Extracellular Signal-Regulated Kinase and p38 Subgroups of Mitogen-Activated Protein Kinases Regulate Inducible Nitric Oxide Synthase and Tumor Necrosis Factor-α Gene Expression in Endotoxin-Stimulated Primary Glial Cultures (original) (raw)
Abstract
Tumor necrosis factor-α (TNFα) and nitric oxide (NO), the product of inducible NO synthase (iNOS), mediate inflammatory and immune responses in the CNS under a variety of neuropathological situations. They are produced mainly by “activated” astrocytes and microglia, the two immune regulatory cells of the CNS. In this study we have examined the regulation of TNFα and iNOS gene expression in endotoxin-stimulated primary glial cultures, focusing on the role of mitogen-activated protein (MAP) kinase cascades. The bacterial lipopolysaccharide (LPS) was able to activate extracellular signal-regulated kinase (ERK) and p38 kinase subgroups of MAP kinases in microglia and astrocytes. ERK activation was sensitive to PD98059, the kinase inhibitor that is specific for ERK kinase. The activity of p38 kinase was inhibited by SB203580, a member of the novel class of cytokine suppressive anti-inflammatory drugs (CSAIDs), as revealed by blocked activation of the downstream kinase, MAP kinase-activated protein kinase-2. The treatment of glial cells with either LPS alone (microglia) or a combination of LPS and interferon-γ (astrocytes) resulted in an induced production of NO and TNFα. The two kinase inhibitors, at micromolar concentrations, individually suppressed and, in combination, almost completely blocked glial production of NO and the expression of iNOS and TNFα, as determined by Western blot analysis. Reverse transcriptase-PCR analysis showed changes in iNOS mRNA levels that paralleled iNOS protein and NO while indicating a lack of effect of either of the kinase inhibitors on TNFα mRNA expression. The results demonstrate key roles for ERK and p38 MAP kinase cascades in the transcriptional and post-transcriptional regulation of iNOS and TNFα gene expression in endotoxin-activated glial cells.
Keywords: astrocytes, microglia, TNFα, inducible nitric oxide synthase, p38 MAP kinase, extracellular signal-regulated kinase
The pro-inflammatory cytokine tumor necrosis factor-α (TNFα) and nitric oxide (NO), a short-lived and highly reactive free radical, have been implicated in several CNS disorders, including inflammatory, infectious, traumatic, and degenerative diseases (Ott et al., 1994; Benveniste, 1995; Dawson and Dawson, 1996; Bolanos et al., 1997). Both TNFα and NO are thought to contribute significantly to the pathogenesis of inflammatory demyelinating diseases such as multiple sclerosis (MS) (Raine, 1995;Parkinson et al., 1997). There is evidence for the transcriptional induction of inducible nitric oxide synthase (iNOS, the high-output isoform of NOS) in the CNS that is associated with autoimmune reactions, acute infection, and traumatic injury (Koprowski et al., 1993; Bagasra et al., 1995; Adamson et al., 1996; Oleszak et al., 1997;Samdani et al., 1997). In vitro studies have suggested that TNFα and NO may mediate oligodendrocyte and neuronal injury (Chao and Hu, 1994; Dawson et al., 1994; Raine, 1995; Parkinson et al., 1997).
In the CNS, TNFα and iNOS are expressed mainly by activated astrocytes and microglia, the two glial cell types involved in intracerebral immune regulation (Mucke and Eddleston, 1993; Perry et al., 1993; Merrill and Jonakait, 1995; Kruetzberg, 1996). As documented in several in vitro studies, they typically are induced by cytokines [i.e., IL-1, interferon-γ (IFN-γ), and TNFα] and by microbial products, such as bacterial lipopolysaccharide (LPS), or by a combination of the two (Lieberman et al., 1989; Lee et al., 1993;Murphy et al., 1993; Benveniste, 1995). The details of the signals and the mechanisms that regulate TNFα and iNOS gene expression in glial cells, however, are not well understood. Studies with various immune cell systems have suggested multiple levels of regulation: transcriptional, post-transcriptional, and post-translational (Beutler, 1992; Lowenstein et al., 1993; Xie et al., 1993). Transcriptional regulation of TNFα and iNOS is complex, involving a number of factors (TFs), including NFκB, AP-1, and various members of the C/EBP, ATF/CREB, and STAT family (Lowenstein et al., 1993; Xie et al., 1993;Jongeneel, 1995). Intracellularly, both second messenger-dependent and second messenger-independent mechanisms of cell signaling seem to participate in iNOS gene expression. Various activators and/or inhibitors of signaling kinases, including protein kinase C (Díaz-Guerra et al., 1996; Hellendall and Ting, 1997), protein kinase A (Imai et al., 1994; Hellendall and Ting, 1997; Mullet et al., 1997), and protein tyrosine kinases (Kong et al., 1996; Hellendall and Ting, 1997; Lee et al., 1997), have been shown to alter iNOS induction in cytokine and LPS-stimulated cells. Although previously it was shown that tyrosine kinase inhibitors inhibit NO production in glia (Kong et al., 1996; Hellendall and Ting, 1997), the identities of the specific kinases that are involved have not been clear.
In contrast to the predominantly transcriptional activation of the iNOS gene, post-transcriptional control accounts for most of the increase in TNFα expression, as demonstrated in LPS-activated monocytes/macrophages (Beutler, 1990; Han et al., 1991). Lee et al. (1994) found that a member of the mitogen-activated protein kinase (MAPK) family, i.e., p38 kinase, which acts as a specific target for a novel class of cytokine suppressive anti-inflammatory drugs (CSAIDs), plays a key role in this regulation. The p38 MAPK is one of at least three mammalian MAPKs [the other two being extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK)] that are activated by three homologous but distinct signaling pathways (Davis, 1994; Cano and Mahadevan, 1995; Cobb and Goldsmith, 1995; Kyriakis and Avruch, 1996). The activation is effected by dual Ser/Thr and tyrosine phosphorylation that is catalyzed by a specific upstream MAPK kinase. The JNK and p38 kinases are activated in response to inflammatory agents and environmental stress, whereas ERK, the “classic” MAPK, is stimulated primarily by growth factors and tumor promoters; however, activation by TNF or IL-1 also has been demonstrated. Each of the three MAPK modules has the potential to elicit transcriptional activation via phosphorylation of different sets of TFs (Hill and Treisman, 1995;Karin, 1995).
In the present study we show that the bacterial LPS activates multiple MAPK cascades in brain microglia and astrocytes and that specific inhibitors of MAPK subgroups, i.e., ERK and p38 kinase, block glial expression of iNOS and TNFα involving transcriptional and post-transcriptional control mechanisms.
Portions of this work have been reported in abstract form (Bhat et al., 1997).
