted of neural progenitors of mixed lineage potential. Approximately 20% of all cells isolated were able to generate both neuronal and glial progeny, suggesting that both cortex and hippocampus contained multipotent precursors.
A novel role for FGF-2 in neurogenesis
Neurogenesis is not detected in the adult cortex, yet a significant number of the progenitors from cortical tissues were competent to generate neurons once removed from their in vivo environment. Activation of this neurogenic program could be triggered by several mechanisms. One possibility is that multipotent precursors may yield progeny that are competent to differentiate into neurons but are suppressed by cell-extrinsic signals in vivo. If so, the simple act of removing them from the in vivo environment may disinhibit or activate a latent neuronal differentiation program. To test this possibility, low-buoyancy cells were isolated from adult tissue and immediately plated into differentiation medium. The presence of β-tubulin-positive neurons was scored after 10 d [0 days in vitro (DIV) in FGF-2] (Fig.4). Both cortical and hippocampal populations generated abundant populations of glia, but only the hippocampal preparations generated β-tubulin-positive neurons. The absence of neurons in cortical preparations suggested that precursors from non-neurogenic tissues require signals provided in culture to acquire the competence to differentiate into neurons.
Fig. 4.

Exposure to FGF-2 activates a neurogenic potential in progenitors from cortex. Cells from the hippocampus or cortex were cultured for various times in FGF-2 and then allowed to differentiate for 7 d. Without previous exposure to FGF-2, cortex-derived cells were unable to generate β-tubulin-positive neurons (<0.001% at day 0). With as little as 3 d of FGF-2 exposure, some cortical progenitors began to generate neurons and, with increasing passage, cortical and hippocampal cultures normalized to a steady-state population, ∼10% of which were capable of differentiating into neurons.
To determine how rapidly cortical cells acquired the competence to differentiate into neurons in culture, cells were isolated and cultured in growth medium for various times, allowed to differentiate in differentiation medium, and then evaluated for the presence of neurons (Fig. 4). In control cultures, cells were cultured in 10% fetal bovine serum or 5 ng/ml recombinant platelet-derived growth factor (PDGF) instead of FGF-2. After 3 d in FGF-2, the occasional neuron was observed in cortical cultures, and this number gradually increased with lengthening exposure to FGF-2. By 4 weeks, both cultures had been passaged three times, and ∼10% of the cells formed neurons when induced to differentiate. Although cells were rapidly recruited into cell cycle in serum or PDGF, no neurons were formed (data not shown). Cultures were also treated with BrdU for the first 10 d, and the newly generated neurons were evaluated for evidence of cell division. All neurons scored were labeled (>99%). This demonstrated that exposure to FGF-2 was necessary for the recruitment of neuron-competent precursors and that proliferation invariably accompanied the recruitment process.
Neuron-competent progenitors are found in areas distant from the proliferative zones of the anterior SVZ
Cortical gray matter contains a small population of endogenously dividing glial progenitors (Mares et al., 1975; Kaplan and Hinds, 1980), but the underlying subcortical white matter and ventricular zone have relatively abundant populations of dividing cells (Fig.1A,E). Although we expect that many of the “cortical” progenitors were derived from this underlying proliferative zone, it was also possible that contaminating cells from the more rostral neurogenic areas of the lateral ventricle may have been present in the cortical preparations. Noble, Raff, and others have shown that the adult optic nerve retains an active population of glial progenitors (Raff et al., 1987; Miller et al., 1989; Wolswijk and Noble, 1989), and the optic nerve rostral to the optic chiasma can be easily harvested without risk of contamination from the SVZ of the lateral ventricle.
