Virus-like particle vaccines: immunology and formulation for clinical translation (original) (raw)
ABSTRACT
Introduction: Virus-like particle (VLP) vaccines face significant challenges in their translation from laboratory models, to routine clinical administration. While some VLP vaccines thrive and are readily adopted into the vaccination schedule, others are restrained by regulatory obstacles, proprietary limitations, or finding their niche amongst the crowded vaccine market. Often the necessity to supplant an existing vaccination regimen possesses an immediate obstacle for the development of a VLP vaccine, despite any preclinical advantages identified over the competition. Novelty, adaptability and formulation compatibility may prove invaluable in helping place VLP vaccines at the forefront of vaccination technology.
Areas covered: The purpose of this review is to outline the diversity of VLP vaccines, VLP-specific immune responses, and to explore how modern formulation and delivery techniques can enhance the clinical relevance and overall success of VLP vaccines.
Expert commentary: The role of formation science, with an emphasis on the diversity of immune responses induced by VLP, is underrepresented amongst clinical trials for VLP vaccines. Harnessing such diversity, particularly through the use of combinations of select excipients and adjuvants, will be paramount in the development of VLP vaccines.
KEYWORDS: Clinical translation, formulation, immunology, vaccine, virus-like particle, VLP
1. Introduction
Virus-like particles (VLP) are a type of subunit vaccine based on virus-derived proteins, assembled to form a particle. VLP hold several advantages over other particulate subunit vaccines. These include a morphological resemblance to their parent virus, a highly repetitive immunogenic surface structure, and the retention of cell uptake and immune processing pathways associated with their parent virus [1]. VLP themselves are nonpathogenic, devoid of an intact virus genome, and are incapable of infection or replication. The noninfectious nature of VLP significantly improves their safety profile over live-attenuated vaccines, while also possessing advantages when compared to other forms of subunit, killed, or particulate vaccines. Commercially successful VLP vaccines include hepatitis B virus (HBV) VLP, such as Recombivax HB (Merck), Engerix-B (GlaxoSmithKline), Elovac B (Human Biologicals Institute), Genevac B (Serum Institute) and Shanvac B (Shantha Biotechnics), hepatitis E virus (HEV) VLP, such as Hecolin (Innovax), and human papillomavirus (HPV) VLP, including Gardasil (Merck) and Cervarix (GlaxoSmithKline). VLP vaccines currently under investigation in clinical trials include influenza A virus (IAV) VLP (NCT02768805, NCT02233816) [2,3], Chikungunya virus VLP (NCT02562482) [4], human cytomegalovirus (HCMV) VLP (NCT02826798) [5], and human norovirus (HuNV) VLP (NCT02669121, NCT03039790, NCT02661490) [6–8]. Recent and current VLP clinical trials registered in the United States of America and the European Union are summarized in Table 1.
Table 1.
VLP vaccines in clinical trials.
| Status | VLP Vector | Antigen | Disease | Formulation | Route | Phase | Trial Number | Company |
|---|---|---|---|---|---|---|---|---|
| Active | Enterovirus 71 | EV-A71 | Hand, foot and mouth disease | Alhydrogel and aluminum hydroxide in Bis-Tris buffer with 150 mM NaCl at pH 6.5 | Intramuscular | I | ACTRN12617001027303 | Sentinext Therapeutics Sdn Bhd |
| Active | Cytomegalovirus | VBI-1501 | Cytomegalovirus | VBI-1501A with alum | Intramuscular | I | NCT02826798 | VBI Vaccines Inc. |
| Active | Chikungunya virus | E1, E2 and CHIKV capsid proteins | Chikungunya virus | VRC-CHKVLP059-00-VP VLP in phosphate buffered saline, no adjuvant specified | Intramuscular | II | NCT02562482 | National Institute of Allergy and Infectious Disease |
| Active | Influenza A virus | Unspecified | Influenza A virus | Quadrivalent VLP combination in phosphate buffer with sodium chloride and Tween-80, no adjuvant specified | Intramuscular | II | NCT02768805/NCT02233816/NCT02236052 | Medicago |
| Active | Influenza A virus | H1, H3, B hemagglutinin | Influenza A virus | Quadrivalent VLP combination in phosphate buffer with sodium chloride and Tween-80, no adjuvant specified | Intramuscular | III | NCT03301051 | Medicago |
| Active | GI.1 Norovirus and GII.4 Norovirus | VP1 Capsid Protein | Norovirus | Bivalent VLP combinationwith aluminum hydroxide | Intramuscular | II | NCT02669121, NCT03039790 | Takeda |
| Active | HPV 16 and 18 | L1 CapsidProtein | Humanpapillomavirus | Bivalent VLP combination withaluminum phosphate, in a histidine buffer with sodium chloride and Tween-80 | Intramuscular | II/III | NCT02740777, NCT02733068 | Shanghai ZerunBiotechnology Co., Ltd |
| Active | HPV 6, 11, 16, 18, 31, 33, 45, 52, 58 | L1 Capsid Protein | Human papillomavirus | Aluminum hydroxyphosphate sulfate, yeast protein, NaCl, polysorbate 80, sodium borate | Intramuscular | III | EudraCT 2015–005093-38, NCT03158220 | Merck Sharp and Dohme Corp |
| Active | HPV 6, 11, 16, 18, 31, 33, 45, 52 and 58 | L1 CapsidProtein | High-grade squamous intraepithelial lesions | Nonavalent HPV VLP (Gardasil 9), NaCl, aluminum hydroxyphosphate sulfate, L-histidine, polysorbate 80, sodium borate | Intramuscular | IV | NCT03051516 | Fred Hutchinson Cancer Research Center |
| Completed (2017) | Alfalfa mosaic virus | Pfs25 | Malaria | Pfs25-CP VLP with alhydrogel | Intramuscular | I | NCT02013687 | Fraunhofer Centerfor MolecularBiotechnology |
| Completed (2017) | HPV 6, 11, 16, 18, 31, 33, 45, 52 and 58 | L1 CapsidProtein | Humanpapillomavirus | Nonavalent HPV VLP (V503), no adjuvant specified | Intramuscular | III | NCT00943722/NCT01984697 | Merck Sharp & Dohme Corp. |
| Completed (2016) | Hepatitis E virus | HEV 239 | Hepatitis E virus | Monovalent HEV VLP (HEV 239), phosphate-buffered saline, aluminum hydroxide, thiomersal | Intramuscular | IV | NCT02417597 | Xiamen Innovax Biotech Co., Ltd |
| Completed (2016) | Influenza A virus | H7, N9 | Influenza A virus | Monovalent H7N9 influenza VLP with Matrix-M1TM adjuvant | Intramuscular | II | NCT02078674 | Novavax |
| Completed (2016) | Norwalk virus | VP1 Capsid Protein | Norwalk virus | Norwalk VLP in dry power | Intranasal | II | NCT00973284 | Takeda |
VLP can be bulk expressed in bioreactor cultures, utilizing advanced in vitro protein expression systems for producing vaccine-grade VLP suitable for clinical administration. The manufacture of some VLP also includes additional processing steps, such as disassembly and reassembly of VLP. The manufacture of the HPV VLP utilizes disassembly and reassembly to improve VLP morphology and stability [9]. Similarly, the manufacture of Qβ VLP includes this method [10]. Different VLP expression systems and the applications of VLP disassembly and reassembly was explored in a recent review [1]. Compatibility with commercial upscaling technology, GMP production, and with minimal post-production manipulation or modification supports large-scale use of VLP vaccines; however, some VLP vaccines struggle in their translation from laboratory research and development, to clinical trials and routine public access [11–13]. Open accessibility to the molecular or genetic components of derivative constituents may place some limitations on the commercialization of specific VLP vaccines. This may be circumvented through tactical use of proprietary modification, or by utilizing the vaccine as a constituent within a composite formulation [14]. The development of some VLP vaccines can be challenged by issues with stability and longevity, which may be alleviated with formulation excipients, or other vaccine additives that facilitate vaccine distribution and storage [15,16]. The purpose of this review is to explore some of the challenges in the translation of VLP from the laboratory to the clinic, including the immune response to VLP vaccines, and an exploration of vaccine formulation techniques used to enhance the stability, immunogenicity, and efficacy of VLP vaccines.
1.1. VLP biodiversity
VLP possess a variety of shapes and structures, representative of the inherently vast diversity of the virus taxon. Examples of VLP can be identified within each of the seven groups defined by the Baltimore classification [17], including VLP derived from double-stranded DNA viruses such as Epstein-Barr virus [18], positive-sense RNA viruses such as Chikungunya virus [19], and negative-sense RNA viruses such as Human parainfluenza virus type 3 [20]. Variety can be observed in VLP size, ranging from MS2 bacteriophage VLP at around 27.5 nm in diameter [21], to HPV VLP at around 60 nm [22], and influenza VLP at around 100 nm [23,24]. VLP also vary in structural complexity, as illustrated in Figure 1, including mono-layer VLP such as HBV VLP formed from HBV surface antigen (HBsAg) or HBV core antigen (HBcAg) [25–27], and multi-layer VLP such as rotavirus VLP [28,29]. VLP can be encapsulated within a phospholipid bilayer envelope to resemble their parent virus, such as HIV [30] or Sendai virus VLP [31]. The envelope itself can also form the primary particle structure of some VLP, with recombinantly expressed virus envelope-stabilizing proteins embedded within the membrane, such as with IAV virosomes [32].
