Cerebral Amyloid Induces Aberrant Axonal Sprouting and Ectopic Terminal Formation in Amyloid Precursor Protein Transgenic Mice (original) (raw)
Abstract
A characteristic feature of Alzheimer’s disease (AD) is the formation of amyloid plaques in the brain. Although this hallmark pathology has been well described, the biological effects of plaques are poorly understood. To study the effect of amyloid plaques on axons and neuronal connectivity, we have examined the axonal projections from the entorhinal cortex in aged amyloid precursor protein (APP) transgenic mice that exhibit cerebral amyloid deposition in plaques and vessels (APP23 mice). Here we report that entorhinal axons form dystrophic boutons around amyloid plaques in the entorhinal termination zone of the hippocampus. More importantly, entorhinal boutons were found associated with amyloid in ectopic locations within the hippocampus, the thalamus, white matter tracts, as well as surrounding vascular amyloid. Many of these ectopic entorhinal boutons were immunopositive for the growth-associated protein GAP-43 and showed light and electron microscopic characteristics of axonal terminals. Our findings suggest that (1) cerebral amyloid deposition has neurotropic effects and is the main cause of aberrant sprouting in AD brain; (2) the magnitude and significance of sprouting in AD have been underestimated; and (3) cerebral amyloid leads to the disruption of neuronal connectivity which, in turn, may significantly contribute to AD dementia.
Keywords: Alzheimer’s disease, hippocampus, PHAL, tracing, entorhinal cortex, axon, synapse, sprouting, CNS, neurodegeneration, vasculature, APP, mouse, aging
The deposition of amyloid throughout the brain is a characteristic pathology of Alzheimer’s disease (AD), although its pathophysiological significance remains unclear (Selkoe, 1991; Yankner, 1996). It has been proposed that neuronal circuits are compromised in AD (Hyman et al., 1984; Kowall and Kosik, 1987; Geula, 1998), and one hypothesis is that amyloid plaques disrupt neuronal connectivity in the brain, resulting in loss of function and dementia (Knowles et al., 1998). This hypothesis is difficult to test because of the presence of other lesions, such as neurofibrillary tangles, along with the deposition of amyloid in tissue of AD patients. In contrast, mouse models of cerebral amyloidosis have opened new avenues into the investigation of the specific role of amyloid deposition in the pathogenesis of AD (Games et al., 1995; Hsiao et al., 1996;Sturchler-Pierrat et al., 1997; Hsia et al., 1999).
To investigate the effect of amyloid deposition on axons and neuronal connectivity, we studied the axonal projections from the entorhinal cortex to the hippocampus in APP23 mice that overexpress mutated human amyloid precursor protein (APP) and form amyloid plaques progressively with age (Sturchler-Pierrat et al., 1997; Calhoun et al., 1998). The entorhinal cortex and the hippocampus have a well known function in learning and memory (Wallenstein et al., 1998) and exhibit early and severe pathology in AD (Hyman et al., 1984; Braak and Braak, 1991). To visualize the entorhinal axons, the sensitive anterograde tracer_Phaseolus vulgaris_ leucoagglutinin (PHAL), which allows for the analysis of projections at the level of single axons (Gerfen and Sawchenko, 1984), was injected into the entorhinal cortex of aged and young APP23 mice as well as nontransgenic controls.
MATERIALS AND METHODS
Animals. The generation of APP23 transgenic mice is described in detail elsewhere (Sturchler-Pierrat et al., 1997). Briefly, a murine Thy-1 promoter element was used to drive neuron-specific expression of human mutated APP751 (Swedish double mutation 670/671 KM→NL) in B6D2 mice. In this study we used six aged (18–20 month) hemizygous APP23 mice and two age-matched nontransgenic controls. In addition, two young (5 month) hemizygous APP23 mice were used. The mice are from the F8-F10 generation of backcrossing to B6 mice and thus can be considered C57BL/6J-TgN(Thy1-APP670/671 KM→NL) mice.
Anterograde tracing. For anterograde tracing of entorhinal axons, mice received a unilateral injection of PHAL into the entorhinal cortex (Gerfen and Sawchenko, 1984; Deller et al., 1999). In brief, mice were deeply anesthetized using a combination of ketamine (10 mg/kg body weight; Ketalar; Parke-Davis, Ann Arbor, MI) and xylazine (20 mg/kg; Rompun, Bayer, Germany) in saline. PHAL (2.5% in 10 mm PBS, pH 7.8; Vector Laboratories, Burlingame, CA) was delivered by iontophoretic injection via a stereotactically positioned glass micropipette (tip diameter, 15–30 μm). A 5 μA positive current was applied for 15 min in a 5 sec on–5 sec off cycle. Two adjacent injections were administered to the left entorhinal cortex at the following stereotaxic coordinates: (for mice up to 26 gm body weight) anteroposterior (bregma), −4.4; lateral, 2.9; dorsoventral (skull), −4.2; and anteroposterior, −4.6; lateral, 2.9; dorsoventral, −3.9; for mice with body weight >26 gm, coordinates were adjusted to +0.1 anteroposterior, +0.1 lateral, and +0.1 dorsoventral.