MATERIALS AND METHODS
[γ32P]ATP and [α32P]dCTP were purchased from DuPont-NEN (Boston, MA). Calf serum (CS, supplemented), fetal calf serum (FCS), DMEM, and Moloney murine leukemia virus (MLV) reverse transcriptase were obtained from Life Technologies (Grand Island, NY). Antibiotic–antimycotic mixture, recombinant heat shock protein (hsp27), and LPS were obtained from Sigma (St. Louis, MO). Interferon-γ was purchased from Genzyme Diagnostics (Cambridge, MA). Recombinant TNFα and IL-1 were obtained from R & D Systems (Minneapolis, MN). The “Phosphoplus” kit containing phospho-specific p38 MAPK (Tyr 182) antibody and total p38 antibody and PD98059, a specific inhibitor of MAPK kinase or ERK kinase (MEK), were purchased from New England Biolabs (Beverly, MA). Anti-active MAPK pAb was obtained from Promega (Madison, WI). Polyclonal anti-TNFα was purchased from BioSource (Camarillo, CA). Monoclonal anti-ERK2 and iNOS antibodies were obtained from Transduction Laboratories (Lexington, KY). Polyclonal anti-MAPKAP K-2 antibodies were purchased from Upstate Biotechnology (Lake Placid, NY).Taq DNA polymerase was obtained from Boehringer Mannheim (Indianapolis, IN). Six-well culture dishes and 75 cm2 T-flasks were purchased from Corning–CoStar (Cambridge, MA). Pregnant rats (Sprague Dawley strain) were procured from either Harlan Sprague Dawley (Indianapolis, IN) or Charles Rivers Laboratories (Boston, MA).
Cell culture. Primary cultures of astrocytes and microglia were prepared as described previously (Bhat et al., 1995a,b). First, mixed glial cultures used as the source of these cells were established from newborn rat brain as follows. Cerebra were dissected and placed temporarily in DMEM supplemented with glucose (5 gm/l), sodium bicarbonate (3.7 gm/l), and antibiotic–antimycotic mixture. Meninges and blood vessels were removed under a dissecting microscope. Cerebra then were dissociated mechanically, and the cell suspension (in DMEM containing 10% FCS) was passed through a sterilized nylon mesh (78 μm). The dissociated cells were pelleted by centrifugation at a low speed (90 × g) and washed twice with the medium; the final cell suspension (10 ml, representing three cerebra) was seeded in 75 cm2 T-flasks (Falcon, Oxnard, CA) and incubated at 37°C in an atmosphere of 5% CO2 in air. The medium was changed after 3–4 d and twice a week thereafter. After 7–10 d, microglia that grow loosely attached on top of mixed glial cultures were isolated by a mechanical shaking of the culture flasks for 30 min at 200 rpm on a gyratory shaker. Harvested cells were transferred to fresh culture dishes; after 2 hr, any contaminating oligodendrocyte progenitors were detached with Tris-buffered saline (TBS) containing 1 mm EDTA. This procedure leaves behind firmly attached microglial cell populations. The purity of the cultures, as tested by morphological criteria and by their reactivity toward OX-42 and RCA lectin (determined by immunocytochemistry), was >95%.
The original “source” cultures were fed fresh medium, equilibrated in the CO2 incubator, and shaken for an additional 16 hr at 250 rpm to separate the phase-dark, round oligodendrocyte progenitors that grow on top of a confluent layer of astrocytes. The procedure was repeated as needed. The remaining source cultures, substantially depleted of oligodendrocyte progenitors and microglia, were subcultured into six-well culture dishes and used as astrocyte-enriched cultures. The purity (>95%) was confirmed by labeling with anti-glial fibrillary acidic protein (GFAP, an astrocyte marker) antibodies.
Western blot analysis. Western blot was performed for the analysis of ERK and p38 MAPK activation (using antibodies specific for the phosphorylated forms of the two kinases) and for the estimation of iNOS and TNFα expression. Briefly, protein samples (cell extracts, 25 μg of protein, and/or spent medium) were separated by SDS-PAGE and blotted onto polyvinylidene difluoride membranes. The membrane was blocked with 5% BSA and 1% milk powder in 10 mm Tris-HCl containing 150 mm NaCl and 0.5% Tween 20 (TBST) for 1 hr and incubated overnight with suitably diluted primary antibodies. After extensive washing with TBST, the membranes were incubated with anti-IgG–alkaline phosphatase conjugate. Finally, the blots were developed with the alkaline phosphatase substrate 5-bromo-4-chloro-3-indolyl phosphate (BCIP; 50 μg/ml) along with nitroblue tetrazolium (NBT; 100 μg/ml) in sodium glycinate buffer, pH 9.6, in the presence of 4 mm MgCl2.
In-gel assay of ERK. The substrate gel assay was performed as described previously (Bhat et al., 1995b; Bhat and Zhang, 1996), using SDS gels polymerized in the presence of myelin basic protein (MBP, 0.1 mg/ml). After electrophoresis of the samples to be assayed, the gel was washed off SDS with 2-propanol (20%) in 50 mmTris-HCl, pH 8.0, and the separated proteins were denatured completely with 6 m guanidine-HCl, followed by their renaturation in a buffer containing 0.04% Tween-40. The kinase assay was performed by incubation of the gel for 1 hr at 30°C in the kinase assay buffer [40 mm HEPES-HCl, pH 8.0, 2.0 mm DTT, 0.1 mm EGTA, and 10 mm magnesium chloride) containing [γ-32P]ATP (10 μm, 50 μCi/ml). Then the gel was washed extensively with 5% TCA containing 1% sodium pyrophosphate, dried under vacuum, and exposed to x-ray film. The kinase activities were visualized as radioactive bands of phosphorylated MBP.
Immunoprecipitation and immune complex kinase assay. These procedures were performed as described previously (Bhat and Zhang, 1996). Cultures were lysed with cold RIPA buffer (50 mmTris-Cl, pH 8.0, containing 0.1% SDS, 1 mm EDTA, 150 mm NaCl, 0.5% sodium deoxycholate, 1 mm sodium vanadate, 1 mm phenylmethylsulfonyl fluoride, and 1% Nonidet P-40) and spun in an Eppendorf microfuge for 30 min to remove the insoluble material. The supernatant was incubated with 3 μg of anti-ERK2 antibodies for 1 hr at 4°C, followed by 30 min of incubation with 7.5 μg of affinity-purified goat anti-rabbit IgG. Then protein A-Sepharose was added, and the mixture was incubated for 1 hr at 4°C. The immunoprecipitates were collected by centrifugation and used for MAPK assay after being washed three times with RIPA buffer and twice with MAPK assay buffer (see above). MAPK assay was performed by incubating the suspension of the immune complex with 0.33 mg/ml MBP and 0.5 μCi/ml [γ-32P]ATP for 30 min at 30°C. The reaction mixture was mixed with 5× electrophoresis sample buffer, boiled, and subjected to SDS-PAGE on a 15% Laemmli gel. The radioactive band was visualized by autoradiography of the dried gel.
MAPKAP kinase-2 was assayed by using hsp27 as the substrate (Foltz et al., 1997). The lysates were incubated with 5 μl of G-protein conjugated to 2 μg of MAPKAP kinase-2 antibody for 90 min 4°C on a shaking platform. The immunoprecipitates were washed twice with the lysis buffer containing 0.5 m NaCl and twice with the lysis buffer as such, and then MAPKAPK-2 activity was assayed by using hsp27 as the substrate in the presence of 32P-ATP. The labeled product was resolved by SDS-PAGE and autoradiographed.
Measurement of nitrite production. NO production was determined by measurement of nitrite in the medium, based on the Griess reaction (Stuehr and Nathan, 1989). An aliquot of the spent medium was mixed with an equal volume of 1:1 mixture of 1% sulfanilamide in water and 0.1% _N_-1-naphthylethylenediamine dihydrochloride in 5% phosphoric acid. Then the absorbance was read at 570 nm. Sodium nitrite dissolved in the culture medium was used as the standard.
