Traumatic Spinal Cord Injury Induces Nuclear Factor-κB Activation (original) (raw)
Abstract
Inflammatory responses are a major component of secondary injury and play a central role in mediating the pathogenesis of acute and chronic spinal cord injury (SCI). The nuclear factor-κB (NF-κB) family of transcription factors is required for the transcriptional activation of a variety of genes regulating inflammatory, proliferative, and cell death responses of cells. In this study we examined the temporal and cellular expression of activated NF-κB after traumatic SCI. We used a contusion model (N.Y.U. Impactor) to initiate the early biochemical and molecular changes that occur after traumatic injury to reproduce the pathological events associated with acute inflammation after SCI. The activation and cellular distribution of activated NF-κB was evaluated by using a monoclonal antibody that selectively recognizes activated p65 in a NF-κB dimer. Immunohistochemical and Western blot analyses demonstrated that NF-κB activation occurred as early as 0.5 hr postinjury and persisted for at least 72 hr. Using electrophoretic mobility shift assays (EMSA), we demonstrate that NF-κB is activated after SCI. In our immunohistochemical, Western, and EMSA experiments there are detectable levels of activated NF-κB in our control animals. Using double-staining protocols, we detected activated NF-κB in macrophages/microglia, endothelial cells, and neurons within the injured spinal cord. Colocalization of activated NF-κB with the NF-κB-dependent gene product, inducible nitric oxide synthase (iNOS), suggests functional implications for this transcription factor in the pathogenesis of acute spinal cord injury. Although there is considerable evidence for the involvement of an inflammatory reaction after traumatic SCI, this is the first evidence for the activation of NF-κB after trauma. Strategies directed at blocking the initiation of this cascade may prove beneficial as a therapeutic approach for the treatment of acute SCI.
Keywords: nuclear factor-κB, spinal cord injury, inflammation, secondary injury, nitric oxide synthase, CNS, EMSA
Traumatic injury of the spinal cord initiates a series of cellular and molecular events that include both primary and secondary injury cascades (Blight, 1992; Dusart and Schwab, 1993; Popovic et al., 1994; Blight et al., 1995, 1997). Secondary injury may contribute significantly to the neuropathology associated with the initial injury (Blight, 1992; Dusart and Schwab, 1993; Popovic et al., 1994; Yakovlev and Faden, 1994; Blight et al., 1995, 1997;Zhang et al., 1995). Inflammatory responses are a major component of secondary injury and play a central role in regulating the pathogenesis of acute and chronic spinal cord injury (SCI) (Blight, 1992; Dusart and Schwab, 1993; Popovic et al., 1994; Blight et al., 1995, 1997). Many inflammatory responses are mediated by enhanced and/or induced gene expression. A principal player in the regulation of inflammatory gene expression is the nuclear factor-κB (NF-κB) family (cRel, RelA/p65, RelB, p50, and p52) of transcription factors (Baeuerle, 1991; Baeuerle and Henkel, 1994; Baeuerle and Baltimore, 1996). NF-κB transcription factors regulate the expression of many genes mediating the inflammatory responses in the CNS and may be important determinants of cell death and disease of the CNS (Baeuerle, 1991; Kaltschmidt et al., 1993, 1994a,b; Baeuerle and Henkel, 1994; Salminen et al., 1995;Baeuerle and Baltimore, 1996). NF-κB has been shown to activate transcriptionally the genes encoding cytokines (Benveniste, 1992;Rothwell and Relton, 1993; Feuerstein et al., 1994; Shohami et al., 1994; Hopkins and Rothwell, 1995; Merrill and Benveniste, 1996), prostaglandin synthase-2 (Shohami et al., 1988; Yamamoto et al., 1995;Adams et al., 1996; Nogawa et al., 1997), cell adhesion molecules (CAM) (Kaltschmidt et al., 1993; Jander et al., 1996; Shrikant et al., 1996), and inducible nitric oxide synthase (iNOS) (Ransohoff and Benveniste, 1996; O’Neil and Kaltschmidt, 1997). NF-κB was detected in degenerating hippocampal neurons after global ischemia, although it was not present in nondegenerating neurons (Clemens et al., 1997). In other studies that used PC12 cells it was demonstrated that inhibition of NF-κB activation induced apoptosis (Taglialatela et al., 1997). Thus, NF-κB may be a regulator of cell death programs in CNS neurons (Baeuerle and Baltimore, 1996; Grilli et al., 1996; Clemens et al., 1997). Additionally, excitotoxic neuronal death was blocked by pharmacological agents shown to inhibit NF-κB activation (Grilli et al., 1996), supporting the hypothesis that NF-κB activation in neurons may be an initiator of cell death. In addition to its possible role in regulating apoptotic programs in neurons, constitutively active NF-κB has been detected in a small population of cortical neurons (Kaltschmidt et al., 1994b). These data suggest that NF-κB activation may play an important role in normal neuronal signal transduction.
In this report we have used an in vivo model of SCI (N.Y.U. Impactor) to induce acute SCI and reproduce the acute pathological events associated with inflammation after traumatic SCI in rats. Cellular and molecular events regulating secondary injury and associated with the pathogenesis of acute SCI were studied by immunohistochemical procedures using a monoclonal antibody, designated α-p65 mAb, which recognizes the nuclear localization signal of the p65 DNA binding subunit of activated NF-κB. Using immunohistochemical and Western blot analysis, we evaluated the activation and distribution of NF-κB in the acutely injured spinal cord. Our results demonstrate that NF-κB is activated after contusion injury of the spinal cord and that NF-κB is coexpressed with iNOS in macrophages/microglia and neurons. This is the first in vivo demonstration of NF-κB activation and iNOS expression in neurons after spinal cord injury and may be useful toward understanding the molecular mechanisms responsible for secondary pathological changes after acute SCI.
MATERIALS AND METHODS
Contusion injury. Traumatic injury was induced by the weight drop device developed at New York University (Gruner, 1992). Sixty adult female (250–300 gm) Sprague Dawley rats (12 per group) were anesthetized with a mixture of 1% halothane and a mixture of 70% nitrous oxide and 30% oxygen. The dorsal aspect of the back was shaved and scrubbed with Betadine solution. An adequate amount of anesthesia was determined by monitoring the corneal reflex and withdrawal to painful stimuli. A laminectomy was performed at vertebral levels T9–T10, exposing the cord underneath without disrupting the dura. To maintain consistency within each experiment, we induced a “moderate” injury by adjusting the height of the weight drop (10.0 gm) to 12.5 mm above the exposed spinal cord. After injury, muscles were closed in layers, the incision was closed with wound clips, and the animals were returned to their home cages. Appropriate care was provided by the technical staff and veterinary services to ensure that the animals did not develop any postoperative infections or experience discomfort. Animals were killed at different time intervals postinjury (see below). All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Miami.