Figure 1.
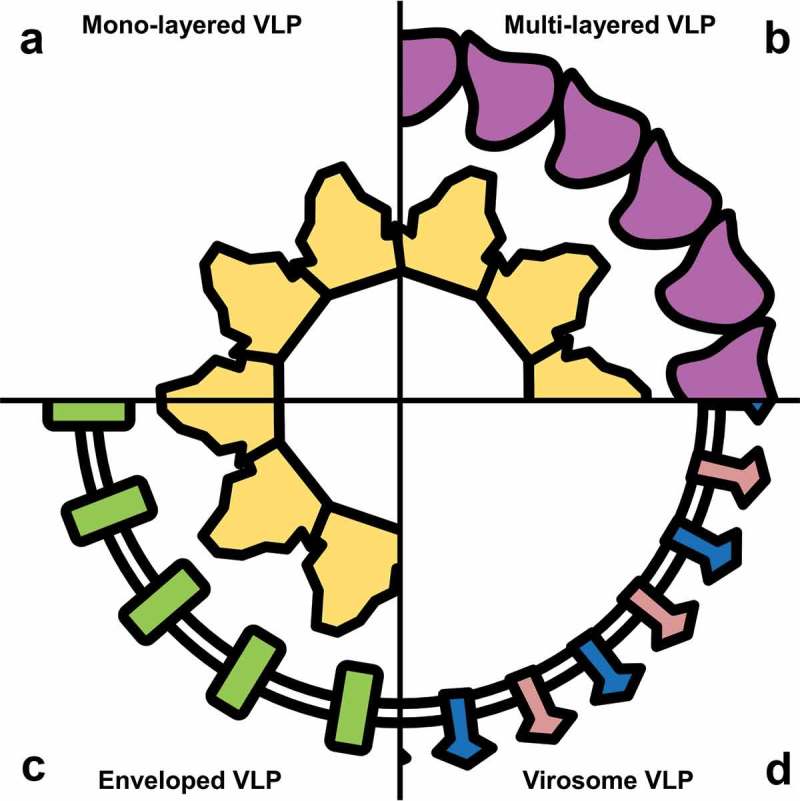
Structural biodiversity of VLP.
VLP can be produced with a variety of structural morphologies defined by the structure of their parent virus. These morphologies include: (a) mono-layered VLP, usually consisting of a single virus capsid protein; (b) multi-layered VLP, formed from multiple concurrently expressed capsid proteins; (c) enveloped VLP, with a lipid bilayer formed over the VLP capsid; and (d) virosomes, consisting of proteins embedded within a lipid bilayer envelope.
Some VLP are compatible with the formation of polyvalent or mosaic VLP, derived from multiple virus strains [33]. While this increases the diversity of VLP vaccines beyond the variety of parent viruses, the increased complexity of polyvalent or mosaic VLP may require post-production manipulation to facilitate stable particle formation [34,35]. In addition to facilitating the development of complex VLP vaccine constructs, introduction of postproduction manipulation has also been demonstrated to improve the consistency and stability of some standard structure VLP vaccines [9,36]. The diversity of VLP can also be characterized based on the variety of diseases these vaccines have been developed to prevent or treat. These include VLP vaccines developed for both human and veterinary pathologies, with some examples including an HBV HBcAg core particle-based vaccine for HER2+ cancer [37], an adenovirus VLP-based vaccine for placental malaria [38], and a Qβ VLP-based vaccine for Leishmania infection [39]. A selection of recently developed and investigated novel VLP vaccines are provided in Table 2.
Table 2.
Novel VLP vaccines.
| Disease | VLP Vector | Antigen | Expression System | Model | Formulation | Route | Reference |
|---|---|---|---|---|---|---|---|
| Japanese encephalitis | Japanese encephalitis virus | JEV E protein | Bm-N cells/Silkworm | Mouse, Rabbit | JEV-NVLP with complete or incomplete Freund’s adjuvant | NS | Matsuda et al 2017 |
| Zika fever | Zika virus | Zika E protein | Expi293 cells | Mouse | Zika VLP 1:1 with AddaVax (InvivoGen, CA) | Intramuscular | Boigard et al 2017 |
| _M. rosenbergii w_hite tail disease | Macrobrachiumrosenbergii nodavirus | MrNV capsid protein | Baculovirus/E. coli | ND | ND | ND | Kueh et al 2017 |
| Leishmania infection | Qβ VLP | α-Gal trisaccharide | E. coli | Mouse | Qβ VLP chemically conjugated with Galα(1,3)Galβ(1,4)GlcNAcβ | Subcutaneous | Moura et al 2017 |
| Placental malaria | Adenovirus | ID1-ID2a domain of VAR2CSA | Baculovirus | Mouse | ID1-ID2a VLP in phosphate buffered saline, no adjuvant specified | Intramuscular | Andersson et al 2017 |
| Crimean-Congo hemorrhagic fever | Crimean-Congo hemorrhagic fever virus | CCHFV nucleoprotein and glycoprotein | HuH-7 cells | Mouse | CCHFV VLP in phosphate buffered saline, no adjuvant specified | Intraperitoneal | Hinkula et al 2017 |
| Porcine parvovirus | Porcine parvovirus | PPV VP2 protein | E. coli | Mouse, Guinea pig | PPV VLP in phosphate buffered saline with complete Freund’s adjuvant or Montanide ISA71TM VG | Subcutaneous or intramuscular | Pengchao et al 2017 |
| HER2-positive cancer | Hepatitis B virus | HER2 receptor | E. coli | Mouse | HER2-HBV VLP in phosphate buffered saline with BM MatrigelTM | Intraperitoneal | Suffian et al 2017 |
| Middle East respiratory syndrome | MERS coronavirus | Coronavirus proteins | Baculovirus | Rhesus macaque | MERS-CoV VLP with alum | Intramuscular | Wang et al 2016 |
| Porcine reproductive and respiratory syndrome | PRRS virus | GP5-4–3-2s-M, GP5-M | Baculovirus | Pig | PRRS VLP coated with PLGA | Intranasal | Binjawadagi et al 2016 |
| Equua caballus papillomavirus | Equua caballus papillomavirus | L1 capsid protein | Baculovirus | Rabbit, Mouse | EcPV2-L1 VLP | NS | Schellenbache et al 2015 |
| Ebola virus | Ebola virus and Sudan virus | Glycoprotein,nucleoprotein and VP40 matrix protein | Baculovirus | Rhesus macaque | EBOV and SUDV VLP with QS-21 | Intramuscular | Warfield et al 2015 |
| Red-spotted grouperNervous necrosis | Nervous necrosis virus | RGNNV capsid protein | Yeast | Convict grouper | NNV VLP | Intraperitoneal and oral | Wi et al 2015 |
1.2. Modification of VLP
VLP can be produced using a variety of expression systems, usually involving VLP assembly by spontaneous polymerization upon the expression of virus protein constituents. VLP can be expressed in cells derived from bacteria, yeast, insects and mammals, in cell-free expression systems, and in live organisms such as silkworm pupae and various plants [40–42]. Selection of an appropriate expression system is important, as each expression system can differ in their efficacy for expressing specific VLP, their post-translational modification (e.g. phosphorylation, glycosylation), and their potential for contamination with biologically compatible zoonotic viruses. Protein expression for the production of VLP is highly amenable to modification, such as substituting strain-specific amino acid sequences [43], or inserting foreign peptide sequences or proteins [44,45]. Manipulation of this process can facilitate the engineering of recombinant chimeric VLP containing xenogeneic antigens, inducing an immune response against targets other than the parent virus [46]. VLP can also present self-antigens in an immunogenic context, which can be particularly useful in immunotherapeutic vaccination for cancer [46]. A diverse range of applications for this type of modification have been investigated prophylactically and therapeutically for various conditions, including protection against infection from other organisms [47], auto-immune inflammatory disease [48], and as an antitumor immunotherapy [46,49,50]. Chimeric VLP have been investigated as a potential therapy for conditions not usually associated with vaccination, such as atherosclerosis [51], type II diabetes mellitus [52], and nicotine addiction [53]. Some of these VLP vaccines utilize chemical conjugation for incorporation of antigenic sequences as opposed to recombinant insertion. VLP can serve as an effective immunogenic scaffold for chemical conjugation, compatible with a broad range of conjugation chemistries. The applications of chemical conjugation with VLP has been comprehensively reviewed in recent articles [1,54–56]; however, it is worth noting that introducing unique forms of chemical conjugation may provide some proprietary claim over methods involving VLP vaccines already in the public domain.