To determine whether there were progenitors in the optic nerve with a latent ability to generate neurons, optic nerve was harvested, dissociated, and fractionated. Low-buoyancy cells were cultured for 14 d in the presence of high FGF-2 and then allowed to differentiate for an additional 14 d. BrdU was added during the last 72 hr of FGF-2 treatment (day 14). On day 28, cultures were evaluated for the presence of neurons (β-tubulin), and those neurons present were scored for BrdU immunoreactivity. Figure5A–C shows a typical culture from the optic nerve after 1, 3, or 7 d in vitro. Although many glia rapidly differentiated in the primary culture (Fig.5A, arrows), clusters of proliferative precursor-like cells were readily detected (Fig.5B,C). After several weeks in culture, a small but significant minority of these responding cells were able to generate β-tubulin-positive neurons when induced to differentiate (0.8 ± 0.3% of the total population; mean ± SEM; n = 5) (Fig. 5). Neuronal markers, such as β-tubulin, 200 kDa neurofilament, and tau, were never detected in GFAP-positive or O4-positive glia, suggesting that the neuron-like cells were authentic neurons rather than glia that inappropriately expressed neuronal markers. Neurons were often found in small clusters, suggesting a clonal derivation, and all cells, including neurons, were labeled with BrdU during the last 72 hr of FGF-2 treatment (less than one unlabeled cell per 50,000 total nuclei) (Figs. 2,5D–K). This data indicates that all neurons were derived from proliferative precursors. The fact that these neurons are generated from cells isolated from the optic nerve dispels any concerns of contamination from known neurogenic zones and demonstrates that a latent neurogenic potential is retained by precursors from divergent regions of the adult brain.
Fig. 5.
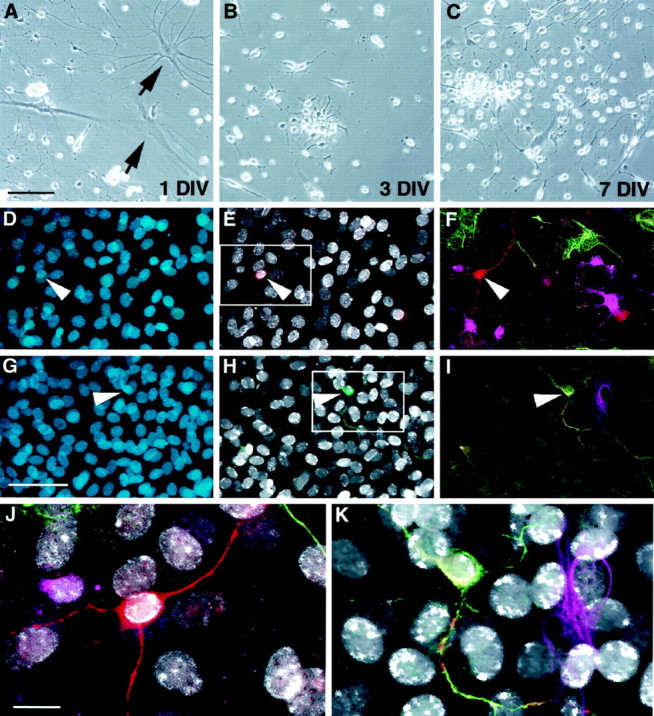
Neurogenesis in cultures initiated from adult optic nerve. Low-buoyancy cells were isolated from adult optic nerve and placed in culture. At 1 DIV, a mixture of immature and differentiated cell morphologies was present (A).Arrows in A show typical glial profiles present at 1 DIV. On days 3 (B) and 7 (C), small proliferating clusters became apparent. After 3 weeks in the presence of FGF-2, a small but significant proportion of cells (0.8%) were able to differentiate into β-tubulin-positive neurons. In D–K, cells were treated with BrdU for 72 hr (days 12–14) and then switched to differentiation medium on day 14. Cells were fixed on day 28 and evaluated for BrdU and lineage-specific markers. Total nuclei are shown in blue (D, G). BrdU-positive nuclei (white) in the same fields are shown along with β-tubulin (red, E) or 200 kDa neurofilament (green, H).F and I, respectively, show multiple labeling for β-tubulin (red), GFAP (green), and O4 (magenta) or 200 kDa neurofilament (green), tau (red), and GFAP (magenta). Enlargements of the areas boxed in E and_H_ clearly show that the neuronal nuclei are BrdU-positive (white in J and_K_). Scale bars: A–C, 60 μm;D–I, 35 μm; J, K, 15 μm.