Tissue preparation and immunocytochemistry. All mice were killed 9 d after PHAL injections by an overdose of pentobarbital (50 mg/ml Nembutal; Abbott Laboratories, Chicago, IL) and subsequent transcardial perfusion with 0.01 m PBS followed by 4% paraformaldehyde (PFA) in PBS. Some mice were perfused with PFA plus 1% glutaraldehyde and 0.2% picric acid. Brains were removed and post-fixed in PFA for 24 hr. Brains fixed with glutaraldehyde were used for combined light microscopic and ultrastructural analysis and were cut on a vibratome (50 μm section thickness) and then processed for electron microscopy according to a previously published protocol (Deller et al., 1996). Brains for light microscopic analysis only were placed in 30% sucrose for 24 hr, frozen in 2-methylbutane at −30°C, and sectioned on a freezing-sliding microtome (25–100 μm section thickness).
Immunocytochemistry was performed on free-floating sections using the avidin–biotin peroxidase method (ABC Elite kit; Vector Laboratories) according to previously published protocols (Jucker et al., 1994;Deller et al., 1996). In brief, sections were incubated in 1% H2O2 followed by 0.3% Triton X-100 and blocked in 5% goat or horse serum, all in Tris-buffered saline (TBS). After an overnight incubation at 4°C with the primary antibody in 2% serum and 0.3% Triton X-100, sections were incubated in biotinylated secondary antibody (except for sections incubated with biotinylated PHAL), followed by the avidin—biotin peroxidase complex solution (ABC Elite kit). The chromogen was 3′,3-diaminobenzidine-dihydrochloride (DAB; 0.08%; Sigma, St. Louis, MO). For double immunolabeling, sections were processed in a sequential manner. Immediately after visualization of the first primary antibody by DAB (brown reaction product), sections were incubated for 30 min in 1% H2O2, rinsed extensively in TBS, and immunoreacted as described above with the second primary antibody. The peroxidase substrate for the second peroxidase reaction was Vector-SG (blue–gray reaction product; Vector Laboratories). Additional immunolabeling was performed on paraffin-embedded sections that were prepared according to standard protocols (Phinney et al., 1999).
For fluorescence immunocytochemistry, free-floating sections were labeled using a protocol similar to that described above with the following changes. TBS containing 0.5% bovine serum albumin was used for all steps. The secondary antibodies were Cy2-conjugated goat anti-rabbit IgG and Cy3-conjugated goat anti-mouse IgG (diluted 1:100 and 1:500, respectively; Dianova, Hamburg, Germany). For double immunofluorescence labeling, sections were incubated in both primary antibodies simultaneously while secondary antibodies were added sequentially. For all immunocytochemistry, control sections were processed in the absence of one or both primary antibodies to affirm specificity.
The following antibodies were used: polyclonal biotinylated goat anti-PHAL (for peroxidase immunocytochemistry; diluted 1:800; Vector Laboratories); polyclonal rabbit anti-PHAL (for immunofluorescence; 1:800; Vector Laboratories); polyclonal anti-Aβ (NT-11; 1:2000) (Sturchler-Pierrat et al., 1997); polyclonal and monoclonal antibodies to growth associated protein-43 (GAP-43) (1:1000 and 1:10, respectively; gift of P. Caroni) (Aigner et al., 1995); polyclonal anti-calretinin (1:10,000; SWant, Bellinzona, Switzerland); polyclonal anti-synaptophysin (1:2000; Dako, Glostrup, Denmark); monoclonal anti-MAP2 (AP14; 1:500; gift of L. Binder) (Caceres et al., 1983); and polyclonal antibody to spinophilin (1:2000; gift of P. Greengard) (Allen et al., 1997).
RESULTS
PHAL tracing revealed a typical entorhinohippocampal projection in young APP23 mice and nontransgenic controls, and single entorhinal axons showed a normal morphology (Stanfield et al., 1979). No immunoreactivity for amyloid-β peptide (Aβ) could be detected in these animals (Fig. 1a,c). Consistent with previous reports, aged APP23 mice showed a significant amyloid load in brain (Fig. 1b,d) that is largely congophilic in nature (Sturchler-Pierrat et al., 1997; Calhoun et al., 1998). In areas free of amyloid plaques, as well as in areas of dif fuse amyloid deposition, PHAL-labeled entorhinal fibers in these mice were indistinguishable from controls. However, fibers in the vicinity of compact amyloid deposits often appeared swollen and tortuous and formed large balloon-like structures around the plaques (Fig. 1d,e). Typically, axons maintained a normal morphology until in the vicinity of a plaque. At this point, axons lined the periphery of the plaque and characteristically formed one or more small swellings followed by one or more larger, terminal swellings. Such terminal swellings usually formed facing away from the plaque (Fig.1e).