2. The immune response to VLP vaccines
VLP are a form of subunit vaccine, inducing a characteristic immune response shared amongst exogenous antigens. While VLP are taken up by cells through non-specific pathways such as phagocytosis and macropinocytosis, some VLP can also utilize specialized uptake pathways inherited from their parent virus. In general, protein-based subunit vaccines like VLP are internalized into antigen-presenting cells (APCs), digested within the phagolysosome, and the resulting antigen peptides are presented loaded on major histocompatibility complex (MHC) II molecules to CD4+ T helper cells (TH cells). TH cells recognize epitopes presented on MHC-II through their specific T cell receptor (TCR), and activate dependent on the strength of this interaction, in addition to the degree of signaling through costimulatory receptors and cytokines. These additional signals are usually supplied by the APC, which is activated to increase the presentation of costimulatory receptors and the release of cytokines in response to a variety of stimulatory signals. These stimulatory signals correspond with the activation of pattern recognition receptors (PRRs), such as toll-like receptors (TLRs), nod-like receptors (NLRs), and RIG1-like receptors (RLRs), which each recognize specific pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). As VLP vaccines usually contain minimal or negligible amounts of these stimulatory molecules, these signals are instead induced by vaccine adjuvants.
2.1. Cellular targeting of VLP
Manipulation and selective targeting of specific APCs or other target cell populations may be an effective means of developing a novel proprietary VLP vaccine. Chemical conjugation of receptor ligands onto the surface of compatible VLP should promote binding and uptake into cells expressing the corresponding receptor, while primary VLP uptake and processing pathways should continue unperturbed. This type of modification may be particular effective when targeting a specific APC population is desired for the induction of a specific type of immune response. For example, chemical conjugation of mannoside-based saccharides on the surface of Rabbit hemorrhagic disease virus (RHDV) VLP selectively targets the mannose receptor expressed on the surface of APCs, inducing increased uptake and alteration of antigen cross-presentation in murine dendritic cells [57]. Chemical conjugation of unmethylated CpG oligonucleotides on the surface of RHDV VLP has also been investigated for targeting uptake through the DEC205 receptor [58]. Similarly, coating of HIV VLP with phosphatidylserine-laced liposomes enhanced uptake into macrophages by mimicking apoptotic bodies [59].
2.2. VLP internalization and processing
Although the immune response to VLP may be generalized due to similarities shared with other subunit vaccines, there are some unique components of a VLP-induced immune response. The pathways for uptake of VLP into APCs vary depending on the size and morphology of each VLP, including the potential retention of receptor binding motifs. Particles smaller than 20 nm, such as norovirus P particles [60], can drain directly from the vaccination site into the lymph through pores in the fenestrated lymphatic vessels [61]. Trafficking of molecules within the lymphatics is likewise size dependent, with soluble proteins smaller than 70 kDa or 5 nm transported by specialized small antigen conduits [62,63]. VLP are larger than 5 nm, and many are instead transported passively to the lymph nodes bound on the surface of myeloid immune cells [64–66]. VLP larger than 200 nm cannot drain directly through the fenestration pores into the lymph, and instead require active transport following internalization by APCs at the vaccination site, such as in dendritic cells (DCs), macrophages and B cells [67].
Uptake of virus antigens into antigen presenting cells can occur through multiple pathways, including phagocytosis, macropinocytosis, caveolae-mediated uptake, clathrin-mediated uptake and clathrin noncaveolae-mediated uptake [68]. VLP have been identified to utilize a similar repertoire of uptake pathways, including phagocytosis [69], size-dependent macropinocytosis [70], and clathrin-dependent or independent forms of receptor-mediated endocytosis [71–73]. The retention of receptor-mediated endocytosis indicates that some VLP can inherit functional receptor binding domains, and are compatible with the corresponding uptake pathways derived from their parent virus. These mechanisms of receptor-mediated uptake can also be introduced to VLP through post-expression modification, such as the chemical conjugation of superantigen on HBV VLP for internalization through MHC-II molecules [74], or the chemical conjugation of transferrin on Qβ VLP for internalization through the transferrin receptor, which is upregulated in some cancer cells [75].
Non-specific uptake into APCs subjects VLP to standard components of exogenous antigen processing pathways, including lysosomal fusion, enzymatic digestion, MHC-II loading and antigen presentation (Figure 2(a)). Parent viruses of VLP internalized through these pathways engage additional mechanisms to progress toward virus replication, including capsid disassembly or uncoating, exportation of the virus genomic material, and disruption of vesicular fusion. While the retention of intracellular processing mechanisms resembling those utilized by parent viruses has been investigated for some VLP [76,77], it remains unclear whether VLP universally mimic the intracellular processing of their parent virus. VLP can retain access to cross-presentation pathways, facilitating presentation of antigens on MHC Class I (MHC-I) for induction of a CD8+ T cell-mediated cytotoxic immune response. There are several known mechanisms of cross-presentation in APCs, including antigen escape from the early endosome, fusion of the endosome with the endoplastic reticulum, and recycling of MHC-I receptors from the cell surface. RHDV VLP are known to utilize the MHC-I receptor recycling pathway of cross-presentation [70] (Figure 2(b)). Additional examples of VLP known to induce cross-presentation of antigens include VLP derived from HBV [78,79], Hepatitis C virus (HCV) [80], HPV [73], papaya mosaic virus [81], and parvovirus [82]. The ability to induce cross-presentation is important when a cell-mediated cytotoxic immune response is the primary desired outcome of a VLP vaccine.
Figure 2.
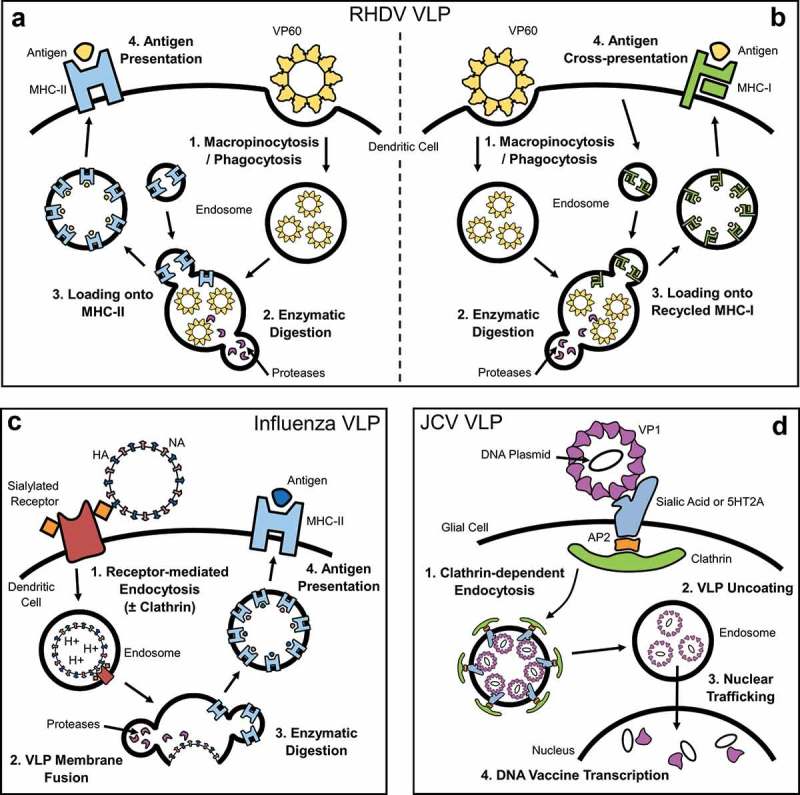
Intracellular processing of VLP.
VLP can be internalized and processed through a variety of intracellular pathways. (a) VLP internalized through non-specific pathways such as phagocytosis and macropinocytosis can be processed, with peptides presented on MHC-II as exogenous antigen. (b) Some VLP can also utilize cross-presentation pathways, facilitating the presentation of peptides on MHC-I [70]. (c) Influenza VLP, in this example an influenza virosome, is internalized through receptor-mediated endocytosis prior to fusion of its envelope with the endosomal membrane [90,92,93]. (d) JCV VLP is thought to utilize the processing pathway of its parent virus to facilitate delivery of exogenous nucleic acids, including clathrin-dependent endocytosis, nuclear trafficking, and uncoating of the capsid within the nucleus [99,100].
The retention of receptor-mediated internalization in VLP illustrates that these mechanisms of uptake in their parent virus are autonomous, independent of the integrity of the complete virion. VLP derived from influenza A or B viruses are prime examples of this process. Influenza VLP can be produced in several forms, including enveloped VLP formed by expressing the influenza matrix 1 (M1) protein [24], retroviral Gag proteins [83], or as independent virosome particles without a traditional protein core [84,85]. Each of these constructs includes the expression of influenza hemagglutinin (HA) with or without neuraminidase (NA). These influenza proteins are crucial for the induction of an influenza-specific immune response, and they also play essential roles the budding of influenza virus-like particles [23,86,87], and in receptor-mediated endocytosis. Influenza VLP can bind to sialylated glycoproteins and glycolipids on the surface of cells from the inherent activity of HA, and are internalized through clathrin-mediated endocytosis [88,89] (Figure 2(c)); however, alternative endocytic pathways have also been identified [90–93]. As the early endosomal pH lowers, the envelope fuses with the endosome membrane. HA and NA proteins distributed along the internal surface of the endosome membrane are enzymatically degraded, with peptides presented as exogenous antigens on MHC-II (Figure 2(c)).