Paraffin histopathology. Injured animals were allowed to survive for 0.5–72 hr and then prepared for histopathological and morphological analysis. Sham-operated rats were processed by using the same protocol but were not traumatized. Rats were anesthetized and perfused transcardially with isotonic saline for 5 min. This was followed by fixative for 20 min with a mixture of 40% formaldehyde, glacial acetic acid, and methanol (FAM) 1:1:8 by volume. After perfusion, the vertebral columns with the cord were immersed in FAM at 4°C for 24 hr; then the cord was removed and placed in 20% sucrose for 24 hr. The spinal column was blocked and embedded in paraffin for tissue sectioning. Serial longitudinal sections 23–25 mm in length (10 μm thickness) were taken through the full dorsoventral dimension of the cord. Alternating sections were stained with hematoxylin and eosin for morphological and histopathological analyses.
Immunostaining. Spinal cords from injured and uninjured animals were prepared as described above. To neutralize the endogenous peroxidase activity before antibody application, we incubated the sections for 30 min at room temperature in 0.1 mTris-buffered saline (TBS), pH 7.4, that contained 0.3% hydrogen peroxide and then rinsed the sections several times in TBS. TBS or TBS plus 0.25% Triton X-100 was used as a rinse, and nonspecific binding was blocked with 0.1% BSA (Sigma, St. Louis, MO). Nonspecific binding was evaluated by performing controls with mouse or rabbit immunoglobulins that were applied in the absence of primary antibodies. These controls were performed on sections from injured spinal cords at each time point. Incubation with the primary antibody at a dilution of 1:1000 (mouse monoclonal NF-κB; Boehringer Mannheim, Indianapolis, IN) was performed overnight at 4°C. This antibody allows for the exclusive identification of activated p65 in an NF-κB dimer (Brand et al., 1996). Biotinylated horse anti-mouse immunoglobulin (1:1000; Vector Elite ABC kit, Vector Laboratories, Burlingame, CA) and streptavidin–horseradish peroxidase (HRP) complex were applied, followed by 3–3′-diaminobenzidine (DAB; Sigma) until a brown reaction product was observed. To suppress any remaining peroxidase, we incubated the slides in 3% hydrogen peroxide for 3 min. After being washed three times, the sections were incubated with the second incubation series consisting of primary and secondary antibodies and streptavidin–biotin peroxidase complex, as described above. In our colocalization experiments the sections were incubated with primary antibodies specific for Factor VIII (Sigma), CD11b (Chemicon, Temecula, CA), glial fibrillary acidic protein (GFAP; Dako, Carpinteria, CA), neuron-specific enolase (NSE; Polysciences, Warrington, PA), or iNOS (Transduction Laboratories, Lexington, KY) diluted 1:1000 in 0.1m phosphate buffer plus 0.3% Triton X-100 overnight at 4°C. Next, the sections were rinsed several times in phosphate buffer, followed by incubation in TrueBlue peroxidase substrate (Kirkegaard & Perry Laboratories, Gaithersburg, MD) for the second staining. Double-labeled profiles stained deep purple or black when colocalization occurred, because the TrueBlue reaction product yields a blue color and DAB yields a brown reaction product. Before the colocalization studies we optimized the individual reactions for DAB and TrueBlue. Color slides for Figures 6 and 7 were digitized by a UMAX Astra 600s color scanner, using a single-pass scanning method with color CCD attached to an Apple Macintosh computer.
Fig. 6.

Colocalization of NF-κB immunoreactivity with macrophages/microglia after SCI, using a double immunohistochemical staining procedure 72 hr after SCI. Macrophages/microglia were identified by using an antibody specific for CD11b and by the brown reaction product characteristic of DAB immunohistochemistry. NF-κB immunoreactivity was colocalized with macrophages/microglia (arrows) in the lesion epicenter and adjacent tissue (200× magnification). Cells expressing CD11b and activated NF-κB stained a dark purple or black.
Fig. 7.
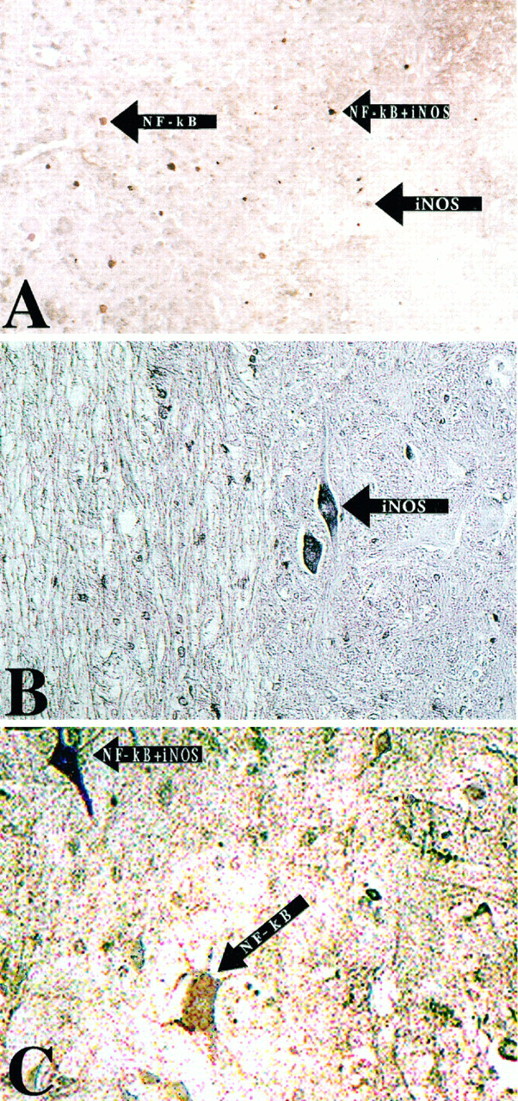
Colocalization of activated NF-κB with the NF-κB target gene product iNOS 72 hr after SCI. NF-κB immunoreactivity was visualized by using DAB immunohistochemistry in the first staining reaction. iNOS was visualized by using TrueBlue immunohistochemistry in the subsequent reaction. A, iNOS immunoreactivity was colocalized with NF-κB in non-neuronal cells in the lesion epicenter (200× magnification). B, Sections were stained for iNOS immunoreactivity, using TrueBlue immunohistochemistry; cells with the morphological appearance of neurons expressed iNOS immunoreactivity (200× magnification).C, Colocalization of iNOS with activated NF-κB in neurons results in a rich purple (200× magnification).