Fig. 1.

Entorhinal axonal projections in APP23 mice.a, PHAL-labeled projection from the medial entorhinal cortex to the middle molecular layer (MML;arrow) of the dentate gyrus (DG) in an 18-month-old nontransgenic control mouse. Minor projections were identified to CA3 and CA1 of the hippocampus and to the thalamus (TH). The same pattern was observed in young APP23 mice that lack amyloid plaque formation. b, PHAL-labeled projection in an 18-month-old APP23 mouse. Main termination pattern in the MML (arrow) is similar to that seen in the control mouse with the most notable difference being the hyperinnervation of the thalamus. Note the amyloid deposits (blue–gray reaction product) throughout the hippocampus, the thalamus, and within the alveus (ALV). c, d, High magnification of the inferior blade of DG from the control mouse shown in a, and APP23 mouse shown in b. Note the thickened PHAL-labeled axons (arrow) and ballooned, spheroidal axon terminals (arrowheads) in the vicinity of amyloid plaques. In the MML, amyloid plaques were completely engulfed by entorhinal dystrophic terminals, whereas outside of the main entorhinal termination zone only a subpopulation of dystrophic boutons were PHAL-labeled. e, High magnification of PHAL-labeled axons with the characteristic dystrophic terminals around an amyloid plaque in the stratum-lacunosum moleculare of CA3. Many PHAL-labeled axons appear normal until directly adjacent to the amyloid (arrow), then typically curve around the amyloid periphery, forming several small swellings followed by a large terminal balloon-shaped swelling that turns away from the plaque (arrowhead). GC, granule cell layer of the dentate gyrus; IML, inner molecular layer;OML, outer molecular layer. Scale bars:a, 300 μm; c, e, 25 μm; panels a and b, and_c_ and d have the same magnification.
To further investigate the nature of the PHAL-labeled spheroid structures surrounding the plaques, correlated light and electron microscopy was performed (Fig. 2). At the ultrastructural level, PHAL-labeled structures could be identified as swollen, or dystrophic, presynaptic boutons of varying sizes that contain large amounts of unstained membranous material and lysosomal dense bodies (Fig. 2b,d). Only a percentage of the swollen boutons associated with the plaques were labeled, as would be expected since the ipsilateral entorhinal neurons provide the major but not exclusive innervation to this area (Stanfield et al., 1979). Many of the labeled terminals were observed maintaining a synapse (Fig.2e). These PHAL-labeled terminals strongly resemble the plaque-associated dystrophic neurites described in AD (Wisniewski and Terry, 1973; Masliah et al., 1991a; Peters et al., 1991) and provide direct evidence that such dystrophic neurites are of axonal origin. Further substantiating this conclusion, immunohistochemistry for the dendritic marker microtubule-associated protein-2 (MAP-2) (Caceres et al., 1983) and the dendritic spine protein spinophilin (Allen et al., 1997) failed to label amyloid-associated dystrophic neurites in APP23 mice (data not shown).
Fig. 2.
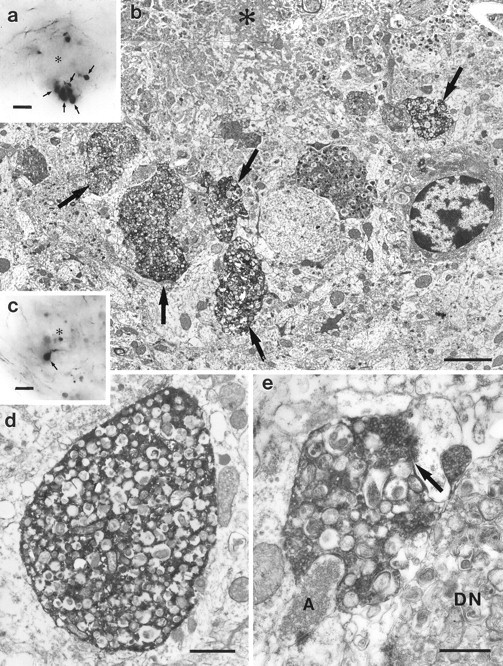
Ultrastructure of PHAL-labeled dystrophic entorhinal boutons in the hippocampus. a, Light micrograph of an amyloid plaque (asterisk) in the molecular layer of the dentate gyrus surrounded by numerous large PHAL-labeled entorhinal boutons (arrows).b, Electron micrograph of the bottom half of the plaque shown in a. The amyloid (asterisk) is surrounded by several dystrophic PHAL-labeled entorhinal boutons (arrows). c, Light micrograph of an amyloid plaque (asterisk) in the molecular layer of the dentate gyrus. The arrow points to a heavily PHAL-labeled entorhinal bouton. d, Electron micrograph of the PHAL-labeled entorhinal bouton illustrated in c. The bouton is filled with numerous multilamellar bodies, characteristic of dystrophic neurites. These structures are surrounded by electron dense immunoprecipitate. e, PHAL-labeled entorhinal bouton in the vicinity of a plaque. This dystrophic bouton contains synaptic vesicles and forms a synapse with a spine. The_arrow_ points to the synaptic cleft. An unlabeled dystrophic neurite (DN), as well as a normal axon terminal (A) are also illustrated. Scale bars:a, c, 10 μm; b, 2.5 μm; d, 1 μm; e, 0.5 μm.