Specialized VLP vaccines designed to deliver a genetic payload add further complications to VLP uptake and processing. Retention of the ability to deliver DNA/RNA requires conservation of intracellular processing pathways utilized by the parent virus for replication, or the induction of an alternative method of nucleic acid translocation. John Cunningham virus (JCV) VLP is a polyomavirus VLP that has received significant attention over its ability to facilitate gene delivery and genetic manipulation [77,94,95]. JCV is internalized by clathrin-dependent endocytosis upon binding to an undefined plethora of receptors and molecules known to include the serotonin receptor and sialic acid [96,97]. The virus then colocalizes to endosomes along with transferin, prior to cytosolic translocation, and subsequent trafficking through nuclear pores, mediated by the N-terminus of the VP1 capsid protein [98]. The virus then uncoats, exposing the genomic material within the capsid. JCV VLP are thought to mimic this process due to their ability to deliver DNA plasmids or other nucleic acids to the nucleus [99,100] (Figure 2(d)). VLP that demonstrate this ability to deliver nucleic acids likely utilize similar processing pathways to their parent virus [101,102], but VLP can also be modified to utilize alternative processing pathways [103,104].
2.3. Humoral immune response
The primary desired outcome of most commercial VLP vaccines is the production of antibodies specific for the VLP parent virus. Anti-VLP antibodies produced by these vaccines correspondingly neutralize the parent virus, protecting against infection. The production of anti-VLP antibodies involves the activation of a humoral immune response. B cells that specifically recognize VLP surface domains through their B cell receptor (BCR) bind and internalize VLP. Binding of VLP to the BCR provides a stimulatory signal that primes the B cell for activation. Most VLP consist of structurally identical capsid proteins arranged in a repetitive quasicrystalline pattern, which can crosslink multiple BCRs to provide a stimulatory advantage over other types of subunit vaccines. Crosslinking of BCRs is important for inducing a robust humoral response, as B cells can ignore some monomeric soluble antigens [105]. Potent stimulation through BCR crosslinking can override inherent tolerogenic mechanisms, and can activate unresponsive or anergic B cells [106]. Highly repetitive structures also promote binding to low-affinity BCRs through multivalent, or high-avidity interactions [107].
Antigen primed B cells receive additional activation signaling induced by PAMPs delivered with the VLP vaccine. These signals are usually provided by vaccine adjuvants, but may also be induced by other virus-associated or expression-derived endogenous molecules associated with the VLP. VLP-derived antigens are presented to TH cells loaded on MHC-II, inducing the cytokine and costimulatory receptor signaling from the TH cell that continues B cell activation. Activated B cells initially become plasmablasts, a transient extrafollicular activation state that can provide some immediate antibody production [108]. Plasmablasts migrate to the follicular region of lymph nodes to form germinal centers, a specialized region where activated B cells proliferate. Plasmablasts within germinal centers under affinity maturation and immunoglobulin class switching, guided by signaling from T follicular helper (TFH) cells [109,110]. This specialized TFH cell guidance results in the development of high-affinity antibody-producing plasma cells, and long-lived memory B cells. Rapid T cell-independent activation can also be induced in marginal zone B cells and B1 cells, providing an alternative pathway for activation and antibody production over the predominant follicular B cell population [111]. Some VLP have been identified utilizing this alternative form of B cell activation [112].
The production of anti-VLP antibodies has been investigated and characterized amongst a broad range of VLP. This process can be influenced by various vaccine components, constituents, and even unexpected molecules, such as those derived from the VLP expression system. For example, the presence of bacterial RNA packed inside Qβ VLP plays a role in the induction of IgG2a/c antibodies in mice, and IgG1 antibodies in humans [113,114]. Qβ VLP can also induce the production of IgA class antibodies in immunocompetent mice when immunized through the intranasal route [115], while TH cell-deficient mice required subcutaneous rather than intranasal vaccination to induce robust IgA production through T-cell independent B cell activation [114]. TLR-7, simulated by single-stranded RNA in endosomes, was also identified as crucial for the induction of both IgG2c and IgA antibodies against Qβ VLP in mice. This further indicates the importance of endogenous bacterial RNA present in these VLP. The induction of anti-HPV VLP antibodies has also been extensively studied. Although the production of secretory IgA antibodies is desired for protection against viral infection at mucosal surfaces, the predominant antibody class found in secretions from the female genital tract following vaccination with HPV VLP is IgG [116–118]. While the presence of IgG in these secretions may be due to transudation from the serum, some active transportation or local production in the mucosa may be possible [119,120]. Vaccination with HPV VLP induces the production of both IgG and IgA antibodies in the serum and cervical secretions of humans [121]. While serum titers of anti-HPV antibodies may initially decline post-vaccination, these levels plateau and remain stable to provide prolonged immunity [122].
2.4. Cell-mediated immune response
While the induction of a potent humoral immune response and the subsequent production of anti-VLP antibodies is the primary desired outcome of most commercial VLP vaccines, these is increasing appreciation for the role of vaccine-induced cell-mediated immunity [123–125]. Measurements of anti-VLP titers can provide an important indication of vaccine efficacy with respect to the neutralization of a virus challenge, a cell-mediated immune response also plays an important role in antivirus immune defense [126,127]. Activation of a cell-mediated immune response can also be the primary desired outcome of VLP vaccines, particularly for chimeric or other modified VLP vaccines that target non-virus pathologies. A potent cell-mediated response to VLP vaccines is dependent upon cross-presentation of VLP-derived antigens on MHC-I. CD8+ T cytotoxic (TC) cells can recognize through MHC-I antigen complexes through their TCR, and become primed for activation. These antigen-primed TC cells require additional signaling for activation, including interaction with costimulatory receptors, and cytokines from TH cells. While many VLP may be capable of cross-presentation, some may be more effective at inducing cross-presentation due to uptake and processing pathways inherited from their parent virus [49,128].
The induction of a potent cell-mediated immune response is particularly important for immunotherapeutic cancer vaccines. Tumor cell-specific antibodies can enhance the inherent cytotoxic activity of natural killer cells through mechanisms such as antibody-dependent cell-mediated cytotoxicity, or directly through complement-dependent cytotoxicity; however these mechanisms tend to be restricted to passive vaccination with monoclonal antibodies, such as the HER2-specific monoclonal trastuzumab [129]. A cell-mediated immune response combines target specificity with the in vivo expansion of effector cell populations, with the capacity to establish prolonged memory. RHDV VLP are particularly effective as a vector for cancer vaccines, capable of inducing the cross-presentation of VLP-derived antigens [70], and compatible with recombinant insertion and chemical conjugation [49]. RHDV VLP vaccines have been investigated in models of HPV-infected cervical cancer [47], melanoma [49], Lewis’ lung carcinoma [130], and colorectal cancer [46]. Additional examples of VLP that can induce cross-presentation and a potent cell-mediated immune response include Qβ VLP [128], MS2 VLP [131], and alphavirus VLP [132].
2.5. Preexisting immunity
Preexisting immunity in the form of preformed anti-VLP antibodies can be an important consideration for some VLP vaccines [133,134]. Binding of anti-VLP antibodies may interfere with the normal uptake and presentation pathways of VLP vaccines. Anti-VLP antibodies may be present in unvaccinated individuals due to environmental exposure to prevalent strains of the parent virus, or induced following VLP vaccination. While the presence of preexisting antibodies may decrease the efficacy or alter the immune recognition of some VLP vaccines [135,136], they can also be associated with measures of enhanced vaccine efficacy [133]. This may be due to accessing Fc receptor-mediated uptake pathways, the formation of antibody-VLP complexes, or antigenic boosting of specific B cells. The presence of anti-VLP antibodies have also been found to have no observable effect on the intended vaccine outcome for several VLP vaccines, such as polyomavirus [137], and RHDV VLP [138].
When anti-VLP antibodies present deleterious interference, alternating between different VLP vaccine vectors may be a suitable solution. Alternating between rotavirus and adenovirus VLP has been found to enhance both the humoral and cell-mediated immune responses against target antigens in comparison to repeat delivery with the same VLP vector [139]. Similarly, alternating between closely related VLP such as RHDV and human norovirus (HuNV) VLP can be sufficient to enhance vaccine immunogenicity [140]. Further complication can arise when VLP vaccines induce a phenomenon referred to as carrier-induced epitopic suppression (CIES), in which the intended immune response is outcompeted by a strong anti-VLP response [135]. CIES may be prevented by masking VLP surface antigens, or by avoiding recognition through recombinant modification. For example, recombinant insertion of the p18 domain of HIV into HEV VLP was identified to avoid recognition by anti-HEV antibodies [141].