Northern blot analysis. Total cellular RNA was extracted with the RNA-STAT kit (Tel-Test, Friendswood, TX) and subjected to Northern analysis as described previously (Bhat et al., 1995a). Briefly, 10–15 μg of total RNA from each sample mixed with ethidium bromide was electrophoresed via a 1% agarose, 2.2 mformaldehyde gel. The gels were treated with 50 mm NaOH for 20 min, neutralized with 0.1 m Tris, pH 7.4, for 20 min, and soaked in 20× SSC for 1 hr. The RNA was transferred to nitrocellulose and hybridized in 50% formamide, 100 μg/ml denatured salmon sperm DNA, 5× Denhardt’s (0.1% each BSA, Ficol, and polyvinyl pyrrolidine), 0.1% SDS, 20 mm sodium phosphate, pH 6.8, and 1 m NaCl with random primer-labeled iNOS and glyceraldehyde phosphate dehydrogenase (GAPDH) cDNA probes (1 × 106 cpm/ml) for 12–16 hr at 42°C. The blots were washed twice in 2× SSC and 0.5× SSC with 0.1% SDS for 30 min each at 68°C before autoradiography.
PCR analysis of iNOS and TNFα gene expression. A semiquantitative reverse transcriptase PCR (RT-PCR) assay was used to determine the mRNA levels of TNFα and iNOS in relation to GAPDH message. Approximately 2 μg of total RNA, extracted as above, was used for cDNA synthesis by reverse transcription with 20 U of MLV reverse transcriptase in RT buffer in the presence of 0.5 mm each of dNTPs, 20 U of RNase inhibitor, and random hexamers as primers. The thermal cycler (Cetus 480) was programmed for 50 min at 42°C, 1 min at 25°C, and 5 min at 95°C. One per cent of each of the cDNA synthesized in the RT reaction was used for PCR amplification in the presence of 1 U Taq DNA polymerase in_Taq_ buffer, 0.2 mm each of dNTPs, and a 1 μm concentration of each primer. Each sample was amplified for 35 cycles, using a three-step program (15 sec at 95°C, 30 sec at 60°C, and 30 sec at 72°C). After amplification, the products were separated on an agarose (1%) gel (cast in the presence of ethidium bromide) and visualized under UV light. In some cases the separated products were transferred to nylon, and the blots were probed with end-labeled oligonucleotides internal to the PCR primer sequences.
The following sequences of the primers were used according to previously published reports (Herskowitz et al., 1995; Kawase et al., 1996): TNFα (upstream), 5′-CACGCTCTTCTGTCTACTGA-3′; TNFα (downstream), 5′-GGACTCCGTGATGTCTAAGT-3′; iNOS (upstream), 5′-CGTGTGCCTGCTGCCTTCCTGCTGT-3′; iNOS (downstream), 5′-GTAATCCTCAACCTGCTCCTCACTC-3′; GAPDH (upstream), 5′-CATTGACCTCAACTACATGGT-3′; GAPDH (downstream), 5′-TTGTCATTACCAGGAAATGAGC-3′.
RESULTS
The activation of ERK and p38 MAPK in microglia
The primary cultures of rat brain microglia were treated with the bacterial LPS, and the cell extracts were analyzed for MAPK activation. The activation of ERK and p38 kinases was determined by Western blot analysis, using commercially available antibodies specific for the activated forms of the two kinases. Figure1A(I) shows the time course and dose–response of LPS activation of ERK. The endotoxin at micromolar concentrations elicited a rapid (detectable at 5 min) Tyr phosphorylation of ERK that increased with time, reaching a maximum between 20 and 30 min, followed by a decline reaching low levels by 60 min. Parallel blots run as controls that used antibodies directed against the total antigen did not show any changes (Fig. 1A, II). Simultaneous inclusion of PD98059 (15 μm), a specific inhibitor of MAPK or ERK kinase (MEK) (Dudley et al., 1995), the upstream dual-specificity kinase that phosphorylates and activates ERK, resulted in an inhibition of ERK activation, as shown in Figure1B. Figure 1C shows the concentration-dependent inhibition of LPS-stimulated ERK phosphorylation by the MEK inhibitor. Maximal inhibition was obtained with a concentration of 25 μm, the concentration used in subsequent experiments to test the role of ERK activation in iNOS and TNFα expression. In a parallel experiment, the activity of ERK in LPS-treated cells was determined by in-gel and immune complex kinase assays. The data given in Figure 1D confirm the relationship between the phosphorylation of ERK (i.e., immunoreactivity with phospho-specific antibodies) and kinase activation, thereby validating the use of the immunoblot procedure to detect MAPK activation.
Fig. 1.

LPS activation of ERK in microglia. Aliquots of cell extracts prepared from microglial cultures treated either with 1 μg/ml LPS for the indicated times or with different concentrations of LPS for 15 min were subjected to immunoblot analysis by using antibodies raised against the active forms of ERK* (A, I). Parallel blots were run, using the antibodies recognizing the total ERK (A, II) proteins. Incubations also were run with LPS added in the presence and absence of 15 μm PD98059 for various times (B) and in the presence of different concentrations of the inhibitor for 15 min (C). In another set of experiments, the cell lysates were used for in-gel (D, I) and immune complex kinases assays (D, II) of activated ERK, using MBP as the substrate, as described in Materials and Methods. The results shown here and elsewhere are representatives from replicate experiments.
Figure 2A shows the dose- and time-dependent activation of p38 MAPK in microglia treated with LPS. That the increased phosphorylation of p38 kinase was associated with an increase in its activity and that the kinase was sensitive to a specific inhibitor SB203580 (Lee et al., 1994) were confirmed by an assay of the downstream target, i.e., MAPKAP kinase-2 in cells treated with LPS in the presence and absence of the inhibitor (15 μm). The activation of MAPKAP kinase-2 was determined by an immune complex kinase assay that used recombinant hsp27 in the presence of [γ32P]ATP (see Materials and Methods). The radioactively labeled hsp27 was resolved on an SDS gel, followed by autoradiography (Fig. 2B, II). Aliquots of the cell extracts also were analyzed for p38 kinase phosphorylation by immunoblot, as above (Fig. 2B, I). The results show a complete inhibition by SB203580 of LPS-activated MAPKAP kinase-2 (hsp27 phosphorylation). As expected of its specificity, the inhibitor did not affect p38 kinase activation or phosphorylation (Fig. 2B, I) but, rather, its_activity_ toward MAPKAP kinase-2.
Fig. 2.

LPS activation of p38 MAPK and its inhibition by SB203580. Cultures of microglial cells treated either with a constant amount (1 μg/ml) of LPS for various times or with different concentrations of LPS for 15 min were lysed and subjected to immunoblot analysis by using antibodies specific for the active (p38*) and total p38 MAPK, as described in Materials and Methods (A). In another set of experiments (B), cultures treated with LPS in the presence and absence of SB203580 (15 μm) were lysed, and the lysates were subjected to immune complex kinase assay of MAPKAP kinase-2, using hsp27 as the substrate (B,II), as described in Materials and Methods. Aliquots of cell lysates also were immunoblotted with anti-Phosphoplus p38 antibodies (B, I).
Microglial production of NO and its inhibition by MAPK inhibitors
NO production in LPS-treated glial cultures was determined as nitrite (a stable oxidation product of NO) released into the culture medium, using a colorimetric method (see Materials and Methods). As reported in the literature, LPS was found to be a potent stimulator of glial NO production. Figure 3 shows a dose-dependent inhibition of NO production by the two MAPK inhibitors, i.e., PD98059 (Fig. 3A) and SB203580 (Fig. 3B), in microglial cultures stimulated with LPS. When added together, the two compounds caused a drastic (80%) inhibition of LPS-stimulated NO synthesis (Fig. 3C).
Fig. 3.