DISCUSSION
The use of acutely isolated cells in this study suggests several possibilities regarding the presence of multipotent progenitors in the adult brain. First, the potential of cells from non-neurogenic areas to generate neurons does not appear to be an artifact of perpetualized cultures because new neurons can be recruited within days of isolating cells from normal adult tissues. Second, this normal progenitor population appears to exist as an early multipotent progenitor or stem cell (Anderson, 1994; Temple and Qian, 1996;Morrison et al., 1997), because the competence to differentiate into neurons is not intrinsic to these cells but is only gained by progeny of the original cell after the instructional influence of exogenous cues (i.e., removal from in vivo cues and the application of high concentrations of FGF-2). Third, because white matter areas rich in proliferative glial progenitors also yield populations of multipotent cells, it seems likely that stem cells continue to participate in ongoing gliogenesis in the adult. This could be an active process, i.e., multipotent cells actively proliferate to generate glial progeny, or an indolent process by which a quiescent population of multipotent cells slowly replenishes the proliferative pool of glial progenitors.
The exact number of multipotent stem cells in adult tissues is still open for debate. The clonal analysis suggests that ∼20% of all proliferative cells present after 1 week of culture were multipotent stem-like cells. However, because progenitors at different stages of commitment may cycle at different rates, it would be difficult to calculate the exact number of multipotent precursors isolated from each tissue. A crude estimate might be made as follows. The number of low-buoyancy cells recovered from a known weight of adult tissue was ∼1000 cells/mg for hippocampus and ∼700 cells/mg for cortex. If 20% are multipotent upon isolation, then ∼200 or 140 multipotent cells are present in each milligram of HC or cortical tissue, respectively. Although this number is exceedingly small relative to the total cell complement, it is probably quite significant given the considerable proliferative capacity of these stem-like cells and the ability to generate neurons. In combination with our past observations, the present findings suggest that stem-like cells can be isolated from very diverse regions of the adult CNS, including septum, striatum, cortex, spinal cord, and optic nerve (Gage et al., 1995a; Palmer et al., 1995; Shihabuddin et al., 1997).
Although multipotent cells may be broadly distributed, this distribution is likely to be nonuniform. Of the total low-buoyancy cell complement from hippocampus, cortex, and optic nerve, 7.7, 2.7, or 0.8%, respectively, differentiate into neurons after 2–3 weeks of proliferation in the presence of FGF-2. Consistent with these results, preliminary experiments on carefully subdissected tissues indicate that the ventricular zone of the lateral ventricle is particularly rich in low-buoyancy multipotent cells, an observation consistent with the numerous works of Weiss, van der Kooy, and others (Cepko, 1988;Reynolds et al., 1992; Morshead et al., 1994) and an observation consistent with the recent report of stem cells within or immediately adjacent to the ependymal layer proper (Doetsch et al., 1999; Johansson et al., 1999). White matter of the corpus callosum and spinal cord also yield abundant but lesser populations of FGF-2-responsive neurogenic cells, and samples of gray matter conservatively trimmed of white matter or ependymal surfaces yield lower, yet still significant, numbers of cells. This finding implies that multipotent precursors are most abundant in ventricular areas, i.e., the residuum of the developmental neurogenic zone, but also suggests that cells with multi-lineage potential may be present within the parenchyma as well. When comparing results from Johansson et al. (1999) to the recent findings of Doetsch et al. (1999), it is unclear what form the multipotent stem cell takes under normal conditions (i.e., ependymal vs astrocyte-like or some undetermined phenotype); however, the implication of both works is that a common and widespread cell type can generate neurons under appropriate conditions, an observation first suggested for optic nerve by Omlin and colleagues (Omlin and Waldmeyer, 1989; Omlin and Riederer, 1992).