3. Formulation
The formulation of a vaccine refers to the constituents that make up the final administrable solution, including the vaccine vector, adjuvants, and excipients. For VLP vaccines, these excipients can include a variety of salts and compounds prepared as an aqueous solution or emulsion, which maintains the physical stability of VLP for enhanced shelf life of the vaccine. Appropriate use of buffers can limit fluctuations in pH, while protection from fluctuations in temperature and desiccation may be provided by thermoprotectants and lyoprotectants, respectively. Formulation science involves investigating the various components of a vaccine under different environmental conditions, with the intention of formulating a stable vaccine product suitable for the route of administration, and with maximized immunogenicity. The combination of formulation components, VLP vaccine preparation states and routes of administration is outlined in Figure 3. The majority of VLP vaccines currently on the market and under clinical evaluation are liquid suspensions, ready for administration. This places strict limitations on VLP vaccine storage and distribution for safe and compliant administration. For example, the commercial HPV vaccine Gardasil (Merck) must be refrigerated at 2–8°C, and protected from light. Gardasil cannot be frozen, and guidelines for the use of Gardasil advise that the vaccine must be used within 72 h when removed from refrigeration at temperatures below 25°C, or when stored at 0–2°C. These guidelines mirror those of many other commercial VLP vaccines, and outlines the primary stability, storage and distribution challenges for formulation science to investigate.
Figure 3.
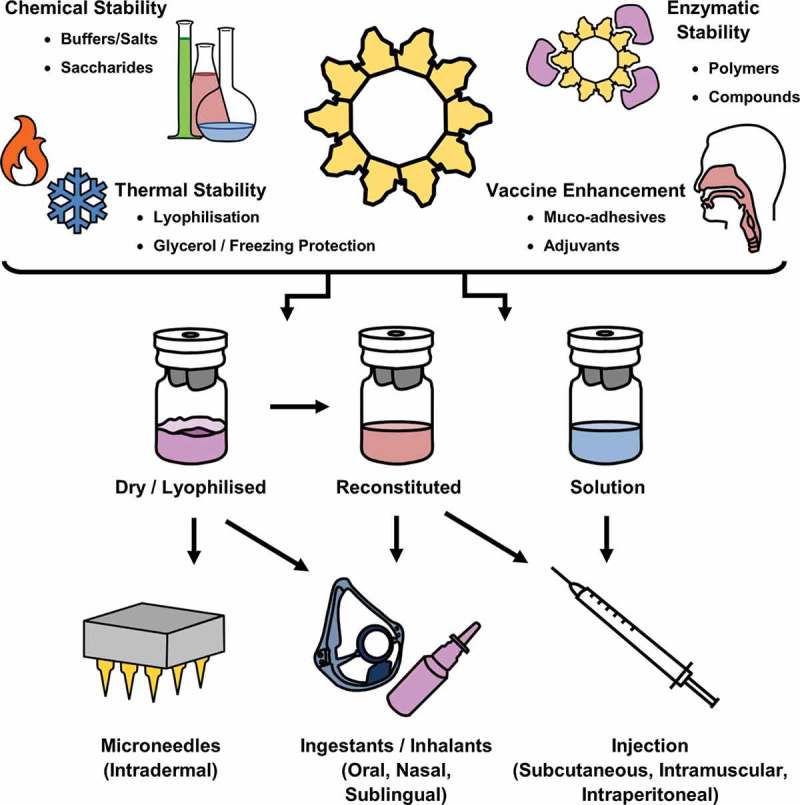
The Roles of Formulation Science in VLP Vaccines.
The role of formulation science in VLP vaccine manufacture includes the chemical composition of buffers, preservatives, additives and other stabilizing compounds for maintaining intact VLP. This includes protecting VLP from chemical or physical instability, and enzymatic degradation. Formulations can also include targeted delivery compounds, such as muco-adhesives, and immunogenic components such as adjuvants. Storage and distribution of VLP vaccines, and the subsequent route of administration are also important considerations in formulation science, critical in determining the efficacy and immunogenicity of the vaccine.
3.1. Excipients
Maintaining the integrity and stability of VLP in solution is largely dependent on the combination of salts, and the buffering chemicals and compounds used. Optimization of buffer pH, ionic strength, and other stabilizing components is imperative to developing a marketable, stable, liquid VLP vaccine [142]. An investigation into the stability of EV71 VLP identified that sodium phosphate-based buffers were superior to citrate or Tris buffered solutions, with VLP stored in sodium phosphate buffer remaining stable for 1 month at both 25 and 37°C [142]. Such prolonged stability at room and core body temperature suggests that this VLP may be suitable for importation into countries where maintaining cold-chain storage and distribution can be difficult. Other important considerations for the selection of an appropriate buffer include compatibility with downstream applications, such as chemical conjugation, and the availability of scavengable nutrients that may promote the growth of potentially harmful organisms in an improperly stored vaccine. Various additive molecules, particularly carbohydrates such as trehalose, sucrose and glycerol, have been investigated extensively in VLP vaccine formulations. The addition of these molecules to vaccine formulations demonstrated enhancement in VLP stability within a liquid suspension of Norwalk virus VLP [143] and rotavirus VLP [144]. Other than sugar-based formulation additives, polyanionic solutions can also stabilize VLP that would otherwise be unstable at neutral pH, such as Chikungunya virus VLP [145].
The majority of commercial VLP vaccines are distributed as a liquid suspension, requiring a cold chain maintaining 4–8°C throughout distribution and storage. Even under stable cold chain conditions, the longevity of VLP can be limited. This can be prolonged by storage at temperatures at or below −20°C, stabilized with the addition of a cryopreservative such as glycerol or trehalose [146]; however, this only further exacerbates the issue of delivering these vaccines intact where they are needed, such as in developing countries without reliable cold-chain delivery infrastructure. Alternative storage methods such as freeze-drying, or lyophilization, can have variable effects on the stability of VLP. Mechanical damage induced by ice crystallization, or exposure to varying salt concentrations and pH changes during the freezing process may adversely affect the integrity of VLP [147]. Exposure to these factors can be limited through the addition of specific cryoprotectants and lyoprotectants. For example, red-spotted grouper nervous necrosis virus (RGNNV) VLP [148] reportedly retain stable particles and remain immunogenic when freeze-dried in the presence of sorbitol, but were adversely affected in the presence of mannitol. Qβ VLP [147], Murine polyomavirus (MuPyV) VLP [149,150], and HBV VLP [151] have likewise demonstrated some capacity to survive varying freeze-drying methodologies.
Spray-drying has also gained traction as an alternative vaccine storage mechanism, avoiding the potential mechanical damage induced by freeze-drying by instead forming a dry powder formulation through a combination of nebulization and dehumidification [152]; however, spray-drying involves exposure to elevated temperatures, which can be similarly damaging to thermolabile VLP. HPV VLP suspended in a formulation containing mannitol, trehalose, dextran, L-leucine and inositol are capable of surviving this procedure [153–155]. Spray-dried MS2-16L2 VLP stored at room temperature for 34 months were found to induce high titer anti-HPV L2 IgG antibodies, and significantly protected vaccinated mice from challenge with HPV16 [155]. Air or vacuum drying methods have also been reported for coating microneedles with influenza VLP suspended in a formulation containing carboxymethylcellulose (CMC). Microneedle-based delivery of dried H1N1 (A/PR8) [156] and H3N2 (A/Aichi) [157] were found to induce immune responses comparable to intramuscular injection, but with the advantage of prolonged longevity in storage. Investigation of similar formulation and preparation strategies with different types of VLP may uncover some universality in their application, with the potential for these methods to eradicate the cold-chain limitation imposed on many VLP vaccines.
3.2. Adjuvants
As has been previously described, many VLP possess structural or molecular features that can confer some auto-immunostimulatory properties. These properties facilitate the induction of immune responses by VLP without the need for adjuvants; however, the use of adjuvants with VLP vaccines may enhance vaccine immunogenicity, and promote the activation of a specific type of immune response. Currently licensed adjuvants for use with vaccines include aluminum sulfate salts (Alum), proprietary combination adjuvants such as AS03 (GlaxoSmithKline), AS04 (GlaxoSmithKline) and MF59 (Novartis), thermo-reversible oil-in-water immersions, and Montanide ISA51 [158]. While many standard vaccine adjuvants may be suitable for VLP vaccines where the generation of anti-VLP antibodies is the desired outcome, these adjuvants may fail to overcome immunotolerance and induce antitumor immunity in cancer vaccine formulations [159]; however, where traditional adjuvants may fail, novel TLR agonist adjuvants have had success. The TLR7 agonist adjuvant imiquimod was recently approved for use with vaccines for melanoma, and another clinical trial is assessing its candidacy for treatment of bladder cancer [159]. TLR7/8 stimulate anti-viral interferon pathways in APCs, promoting the activation of CD8 + T cells and NK cells [160,161]. The use of the adjuvant gardiquimod in combination with vaccination has been found to induce tumor regression in murine models of melanoma [162], human hepatocellular carcinoma [163] and pancreatic cancer [164].