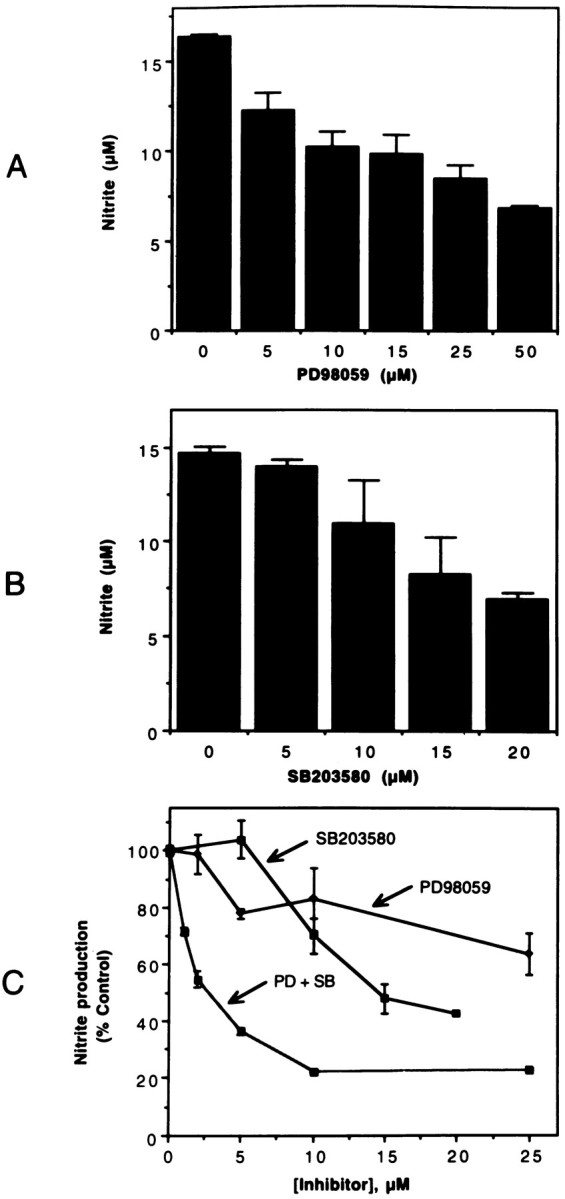
MAPK inhibitors inhibit LPS-induced nitrite production. Microglial cultures were treated with 0.1 μg/ml of LPS in the presence of various concentrations of PD98059 (A), SB203580 (B), and combinations of the two inhibitors (C). After 48 hr, aliquots of the culture medium were mixed with Greiss reagent for the estimation of nitrite produced. The optical density of the colored product was measured at 570 nm. Untreated samples showed minimal (<0.5 μm) nitrite levels. The values shown are mean ± SD of data from triplicate determinations.
Because LPS-induced production of NO is catalyzed by iNOS, we studied the induction of iNOS in microglia by immunoblot. As shown in Figure4A, LPS markedly induced the expression of iNOS, which was inhibited by the kinase inhibitors in parallel to NO production. A combination of the two inhibitors was able to block iNOS induction almost completely. To test the effectiveness of the drugs on iNOS induction when they were present only during the initial kinase activation period and to rule out their indirect effects, if any, on nitrite production during longer incubation (although we did not observe any toxic effects of the drugs), we performed the following experiment. Five sets of cultures in triplicate dishes were used: one control, one treated with LPS alone, and three sets treated with LPS plus a combination of the two inhibitors. After 2 hr, two of the sets treated with LPS plus the inhibitors were washed and were fed fresh medium. LPS was re-added to one set of these “washed” cultures, and, after 48 hr, the medium was tested for nitrite production and the cells were tested for iNOS expression by immunoblot in all of the cultures. As depicted in Figure4B, the inhibitors were effective in preventing LPS-mediated induction of iNOS and the production of nitrite even when they were present only during the kinase activation period.
Fig. 4.

Immunoblot analysis of iNOS induction in microglia and its inhibition by MAPK inhibitors. A, Microglial cultures were treated with LPS in the presence of MAPK inhibitors, i.e., PD98059 (25 μm) and SB203580 (15 μm), individually or in combination for 48 hr. Control cultures, cultures treated with LPS alone, and those treated with LPS with the kinase inhibitors were analyzed for iNOS expression by immunoblot, using anti-iNOS antibodies (bottom). Aliquots of the culture medium were analyzed for nitrite production (top). B, In another experiment, cultures were exposed to LPS plus the inhibitor combination for 2 hr, washed, re-fed without the additives, and divided into two sets: one receiving no further treatment (d) and the other reexposed to LPS (e). These two sets of cultures, untreated controls (a), and those treated with LPS with the inhibitors (c) and without the inhibitors (b) were analyzed after 48 hr for nitrite production (top panel) and iNOS expression by immunoblot (bottom panel). The values for nitrite synthesis are mean ± SD of data from triplicate determinations.
Astroglial production of NO and its inhibition by MAPK inhibitors
Figure 5A shows the production of NO and the induction of iNOS in astroglial cultures. Although in microglial cells the endotoxin acted independently of other factors to stimulate NO release, in astrocytes it required cotreatment with IFN-γ for optimal activity. However, as observed with microglial cells, the kinase inhibitors interfered with the production of NO and iNOS in astrocytes treated with LPS plus IFN-γ. We also have found that the kinase inhibitors block iNOS induction in astrocytes treated with LPS alone to the same extent as that observed in microglia (data not shown). Because it has been reported that in glomerular mesangial cells p38 kinase may regulate cytokine (IL-1)-induced NO synthesis negatively (Guan et al., 1997), we tested the role of MAPKs in glial iNOS expression in response to IL-1. IL-1 did not stimulate iNOS synthesis in microglia. However, as shown in Figure 5B, IL-1 in combination with IFN-γ induced iNOS expression in astrocytes. Once again, this induction was blocked by the kinase inhibitors.
Fig. 5.

Immunoblot analysis of iNOS induction in astrocytes and its inhibition by MAPK inhibitors. A, Triplicate cultures of astrocytes were treated with LPS (100 ng/ml) plus IFN-γ (50 U/ml) in the presence of MAPK inhibitors, i.e., PD98059 (25 μm) and SB203580 (15 μm), individually or in combination for 48 hr. Control cultures, cultures treated with LPS plus IFN-γ alone, and those treated with LPS plus IFN-γ with the kinase inhibitors were analyzed for iNOS expression by immunoblot analysis, using anti-iNOS antibodies (bottom). Aliquots of the culture medium were analyzed for nitrite production (top). B, Cultures also were treated with IL-1 (50 ng/ml) plus IFN-γ either in the presence or absence of PD98059, SB203580, or a combination of the two for 48 hr. These cultures, along with untreated controls and those exposed to individual cytokines, were analyzed for iNOS expression by immunoblot.
Figure 6 shows cytokine and endotoxin activation of ERK and p38 kinase in astrocytes. It is clear that both LPS and IL-1 (but not IFN-γ; results not shown) activated both ERK and p38 kinase. However, cotreatment of astrocytes with IFN-γ did not seem to alter the level of MAPK activation significantly, although the cytokine greatly enhanced LPS- and IL-1-induced astroglial production of NO.
Fig. 6.

LPS activation of ERK and p38 MAPK in astrocytes. Astrocyte cultures were treated with LPS and IL-1 individually or in combination with IFN-γ for various time periods, and the cell lysates were subjected to immunoblot analysis, using antibodies specific for the active (phosphorylated) forms of ERK and p38 MAPK. IFN-γ by itself had no effect on kinase activation.