Preparation of nuclear extracts. Nuclear extracts were prepared as previously described, using a modified method of Dignam and colleagues (Dignam et al., 1983; Bethea et al., 1997). Spinal cords were frozen immediately on dry ice, homogenized in Buffer A [containing (in mm) 10 HEPES, pH 7.5, 10 KCl, 0.1 EDTA, 0.1 EGTA, 1.0 DTT, and 1 phenylmethylsulfonyl fluoride (PMSF) with 10 μg/ml of leupeptin, antipain, aprotinin, and pepstatin A], and placed on ice for 10 min. Then the extracts were treated with 1.0% Nonidet P-40. The nuclei were separated from the cytosolic proteins and lipids by multiple centrifugations at 20,000 × g for 15 min. Then the extracts were resuspended in Buffer C [25% glycerol, 0.4 m NaCl, and (in mm) 20 HEPES, 1.0 EDTA, 1.0 EGTA, 1.0 DTT, and 1 PMSF with 10 μg/ml of leupeptin, antipain, aprotinin, and pepstatin A] and briefly sonicated on ice. Nuclear extracts were obtained by centrifugation at 12,000 × _g_for 10 min. Protein concentration was determined by Coomassie Plus Protein Assay (Pierce, Rockford, IL).
Electrophoretic mobility shift assay (EMSA). EMSA was performed on spinal cord extracts isolated from sham animals or SCI animals at different times after injury (0.5, 1.5, 24.0, and 72.0 hr). For binding reactions, 25 μg of protein was incubated in binding buffer [5.0% glycerol and (in mm) 20 HEPES, 50 KCl, 0.1 EDTA, and 1.0 DTT with 200 μg/ml BSA and 2.5 μg of poly (dI-dC)] for 15 min at room temperature. Double-stranded NK-κB oligonucleotides were end-labeled with T4 polynucleotide kinase and γ-32P ATP. Radiolabeled oligonucleotide (5.0 × 105 cpm) was added to the reaction mixture and incubated for 20 min. In our supershift experiments the antibodies were incubated with the nuclear extracts on ice for 30 min before the binding reaction. The reaction products were analyzed by electrophoresis in a 4% polyacrylamide gel with 0.25× TBE buffer (22.3 mm Tris, 22.2 mm borate, and 0.5 mm EDTA). The dried gels were analyzed by autoradiography after an overnight exposure.
Preparation of spinal cord extracts. Uninjured and injured spinal cords with the lesion epicenter in the middle of the samples were removed at the appropriate times after injury and frozen immediately on dry ice. Spinal cord tissues to be used in immunoblotting were homogenized in cell extraction buffer [1% Triton X-100 and (in mm) 100 HEPES, pH 7.5, 10 DTT, 1 PMSF, and 1 EDTA with 5 μg/ml leupeptin and 1 μg/ml pepstatin A]. Extracts were cleared by centrifugation at 20,000 × g for 15 min, and the protein concentration was determined by Coomassie Plus Protein Assay (Pierce). Samples were stored at −80°C until SDS-PAGE analysis was performed.
Immunoblotting with an NF-κB antibody. For the detection of activated NF-κB in spinal cord extracts, proteins were resolved on 12% SDS-PAGE, transferred to polyvinylidene difluoride membranes, and placed in blocking buffer (Tropix, Bedford, MA). The membranes were incubated with anti-p65 monoclonal antibody (mouse monoclonal NF-κB; Boehringer Mannheim) at a dilution of 1:10,000 in blocking buffer, followed by the secondary antibody, alkaline phosphatase-conjugated goat anti-mouse immunoglobulin (1:5000; Tropix). Visualization of the signal was by enhanced chemiluminescence (Tropix).
RESULTS
Histopathological analysis of injured spinal cords
At 1 d after SCI, a well defined hemorrhagic zone was observed within the central gray matter of the spinal cord (Fig.1A). Within the epicenter of the contusion, gray matter structures appeared necrotic, with polymorphonuclear leukocytes (PMNL) invading the injured parenchyma. Necrotic neurons contained pyknotic nuclei surrounded by an eosinophilic cytoplasm. Severely damaged white matter tracts appeared swollen and edematous (Fig. 1B). In gray areas bordering the contusion, selective neuronal necrosis was observed within parenchyma containing swollen astrocytic cell bodies. In addition, petechial hemorrhages were observed throughout the gray matter and white matter tracts remote from the contusion.
Fig. 1.
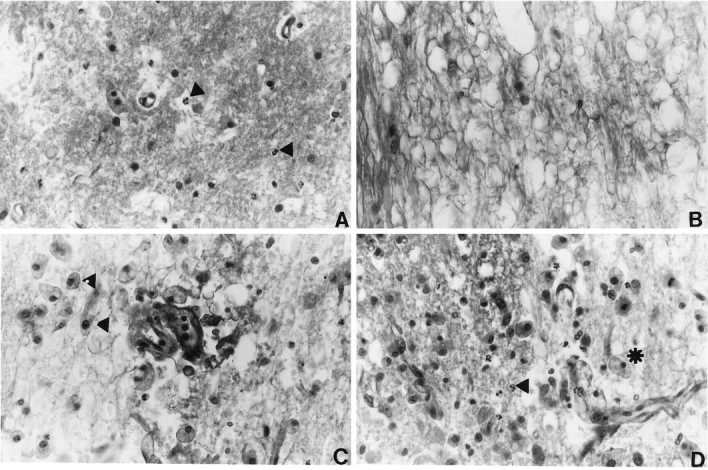
Histopathological analysis of spinal cords at 24 and 72 hr after contusion injury. Photomicrographs show necrosis, infiltration of leukocytes, and white matter vacuolization.A, At 24 hr after SCI, necrotic tissue is present at the epicenter of the lesion. PMNLs (arrowheads) are present in the injured spinal cord. B, Severe vacuolization of the white matter is observed 24 hr after SCI. C, At 72 hr after SCI, large numbers of macrophages (arrowheads) are observed in the white matter. D, Macrophages (asterisk) and PMNLs (arrowheads) are present at the gray/white interface 72 hr after SCI. The data presented in B–D are from regions of the spinal cord adjacent to the lesion areas. All micrographs are shown at 200× magnification.
By 3 d after spinal cord trauma, the lesion remained well defined, and evidence of hemorrhage was observed. A difference between this pathology and that seen at Figure 1D after trauma was the appearance of large numbers of foamy macrophages (Fig.1C). These foamy macrophages were seen within necrotic gray and white matter tracts. PMNLs also were dispersed throughout the necrotic areas (Fig. 1D). Within areas adjacent to the injury, petechial hemorrhages were detected, and blood vessels demonstrated red blood cell stasis. Blood vessels also contained luminal leukocytes.