TLR9 agonist adjuvants such as unmethylated CpG oligonucleotides have drawn considerable interest due to their inherent ability to associate with some VLP [138,165]. Similarly, the presence of other nucleic acids can have downstream ramifications on the immune response induced by VLP. For example, the presence of RNA inside Qβ VLP can skew the humoral response induced in mice to produce IgG2a antibodies, while the removal of this RNA results in the production of IgG1 antibodies [166]. Qβ VLP can also contain additional adjuvant molecules derived from their expression system, encapsidated during particle formation. A recent study in mice investigated the use of various adjuvants in conjunction with a filovirus VLP vaccine, including the TLR3 agonist adjuvant poly-ICLC (Hiltonol), TLR4 agonist adjuvant monophospholipid A (MPLA), CpG ODN2395 and alhydrogel [167]. Poly-ICLC was found to induce a predominantly TH1 response, with an IgG2c subtype antibody production that correlated with protection from virus challenge. In comparison, administration of the VLP vaccine with alhydrogel elicited a strong TH2 response with high antibody titers, but conferred no protection from virus challenge. This study highlighted the importance of carefully considering what kind of adjuvant is suitable to accompany a VLP vaccine, as the most immunogenic adjuvant may not necessarily provide the desired vaccine outcome.
3.3. Routes of administration
VLP vaccines approved for clinical use have utilized various administration modalities, including vaccination subcutaneously, intradermally, intramuscularly or at mucosal surfaces [168]. Live attenuated vaccines are traditionally injected subcutaneously, while inactivated or subunit vaccines are often administered intramuscularly. In infants, intramuscular injection of vaccines tends to confer enhanced immunogenicity over subcutaneous administration [169]. Modern vaccine delivery technologies, such as intradermal microneedle patches, have also been investigated for their potential applications with VLP vaccines [156,157,170]. While the majority of recent clinical trials involving the administration of VLP favor intramuscular injection, alternative routes of administration are being investigated based on differential immunogenicity at different delivery sites. For example, an investigation into the routes of administration of HuNV VLP found that intranasal inoculation of mice induced the production of mucosal anti-HuNV IgG and IgA antibodies, while intramuscular injection only induced IgG production [171]. The study identified that only mucosal IgA was suitable for neutralization of infectious HuNV virions, rendering intramuscular injection unsuitable for administration of HuNV VLP. A similar finding was identified regarding the administration of respiratory syncytial virus (RSV) VLP, with the desired TH1 response, neutralizing mucosal antibodies and CD8+ T cell responses identified following intranasal inoculation, rather than intramuscular injection [172]. Vaccination with VLP at mucosal surfaces also requires higher doses of VLP in comparison to parental administration.
Oral delivery of virus-like particles is also being investigated as an alternative, convenient route of administration. VLP expressed in plant-based systems represents a particularly efficient means of both production and delivery, with expression in an edible plant species potentially forgoing the need for VLP purification and vaccine formulation. Examples of this concept include the expression of HBV and Norwalk virus VLP in Solanum tuberosum potato and Lycopersicon esculentum tomato plants [173,174]. Nicotiana benthamiana tobacco are also used as an efficient plant expression system for production of VLP. HBV VLP purified from N. benthamiana are capable of withstanding a simulation of the acidic environment within the stomach [175]; however, the HBcAg polypeptide of HBV VLP was digested when exposed to porcine pepsin. The protection of protein-based VLP from enzymatic digestion is a significant hurdle for oral delivery, despite the promise of this vaccination route.
4. Conclusion
The translation of VLP vaccines from preclinical research to routine clinical administration is a multifaceted task combining studies into stability, formulation science, immunogenicity, and clinical vaccine efficacy. These issues are not necessarily unique to the development of VLP vaccines, encompassing the breadth of research and development necessary for bringing any vaccine candidate to market. The expression of stable VLP is only the first step toward the development of a novel VLP vaccine, which may eventually include various excipients, adjuvants and other compounds in the administrable form. Advancements in virology and vaccinology that have facilitated the development of novel VLP vaccines seemingly parallel regulatory and proprietary restrictions placed upon new vaccines; however, VLP vaccines also possess unparalleled potential due to their balance between clinical vaccine efficacy and safety, and their versatility as a vaccine vector.
5. Expert commentary
The field of VLP vaccines has continued to experience significant growth over the past decade, both in the diversity of vaccines and in the translation of vaccines toward routine clinical administration. Our research focuses upon the development of RHDV VLP as a versatile vaccine scaffold, particularly for immunotherapeutic vaccination as an alternative treatment option for cancer. The translation of VLP vaccine constructs from proof of concept models toward clinical administration is no menial feat. We have observed a corresponding shift toward focusing on clinical translation amongst our colleagues in the field. Focal points of discussion highlighted by the process include the relevance and translatability of research animal models, adaptation to the established dogma within the field of human vaccination, and the establishment of a developmental and commercial niche conducive to the advancement of novel vaccines throughout the rigors of clinical trials and regulatory approval.
The immune response to VLP vaccines tends to strike a desirable balance between outcomes indicative of optimal vaccine efficacy, such as the induction of high-titer mucosal IgA antibodies, while also maintaining an almost unparalleled vaccine safety profile. The inherent inability of VLP to infect or replicate alleviates potential vaccine risks, such as spontaneous reversion to pathogenesis, or incomplete inactivation. Retention of the ability to facilitate cross-presentation of associated antigens, and the induction of a potent cell-mediated immune response can also have significant implications for VLP vaccines, diversifying the field to cover a broader range of disorders and diseases. The intracellular processing pathways remain largely unexplored for many VLP, and elucidation of the underlying mechanisms involved in some of the more advanced applications of VLP vaccines, such as gene delivery and immunomodulation.
The role of formulation science appears underrepresented amongst clinical trials and clinical reports for VLP vaccines, with at times minimal elucidation of the vaccine administration route, the excipients included, and even exclusion of adjuvant delivered with the vaccine. The importance of formulation science and the relevance of excipients and adjuvants may be expectedly underappreciated during earlier phases of research and development, where establishment of vaccine efficacy may be paramount; however, early adoption of these vital constituents may ease clinical translation, potentially expanding the repertoire of marketable VLP vaccines. Novelty, proprietary design, and versatility are almost as important in the development of VLP vaccines as the actual vaccine efficacy itself. Maintaining compatibility with the upscaling of production and GMP manufacture pipelines is another important component in the development of a VLP vaccine for routine clinical administration. The benefit of such foresight is clear, and the promotion of clinical translation will be advantageous across the field of VLP vaccines.
6. Five-year view
Upon review across the field including current trends in new and emerging research, the progression of established vaccine research, and clinical trial schedules, VLP vaccine research is experiencing a period of rapid growth. This includes both the repertoire of organisms and conditions being targeted with VLP vaccines, and the advancement of established vaccines through clinical trials, translation, and clinical applications. Over the next five years, we predict that there will be a significant increase in the diversity of VLP vaccines in active circulation. We expect that VLP will be used in more complex applications than as a vaccine against the cognate virus from which they were derived. We also predict the development of novel VLP vaccine constructs for human and zoonotic pathogenic organisms and conditions with or without current vaccination options, and the incorporation of advanced vaccination methodologies and techniques into routine VLP vaccine production, distribution and administration. In particular, we predict increased incorporation of alternative vaccine-storage techniques such as freeze-dried or spray-dried methods. The development of VLP vaccines is often focused on facilitating distribution amongst populations and communities where these vaccines are most needed. We predict that over the next five years, the capability to support VLP vaccine distribution will increase by limiting the current reliance on cold-chain delivery. These predictions outline an exciting future over the next five years for VLP vaccine research and development.
Key issues
- Virus-like particles consist of virus-derived proteins and associated molecules that spontaneously form a particulate structure.
- VLP vaccines have the safety profile of a subunit vaccine, but with efficacy and vaccination outcomes that can be comparable to killed or live attenuated vaccines, depending on the particular VLP vaccine.
- While the predominant desired immune response amongst the majority of VLP vaccines is a potent humoral response producing high titer antibodies, some VLP vaccines can induce a potent cytotoxic immune response for selective elimination of cells currently infected by the target virus, or target tumor cells.
- Formulation science plays a major role in the development of VLP vaccines, facilitating the selection of appropriate combinations of excipients and adjuvants. Excipients are used to increase VLP particle stability in the vaccine formulation, while adjuvants enhance vaccine efficacy, and help to select for a specific desired immune response and outcome.
Funding Statement
This research was funded by the University of Otago.
Declaration of interest
B Donaldson was supported by funding from the University of Otago, Freemasonry New Zealand and the Prostate Cancer, Foundation of New Zealand. The rest of the authors are supported by funding from the University of Otago. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Al-Barwani F, Donaldson B, Pelham SJ, et al. Antigen delivery by virus-like particles for immunotherapeutic vaccination. Ther Deliv. 2014. November;5(11):1223–1240. PubMed PMID: 25491672. [DOI] [PubMed] [Google Scholar]; • This review covers the structural diversity of VLP, post-production modification, and the immune response to VLP vaccines with a specific focus on immunotherapeutic vaccine development.