One of the most intriguing observations was that entorhinal fibers were found outside of their normal termination zone in APP23 mice. Such ectopic terminals were most notable in the strictly laminated dentate gyrus (DG): entorhinal axons, normally restricted to the outer molecular layer of the DG (even during regenerative sprouting) (Frotscher et al., 1997), invade the inner molecular layer, which is the zone of commissural fibers (Fig.3a). Similarly, commissural axons, normally restricted to the inner molecular layer of the DG, entered the outer molecular layer which is the zone of entorhinal fibers (Fig. 3b) (Stanfield et al., 1979). Thus, amyloid deposition causes aberrations of both entorhinal and commissural fibers and disrupt the normal laminar organization of the DG.
Fig. 3.
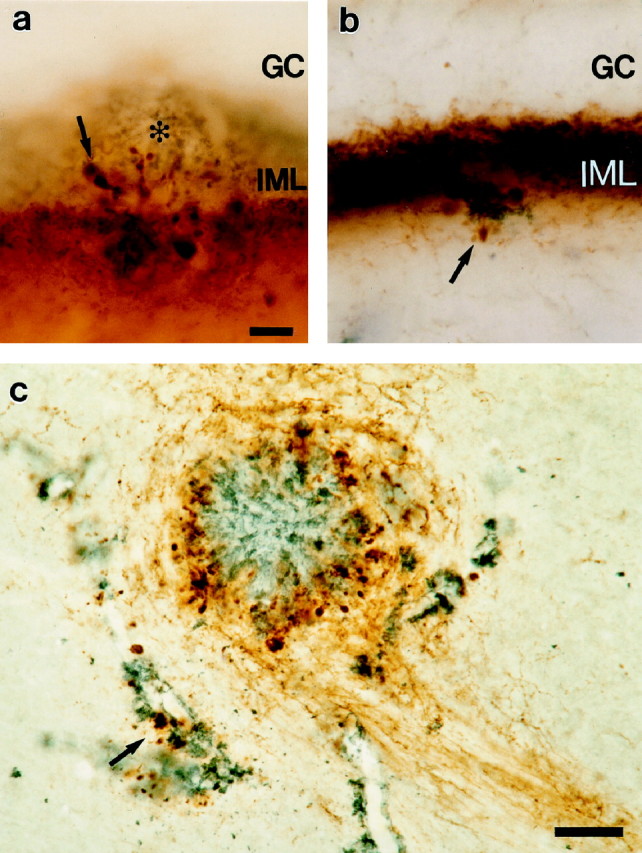
Plaque-associated ectopic axon terminals.a, Disruption of the layer-specific termination of the PHAL-labeled entorhinal axons in the vicinity of an amyloid plaque (asterisk) is evidenced by the invasion of these fibers (brown reaction product) into the inner molecular layer (IML) of the dentate gyrus, which is labeled for calretinin (blue–gray reaction product). Calretinin is specific for the commissural fibers in the mouse and labels specifically the IML (Liu et al., 1996). b, Calretinin-labeled commissural fibers (brown) also form dystrophic terminals (arrow) and deviate from their normally specific termination in the IML when in vicinity of amyloid (blue–gray). c, High magnification of the amyloid plaque (blue–gray) shown in Figure1b. PHAL-labeled entorhinal axons are in_brown_ and reveal a hyperinnervation of the thalamic region around the amyloid plaque (see Fig. 1a for control). Here, entorhinal axons curve around the amyloid and form dystrophic boutons directly adjacent to the amyloid. Interestingly, PHAL-labeled entorhinal axons also formed dystrophic boutons around vascular amyloid deposits (arrow). _GC,Granule cell layer of the dentate gyrus. Scale bars: a, 10 μm; c, 50 μm; panels a and_b have the same magnification.