LPS induction of TNFα synthesis and its inhibition by MAPK inhibitors
The induction of TNFα synthesis in glial cultures was determined by immunoblot analysis of the cell extract and the culture medium with the use of TNFα-specific antibodies. As shown in Figure7, the bacterial LPS induced the production of TNFα, as detected by the immunoreactive band that corresponded to the standard TNFα. As in the case of iNOS, the kinase inhibitors inhibited TNFα production in both microglia and astrocytes. Also, there was an almost complete inhibition of TNFα production in cultures that were exposed to a combination of the two kinase inhibitors. Similar results were obtained with cell extracts and the medium (Fig. 7A, II), thereby ruling out the possibility of an altered TNFα secretion accounting for the inhibition that was observed.
Fig. 7.

Immunoblot analysis of TNFα production in glia and its inhibition by MAPK inhibitors. Aliquots of the medium from cultures of microglia and astrocytes treated with LPS and LPS plus IFN-γ, respectively, in the presence and absence of the two kinase inhibitors were resolved on a 15% SDS gel and immunoblotted with antibodies specific for TNFα (I). Aliquots of the cell extracts from microglial cultures also were subjected to immunoblot to determine the cell-associated TNFα (II). The 17 kDa form of TNF comigrates with the standard cytokine. The top arrow in _B_probably indicates the 26 kDa form of TNFα, and the thick band above that is nonspecific.
RT-PCR analysis of iNOS and TNFα gene expression
The steady-state levels of mRNAs for iNOS, TNFα, and GAPDH (a housekeeping gene product used as internal control) in glial cultures treated with LPS (or a combination of LPS and IFN-γ), in the presence and absence of MAPK inhibitors, were determined by RT-PCR. Southern hybridization that used labeled probes internal to the primers was performed to confirm the identities of all three of the gene products (data not shown). As shown in Figure8A, LPS time-dependently induced the expression of iNOS and TNFα mRNAs in microglial cells. iNOS expression could be seen as early as 2 hr, with peak levels reached by 8 hr, followed by a decline to uninduced levels. In contrast to iNOS, which was not detectable in untreated cultures, there was a basal level of TNFα mRNA in the controls that was increased by LPS treatment. Figure 8B shows iNOS and TNFα mRNA levels in astrocyte cultures exposed to LPS, IFN-γ, and a combination of the two for 4 hr. It is clear that a combination of LPS and IFN-γ resulted in a strong induction of iNOS mRNA. As in the case of microglial cells, there was a basal expression of TNFα mRNA that showed substantial induction on LPS plus IFN-γ treatment.
Fig. 8.

PCR analysis of iNOS and TNFα gene expression in glial cultures stimulated with LPS. RNA samples of isolated microglia treated with LPS for different times (A) and astrocytes treated with LPS, IFN-γ, and a combination of the two for 4 hr (B) were subjected to RT-PCR, using the primers specific for iNOS, TNFα, and GAPDH, as described in Materials and Methods.
We next tested the effects of the two kinase inhibitors on iNOS and TNFα mRNA levels in LPS-treated microglia and in LPS plus IFN-treated astrocytes. As shown in Figure 9, a combination of the kinase inhibitors was effective in inhibiting iNOS mRNA expression both in microglia (Fig. 9A) and astrocytes (Fig. 9B). The data corresponded well to those for iNOS protein (see Figs. 4, 5). Added separately, the kinase inhibitors had marginal or smaller inhibitory effects. The combinatorial inhibitory effects of the drugs on iNOS gene expression were supported further by Northern blot analysis, as shown in Figure 9A(II). In contrast to the situation with iNOS mRNA expression, the inhibitors did not effect a similar reduction in the steady-state levels of TNFα mRNA, thereby suggesting a post-transcriptional regulation of the induction of TNFα in LPS-treated cultures via activation of MAPK cascades. When astrocytes treated with LPS alone in the presence and absence of the kinase inhibitors were analyzed, the results that were obtained were quite similar to those observed in the case of microglia stimulated with LPS (data not shown).
Fig. 9.

Effects of MAPK inhibitors on iNOS and TNFα levels in LPS-treated microglia and astrocytes. RNA samples isolated from glial cultures treated with either LPS or LPS plus IFN-γ in the presence and absence of the kinase inhibitors for 4 hr were analyzed by RT-PCR, using the primer pairs specific for iNOS, TNFα, and GAPDH. The products were run on a 1% agarose gel impregnated with ethidium bromide. The bands were visualized under UV. RNA samples extracted from a set of microglial cultures treated as above also were analyzed by Northern blot for iNOS expression.
DISCUSSION
This study has dealt with the activation of MAPKs in endotoxin-stimulated primary glial cultures (i.e., astrocytes and microglia) in relation to their production of the two pro-inflammatory mediators, TNFα and NO. The bacterial LPS is capable of activating ERK and p38 subgroups of MAPKs and, as demonstrated in a related study, the third MAPK subgroup, JNK/SAPK (Zhang et al., 1998). The treatment of microglia with LPS and of astrocytes with a combination of LPS and IFN-γ resulted in an induced production of NO and TNFα. NO production was the result of an induced expression of iNOS, the high-output isoform of NOS, as revealed by Western blot and RT-PCR analysis of iNOS protein and its mRNA, respectively. Specific inhibitors of ERK kinase and p38 kinase (i.e., PD98059 and SB203580, respectively) inhibited LPS-induced production of NO and the expression of iNOS, thereby suggesting key roles for these kinase cascades in LPS-induced glial cell activation. The inhibitors also interfered with TNFα synthesis and, when added together, elicited a drastic inhibition of the biosynthesis of both TNFα and NO/iNOS. RT-PCR analysis of iNOS mRNA suggested the MAPK regulation of iNOS gene expression at the level of transcription and/or mRNA stability. In contrast to iNOS, a lack of attenuation in TNFα mRNA levels in kinase inhibitor-treated cultures suggested that MAPK regulation of TNFα gene expression involves post-transcriptional control mechanisms.
Although, in general, distinct stimuli activate mitogen- and stress-activated kinase subgroups with distinct cellular effects, certain stimuli, such as LPS and TNFα, activate multiple MAPKs in their target cells. Because each of the three kinase cascades has the potential to elicit transcriptional activation via specific TFs, it is possible that many MAPK responsive TFs may cooperate to regulate single promoter elements, either by forming complexes or by competing for common binding sites (Hill and Treisman, 1995). The cooperativity among MAPK signaling also may occur at a post-transcriptional level via the phosphorylation of key regulatory molecules involved in biosynthetic or metabolic activities. With the availability of specific kinase inhibitors, the importance of individual pathways to cellular responses can be determined. With the use of one of these pharmacological agents, i.e., SB203580, it has been shown that p38 MAPK (specific target for the drug), in particular, plays a key signaling role in inflammatory cytokine biosynthesis (Lee et al., 1994; Lee and Young, 1996). As we have shown in this study, SB203580 inhibits the expression of TNFα and iNOS in glial cells. This inhibitory effect is enhanced further by PD98059, a specific inhibitor of MEK, thereby suggesting a cooperation between ERK and p38 subgroups of MAPK in the induction of iNOS and TNFα gene expression. It should be pointed out, however, that there are varying recent reports on the role of MAPKs in iNOS gene expression. Thus, although our finding of an involvement of ERK pathway in glial expression of iNOS is consistent with previous studies with myocytes and cardiac endothelial cells (Singh et al., 1996), it differs from another study with C-6 glioma cells (Nishiya et al., 1997) in which IFN-γ-induced ERK activation and iNOS expression could be dissociated. It is possible that, being a transformed cell line, C-6 glioma may elicit a different response to IFN-γ from their normal counterparts. In fact, we have not been able to detect an effect of IFN-γ on ERK in primary astrocytes. Our results implicating a positive regulation of p38 kinase in cytokine-mediated iNOS expression are also at odds with another study that reported a p38 kinase-dependent downregulation of iNOS in mesangial cells treated with IL-1 (Guan et al., 1997). These inconsistencies probably stem from the well known, but not well understood, complex regulation of iNOS gene expression, which is cell type-, species-, and stimulus-specific.