- 2.Lachance P, Dionne M, Libman M, et al, Medicago, Inc; Immunogenicity of a quadrivalent virus-like particles (VLP) influenza vaccine in healthy adults. NCT02768805. 2016. [Google Scholar]
- 3.Sheldon E, Seiden DJ, Medicago, Inc; Immunogenicity, safety and tolerability of a plant-derived seasonal virus-like-particle quadrivalent influenza vaccine in adults. NCT02233816. 2016. [Google Scholar]
- 4.Acevedo-Flores M, Diaz C, Hoen B, et al, National Institute of Allergy and Infectious Diseases (NIAID) Trial for safety and immunogenicity of a chikungunya vaccine, VRC-CHKVLP059-00-VP, in healthy adults. NCT02562482. 2016. [Google Scholar]
- 5.Langley J, VBI Vaccines Inc; Study to evaluate safety, tolerability, and immunogenicity of candidate human cytomegalovirus vaccine in healthy adults. NCT02826798. 2016. [Google Scholar]
- 6.Takeda Efficacy and immunogenicity of norovirus GI.1/GII.4 bivalent virus-like particle vaccine in adults. NCT02669121. Takeda; 2017. [Google Scholar]
- 7.Takeda Long-term immunogenicity of the norovirus GI.I/GII.4 bivalent virus-like particle (VLP) vaccine in adults. NCT03039790. Takeda; 2017. [Google Scholar]
- 8.Takeda Safety and immunogenicity of norovirus GI.1/GII.4 bivalent virus-like particle vaccine in an elderly population. NCT02661490. Takeda; 2017. [Google Scholar]
- 9.Zhao Q, Allen MJ, Wang Y, et al. Disassembly and reassembly improves morphology and thermal stability of human papillomavirus type 16 virus-like particles. Nanomedicine. 2012. October;8(7):1182–1189. PubMed PMID: 22306156. [DOI] [PubMed] [Google Scholar]; • This research article investigates the effects of disassembly and reassembly on the morphological consistency and thermal stability of human papillomavirus 16 VLP. Disassembly and reassembly is including as a post-production modification in the manufacture of select commercial VLP vaccines.
- 10.Bachmann MF, Proba KG, Maurer P, et al, inventors; Cytos Biotechnology AG, assignee; VLP-antigen conjugates and their uses as vaccines. 2011. [Google Scholar]
- 11.Fischer R, Schillberg S, Hellwig S, et al. GMP issues for recombinant plant-derived pharmaceutical proteins. Biotechnol Adv. 2012;30(2):434–439. [DOI] [PubMed] [Google Scholar]
- 12.Franco E, Schiffman M, Wacholder S, et al Chapter 3. Methodological issues for trials of vaccine efficacy against HPV types 16 and 18. In: IARC HPV Working Group. Primary End-points for Prophylactic HPV Vaccine Trials. Lyon, France: International Agency for Research on Cancer; 2014. [PubMed] [Google Scholar]
- 13.Schiller JT, Lowy DR.. Raising expectations for subunit vaccine. J Infect Dis. 2014;211(9):1373–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevens H, Van Overwalle G, Van Looy B, et al. Intellectual property policies in early-phase research in public-private partnerships. Nat Biotechnol. 2016. May 06;34(5):504–510. PubMed PMID: 27153280. [DOI] [PubMed] [Google Scholar]
- 15.Kumru OS, Joshi SB, Smith DE, et al. Vaccine instability in the cold chain: mechanisms, analysis and formulation strategies. Biologicals: Journal Int Assoc Biol Standardization. 2014;42(5):237–259. [DOI] [PubMed] [Google Scholar]
- 16.Tonnis W, Amorij J-P, Vreeman M, et al. Improved storage stability and immunogenicity of hepatitis B vaccine after spray-freeze drying in presence of sugars. Eur J Pharm Sci. 2014;55:36–45. [DOI] [PubMed] [Google Scholar]
- 17.Baltimore D. Expression of animal virus genomes. Bacteriol Rev. 1971. September;35(3):235–241. PubMed PMID: 4329869; PubMed Central PMCID: PMCPMC378387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez EM, Foley J, Tison T, et al. Novel Epstein-Barr virus-like particles incorporating gH/gL-EBNA1 or gB-LMP2 induce high neutralizing antibody titers and EBV-specific T-cell responses in immunized mice. Oncotarget. 2017. March 21;8(12):19255–19273. PubMed PMID: 27926486; PubMed Central PMCID: PMCPMC5386682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saraswat S, Athmaram TN, Parida M, et al. Expression and characterization of yeast derived chikungunya virus like particles (CHIK-VLPs) and its evaluation as a potential vaccine candidate. PLoS Negl Trop Dis. 2016. July;10(7):e0004782 PubMed PMID: 27399001; PubMed Central PMCID: PMCPMC4939942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bracken MK, Hayes BC, Kandel SR, et al. Viral protein requirements for assembly and release of human parainfluenza virus type 3 virus-like particles. J Gen Virol. 2016. June;97(6):1305–1310. PubMed PMID: 26960133; PubMed Central PMCID: PMCPMC5042090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashley CE, Carnes EC, Phillips GK, et al. Cell-specific delivery of diverse cargos by bacteriophage MS2 virus-like particles. ACS Nano. 2011;5(7):5729–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen XS, Garcea RL, Goldberg I, et al. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Mol Cell. 2000. March;5(3):557–567. PubMed PMID: 10882140. [DOI] [PubMed] [Google Scholar]
- 23.Chen BJ, Leser GP, Morita E, et al. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J Virol. 2007. July;81(13):7111–7123. PubMed PMID: 17475660; PubMed Central PMCID: PMCPMC1933269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pushko P, Tumpey TM, Bu F, et al. Influenza virus-like particles comprised of the HA, NA, and M1 proteins of H9N2 influenza virus induce protective immune responses in BALB/c mice. Vaccine. 2005. December 30;23(50):5751–5759. PubMed PMID: 16143432. [DOI] [PubMed] [Google Scholar]
- 25.Almeida FMF, Blanco A, Trujillo H, et al. Dynamic of immune response induced in hepatitis B surface antigen-transgenic mice immunized with a novel therapeutic formulation. Euroasian J Hepatogastroenterol. 2016. Jan-Jun;6(1):25–30. PubMed PMID: 29201720; PubMed Central PMCID: PMCPMC5578554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keating GM, Noble S. Recombinant hepatitis B vaccine (Engerix-B): a review of its immunogenicity and protective efficacy against hepatitis B [Review]. Drugs. 2003;63(10):1021–1051. PubMed PMID: 12699402; eng. [DOI] [PubMed] [Google Scholar]
- 27.Lobaina Y, Palenzuela D, Pichardo D, et al. Immunological characterization of two hepatitis B core antigen variants and their immunoenhancing effect on co-delivered hepatitis B surface antigen. Mol Immunol. 2005. February;42(3):289–294. PubMed PMID: 15589316. [DOI] [PubMed] [Google Scholar]
- 28.Crawford SE, Labbe M, Cohen J, et al. Characterization of virus-like particles produced by the expression of rotavirus capsid proteins in insect cells. J Virol. 1994. September;68(9):5945–5952. PubMed PMID: 8057471; PubMed Central PMCID: PMC237000. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sabara M, Parker M, Aha P, et al. Assembly of double-shelled rotaviruslike particles by simultaneous expression of recombinant VP6 and VP7 proteins. J Virol. 1991. December;65(12):6994–6997. PubMed PMID: 1658389; PubMed Central PMCID: PMC250814. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haglund K, Forman J, Krausslich HG, et al. Expression of human immunodeficiency virus type 1 Gag protein precursor and envelope proteins from a vesicular stomatitis virus recombinant: high-level production of virus-like particles containing HIV envelope. Virology. 2000. March 01;268(1):112–121. PubMed PMID: 10683333. [DOI] [PubMed] [Google Scholar]
- 31.Sugahara F, Uchiyama T, Watanabe H, et al. Paramyxovirus sendai virus-like particle formation by expression of multiple viral proteins and acceleration of its release by C protein. Virology. 2004;325(1):1–10. [DOI] [PubMed] [Google Scholar]
- 32.Wiedermann U, Wiltschke C, Jasinska J, et al. A virosomal formulated Her-2/neu multi-peptide vaccine induces Her-2/neu-specific immune responses in patients with metastatic breast cancer: a phase I study. Breast Cancer Res Treat. 2010. February;119(3):673–683. PubMed PMID: 20092022; eng. [DOI] [PubMed] [Google Scholar]
- 33.Pushko P, Pearce MB, Ahmad A, et al. Influenza virus-like particle can accommodate multiple subtypes of hemagglutinin and protect from multiple influenza types and subtypes. Vaccine. 2011. August 11;29(35):5911–5918. PubMed PMID: 21723354. [DOI] [PubMed] [Google Scholar]
- 34.Cheng F, Mukhopadhyay S. Generating enveloped virus-like particles with in vitro assembled cores. Virology. 2011;413(2):153–160. [DOI] [PubMed] [Google Scholar]
- 35.Goicochea NL, De M, Rotello VM, et al. Core-like particles of an enveloped animal virus can self-assemble efficiently on artificial templates. Nano Lett. 2007;7(8):2281–2290. [DOI] [PubMed] [Google Scholar]