Activated NF-κB in the injured spinal cord
To investigate the expression of activated NF-κB after contusion injury, we examined the temporal expression of activated NF-κB by DAB immunohistochemistry. At four time points (0.5, 1.5, 24, and 72 hr) both injured animals and uninjured sham-operated control animals were evaluated for NF-κB activation. In uninjured sham-operated animals little or no NF-κB immunoreactivity was detected (Fig.2A). DAB staining was absent when the primary antibody was omitted or replaced with a control antibody of an identical isotype (Fig. 2B). At 0.5 and 1.5 hr postinjury, NF-κB immunoreactivity was detected primarily within and adjacent to the lesion epicenter, with little or no detectable immunostaining outside this region. At 24 and 72 hr postinjury, NF-κB immunoreactivity was detected throughout the extent of the spinal cord section (23–25 mm). Figure3 demonstrates the immunoreactivity of activated NF-κB 24 hr after injury. Figure 3A is a schematic representation of the injured spinal cord 24 hr after trauma. There was no detectable NF-κB immunoreactivity in neurons within the epicenter of the lesion (Fig. 3B). Cells with the morphological and size characteristics of neurons were positive for NF-κB immunoreactivity both adjacent to (Fig. 3C,D) and 12 mm from the lesion epicenter (Fig. 3E,F). We analyzed 60 animals, 12 in each group, and consistently observed NF-κB immunoreactivity in the spinal cord after injury.
Fig. 2.
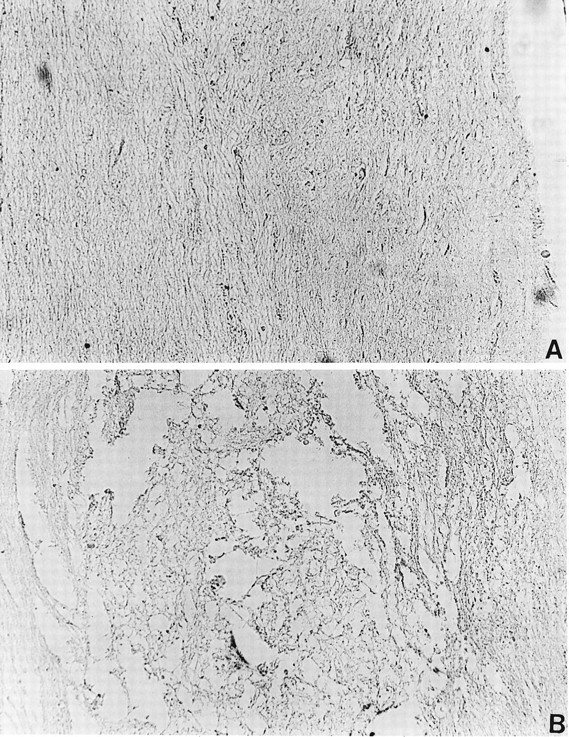
Controls for immunohistochemistry, using sections from sham-operated and SCI animals. A, Spinal cords from sham-operated control animals were stained for activated NF-κB, using DAB immunohistochemistry. Activated NF-κB was not observed in control animals (200× magnification). B, Isotype control for immunohistochemistry, using sections from the lesion epicenter. The primary α-p65 mAb was omitted and replaced with a mouse IgG3 isotype control antibody. No specific staining was observed.
Fig. 3.
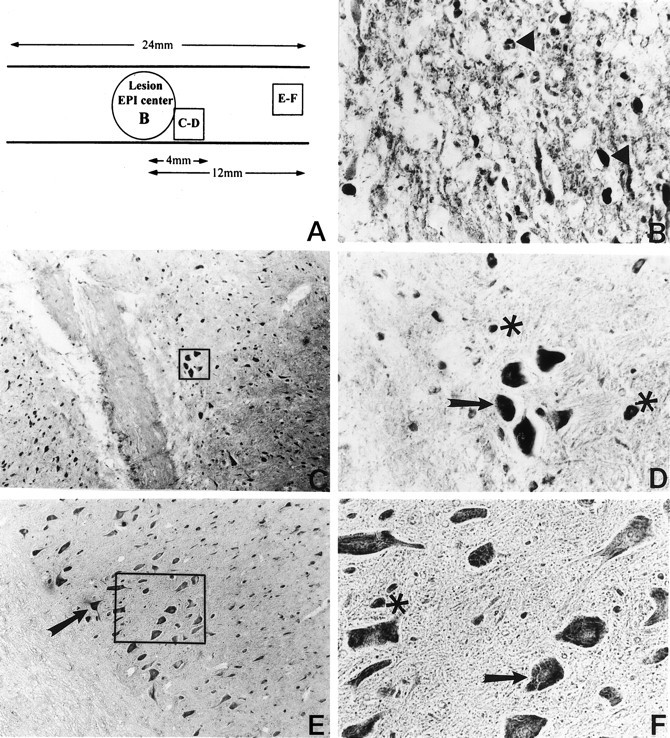
DAB immunohistochemical detection of activated NF-κB 24 hr after traumatic SCI. Activated NF-κB was observed both rostral and caudal to the lesion epicenter throughout the length of the 24 mm sections. A, Schematic diagram describing the expression pattern of activated NF-κB rostral (right side) to the lesion epicenter. B, NF-κB activation was observed within the lesion epicenter (200× magnification; arrowheads). C, Immunohistochemical detection of activated NF-κB within 4 mm of the lesion epicenter (100× magnification). D, Higher magnification (400×) of boxed inset in_C_. E, Activated NF-κB was detected 12 mm away from the lesion epicenter (100× magnification).F, Higher magnification (400×) of the boxed inset in E. Cells positive for NF-κB immunoreactivity have the characteristic size and morphology of neurons (arrows). In C–F, cells that do not have the morphological characteristics of neurons are also positive for activated NF-κB (asterisks).
Detection of NF-κB activation by Western blot analysis
To confirm the immunohistochemical detection of activated NF-κB, we performed Western blot analysis on control and SCI animals. Cellular extracts of spinal cords from control and injured animals (0.5, 1.5, 24, and 72 hr) were prepared and resolved by SDS-PAGE. Western blots that used anti p65 antiserum showed labeling of a single band with an apparent molecular weight of 65 kDa (p65). Increasing levels of activated NF-κB were detected after injury, with maximal levels at 24 and 72 hr after trauma (Fig. 4,lanes 2–5). However, samples showed a weakly detectable band in extracts from uninjured spinal cord (Fig. 4, lane 1). The temporal profile of NF-κB activation detected on Western blots parallels that demonstrated in our immunohistochemical studies. To quantitate changes in NF-κB activation detected by Western analysis, we analyzed the autoradiographs densitometrically. The data in Table 1 show that there was an approximately twofold increase in NF-κB activity after SCI.
Fig. 4.

Western blot demonstrating the temporal expression of activated NF-κB after traumatic SCI. Lane 1, Sham-operated control; lane 2, 0.5 hr; lane 3, 1.5 hr; lane 4, 24 hr; and lane 5, 72 hr postinjury. The arrow points to the position of the 65 kDa activated transcription factor. The antibody used in Western blot analysis was the same as that used in our immunohistochemical studies.
Table 1.