Our results show that microglia in general are more active in producing NO than astrocytes and that the two cell types differ in their responses to LPS, in that astrocytes (but not microglia) require costimulation with IFN-γ for iNOS (and TNFα) expression. This combinatorial effect is consistent with several findings that IFN-γ and LPS or LPS-induced cytokines, such as TNFα and IL-1β, synergize to induce iNOS mRNA expression and NO production in many cell types, including astrocytes (Murphy et al., 1993; Kopnisky et al., 1997). The synergism cannot be explained simply by the level of activation of MAPKs, although specific MAPK inhibitors are able to block iNOS induction in astrocytes that are stimulated with a combination of IFN-γ and LPS, thereby suggesting MAPK activation is required, but not sufficient, under these conditions. Other mechanisms by which the synergistic effect of IFN-γ on LPS induction of iNOS can be elicited include the activation of interferon regulatory factor (IRF-1) that cooperates with LPS-inducible factors (Kamijo et al., 1994) and the action of NO itself. Evidence indicates that iNOS expression is kept suppressed by a low level of preexisting endogenous NO, a product of cNOS, the constitutively expressed Ca2+/calmodulin-activated NOS isoform. In addition, as shown recently in astrocytoma cells (Colasanti et al., 1997), a combination of LPS and IFN-γ elicits a very fast inhibition of cNOS activity, thereby relieving NO-mediated suppression of iNOS expression. The mechanism by which NO inhibits iNOS may involve NFκB (Togashi et al., 1997) and thus also may regulate other NFκB-regulated genes, including TNFα. It would be interesting to see if astrocytes and microglia differ in their expression of cNOS and whether such a difference accounts for their differential response to IFN-γ, as observed in the present studies.
The mechanism of MAPK regulation of iNOS and cytokine gene expression is not yet clear, but it seems to involve both transcriptional and post-transcriptional events. Thus, we have found that a combination of SB203580 and PD98059 blocked the expression of iNOS mRNA in LPS-treated glial cells, whereas the same treatment had no inhibitory effect (in fact, there was an upregulation) on TNFα mRNA levels, thereby suggesting a differential regulation of TNFα and iNOS gene expression by the two MAPKs. Besides transcriptional activation, iNOS gene expression is regulated at the level of mRNA stability and translational efficiency (Lowenstein et al., 1993; Xie et al., 1993). Among the many cis and trans determinants controlling mRNA stabilization is the AU-rich element (ARE) containing one or more AUUUA sequences found in the 3′-untranslated region (3′-UTR) of many short-lived transcripts encoding cytokines (i.e., TNFα), proto-oncogenes, and inducible growth factors (Caput et al., 1986; Jackson, 1993; Chen and Shyu, 1995). The ARE confers instability on the mRNA and is the target for specific AU-binding factors. Several AUUUA motifs are present in the 3′-UTR of iNOS mRNA from rat astrocytes and mouse macrophages (Galea et al., 1994). Interestingly, it has been suggested that, in glial cells (astrocytes), iNOS mRNA stability may be both translation-dependent and transcription-dependent (Park and Murphy, 1996). Future experiments would determine the rate of iNOS mRNA turnover in the presence and absence of the kinase inhibitors and the factors that might regulate mRNA stability.
It is possible that TNFα expression in glia, as in systemic macrophages, is regulated mainly post-transcriptionally by involving translational efficiency in a MAPK-regulated way, because the specific kinase inhibitors were effective in blocking the production of TNFα protein. As suggested by Lee and Young (1996), a block in cytokine mRNA translation in unstimulated cells exerted by a regulatory protein could be relieved by phosphorylation of this protein via the p38 MAPK pathway on cell stimulation. The CSAID class of kinase inhibitors would, therefore, prevent this translational derepression. Besides inducing mRNA instability, the regulatory protein also can interact with the 5′ end of mRNA and in analogy with murine PHAS-I and human homologs 4E-BP-1 and 4E-BP-2 (Lin et al., 1994; Pause et al., 1994), when phosphorylated by MAPK, it may dissociate from the eukaryotic initiation factor eIF-4E, relieving translational inhibition. It is interesting that a Ser/Thr kinase termed MAPK-interacting kinase 1 (MNK1) recently has been shown to phosphorylate eIF-4E at Ser209 (Waskiewicz et al., 1997), an event that enhances its affinity for the 5′ cap structure of mRNAs, and that MNK1 is phosphorylated and activated by both ERK and p38 MAPK, thereby defining a convergence point between the growth factor-activated and one of the stress-activated protein kinase cascades (Fukunaga and Hunter, 1997;Waskiewicz et al., 1997). Simultaneous activation of the two MAPK cascades, as in the case of LPS-stimulated cells, would result in the full activation of MNK1 and an enhancement of protein synthesis. It is possible that the additive inhibitory effects of the two kinase inhibitors on TNFα (and iNOS) synthesis that have been observed in the present studies might have resulted from a blocked activation of MNK1.
Finally, the results of the present investigation show that MAPK plays an important role in glial cell activation, suggesting that pharmacological control of MAPK signaling pathways should prove useful against a variety of CNS inflammatory conditions involving glial cell activation or gliosis, including trauma, stroke, AIDS, and demyelinating diseases such as MS. In this regard, the p38 kinase inhibiting bicyclic imidazoles such as SB203580 have, indeed, been shown to exhibit activity in a number of animal models of acute and chronic inflammation by virtue of their ability to inhibit the synthesis of pro-inflammatory molecules (Badger et al., 1996).
Footnotes
This study was supported by Grants RG2481 and RG2849 from the National Multiple Sclerosis Society and NS31767 from National Institutes of Health. We thank Ms. Aruna Bhat and Ms. Elaine Terry for their technical help and Mr. George Ohlandt for his help with RT-PCR. We also thank Ms. Sallie Bendt for excellent secretarial assistance.
Correspondence should be addressed to Dr. Narayan R. Bhat, Department of Neurology, Medical University of South Carolina, 171 Ashley Avenue, Charleston, SC 29425.