The fact that the ependymal zone retains a population of stem cells may provide an explanation for our ability to isolate such cells from so many different areas of the adult brain, especially if these cells indeed turn out to be astrocyte-like cells. However, a remaining question is why multipotent progenitors are not used to make neurons in all regions. The naturally elicited neurogenic potential in the SGZ and SVZ may involve either escape from several layers of glial-directed cues present in the other regions or simple activation of a dormant program. In our studies, simple removal of cells from in vivo cues does not allow cells from cortical tissues to differentiate into neurons, suggesting that active local inhibition alone does not account for the lack of neurogenesis. Neuronal competence is only acquired after one or more divisions in the presence of high concentrations of FGF-2. The replication of the DNA of a cell may be essential for chromatin restructuring associated with alterations in gene expression patterns, particularly when overcoming the relatively stable chromatin structure associated with silenced regions of the genome (Aparicio and Gottschling, 1994). Replication alone, however, does not appear to be sufficient to activate a neuronal potential. Most of the cells proliferate in the presence of serum or recombinant PDGF, but the responding cells were limited to glial precursors. A high concentration of FGF-2 appears to be an essential component of the environment that allows stem cell progeny to acquire the competence to form neurons.
FGF-2 has long been known for its pleiotropic effects on neural progenitors (Walicke, 1988; McKinnon et al., 1990; Anderson, 1993;Sensenbrenner et al., 1994), and many groups have noted that FGF-2 is a necessary mitogen for maintaining proliferative multipotent precursors_in vitro_ (Richards et al., 1992; Vescovi et al., 1993;Kilpatrick and Bartlett, 1995; Gritti et al., 1999; Tropepe et al., 1999), but little is known regarding the potential of FGF-2 to alter the lineage potential of a cell. Work by Qian et al. (1997) suggests that a developmental change in the concentration of FGF-2 may play a role in regulating the fate of neural stem cells. FGF-2 expression_in vivo_ is upregulated concurrently with the stem cells switch from a neuron-only program to one that also generates glia.In vitro, treatment of embryonic day 10 (E10) cortical progenitors with low levels of FGF-2 (0.1 ng/ml) retains cells in a neuron-only program. Treatment of these same cells with higher concentrations of FGF-2 (10 μg/ml) stimulates stem cell proliferation (i.e., the average clone size increases) and encourages progeny to generate glia in addition to neurons. Although the authors argue that FGF-2 acts to activate a gliogenic program, the observation that FGF-2 stimulates the production of multiple lineages in a population ordinarily limited to generating one cell type may actually be very similar to our findings. The obvious difference is that the predominant differentiation program of “adult” precursors is to generate glia and not neurons. If stem cells are involved in adult gliogenic processes, perhaps both adult and embryonic precursors are “normalized” to a multi-lineage program by high concentrations of FGF-2 (Fig. 6), e.g., FGF-2 may activate both neuronal and glial programs in responding cells. Because the neuronal pathway is fully active in E10 stem cells, only changes in glial production would be measured. In fact, treatment of E10 progenitors with FGF-2 had very little effect on the number of neurons generated by each clone, although the average number of total cells in each colony increased dramatically. In a similar sense, the glial pathway may be fully activated in adult stem cells, and the only measurable effect of FGF-2 would be to stimulate neuron production. Whether this is through a recruitment process in which a latent potential is activated (our favored hypothesis because of the rapidity of neuronal recruitment in vitro) or because of abrupt FGF-2-mediated reprogramming of progenitors normally committed to the glial lineage remains to be determined.
Fig. 6.

Potential influence of FGF-2 on the adult neural stem cell. If multipotent precursors or stem cells (SC) exist in non-neurogenic areas of the brain, they may persist in one of two states. Multipotent cells may actively participate in the gliogenic program by replenishing a pool of committed progenitors (P). Removal of cells from this environment and treatment with FGF-2 may de-repress or stimulate a latent neurogenic program. Alternatively, stem cells may represent a vestigial population that is inactive in most areas of the brain. Native gliogenesis (and/or neurogenesis) would then be mediated by committed progenitors, and the primary effects of FGF-2 on stem cells isolated in culture would be mitogenic. Once actively dividing in culture, stem cells may generate glia (G) or neurons (N) in a stochastic manner.