Fold increase in densitometric units of NF-κB immunoreactivity as detected by Western blot analysis
| Sham | 0.5 hr | 1.5 hr | 24.0 hr | 72.0 hr |
|---|---|---|---|---|
| 1.0 | 1.84 | 1.4 | 2.1 | 2.0 |
EMSA further demonstrate that SCI induces NF-κB activation
Nuclear extracts were isolated from the spinal cords of sham and SCI rats at 0.5, 1.5, 24, and 72 hr after injury. EMSA was performed with a radiolabeled double-stranded oligonucleotide containing the NF-κB consensus sequence (Fig. 5). Our data demonstrate that after SCI the nuclear extracts contain a protein complex that binds to the NF-κB oligonucleotide (Fig. 5, lanes 1–5). Although there is modest binding in extracts isolated from sham-injured animals (Fig. 5, lane 1), in SCI-injured animals there is a large increase in binding relative to the controls. To identify the proteins that were binding in our SCI extracts, we performed supershift experiments, using the same antibody that was used in our immunohistochemical and Western blot experiments. The p65 antibody retarded the migration of the proteins interacting with the NF-κB oligonucleotide (Fig. 5, lanes 6–10), whereas an antibody to STAT-1 had no effect on the migration of the protein complex (Fig. 5, lanes 11–15). These data demonstrate that p65 is activated after SCI.
Fig. 5.

EMSA analysis of NF-κB activation after SCI.Lanes 1, 6, 11, Sham-operated control; lanes 2, 7, 12, 0.5 hr; lanes 3, 8, 13, 1.5 hr;lanes 4, 9, 14, 24 hr; and lanes 5, 10, 15, 72 hr postinjury. In lanes 1–5 and_lanes 11–15_ there is a prominent band that interacts with our NF-κB oligonucleotide (arrowhead). Supershift experiments with anti-p65 demonstrate that the protein complex interacting with the NF-κB oligonucleotide contains the p65 subunit (lanes 6–10). When a nonspecific antibody (STAT-1) was used in our supershift experiments, there was no change in the migration pattern of the bands (lanes 11–15).
Identification of cell types expressing activated NF-κB
The cell types in which NF-κB was activated after traumatic SCI were identified by using double immunohistochemical staining procedures (Miao and Lee, 1990). The detection of NF-κB activation by using α-p65 mAb was performed in the first staining step, which was visualized with DAB (diffuse brown reaction product). DAB staining was inhibited when the primary antibody was omitted or replaced with a control antibody of the same isotype. Specific cell types expressing activated NF-κB were identified by using TrueBlue immunohistochemistry staining, which produces a blue reaction product that was easily distinguishable from the brown DAB reaction product. No TrueBlue staining was visualized in control experiments when the primary antibody was omitted or replaced with the appropriate isotype control (mouse or rabbit). Before performing the colocalization studies, we optimized the conditions for each antibody and detection method. When colocalization occurred, the combination of DAB and TrueBlue reaction products resulted in a dark purple or almost black reaction product.
Cellular localization of activated NF-κB after SCI is summarized in Table 2. Activated NF-κB was present in macrophages/microglia, neurons, and endothelial cells. Activated microglia and macrophages in the CNS were identified by using a monoclonal antibody that recognizes the CD11b integrin. NF-κB colocalized with microglia and macrophages at all time points that were evaluated (Table 2). The majority of CD11b-positive cells expressing activated p65 was found in the area of the lesion (Fig.6). Activated p65 also was observed in neurons and endothelial cells using antibodies specific for NSE and Factor VIII, respectively (Table 2). Cells with the morphological characteristics of neurons expressed activated NF-κB after SCI (see Fig. 3C–F). Using a GFAP monoclonal antibody that recognizes reactive astrocytes, we were unable to detect activated p65 in this cell type after contusion injury.
Table 2.
Temporal and cellular localization of activated NF-κB after traumatic SCI
| 0.5 hr | 1.5 hr | 24 hr | 72 hr | |
|---|---|---|---|---|
| Macrophage/microglia | + | + | + | + |
| Endothelial cells | + | + | + | + |
| Neurons | − | − | + | + |
| Astrocytes | − | − | − | − |
Functional implications of activated NF-κB after SCI
To demonstrate the functional nature of NF-κB activation after SCI, we examined the colocalization of the activated transcription factor with the expression of an established NF-κB gene product, iNOS (Kaltschmidt et al., 1993; Baeuerle and Henkel, 1994). NF-κB activation was labeled in the first reaction and iNOS in the subsequent step. Colocalization of activated NF-κB with iNOS was detected only at 72 hr after trauma (Fig. 7). Interestingly, although we were able to colocalize NF-κB with iNOS in macrophages within the lesion epicenter (Fig. 7A), we also were able to detect neuronal colocalization of iNOS and NF-κB (Fig.7C). In Figure 7B, we demonstrate iNOS immunoreactivity in cells that have the morphological characteristics of neurons. Thus, colocalization of activated NF-κB with iNOS within the same cells indicates that the transcription factor possibly is involved in gene expression after traumatic SCI.
DISCUSSION
This study is the first demonstration of the activation of NF-κB after traumatic SCI. Additionally, we show that iNOS, an important mediator of CNS inflammatory responses and neuropathology, is colocalized with activated NF-κB after SCI. NF-κB is activated within 30 min of injury and is still present 72 hr after injury. Activated NF-κB was detected within macrophages, endothelial cells, and neurons, but this transcription factor was not observed in astrocytes. The expression of activated NF-κB in these cells may play a key role in CNS inflammatory responses. For example, in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis, activated NF-κB was detected in microglia/macrophages at the peak of clinical disease, but not in astrocytes or in nondiseased animals (Kaltschmidt et al., 1994a). Additionally, in atherosclerosis, a disease that also is believed to have an inflammatory component, activated NF-κB is present in endothelial cells and macrophages (Brand et al., 1996).
The early CNS inflammatory responses after SCI may be initiated by neutrophils that infiltrate the lesion site after injury. In support of our observations, Dusart and Schwab (1993) demonstrate that neutrophils and macrophages enter the spinal cord after SCI in a orchestrated temporal sequence. Neutrophils begin to accumulate within 1 hr, are most abundant at 24 hr, and begin to decline at 48 hr. Neutrophils are able to release reactive oxygen and nitrosyl radicals as well as cytokines, chemokines, and a variety of enzymes. Therefore, they have been proposed to participate in enlargement of the lesion and promote tissue destruction (Dusart and Schwab, 1993). _In vitro_studies have shown that cytokine and chemokine gene expression in neutrophils is dependent on NF-κB activation (McDonald et al., 1997). Macrophages and microglia contribute to the secondary pathological and inflammatory response via the release of cytokines and neurotoxins that accompany traumatic SCI (Blight, 1992, 1994; Popovic et al., 1994). Using hematoxylin and eosin histopathology, we demonstrated macrophage accumulation within 72 hr after SCI. However, immunohistochemical studies with the macrophage/microglial marker CD11b detected macrophages as early as 0.5 hr postinjury. This macrophage staining is probably attributable to the extravasation of blood-borne macrophages into the injured cord after disruption of the spinal cord blood barrier. Currently, antibodies are not available that distinguish between activated macrophages and microglia.