REFERENCES
- 1.Adamson DC, Wildemann B, Sasaki M, Glass JD, McArthur JC, Christov VI, Dawson TD, Dawson VL. Immunologic nitric oxide synthase: elevation in severe AIDS dementia and induction by HIV-1 gp41. Science. 1996;274:1917–1921. doi: 10.1126/science.274.5294.1917. [DOI] [PubMed] [Google Scholar]
- 2.Badger AM, Bradbeer JN, Votta B, Lee JC, Adams JL, Griswold DE. Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock, and immune function. J Pharmacol Exp Ther. 1996;279:1453–1461. [PubMed] [Google Scholar]
- 3.Bagasra O, Michaels FH, Zheng YM, Borroski LE, Spitsin SV, Fu ZF, Tawadros R, Koprowski H. Activation of the inducible form of nitric oxide synthase in the brains of patients with multiple sclerosis. Proc Natl Acad Sci USA. 1995;92:12041–12045. doi: 10.1073/pnas.92.26.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benveniste EN. Role of cytokines in multiple sclerosis, autoimmune encephalitis, and other neurological disorders. In: Aggarwal B, Puri R, editors. Human cytokines: their role in research and therapy. Blackwell Scientific; Boston: 1995. p. 195. [Google Scholar]
- 5.Beutler B. Regulation of cachectin biosynthesis occurs at multiple levels. Prog Clin Biol Res. 1990;349:229–240. [PubMed] [Google Scholar]
- 6.Beutler B. Tumor necrosis factors. Raven; New York: 1992. [Google Scholar]
- 7.Bhat NR, Zhang P. Activation of MAP kinases in oligodendrocytes. J Neurochem. 1996;66:1986–1994. doi: 10.1046/j.1471-4159.1996.66051986.x. [DOI] [PubMed] [Google Scholar]
- 8.Bhat NR, Zhang P, Bhat AN. Expression of MARCKS in oligodendrocytes is developmentally regulated. Dev Neurosci. 1995a;17:256–263. doi: 10.1159/000111294. [DOI] [PubMed] [Google Scholar]
- 9.Bhat NR, Zhang P, Hogan EL. Thrombin activates MAP kinase in astrocytes. J Cell Physiol. 1995b;165:417–424. doi: 10.1002/jcp.1041650223. [DOI] [PubMed] [Google Scholar]
- 10.Bhat NR, Lee JC, Zhang P. MAP kinase cascades and microglial activation. J Neurochem. 1997;69:S206D. [Google Scholar]
- 11.Bolanos JP, Almeida A, Stewart V, Peuchen S, Land JM, Clark JB, Heales SJR. Nitric oxide-mediated mitochondrial damage in the brain—mechanisms and implications for neurodegenerative diseases. J Neurochem. 1997;68:2227–2240. doi: 10.1046/j.1471-4159.1997.68062227.x. [DOI] [PubMed] [Google Scholar]
- 12.Cano E, Mahadevan LC. Parallel signal processing among mammalian MAPKs. Trends Biochem Sci. 1995;20:117–122. doi: 10.1016/s0968-0004(00)88978-1. [DOI] [PubMed] [Google Scholar]
- 13.Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. Identification of a common nucleotide sequence in the 3′-untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci USA. 1986;83:1670–1674. doi: 10.1073/pnas.83.6.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chao CC, Hu S. Tumor necrosis factor-α potentiates glutamate neurotoxicity in human fetal brain cell cultures. Dev Neurosci. 1994;16:172–179. doi: 10.1159/000112104. [DOI] [PubMed] [Google Scholar]
- 15.Chen CYA, Shyu AB. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 16.Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 17.Colasanti M, Cavalieri E, Persichini T, Mollace V, Mariotto S, Suzuki H, Lauro GM. Bacterial lipopolysaccharide plus interferon-γ elicit a very fast inhibition of a Ca2+-dependent nitric oxide synthase activity in human astrocytoma cells. J Biol Chem. 1997;272:7582–7585. doi: 10.1074/jbc.272.12.7582. [DOI] [PubMed] [Google Scholar]
- 18.Davis RJ. MAPKs: new JNK expands the group. Trends Biochem Sci. 1994;19:170–173. doi: 10.1016/0968-0004(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 19.Dawson VL, Dawson TM. Nitric oxide in neuronal degeneration. Proc Soc Exp Biol Med. 1996;211:33–40. doi: 10.3181/00379727-211-43950e. [DOI] [PubMed] [Google Scholar]
- 20.Dawson VL, Brahbhatt HP, Mong JA, Dawson TM. Expression of inducible nitric oxide synthase causes delayed neurodegeneration in primary neuronal–glial cortical cultures. Neuropharmacology. 1994;33:1425–1430. doi: 10.1016/0028-3908(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 21.Díaz-Guerra MJM, Bodelón OG, Velasco M, Whelan R, Parker PJ, Boscá L. Up-regulation of protein kinase C-epsilon promotes the expression of cytokine-inducible nitric oxide synthase in RAW 264.7 cells. J Biol Chem. 1996;271:32028–32033. doi: 10.1074/jbc.271.50.32028. [DOI] [PubMed] [Google Scholar]
- 22.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foltz IN, Lee JC, Young PR, Schrader JW. Hemopoietic growth factors with the exception of interleukin-4 activate the p38 mitogen-activated protein kinase pathway. J Biol Chem. 1997;272:3296–3301. doi: 10.1074/jbc.272.6.3296. [DOI] [PubMed] [Google Scholar]
- 24.Fukunaga R, Hunter T. MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 1997;16:1921–1933. doi: 10.1093/emboj/16.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galea E, Reis DJ, Feinstein DL. Cloning and expression of inducible nitric oxide synthase from rat astrocytes. J Neurosci Res. 1994;266:3172–3177. doi: 10.1002/jnr.490370313. [DOI] [PubMed] [Google Scholar]
- 26.Guan Z, Baier LD, Morrison AR. p38 mitogen-activated protein kinase down-regulates nitric oxide and up-regulates prostaglandin E2 biosynthesis stimulated by interleukin-1β. J Biol Chem. 1997;272:8083–8089. doi: 10.1074/jbc.272.12.8083. [DOI] [PubMed] [Google Scholar]
- 27.Han J, Huez G, Beutler B. Interactive effects of the tumor necrosis factor promoter and 3′-untranslated regions. J Immunol. 1991;146:1843–1848. [PubMed] [Google Scholar]
- 28.Hellendall RP, Ting JP. Differential regulation of cytokine-induced major histocompatibility complex class II expression and nitric oxide release in rat microglia and astrocytes by effectors of tyrosine kinase, protein kinase C, and cAMP. J Neuroimmunol. 1997;74:19–29. doi: 10.1016/s0165-5728(96)00202-0. [DOI] [PubMed] [Google Scholar]
- 29.Herskowitz A, Choi S, Ansari AA, Wesselingh S. Cytokine mRNA expression in postischemic/reperfused myocardium. Am J Pathol. 1995;146:419–428. [PMC free article] [PubMed] [Google Scholar]
- 30.Hill CS, Treisman R. Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell. 1995;80:199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- 31.Imai T, Hirata Y, Kanno K, Marumo F. Induction of nitric oxide synthase by cyclic AMP in rat vascular smooth muscle cells. J Clin Invest. 1994;93:543–549. doi: 10.1172/JCI117005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson RJ. Cytoplasmic regulation of mRNA function: the importance of the 3′-untranslated region. Cell. 1993;74:9–14. doi: 10.1016/0092-8674(93)90290-7. [DOI] [PubMed] [Google Scholar]
- 33.Jongeneel CV. Transcriptional regulation of the tumor necrosis factor alpha gene. Immunobiology. 1995;193:210–216. doi: 10.1016/s0171-2985(11)80545-0. [DOI] [PubMed] [Google Scholar]