Additional evidence for amyloid-induced axonal aberration of entorhinal fibers was found in the thalamus. In control mice, very few entorhinal fibers are normally found in this region (Fig. 1a). In contrast, in all APP23 mice that had significant amyloid deposition in the thalamus, a dense plexus of labeled fibers was observed (Figs.1b, 3c). Entorhinal fibers surrounding the amyloid plaques formed the characteristic swollen boutons that were also observed in the hippocampus (Fig. 3c). Thus, the normally weak entorhinal projection to the thalamus was not only found to be appreciably increased but also abnormal (dystrophic) whenever amyloid deposition occurred in this region.
Another intriguing finding was the observation of PHAL-labeled fibers projecting toward and abutting blood vessels containing vascular amyloid (Fig. 3c). In the thalamus, an area heavily affected by vascular amyloid in APP23 mice (Calhoun et al., 1998; Jucker et al., 1998), entorhinal fibers frequently formed dystrophic boutons adjacent to the vascular amyloid deposits. In control mice, entorhinal fibers are not usually associated with blood vessels, and if they are found in the vicinity of a blood vessel, for example in the outer molecular layer of the hippocampus, no dystrophic boutons are formed. This shows that the deposition of vascular amyloid also leads to ectopic axon terminals and abnormal axonal structures and is thus contributing to the disruption of neuronal circuits.
To better understand the process that leads to the entorhinal fiber disruption and ectopic terminal formation in the vicinity of amyloid plaques, we studied markers for axonal growth that are believed to play a role in AD (Geddes et al., 1985, 1986; Masliah et al., 1991b). For this purpose we focused on compact amyloid plaques in the white matter, which are frequently found in the alveus of the hippocampus in aged APP23 mice. Entorhinal fibers travel through the alveus to reach the hippocampus, but they have no known targets in this typically asynaptic white matter tract. Yet, in the alveus, PHAL-labeled entorhinal axons that lie in the vicinity of a plaque form numerous swollen boutons around it (Fig.4a). Immunostaining for GAP-43 (Benowitz et al., 1990) and Aβ revealed amyloid-associated GAP-43-positive boutons similar to the spheroid structures formed by entorhinal fibers around plaques (Fig. 4b). Furthermore, similar to that reported in AD (Masliah et al., 1991b), GAP-43 labeled more fibers throughout the neuropil of APP23 mice than in controls (data not shown). Double-labeling for PHAL and GAP-43 revealed that indeed many dystrophic entorhinal terminals were GAP-43-immunopositive (Fig. 4d-f), supporting the idea that entorhinal fibers found in ectopic locations are the result of amyloid-associated aberrant sprouting.
Fig. 4.

Axonal growth associated with amyloid plaques in the white matter. a, PHAL-labeled entorhinal axons form dystrophic terminals around an amyloid plaque (asterisk) in the alveus, b, Immunohistochemistry with an antibody against GAP-43 reveals that throughout the neuropil and interestingly, as shown here, within the alveus, many dystrophic boutons (brown) associated with amyloid deposits (blue, asterisk) were GAP-43 positive. c, Similar dystrophic neurites were also identified around plaques (asterisk), shown here within the alveus, when sections were immunolabeled for the synaptic marker synaptophysin. d–f, Double fluorescent immunolabeling for PHAL (d,green) and GAP-43 (e, red) revealed that a percentage of PHAL-labeled entorhinal terminals are positive for the growth-associated marker GAP-43 (d–f, arrows). Scale bars: a, 20 μm; b, 20 μm;d, 5 μm; panels b and c, and d–f have the same magnification.
To assess whether the observed aberrant fibers form terminals, immunostaining for the synaptic vesicle protein synaptophysin was performed. Amyloid plaques, even those located in the white matter, were typically engulfed by synaptophysin-positive terminals (Fig.4c). Electron microscopy of white matter plaques confirmed that dystrophic entorhinal terminals form around amyloid deposits (Fig.5a,b). These dystrophic terminals are identical to those observed in the hippocampus, and some of them show elements of presynaptic specialization, such as clustered synaptic vesicles (Fig. 5c,d). These data suggest that cerebral amyloid deposition can induce aberrant axonal growth of entorhinal fibers, and also that it induces synaptic differentiation of these axons in ectopic locations.
Fig. 5.