The infiltration of leukocytes into the CNS is orchestrated by specific adhesion proteins on both endothelial cells and leukocytes. Within the CNS, ICAM-1 and VCAM-1 facilitate cell-to-cell interactions among astrocytes, endothelium, microglia, and effector cells of the peripheral immune system such as T-cells, macrophages, and neutrophils. On entering the CNS, leukocytes can initiate immune responses by releasing proinflammatory molecules such as cytokines, prostaglandins, and matrix metalloproteinases. The physical interaction mediated by ICAM-1, VCAM-1, and other cell adhesion molecules forms an integral component of the effector phase of immunological responses in the CNS. Supporting this idea is the observation that antibodies specific for cell adhesion molecules reduce the level of ischemic injury to the CNS when administered in vivo (Clark et al., 1991; Mori et al., 1992; Chopp et al., 1994; Lindsberg et al., 1995; Zhang et al., 1995). NF-κB is an important mediator of cell adhesion molecule gene expression, and studies are underway to investigate the expression of these molecules after SCI.
CNS inflammatory responses after trauma or diseases of the CNS are mediated in part by infiltrating leukocytes, astrocytes, microglia, and brain endothelium (Rosenberg, 1995; Goetzl et al., 1996; Ransohoff and Benveniste, 1996). Cytokine production is regulated primarily at the transcriptional level. An important intermediate in the transcriptional regulation of cytokine gene expression is the NF-κB family of transcription factors (Kaltschmidt et al., 1993; Baeuerle and Henkel, 1994). Tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), and interleukin-6 (IL-6) are prototypic inflammatory cytokines that are produced in the CNS after injury (Benveniste, 1992; Hopkins and Rothwell, 1995; Merrill and Benveniste, 1996; Ransohoff and Benveniste, 1996). Recently, TNF-α was detected in the spinal cord after traumatic injury in rats (Wang et al., 1996). Most, if not all, inflammatory responses induced by TNF-α are mediated at the transcriptional level by NF-κB (Kaltschmidt et al., 1993; Baeuerle and Henkel, 1994). Both in vitro and _in vivo_studies demonstrate that TNF-α is a potent mediator of microgliosis, astrogliosis, and cell death (Feuerstein et al., 1994). In a transgenic model of chronic CNS inflammation, mice overexpressing TNF-α in astrocytes, both in the spinal cord and cortex, exhibited breakdown of the blood–brain barrier, infiltration of leukocytes, expression of cell adhesion molecules, demyelination, hind limb paralysis, and neuronal cell death (Stalder et al., 1996). Taken together, these data demonstrate that TNF-α is a potent activator of inflammatory responses in the CNS and may contribute to the neuropathology associated with trauma to the CNS by activation of NF-κB. We have demonstrated recently that monocytes isolated from SCI rats secrete TNF-α in a time-dependent manner, whereas monocytes from sham animals do not (our unpublished data). Therefore, NF-κB activation can set in motion a cascade of inflammatory and possibly cell death programs that may participate in the injury process and exacerbate the initial injury.
Although NF-κB-induced responses are associated most commonly with immunological and inflammatory processes, the role of NF-κB in normal or pathological neuronal functions has not been established (O’Neil and Kaltschmidt, 1997). Recent studies have demonstrated constitutive activation of NF-κB in a small subset of cortical neurons (Kaltschmidt et al., 1994b). NF-κB activation may contribute to many neuropathological disorders, such as multiple sclerosis, and its expression has been detected in the cortex and within hippocampal neurons after ischemic injury (Kaltschmidt et al., 1994a; Salminen et al., 1995; Clemens et al., 1997). Clemens et al. (1997) demonstrated that activated NF-κB is present in degenerating CA1 hippocampal pyramidal neurons and is absent in nondegenerating neurons. Apoptosis occurred at the same time as NF-κB expression in these neurons. Although colocalization of NF-κB with cell death markers does not prove that it is involved in this pathway, it does suggest an involvement in this process. Activation of NF-κB in neurons ex vivo has been shown to be linked to excitotoxic cell death in the CNS (Grilli et al., 1996). In these studies the inhibition of NF-κB activation prevented neuronal cell death (Grilli et al., 1996). Consistent with this idea is the finding that NF-κB activation is detected in cerebellar granule cells and within the spinal cord after exposure to glutamate and quisqualic acid, respectively (O’Neil and Kaltschmidt, 1997; J. R. Bethea and R. P. Yezierski, unpublished observations).
Mice that have had the p65 gene deleted by homologous recombination die because of massive apoptosis of liver cells (Beg et al., 1995). This study suggests that p65 may participate in anti-cell death programs, at least in hepatogenesis. Consistent with the concept that NF-κB activation may prevent cell death, it was demonstrated that TNF, a potent activator of NF-κB, prevented glutamate-induced cell death in pure hippocampal cultures (Cheng et al., 1994). However, when microglia were present in these cultures, TNF induced neuronal cell death (M. Mattson, personal communication). Therefore, NF-κB may be an important signaling molecule after injury to the nervous system; depending on the mechanisms through which activation occurs and depending on the surrounding cellular environment, this transcription factor may promote either apoptotic or antiapoptotic genetic programs. In Figure 3, we demonstrate that cells having the morphology and size characteristics of neurons are immunoreactive for NF-κB. However, not all of the cells contain NF-κB immunoreactivity exclusively in the nucleus. This could be explained in part because transcription factor binding to its _cis_-regulatory sequence is a transient event. Our EMSA data suggest that some detectable NF-κB binding activity occurs in sham-injured animals but to a much greater degree after SCI. The presence of activated NF-κB in our EMSA experiments suggests that activated NF-κB may play a role in regulating basal levels of transcription in the CNS. These studies support earlier findings byKaltschmidt et al. (1994b) in which activated NF-κB was detected in a subset of cortical neurons. These data support our immunohistochemical and Western blot studies.
Another important effector of inflammation in the CNS is iNOS. In murine models of CNS inflammation or injury, iNOS immunoreactivity and enzyme activity have been detected in macrophages, glial cells, and neurons (Minc-Golomb et al., 1996; Sato et al., 1996). However, in humans iNOS has not been detected in macrophages, suggesting that neuronal pathology attributed to iNOS-generated NO toxicity results from either glial or neuronal sources. The antibody used in our studies to detect iNOS does not recognize either neuronal cNOS or endothelial NOS (Van Voorhis et al., 1994; Lloyd et al., 1995). In a recent study the neurons that expressed NOS activity after SCI underwent cell death, suggesting a causal relationship between NOS expression and neuronal cell death (Wu, 1993). In an in vivo model of CNS inflammation, iNOS immunoreactivity was detected in cerebellar neurons after direct administration of interferon-γ and lipopolysaccharide (Sato et al., 1996). Because NOS gene expression is dependent, in part, on NF-κB activation and because iNOS immunoreactivity is colocalized with activated NF-κB in neurons, it is suggested that the activation of this transcription factor in neurons may be an important effector in trauma-induced neuropathology.