- 34.Kamijo R, Harada H, Matsuyama T, Bosland M, Gerecitano J, Shapiro D, Le J, Koh SI, Kimura T, Green SJ, Mak TW, Taniguchi T, Vilcek J. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. 1994;263:1612–1615. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- 35.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 36.Kawase M, Kinouchi H, Kato I, Akabane A, Kondo T, Arai S, Fujimura M, Okamoto H, Yoshimoto T. Inducible nitric oxide synthase following hypoxia in rat cultured glial cells. Brain Res. 1996;738:319–322. doi: 10.1016/s0006-8993(96)00924-9. [DOI] [PubMed] [Google Scholar]
- 37.Kong LY, McMillian MK, Maronpot R, Hong JS. Protein tyrosine kinase inhibitors suppress the production of nitric oxide in mixed glia, microglia-enriched or astrocyte-enriched cultures. Brain Res. 1996;729:102–109. [PubMed] [Google Scholar]
- 38.Kopnisky KL, Sumners C, Chandler LJ. Cytokine- and endotoxin-induced nitric oxide synthase in rat astroglial cultures: differential modulation by angiotensin II. J Neurochem. 1997;68:935–944. doi: 10.1046/j.1471-4159.1997.68030935.x. [DOI] [PubMed] [Google Scholar]
- 39.Koprowski H, Zheng YM, Heber-Katz E, Fraser N, Rorke L, Fu ZF, Hanlon C, Dietzschold B. In vivo expression of nitric oxide synthase in experimentally induced neurological diseases. Proc Natl Acad Sci USA. 1993;90:3024–3027. doi: 10.1073/pnas.90.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kruetzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 41.Kyriakis JM, Avruch J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J Biol Chem. 1996;271:24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- 42.Lee BS, Kang HS, Pyun KH, Choi I. Roles of tyrosine kinases in the regulation of nitric oxide synthesis in murine liver cells: modulation of NF-κB activity by tyrosine kinases. Hepatology. 1997;25:913–919. doi: 10.1002/hep.510250421. [DOI] [PubMed] [Google Scholar]
- 43.Lee JC, Young PR. Role of CSBP/p38/RK stress response kinase in LPS and cytokine signaling mechanisms. J Leukoc Biol. 1996;59:152–157. doi: 10.1002/jlb.59.2.152. [DOI] [PubMed] [Google Scholar]
- 44.Lee JC, Laydon JT, McDonnel PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 45.Lee SC, Liu W, Dickson DW, Brosnan CF, Berman JW. Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1β. J Immunol. 1993;150:2659–2667. [PubMed] [Google Scholar]
- 46.Lieberman AP, Pitha PM, Shin HS, Shin ML. Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc Natl Acad Sci USA. 1989;86:6348–6352. doi: 10.1073/pnas.86.16.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin T, Kong X, Haystead TAJ, Pause A, Belsham G, Sonenberg N, Lawrence JC. PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science. 1994;260:653–656. doi: 10.1126/science.7939721. [DOI] [PubMed] [Google Scholar]
- 48.Lowenstein CJ, Alley EW, Rava LP, Snowman AM, Snyder SH, Russell SW, Murphy WJ. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon-γ and lipopolysaccharide. Proc Natl Acad Sci USA. 1993;90:9730–9734. doi: 10.1073/pnas.90.20.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Merrill JE, Jonakait GM. Interactions of the nervous system and immune system development, normal brain homeostasis and disease. FASEB J. 1995;9:611–618. doi: 10.1096/fasebj.9.8.7768352. [DOI] [PubMed] [Google Scholar]
- 50.Mucke L, Eddleston M. Astrocytes in infectious and immune-mediated diseases of the central nervous system. FASEB J. 1993;7:1226–1232. doi: 10.1096/fasebj.7.13.8405808. [DOI] [PubMed] [Google Scholar]
- 51.Mullet D, Fertel RH, Kniss D, Cox GW. An increase in intracellular cyclic AMP modulates nitric oxide production in IFN-γ-treated macrophages. J Immunol. 1997;158:897–904. [PubMed] [Google Scholar]
- 52.Murphy S, Simmons LA, Garcia A, Feinstein DL, Galea E, Reis DJ, Minc-Golomb D, Schwartz JP. Synthesis of nitric oxide in glial cells. Trends Neurosci. 1993;16:323–328. doi: 10.1016/0166-2236(93)90109-y. [DOI] [PubMed] [Google Scholar]
- 53.Nishiya T, Uehara T, Edamatsu H, Kaziro Y, Itoh H, Nomura Y. Activation of Stat1 and subsequent transcription of inducible nitric oxide synthase gene in C6 glioma cells is independent of interferon-γ-induced MAPK activation that is mediated by p21. FEBS Lett. 1997;408:33–38. doi: 10.1016/s0014-5793(97)00383-9. [DOI] [PubMed] [Google Scholar]
- 54.Oleszak EL, Katsetos CD, Kuzmak J, Varadhachary A. Inducible nitric oxide synthase in Theiler’s murine encephalitis virus infection. J Virol. 1997;71:3228–3235. doi: 10.1128/jvi.71.4.3228-3235.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ott L, McClain CJ, Gillespie M, Young B. Cytokines and metabolic dysfunction after severe head injury. J Neurotrauma. 1994;11:447–472. doi: 10.1089/neu.1994.11.447. [DOI] [PubMed] [Google Scholar]
- 56.Park SK, Murphy S. Nitric oxide synthase type II mRNA stability is translation- and transcription-dependent. J Neurochem. 1996;67:1766–1769. doi: 10.1046/j.1471-4159.1996.67041766.x. [DOI] [PubMed] [Google Scholar]
- 57.Parkinson JF, Mitrovic B, Merrill JE. The role of nitric oxide in multiple sclerosis. J Mol Med. 1997;75:174–186. doi: 10.1007/s001090050102. [DOI] [PubMed] [Google Scholar]
- 58.Pause A, Belsham GJ, Gingra A, Donze O, Lin T, Lawrence JC, Sonenberg N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature. 1994;371:762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- 59.Perry VH, Andersson PB, Gordon S. Macrophages and inflammation in the central nervous system. Trends Neurosci. 1993;16:268–273. doi: 10.1016/0166-2236(93)90180-t. [DOI] [PubMed] [Google Scholar]
- 60.Raine CS. Multiple sclerosis: TNF revisited, with promise. Nat Med. 1995;1:211–214. doi: 10.1038/nm0395-211. [DOI] [PubMed] [Google Scholar]
- 61.Samdani AF, Dawson TM, Dawson VL. Nitric oxide synthase in models of focal ischemia. Stroke. 1997;28:1283–1288. doi: 10.1161/01.str.28.6.1283. [DOI] [PubMed] [Google Scholar]
- 62.Singh K, Balligand JL, Fischer TA, Smith TW, Kelly RA. Regulation of cytokine-inducible nitric oxide synthase in cardiac myocytes and microvascular endothelial cells. Role of extracellular signal-regulated kinases 1 and 2 (ERK1/ERK2) and STAT1 alpha. J Biol Chem. 1996;271:1111–1117. doi: 10.1074/jbc.271.2.1111. [DOI] [PubMed] [Google Scholar]
- 63.Stuehr DJ, Nathan CF. Nitric oxide: a macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med. 1989;169:1543–1555. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Togashi H, Sasaki M, Frohman E, Taira E, Ratan RR, Dawson TM, Dawson VL. Neuronal (type I) nitric oxide synthase regulates nuclear factor κB activity and immunologic (type II) nitric oxide synthase expression. Proc Natl Acad Sci USA. 1997;94:2676–2680. doi: 10.1073/pnas.94.6.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997;16:1909–1920. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xie QW, Wishnan R, Nathan C. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon-γ and bacterial lipopolysaccharide. J Exp Med. 1993;177:1779–1784. doi: 10.1084/jem.177.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang P, Hogan EL, Bhat NR. Activation of JNK/SAPK in primary glial cultures. II. Differential activation of kinase isoforms corresponds to their differential expression. Neurochem Res. 1998;23:219–225. doi: 10.1023/a:1022489127107. [DOI] [PubMed] [Google Scholar]