An alternative hypothesis not addressed by our paradigm is that multipotent precursors might not participate in adult gliogenesis at all, and FGF-2 may simply act as a mitogen to recruit a vestigial population of stem-like cells that cofractionate with committed glial progenitors. Intraventricular administration of FGF-2 seems to argue against a pure mitogenic effect because precursors in the ventricular zone only respond with moderate increases in proliferation (Kuhn et al., 1997). Instead, FGF-2 seems to have a stronger effect on the ratio of newborn neurons and glia that survive after treatment, suggesting that FGF-2 may equally influence proliferation and cell fate in vitro.
From birth to senescence, the brain may exhibit a continuum of “developmental” plasticity retained by stem-like cells that respond to different environments in different manners. This plasticity may be gradually attenuated but persists at some level throughout life. Once resident, it may remain unused throughout life. Alternatively, stem-like cells may play an active role in the ongoing gliogenesis found throughout the adult brain. If the latter is true, the latent potential to generate neurons may simply never be invoked outside of the neurogenic regions of the hippocampus and rostral ventricular zone. In the context of our present work, it seems likely that the multipotent precursor recruited by high concentrations of FGF-2 represents a common precursor to both neuronal and glial progenitor populations generated in the adult brain. Although these cells are probably quite rare relative to the total cellular complement in adult brain tissues, the ability to isolate and enrich for these cells on the basis of buoyant density provides an opportunity to elucidate the regulatory mechanisms governing their activity in vitro. In addition, the rapid isolation of progenitors from adult tissues provides a unique opportunity to evaluate native progenitor populations in grafting models for cell replacement and brain repair.
Footnotes
We thank S. Forbes, L. Moore, B. Miller, and L. Kitabayashi for their excellent technical assistance. Our special thanks to M. L. Gage and E. M. Blackwood for their critical reading of this manuscript. We are grateful for the continued support of the Lookout Fund, the Hollfelder Foundation, Robert J. and Claire Pasarow Foundation, and National Institutes of Health Grants AG06088 and N01-NS-6–2348.
Correspondence should be addressed to Theo D. Palmer, Laboratory of Genetics, The Salk Institute, 10010 North Torrey Pines Road, La Jolla, CA 92037.
REFERENCES
- 1.Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319–335. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]
- 2.Altman J, Das GD. Autoradiographic and histological studies of postnatal neurogenesis. I. A longitudinal investigation of the kinetics, migration and transformation of cells incorporating tritiated thymidine in neonate rats, with special reference to postnatal neurogenesis in some brain regions. J Comp Neurol. 1966;126:337–389. doi: 10.1002/cne.901260302. [DOI] [PubMed] [Google Scholar]
- 3.Anderson DJ. Cell fate determination in the peripheral nervous system: the sympathoadrenal progenitor. J Neurobiol. 1993;24:185–198. doi: 10.1002/neu.480240206. [DOI] [PubMed] [Google Scholar]
- 4.Anderson DJ. Stem cells and transcription factors in the development of the mammalian neural crest. FASEB J. 1994;8:707–713. doi: 10.1096/fasebj.8.10.8050669. [DOI] [PubMed] [Google Scholar]
- 5.Aparicio OM, Gottschling DE. Overcoming telomeric silencing: a trans-activator competes to establish gene expression in a cell cycle-dependent way. Genes Dev. 1994;8:1133–1146. doi: 10.1101/gad.8.10.1133. [DOI] [PubMed] [Google Scholar]