The results presented here demonstrate that NF-κB activation occurs after SCI and may be an important determinant in CNS pathology. Therefore, therapeutic approaches that interfere with NF-κB activation and/or processing could represent potential targets for pharmacological intervention after CNS injury.
Footnotes
This work was supported by State of Florida Specific Appropriations number 224, The Miami Project to Cure Paralysis, and the National Multiple Sclerosis Society.
Correspondence should be addressed to Dr. John R. Bethea, The Miami Project to Cure Paralysis, University of Miami School of Medicine, 1600 N.W. 10th Avenue (R-48), Miami, FL 33136.
REFERENCES
- 1.Adams J, Collaco-Moraes Y, de Belleroche J. Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. J Neurochem. 1996;66:6–13. doi: 10.1046/j.1471-4159.1996.66010006.x. [DOI] [PubMed] [Google Scholar]
- 2.Baeuerle PA. The inducible transcription activator NF-κB: regulation by distinct protein subunits. Biochim Biophys Acta. 1991;1072:63–80. doi: 10.1016/0304-419x(91)90007-8. [DOI] [PubMed] [Google Scholar]
- 3.Baeuerle PA, Baltimore D. NF-kB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 4.Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 5.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 6.Benveniste EN. Inflammatory cytokines within the central nervous system: sources, function, and mechanism of action. Am J Physiol. 1992;263:C1–C16. doi: 10.1152/ajpcell.1992.263.1.C1. [DOI] [PubMed] [Google Scholar]
- 7.Bethea JR, Ohmori Y, Hamilton TA. A tandem GC box motif is necessary for lipopolysaccharide-induced transcription of the type II TNF receptor gene. J Immunol. 1997;158:5815–5823. [PubMed] [Google Scholar]
- 8.Blight AR. Macrophages and inflammatory damage in spinal cord injury. J Neurotrauma. 1992;9:S83–S91. [PubMed] [Google Scholar]
- 9.Blight AR. Effects of silica on the outcome from experimental spinal cord injury: implication of macrophages in secondary tissue damage. Neuroscience. 1994;60:263–273. doi: 10.1016/0306-4522(94)90220-8. [DOI] [PubMed] [Google Scholar]
- 10.Blight AR, Cohen TI, Saito K, Heyes MP. Quinolinic acid accumulation and functional deficits following experimental spinal cord injury. Brain. 1995;118:735–752. doi: 10.1093/brain/118.3.735. [DOI] [PubMed] [Google Scholar]
- 11.Blight AR, Leroy EC, Heyes MP. Quinolinic acid accumulation in injured spinal cord: time course, distribution, and species differences between rat and guinea pig. J Neurotrauma. 1997;14:89–98. doi: 10.1089/neu.1997.14.89. [DOI] [PubMed] [Google Scholar]
- 12.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D. Activated transcription factor nuclear factor-κB is present in the atherosclerotic lesion. J Clin Invest. 1996;97:1715–1722. doi: 10.1172/JCI118598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitoxix insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 14.Chopp M, Zhang RL, Chen H, Li Y, Jiang N, Rusche JR. Postischemic administration of an anti-mac-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in rats. Stroke. 1994;25:869–876. doi: 10.1161/01.str.25.4.869. [DOI] [PubMed] [Google Scholar]
- 15.Clark WM, Madden KP, Rothlein R, Zivin JA. Reduction of central nervous system ischemic injury in rabbits using leukocyte adhesion antibody treatment. Stroke. 1991;22:877–883. doi: 10.1161/01.str.22.7.877. [DOI] [PubMed] [Google Scholar]
- 16.Clemens JA, Stephenson DT, Smalstig EB, Dixon EP, Little SP. Global ischemia activates nuclear factor-κB in forebrain neurons of rats. Stroke. 1997;28:1073–1081. doi: 10.1161/01.str.28.5.1073. [DOI] [PubMed] [Google Scholar]
- 17.Diehl JA, Tong W, Sun G, Hannink M. Tumor necrosis factor-α-dependent activation of a RelA homodimer in astrocytes. J Biol Chem. 1995;270:2703–2707. doi: 10.1074/jbc.270.6.2703. [DOI] [PubMed] [Google Scholar]
- 18.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dusart I, Schwab ME. Secondary cell death and the inflammatory reaction after dorsal hemisection of the rat spinal cord. Eur J Neurosci. 1993;6:712–724. doi: 10.1111/j.1460-9568.1994.tb00983.x. [DOI] [PubMed] [Google Scholar]
- 20.Feuerstein GZ, Liu T, Barone FC. Cytokines, inflammation, and brain injury: role of tumor necrosis factor-α. Cerebrovasc Brain Metab Rev. 1994;6:341–360. [PubMed] [Google Scholar]
- 21.Goetzl EJ, Banda MJ, Leppert D. Matrix metalloproteinases in immunity. J Immunol. 1996;156:1–4. [PubMed] [Google Scholar]
- 22.Grilli M, Pizzi M, Memo M, Spano P-F. Neuroprotection by aspirin and sodium salicylate through blockade of NF-κB activation. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 23.Gruner JA. A monitored contusion model of spinal cord injury in the rat. J Neurotrauma. 1992;9:123–128. doi: 10.1089/neu.1992.9.123. [DOI] [PubMed] [Google Scholar]
- 24.Hopkins SJ, Rothwell NJ. Cytokines and the nervous system. I. Expression and recognition. Trends Neurosci. 1995;18:83–88. [PubMed] [Google Scholar]
- 25.Jander S, Pohl J, Gillen C, Schroeter M, Stoll G. Vascular cell adhesion molecule-1 mRNA is expressed in immune-mediated and ischemic injury of the rat nervous system. J Neuroimmunol. 1996;70:75–80. doi: 10.1016/s0165-5728(96)00109-9. [DOI] [PubMed] [Google Scholar]
- 26.Kaltschmidt B, Baeuerle PA, Kaltschmidt C. Potential involvement of the transcription factor NF-κB in neurological disorders. Mol Aspects Med. 1993;14:171–190. doi: 10.1016/0098-2997(93)90004-w. [DOI] [PubMed] [Google Scholar]