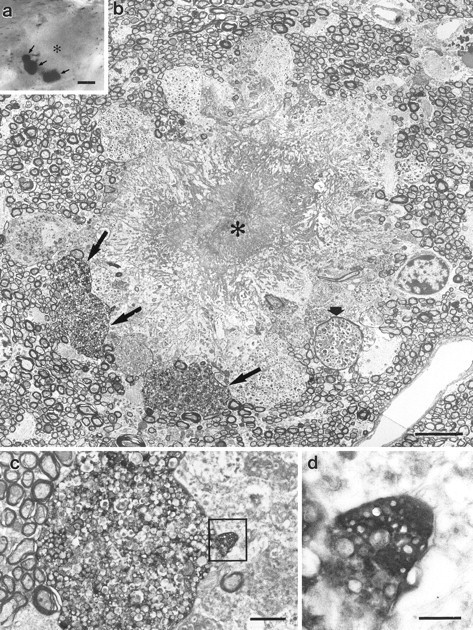
Ultrastructure of entorhinal dystrophic boutons in the white matter. a, Light micrograph of an amyloid plaque (asterisk) in the alveus of the hippocampus. Three PHAL-labeled entorhinal boutons surround the plaque (arrows). b, Electron micrograph of the plaque illustrated in a. The amyloid core (asterisk) is surrounded by numerous dystrophic neurites. The three large entorhinal boutons illustrated in_a_ are indicated by arrows. One of the unlabeled dystrophic neurites is myelinated (short arrow). c, Serial section of the middle PHAL-labeled bouton illustrated in b. The heavily immunolabeled bouton is filled with multilamellar bodies.d, Serial section of the _rectangle_illustrated in c. This part of the dystrophic bouton shows elements of synaptic specialization, i.e., clustered vesicles. Scale bars: a, 10 μm; b, 5 μm;c, 1 μm; d, 0.25 μm.
DISCUSSION
Although the fact that sprouting occurs in AD brain has been recognized earlier (Geddes et al., 1985, 1986; Masliah et al., 1991b), the cause, magnitude, and aberrant nature of the sprouting has remained unclear. In the present study, we examined the effect of amyloid deposition on a well defined axonal projection in APP transgenic mice. Our results revealed that (1) aged APP23 mice show axonal sprouting directly related to amyloid, demonstrating that amyloid deposition and sprouting are intimately linked; (2) axons grow into ectopic locations, and moreover sprout within the white matter and toward vascular amyloid, indicating that amyloid deposition exerts a neurotropic effect on axons not only in vitro (Koo et al., 1993) but also in vivo; and (3) young mice that overexpress APP do not exhibit plaques, dystrophic boutons, or axonal sprouting, demonstrating that APP overexpression alone does not underlie the aberrant sprouting process. Our data therefore suggest that the primary factor underlying abnormal axonal sprouting is the deposition of amyloid.
The well defined and specific laminar organization of the entorhinohippocampal projection, in conjunction with the amyloid deposition in this transgenic model, allowed us to show that not only is amyloid central to the observed sprouting, but that such sprouting is indeed aberrant. It may be argued that the observed sprouting is induced by neurodegeneration or glia activation previously observed in these mice (Calhoun et al., 1998; Stalder et al., 1999). However, if such factors govern the observed sprouting, the sprouting of entorhinal axon should respect the laminar borders of the DG as they do in experimental lesion paradigms (Frotscher et al., 1997). It is the unprecedented nature of the observed sprouting of entorhinal axons into the inner molecular layer of the DG, around blood vessels and within white matter, that strongly supports a primary role for cerebral amyloidosis in the observed axonal sprouting. However, it is possible that other constituents of amyloid deposits, such as perlecan, agrin, and laminin (Snow et al., 1988; Perlmutter et al., 1991; Donahue et al., 1999) also play an important role in the observed sprouting response. It must also be considered that amyloid deposition is a consequence rather then cause of the axonal sprouting in mice that overexpress APP. Yet, the absence of aberrant axons or ectopic terminals in young APP23 mice does not support such a conclusion. The possibility that sprouting/dystrophic terminals release APP/Aβ and thus initiate/accelerate amyloid deposition in a feedforward manner remains open.
The present study has focused on the entorhinohippocampal system, one of many axonal projections in the brain. The vastness of the sprouting and ectopic terminal formation observed in the present study was unexpected. The labeling of amyloid-associated dystrophic terminals throughout the brain using synaptophysin and GAP-43 immunostaining indicates that axon terminal disruption and sprouting is not restricted to the entorhinal–hippocampal projection and occur in all areas that develop amyloidosis. Thus, our results suggest that both the magnitude and significance of alterations to neuronal circuits in AD brain caused by such dystrophic and ectopic axons have been greatly underestimated.
Finally, it should be noted that all PHAL-labeled entorhinal terminals, even those identified as sprouting axon terminals, had dystrophic morphologies. This suggests that axon growth in response to the deposition of amyloid and the formation of abnormal presynaptic terminals, ectopic or otherwise, are not likely to result in functional or restorative neuronal connections. These morphological abnormalities are likely the anatomical correlate of changes in electrophysiological characteristics reported in APP transgenic mouse models with cerebral amyloidosis (Chapman et al., 1999; Hsia et al., 1999).