- 6.Bayer SA. Changes in the total number of dentate granule cells in juvenile and adult rats: a correlated volumetric and 3H-thymidine autoradiographic study. Exp Brain Res. 1982;46:315–323. doi: 10.1007/BF00238626. [DOI] [PubMed] [Google Scholar]
- 7.Burns JC, Friedmann T, Driever W, Burrascano M, Yee JK. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci USA. 1993;90:8033–8037. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cepko C. Immortalization of neural cells via oncogene transduction. Trends Neurosci. 1988;11:6–8. doi: 10.1016/0166-2236(88)90039-2. [DOI] [PubMed] [Google Scholar]
- 9.Doetsch F, Caillé I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neuronal stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- 10.Emi N, Friedmann T, Yee JK. Pseudotype formation of murine leukemia virus with the G protein of vesicular stomatitis virus. J Virol. 1991;65:1202–1207. doi: 10.1128/jvi.65.3.1202-1207.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gage FH, Coates PW, Palmer TD, Kuhn HG, Fisher LJ, Suhonen JO, Peterson DA, Suhr ST, Ray J. Survival and differentiation of adult neuronal progenitor cells transplanted to the adult brain. Proc Natl Acad Sci USA. 1995a;92:11879–11883. doi: 10.1073/pnas.92.25.11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gage FH, Ray J, Fisher LJ. Isolation, characterization, and use of stem cells from the CNS. Annu Rev Neurosci. 1995b;18:159–192. doi: 10.1146/annurev.ne.18.030195.001111. [DOI] [PubMed] [Google Scholar]
- 13.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grinspan JB, Stern JL, Pustilnik SM, Pleasure D. Cerebral white matter contains PDGF-responsive precursors to O2A cells. J Neurosci. 1990;10:1866–1873. doi: 10.1523/JNEUROSCI.10-06-01866.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gritti A, Frolichsthal-Schoeller P, Galli R, Parati EA, Cova L, Pagano SF, Bjornson CR, Vescovi AL. Epidermal and fibroblast growth factors behave as mitogenic regulators for a single multipotent stem cell-like population from the subventricular region of the adult mouse forebrain. J Neurosci. 1999;19:3287–3297. doi: 10.1523/JNEUROSCI.19-09-03287.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johansson CB, Momma S, Clarke DL, Risling M, Lendahl U, Frisen J. Identification of a neural stem cell in the adult mammalian central nervous system. Cell. 1999;96:25–34. doi: 10.1016/s0092-8674(00)80956-3. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan MS, Bell DH. Mitotic neuroblasts in the 9-day-old and 11-month-old rodent hippocampus. J Neurosci. 1984;4:1429–1441. doi: 10.1523/JNEUROSCI.04-06-01429.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaplan MS, Hinds JW. Gliogenesis of astrocytes and oligodendrocytes in the neocortical grey and white matter of the adult rat: electron microscopic analysis of light radioautographs. J Comp Neurol. 1980;193:711–727. doi: 10.1002/cne.901930309. [DOI] [PubMed] [Google Scholar]
- 19.Kilpatrick TJ, Bartlett PF. Cloned multipotential precursors from the mouse cerebrum require FGF-2 whereas glial restricted precursors are stimulated with either FGF-2 or EGF. J Neurosci. 1995;15:3653–3661. doi: 10.1523/JNEUROSCI.15-05-03653.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuhn HG, Winkler J, Kempermann G, Thal LJ, Gage FH. Epidermal growth factor and fibroblast growth factor-2 have different effects on neural progenitors in the adult rat brain. J Neurosci. 1997;17:5820–5829. doi: 10.1523/JNEUROSCI.17-15-05820.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- 22.Levison SW, Goldman JE. Both oligodendrocytes and astrocytes develop from progenitors in the subventricular zone of postnatal rat forebrain. Neuron. 1993;10:201–212. doi: 10.1016/0896-6273(93)90311-e. [DOI] [PubMed] [Google Scholar]