- 27.Kaltschmidt C, Kaltschmidt B, Lannes-Vieira J, Kreutzberg GW, Wekerle H, Baeuerle PA, Gehrmann J. Transcription factor NF-κB is activated in microglia during experimental autoimmune encephalomyelitis. J Neuroimmunol. 1994a;55:99–106. doi: 10.1016/0165-5728(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 28.Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA. Constitutive NF-κB activity in neurons. Mol Cell Biol. 1994b;14:3981–3992. doi: 10.1128/mcb.14.6.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindsberg PJ, Sirén A-L, Feuerstein GZ, Hallenbeck JM. Antagonism of neutrophil adherence in the deteriorating stroke model in rabbits. J Neurosurg. 1995;82:269–277. doi: 10.3171/jns.1995.82.2.0269. [DOI] [PubMed] [Google Scholar]
- 30.Lloyd RV, Jin L, Qian X, Zhang S, Scheitauer BW. Nitric oxide synthase in the human pituitary gland. Am J Pathol. 1995;146:86–94. [PMC free article] [PubMed] [Google Scholar]
- 31.McDonald PP, Bald A, Cassatella MA. Activation of the NF-κB pathway by inflammatory stimuli in human neutrophils. Blood. 1997;89:3421–3433. [PubMed] [Google Scholar]
- 32.Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- 33.Miao FJ, Lee TJ. Cholinergic and VIPergic innervation in cerebral arteries: a sequential double-labeling immunohistochemical study. J Cereb Blood Flow Metab. 1990;10:32–37. doi: 10.1038/jcbfm.1990.4. [DOI] [PubMed] [Google Scholar]
- 34.Minc-Golomb D, Yadid G, Tsarfaty I, Resau JH, Schwartz JP. In vivo expression of inducible nitric oxide synthase in cerebellar neurons. J Neurochem. 1996;66:1504–1509. doi: 10.1046/j.1471-4159.1996.66041504.x. [DOI] [PubMed] [Google Scholar]
- 35.Mori E, del Zoppo GJ, Chambers JD, Copeland BR, Arfors KE. Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke. 1992;23:712–718. doi: 10.1161/01.str.23.5.712. [DOI] [PubMed] [Google Scholar]
- 36.Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclooxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Neill LAJ, Kaltschmidt C. NF-κB: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- 38.Popovic PG, Reinhard J, Flanagan EM, Stokes BT. Elevation of the neurotoxin quinolinic acid occurs following spinal cord trauma. Brain Res. 1994;633:348–352. doi: 10.1016/0006-8993(94)91560-1. [DOI] [PubMed] [Google Scholar]
- 39.Ransohoff RM, Benveniste EN. Cytokines and the CNS. CRC; Boca Raton, FL: 1996. [Google Scholar]
- 40.Rosenberg GA. Matrix metalloproteinases in brain injury. J Neurotrauma. 1995;12:833–842. doi: 10.1089/neu.1995.12.833. [DOI] [PubMed] [Google Scholar]
- 41.Rothwell NJ, Relton J. Involvement of interleukin-1 and lipocortin-1 in ischemic brain damage. Cerebrovasc Brain Metab Rev. 1993;5:178–198. [PubMed] [Google Scholar]
- 42.Salminen A, Liu PK, Hsu CY. Alteration of transcription factor binding activities in the ischemic rat brain. Biochem Biophys Res Commun. 1995;212:939–944. doi: 10.1006/bbrc.1995.2060. [DOI] [PubMed] [Google Scholar]
- 43.Sato I, Himi T, Murota S. Lipopolysaccharide-induced nitric oxide synthase activity in cultured cerebellar granule neurons. Neurosci Lett. 1996;205:45–48. doi: 10.1016/0304-3940(96)12377-6. [DOI] [PubMed] [Google Scholar]
- 44.Shohami E, Shapira Y, Cotev S. Experimental closed head injury in rats: prostaglandin production in a noninjured zone. Neurosurgery. 1988;22:859–863. [PubMed] [Google Scholar]
- 45.Shohami E, Novikov M, Bass R, Yamin A, Gallily R. Closed head injury triggers early production of TNF-α and IL-6 by brain tissue. J Cereb Blood Flow Metab. 1994;14:615–619. doi: 10.1038/jcbfm.1994.76. [DOI] [PubMed] [Google Scholar]
- 46.Shrikant P, Benveniste EN. The central nervous system as an immunocompetent organ: role of glial cells in antigen presentation. J Immunol. 1996;157:1819–1822. [PubMed] [Google Scholar]
- 47.Shrikant P, Benos DJ, Tang LP, Benveniste EN. HIV glycoprotein 120 enhances intercellular adhesion molecule-1 gene expression in glial cells. J Immunol. 1996;156:1307–1314. [PubMed] [Google Scholar]
- 48.Stalder AK, Pagenstecher A, Campbell IL. Lymphocytic meningoencephalomyelitis induced by transgenic expression of TNF-α in the CNS. Soc Neurosci Abstr. 1996;22:1455. [Google Scholar]
- 49.Taglialatela G, Robinson R, Perez-Polo JR. Inhibition of nuclear factor κB (NF-κB) activity induces nerve growth factor-resistant apoptosis in PC12 cells. J Neurosci Res. 1997;47:155–162. [PubMed] [Google Scholar]
- 50.Van Voorhis BJ, Dunn MS, Snyder GD, Weiner CP. Nitric oxide: an autocrine regulator of human granulosaluteal cell steroidogenesis. Endocrinology. 1994;135:1799–1806. doi: 10.1210/endo.135.5.7525252. [DOI] [PubMed] [Google Scholar]
- 51.Wang CX, Nuttin B, Heremans H, Dom R, Gybels J. Production of tumor necrosis factor in spinal cord following traumatic injury in rats. J Neuroimmunol. 1996;69:151–156. doi: 10.1016/0165-5728(96)00080-x. [DOI] [PubMed] [Google Scholar]
- 52.Wu W. Expression of nitric oxide synthase (NOS) in injured CNS neurons as shown by NADPH diaphorase histochemistry. Exp Neurol. 1993;120:153–159. doi: 10.1006/exnr.1993.1050. [DOI] [PubMed] [Google Scholar]
- 53.Yakovlev AG, Faden AI. Sequential expression of c-fos protooncogene, TNF-α, and dynorphin genes in spinal cord following experimental traumatic injury. Mol Chem Neuropathol. 1994;23:179–190. doi: 10.1007/BF02815410. [DOI] [PubMed] [Google Scholar]
- 54.Yamamoto K, Arakawa T, Ueda N, Yamamoto S. Transcriptional roles of nuclear factor-κB and nuclear factor-interleukin-6 in the tumor necrosis factor-α-dependent induction of cyclooxygenase-2 in MC3T3–E1 cells. J Biol Chem. 1995;270:31315–31320. doi: 10.1074/jbc.270.52.31315. [DOI] [PubMed] [Google Scholar]
- 55.Zhang RL, Chopp M, Jiang N, Tang WX, Prostak J, Manning AM, Anderson DC. Anti-intercellular adhesion molecule-1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the Wistar rat. Stroke. 1995;26:1438–1442. doi: 10.1161/01.str.26.8.1438. [DOI] [PubMed] [Google Scholar]