In conclusion, our results demonstrate that the deposition of cerebral amyloid induces dystrophic boutons, aberrant axonal growth, disrupted fiber tracts, as well as the formation of ectopic terminals. Such changes disrupt normal neuronal connectivity and are a likely candidate for the morphological correlate of cognitive decline in AD.
Footnotes
This study was supported by grants to M.J. from the Swiss National Foundation and the VerUm Foundation (Munich, Germany) and by grants to T.D. and M.F. from the Deutsche Forschungsgemeinschaft (SFB 505). We thank A. Schneider and M. Winter (Institute of Anatomy, Freiburg, Germany), E. Billy (FMI, Basel, Switzerland), C. Sturchler-Pierrat, D. Abramowski, and K.-H. Wiederhold (Novartis, Basel, Switzerland) for excellent technical support, J. Geddes (University of Kentucky, Lexington, KY), L. Walker (Parke-Davis, Ann Arbor, MI), M. Tolnay, and A. Probst (Institute of Pathology, Basel, Switzerland) for helpful comments on this manuscript, and P. Caroni (FMI, Basel, Switzerland), L. Binder (Northwestern University Medical School, Chicago, IL), and P. Greengard (Rockefeller University, New York, NY) for antibody gifts.
A. L. Phinney and T. Deller contributed equally to this work.
Correspondence should be addressed to Dr. M. Jucker, Department of Neuropathology, Institute of Pathology University of Basel, Schönbeinstrasse 40, CH-4003 Basel, Switzerland.
REFERENCES
- 1.Aigner L, Arber S, Kapfhammer JP, Laux T, Schneider C, Botteri F, Brenner HR, Caroni P. Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell. 1995;83:269–278. doi: 10.1016/0092-8674(95)90168-x. [DOI] [PubMed] [Google Scholar]
- 2.Allen PB, Ouimet CC, Greengard P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc Natl Acad Sci USA. 1997;94:9956–9961. doi: 10.1073/pnas.94.18.9956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benowitz LI, Rodriguez WR, Neve RL. The pattern of GAP-43 immunostaining changes in the rat hippocampal formation during reactive synaptogenesis. Brain Res Mol Brain Res. 1990;8:17–23. doi: 10.1016/0169-328x(90)90004-w. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 5.Caceres A, Payne MR, Binder LI, Steward O. Immunocytochemical localization of actin and microtubule-associated protein MAP2 in dendritic spines. Proc Natl Acad Sci USA. 1983;80:1738–1742. doi: 10.1073/pnas.80.6.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Neuron loss in APP transgenic mice. Nature. 1998;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- 7.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 8.Deller T, Martinez A, Nitsch R, Frotscher M. A novel entorhinal projection to the rat dentate gyrus: direct innervation of proximal dendrites and cell bodies of granule cells and GABAergic neurons. J Neurosci. 1996;16:3322–3333. doi: 10.1523/JNEUROSCI.16-10-03322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deller T, Drakew A, Frotscher M. Different primary target cells are important for fiber lamination in the fascia dentata: a lesson from reeler mutant mice. Exp Neurol. 1999;156:239–253. doi: 10.1006/exnr.1999.7020. [DOI] [PubMed] [Google Scholar]
- 10.Donahue JE, Berzin TM, Rafii MS, Glass DJ, Yancopoulos GD, Fallon JR, Stopa EG. Agrin in Alzheimer’s disease: altered solubility and abnormal distribution within microvasculature and brain parenchyma. Proc Natl Acad Sci USA. 1999;96:6468–6472. doi: 10.1073/pnas.96.11.6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frotscher M, Heimrich B, Deller T. Sprouting in the hippocampus is layer-specific. Trends Neurosci. 1997;20:218–223. doi: 10.1016/s0166-2236(96)01018-1. [DOI] [PubMed] [Google Scholar]
- 12.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Hagopian S, Johnson-Wood K, Khan K, Lee M, Liebowitz P, Lieberburg I, Little S, Masliah E, McConglogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Sorlano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 13.Geddes JW, Monaghan DT, Cotman CW, Lott IT, Kim RC, Chui HC. Plasticity of hippocampal circuitry in Alzheimer’s disease. Science. 1985;230:1179–1181. doi: 10.1126/science.4071042. [DOI] [PubMed] [Google Scholar]
- 14.Geddes JW, Anderson KJ, Cotman CW. Senile plaques as aberrant sprout-stimulating structures. Exp Neurol. 1986;94:767–776. doi: 10.1016/0014-4886(86)90254-2. [DOI] [PubMed] [Google Scholar]