- 23.Levison SW, Chuang C, Abramson BJ, Goldman JE. The migrational patterns and developmental fates of glial precursors in the rat subventricular zone are temporally regulated. Development. 1993;119:611–622. doi: 10.1242/dev.119.3.611. [DOI] [PubMed] [Google Scholar]
- 24.Lisak RP, Pleasure DE, Silberberg DH, Manning MC, Saida T. Long term culture of bovine oligodendroglia isolated with a Percoll gradient. Brain Res. 1981;223:107–122. doi: 10.1016/0006-8993(81)90809-x. [DOI] [PubMed] [Google Scholar]
- 25.Lois C, Alvarez-Buylla A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc Natl Acad Sci USA. 1993;90:2074–2077. doi: 10.1073/pnas.90.5.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luskin MB. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron. 1993;11:173–189. doi: 10.1016/0896-6273(93)90281-u. [DOI] [PubMed] [Google Scholar]
- 27.Luskin MB, Zigova T, Soteres BJ, Stewart RR. Neuronal progenitor cells derived from the anterior subventricular zone of the neonatal rat forebrain continue to proliferate in vitro and express a neuronal phenotype. Mol Cell Neurosci. 1997;8:351–366. doi: 10.1006/mcne.1996.0592. [DOI] [PubMed] [Google Scholar]
- 28.Mares V, Lodin Z, Jilek M. An estimate of the number of cells arising by division in mouse cerebral hemispheres from age one to 12 months: an autoradiographic study of DNA synthesis. J Comp Neurol. 1975;161:471–482. doi: 10.1002/cne.901610311. [DOI] [PubMed] [Google Scholar]
- 29.Maric O, Maric I, Ma W, Lahojuji F, Somogyi R, Wen X, Sieghart W, Fritschy JM, Barker JL. Anatomical gradients in proliferation and differentiation of embryonic rat CNS accessed by buoyant density fractionation: α3, β3 and γ2 GABAA receptor subunit co-expression by post-mitotic neocortical neurons correlates directly with cell buoyancy. Eur J Neurosci. 1997;9:507–522. doi: 10.1111/j.1460-9568.1997.tb01628.x. [DOI] [PubMed] [Google Scholar]
- 30.McKay R. Stem cells in the central nervous system. Science. 1997;276:66–71. doi: 10.1126/science.276.5309.66. [DOI] [PubMed] [Google Scholar]
- 31.McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA. FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron. 1990;5:603–614. doi: 10.1016/0896-6273(90)90215-2. [DOI] [PubMed] [Google Scholar]
- 32.Miller RH, ffrench-Constant C, Raff MC. The macroglial cells of the rat optic nerve. Annu Rev Neurosci. 1989;12:517–534. doi: 10.1146/annurev.ne.12.030189.002505. [DOI] [PubMed] [Google Scholar]
- 33.Morrison SJ, Shah NM, Anderson DJ. Regulatory mechanisms in stem cell biology. Cell. 1997;88:287–298. doi: 10.1016/s0092-8674(00)81867-x. [DOI] [PubMed] [Google Scholar]
- 34.Morshead CM, Reynolds BA, Craig CG, McBurney MW, Staines WA, Morassutti D, Weiss S, van der Kooy D. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron. 1994;13:1071–1082. doi: 10.1016/0896-6273(94)90046-9. [DOI] [PubMed] [Google Scholar]
- 35.Omlin FX, Riederer BM. Cells with neuronal characteristics differentiate and persist for one year in rat optic nerve explants cultures. Brain Res Bull. 1992;28:9–16. doi: 10.1016/0361-9230(92)90225-m. [DOI] [PubMed] [Google Scholar]
- 36.Omlin FX, Waldmeyer J. Differentiation of neuron-like cells in cultured rat optic nerves: a neuron or common neuron–glia progenitor? Dev Biol. 1989;133:247–253. doi: 10.1016/0012-1606(89)90315-1. [DOI] [PubMed] [Google Scholar]
- 37.Palmer TD, Ray J, Gage FH. FGF-2-responsive neuronal progenitors reside in proliferative and quiescent regions of the adult rodent brain. Mol Cell Neurosci. 1995;6:474–486. doi: 10.1006/mcne.1995.1035. [DOI] [PubMed] [Google Scholar]
- 38.Palmer TD, Takahashi J, Gage FH. The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci. 1997;8:389–404. doi: 10.1006/mcne.1996.0595. [DOI] [PubMed] [Google Scholar]
- 39.Pertoft H, Laurent TC. Isopycnic separation of cells by centrifugation in percoll gradients. Prog Clin Biol Res. 1982;102:95–104. [PubMed] [Google Scholar]
- 40.Poduslo SE, Norton WT. Isolation of specific brain cells. Methods Enzymol. 1975;35:561–579. doi: 10.1016/0076-6879(75)35185-9. [DOI] [PubMed] [Google Scholar]
- 41.Qian X, Davis AA, Goderie SK, Temple S. FGF2 concentration regulates the generation of neurons and glia from multipotent cortical stem cells. Neuron. 1997;18:81–93. doi: 10.1016/s0896-6273(01)80048-9. [DOI] [PubMed] [Google Scholar]
- 42.Raff MC, Temple S, ffrench-Constant C. Glial cell development and function in the rat optic nerve. Prog Brain Res. 1987;71:435–438. doi: 10.1016/s0079-6123(08)61844-0. [DOI] [PubMed] [Google Scholar]
- 43.Reynolds BA, Tetzlaff W, Weiss S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J Neurosci. 1992;12:4565–4574. doi: 10.1523/JNEUROSCI.12-11-04565.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richards LJ, Kilpatrick TJ, Bartlett PF. De novo generation of neuronal cells from the adult mouse brain. Proc Natl Acad Sci USA. 1992;89:8591–8595. doi: 10.1073/pnas.89.18.8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sensenbrenner M, Deloulme JC, Gensburger C. Proliferation of neuronal precursor cells from the central nervous system in culture. Rev Neurosci. 1994;5:43–53. doi: 10.1515/revneuro.1994.5.1.43. [DOI] [PubMed] [Google Scholar]
- 46.Shank RP, Campbell GL. Amino acid uptake, content, and metabolism by neuronal and glial enriched cellular fractions from mouse cerebellum. J Neurosci. 1984;4:58–69. doi: 10.1523/JNEUROSCI.04-01-00058.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shihabuddin LS, Ray J, Gage FH. FGF-2 is sufficient to isolate progenitors found in the adult mammalian spinal cord. Exp Neurol. 1997;148:577–586. doi: 10.1006/exnr.1997.6697. [DOI] [PubMed] [Google Scholar]
- 48.Sommer I, Schachner M. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. Dev Biol. 1981;83:311–327. doi: 10.1016/0012-1606(81)90477-2. [DOI] [PubMed] [Google Scholar]
- 49.Sommer I, Schachner M. Cell that are O4 antigen-positive and O1 antigen-negative differentiate into O1 antigen-positive oligodendrocytes. Neurosci Lett. 1982;29:183–188. doi: 10.1016/0304-3940(82)90351-2. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi J, Palmer TD, Gage FH. Retinoic acid and neurotrophins collaborate to regulate neurogenesis in adult-derived neural stem cell cultures. J Neurobiol. 1999;38:65–81. [PubMed] [Google Scholar]
- 51.Temple S, Qian X. Vertebrate neural progenitor cells: subtypes and regulation. Curr Opin Neurobiol. 1996;6:11–17. doi: 10.1016/s0959-4388(96)80003-1. [DOI] [PubMed] [Google Scholar]
- 52.Tropepe V, Sibilia M, Ciruna BG, Rossant J, Wagner EF, van der Kooy D. Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev Biol. 1999;208:166–188. doi: 10.1006/dbio.1998.9192. [DOI] [PubMed] [Google Scholar]
- 53.Vescovi AL, Reynolds BA, Fraser DD, Weiss S. bFGF regulates the proliferative fate of unipotent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells. Neuron. 1993;11:951–966. doi: 10.1016/0896-6273(93)90124-a. [DOI] [PubMed] [Google Scholar]
- 54.Walicke PA. Basic and acidic fibroblast growth factors have trophic effects on neurons from multiple CNS regions. J Neurosci. 1988;8:2618–2627. doi: 10.1523/JNEUROSCI.08-07-02618.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weiss S, Reynolds BA, Vescovi AL, Morshead C, Craig CG, van der Kooy D. Is there a neural stem cell in the mammalian forebrain? Trends Neurosci. 1996;19:387–393. doi: 10.1016/s0166-2236(96)10035-7. [DOI] [PubMed] [Google Scholar]
- 56.Wolswijk G, Noble M. Identification of an adult-specific glial progenitor cell. Development. 1989;105:387–400. doi: 10.1242/dev.105.2.387. [DOI] [PubMed] [Google Scholar]