- 15.Gerfen CR, Sawchenko PE. An anterograde neuroanatomical tracing method that shows the detailed morphology of neurons, their axons and terminals: immunohistochemical localization of an axonally transported plant lectin, Phaseolus vulgaris leucoagglutinin (PHA-L). Brain Res. 1984;290:219–238. doi: 10.1016/0006-8993(84)90940-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geula C. Abnormalities of neural circuitry in Alzheimer’s disease: hippocampus and cortical cholinergic innervation. Neurology. 1998;51:S18–29. doi: 10.1212/wnl.51.1_suppl_1.s18. [DOI] [PubMed] [Google Scholar]
- 17.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 19.Hyman BT, Van Horsen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 20.Jucker M, Walker LC, Schwarb P, Hengemihle J, Kuo H, Snow AD, Bamert F, Ingram DK. Age-related deposition of glia-associated fibrillar material in brains of C57BL/6 mice. Neuroscience. 1994;60:875–889. doi: 10.1016/0306-4522(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 21.Jucker M, Stalder M, Tolney M, Wiederhold KH, Abramowski D, Sommer B, Staufenbiel M, Calhoun ME. Cerebral amyloid angiopathy in APP transgenic mice with amyloid plaque formation. Soc Neurosci Abstr. 1998;24:1502. [Google Scholar]
- 22.Knowles RB, Gomez-Isla T, Hyman BT. Abeta associated neuropil changes: correlation with neuronal loss and dementia. J Neuropathol Exp Neurol. 1998;57:1122–1130. doi: 10.1097/00005072-199812000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Koo EH, Park L, Selkoe DJ. Amyloid β-protein as a substrate interacts with extracellular matrix to promote neurite outgrowth. Proc Natl Acad Sci USA. 1993;90:4748–4752. doi: 10.1073/pnas.90.10.4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kowall NW, Kosik KS. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann Neurol. 1987;22:639–643. doi: 10.1002/ana.410220514. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Fujise N, Kosaka T. Distribution of calretinin immunoreactivity in the mouse dentate gyrus. I. General description. Exp Brain Res. 1996;108:389–403. doi: 10.1007/BF00227262. [DOI] [PubMed] [Google Scholar]
- 26.Masliah E, Hansen L, Albright T, Mallory M, Terry RD. Immunoelectron microscopic study of synaptic pathology in Alzheimer’s disease. Acta Neuropathol (Berl) 1991a;81:428–433. doi: 10.1007/BF00293464. [DOI] [PubMed] [Google Scholar]
- 27.Masliah E, Mallory M, Hansen L, Alford M, Albright T, DeTeresa R, Terry R, Baudier J, Saitoh T. Patterns of aberrant sprouting in Alzheimer’s disease. Neuron. 1991b;6:729–739. doi: 10.1016/0896-6273(91)90170-5. [DOI] [PubMed] [Google Scholar]
- 28.Perlmutter LS, Barron E, Saperia D, Chui HC. Association between vascular basement membrane components and the lesions of Alzheimer’s disease. J Neurosci Res. 1991;30:673–681. doi: 10.1002/jnr.490300411. [DOI] [PubMed] [Google Scholar]
- 29.Peters A, Palay S, Webster H. The fine structure of the nervous system. Oxford UP; New York: 1991. [Google Scholar]
- 30.Phinney AL, Calhoun ME, Wolfer DP, Lipp HP, Zheng H, Jucker M. No hippocampal neuron or synaptic bouton loss in learning-impaired aged β-amyloid precursor protein-null mice. Neuroscience. 1999;90:1207–1216. doi: 10.1016/s0306-4522(98)00645-9. [DOI] [PubMed] [Google Scholar]
- 31.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 32.Snow AD, Mar H, Nochlin D, Kimata K, Kato M, Suzuki S, Hassell J, Wight TN. The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer’s disease. Am J Pathol. 1988;133:456–463. [PMC free article] [PubMed] [Google Scholar]
- 33.Stalder M, Phinney AL, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–1684. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stanfield BB, Caviness VS, Jr, Cowan WM. The organization of certain afferents to the hippocampus and dentate gyrus in normal and reeler mice. J Comp Neurol. 1979;185:461–483. doi: 10.1002/cne.901850304. [DOI] [PubMed] [Google Scholar]
- 35.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wallenstein GV, Eichenbaum H, Hasselmo ME. The hippocampus as an associator of discontiguous events. Trends Neurosci. 1998;21:317–323. doi: 10.1016/s0166-2236(97)01220-4. [DOI] [PubMed] [Google Scholar]
- 37.Wisniewski H, Terry D. Reexamination of the pathogenesis of the senile plaque. In: Zimmerman H, editor. Progress in neuropathology. Grune and Stratton; New York: 1973. pp. 1–26. [Google Scholar]
- 38.Yankner BA. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]