- 36.Sánchez-Rodríguez SP, Münch-Anguiano L, Echeverría O, et al. Human parvovirus B19 virus-like particles: in vitro assembly and stability. Biochimie. 2012;94(3):870–878. [DOI] [PubMed] [Google Scholar]
- 37.Suffian IFBM, Wang JT-W, Hodgins NO, et al. Engineering hepatitis B virus core particles for targeting HER2 receptors in vitro and in vivo. Biomaterials. 2017;120:126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andersson A-MC, Resende M, Salanti A, et al. Novel adenovirus encoded virus-like particles displaying the placental malaria associated VAR2CSA antigen. Vaccine. 2017;35(8):1140–1147. [DOI] [PubMed] [Google Scholar]
- 39.Moura APV, Santos LC, Brito CRN, et al. Virus-like particle display of the α-Gal carbohydrate for vaccination against Leishmania infection. ACS Cent Sci. 2017;3(9):1026–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marsian J, Lomonossoff GP. Molecular pharming—vLPs made in plants. Curr Opin Biotechnol. 2016;37:201–206. [DOI] [PubMed] [Google Scholar]
- 41.Nerome K, Sugita S, Kuroda K, et al. The large-scale production of an artificial influenza virus-like particle vaccine in silkworm pupae. Vaccine. 2015. January 01;33(1):117–125. PubMed PMID: 25448101. [DOI] [PubMed] [Google Scholar]
- 42.Rodríguez-Limas WA, Sekar K, Tyo KE. Virus-like particles: the future of microbial factories and cell-free systems as platforms for vaccine development. Curr Opin Biotechnol. 2013;24(6):1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Debbink K, Lindesmith LC, Donaldson EF, et al. Chimeric GII. 4 Norovirus Virus-Like-Particle-Based Vaccines Induce Broadly Blocking Immune Responses. J Virology. 2014;88(13):7256–7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noh J-Y, Park J-K, Lee D-H, et al. Chimeric bivalent virus-like particle vaccine for H5N1 HPAI and ND confers protection against a lethal challenge in chickens and allows a strategy of differentiating infected from vaccinated animals (DIVA). PloS one. 2016;11(9):e0162946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao H, Li H-Y, Han J-F, et al. Novel recombinant chimeric virus-like particle is immunogenic and protective against both enterovirus 71 and coxsackievirus A16 in mice Scientific reports. 2015;5:7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donaldson B, Al-Barwani F, Pelham SJ, et al. Multi-target chimaeric VLP as a therapeutic vaccine in a model of colorectal cancer. J Immunother Cancer. 2017. August 15;5(1):69 PubMed PMID: 28806910; PubMed Central PMCID: PMCPMC5556368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jemon K, Young V, Wilson M, et al. An enhanced heterologous virus-like particle for human papillomavirus type 16 tumour immunotherapy. PloS one. 2013;8(6):e66866 PubMed PMID: 23799135; PubMed Central PMCID: PMCPMC3682997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dallenbach K, Maurer P, Röhn T, et al. Protective effect of a germline, IL‐17‐neutralizing antibody in murine models of autoimmune inflammatory disease. Eur J Immunol. 2015;45(4):1238–1247. [DOI] [PubMed] [Google Scholar]
- 49.Peacey M, Wilson S, Perret R, et al. Virus-like particles from rabbit hemorrhagic disease virus can induce an anti-tumor response [Research Support, Non-U.S. Gov’t]. Vaccine. 2008. October 3;26(42):5334–5337. PubMed PMID: 18706958; eng. [DOI] [PubMed] [Google Scholar]
- 50.Speiser DE, Schwarz K, Baumgaertner P, et al. Memory and effector CD8 T-cell responses after nanoparticle vaccination of melanoma patients. J Immunotherapy. 2010;33(8):848–858. [DOI] [PubMed] [Google Scholar]
- 51.Tissot AC, Spohn G, Jennings GT, et al. A VLP‐based vaccine against interleukin‐1α protects mice from atherosclerosis. Eur J Immunol. 2013;43(3):716–722. [DOI] [PubMed] [Google Scholar]
- 52.Cavelti-Weder C, Timper K, Seelig E, et al. Development of an interleukin-1β vaccine in patients with type 2 diabetes. Mol Therapy. 2016;24(5):1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cornuz J, Zwahlen S, Jungi WF, et al. A vaccine against nicotine for smoking cessation: a randomized controlled trial. PloS one. 2008;3(6):e2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Donaldson B, Al-Barwani F, Young V, et al. Virus-like particles, a versatile subunit vaccine platform In: Foged C, Rades T, Perrie Y, et al, editors. Subunit Vaccine Delivery. New York: Springer; 2015. 159–180. [Google Scholar]
- 55.Lua LH, Connors NK, Sainsbury F, et al. Bioengineering virus‐like particles as vaccines. Biotechnol Bioeng. 2014;111(3):425–440. [DOI] [PubMed] [Google Scholar]
- 56.Zeltins A. Construction and characterization of virus-like particles: a review. Mol Biotechnol. 2013. January;53(1):92–107. PubMed PMID: 23001867; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This review covers the breadth of expression systems available for VLP vaccine production, while also discussing VLP structural diversity and characterization.
- 57.Al-Barwani F, Young SL, Baird MA, et al. Mannosylation of virus-like particles enhances internalization by antigen presenting cells. PloS one. 2014;9(8):e104523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pelham SJ. Coupling the adjuvant CpG oligonucleotides to RHDV VLP. Dunedin: University of Otago; 2014. [Google Scholar]
- 59.Gramatica A, Petazzi RA, Lehmann MJ, et al. αEnv-decorated phosphatidylserine liposomes trigger phagocytosis of HIV-virus-like particles in macrophages. Nanomedicine: Nanotechnology, Biol Med. 2014;10(5):e981–e989. [DOI] [PubMed] [Google Scholar]
- 60.Tan M, Jiang X. Norovirus P particle: a subviral nanoparticle for vaccine development against norovirus, rotavirus and influenza virus. Nanomedicine (Lond). 2012. June;7(6):889–897. PubMed PMID: 22734641; PubMed Central PMCID: PMCPMC3514417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manolova V, Flace A, Bauer M, et al. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008. May;38(5):1404–1413. PubMed PMID: 18389478. [DOI] [PubMed] [Google Scholar]
- 62.Roozendaal R, Mempel TR, Pitcher LA, et al. Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity. 2009. February 20;30(2):264–276. PubMed PMID: 19185517; PubMed Central PMCID: PMCPMC2699624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pape KA, Catron DM, Itano AA, et al. The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity. 2007. April;26(4):491–502. PubMed PMID: 17379546. [DOI] [PubMed] [Google Scholar]
- 64.Cubas R, Zhang S, Kwon S, et al. Virus-like particle (VLP) lymphatic trafficking and immune response generation after immunization by different routes. J Immunothe. 2009;32(2):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cinamon G, Zachariah MA, Lam OM, et al. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol. 2008. January;9(1):54–62. PubMed PMID: 18037889; PubMed Central PMCID: PMCPMC2488964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carrasco YR, Batista FD. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity. 2007. July;27(1):160–171. PubMed PMID: 17658276. [DOI] [PubMed] [Google Scholar]
- 67.Jegerlehner A, Tissot A, Lechner F, et al. A molecular assembly system that renders antigens of choice highly repetitive for induction of protective B cell responses. Vaccine. 2002. August 19;20(25–26):3104–3112. PubMed PMID: 12163261; eng. [DOI] [PubMed] [Google Scholar]
- 68.Sieczkarski SB, Whittaker GR. Dissecting virus entry via endocytosis. J Gen Virol. 2002. July;83(Pt 7):1535–1545. PubMed PMID: 12075072. [DOI] [PubMed] [Google Scholar]
- 69.Yan M, Peng J, Jabbar IA, et al. Despite differences between dendritic cells and Langerhans cells in the mechanism of papillomavirus-like particle antigen uptake, both cells cross-prime T cells. Virology. 2004. July 01;324(2):297–310. PubMed PMID: 15207617. [DOI] [PubMed] [Google Scholar]
- 70.Win SJ, Ward VK, Dunbar PR, et al. Cross-presentation of epitopes on virus-like particles via the MHC I receptor recycling pathway [Research Support, Non-U.S. Gov’t]. Immunol Cell Biol. 2011. August;89(6):681–688. PubMed PMID: 21221122; eng. [DOI] [PubMed] [Google Scholar]
- 71.Cureton DK, Massol RH, Whelan SP, et al. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLoS Pathog. 2010. September 30;6(9):e1001127 PubMed PMID: 20941355; PubMed Central PMCID: PMCPMC2947997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ewers H, Smith AE, Sbalzarini IF, et al. Single-particle tracking of murine polyoma virus-like particles on live cells and artificial membranes. Proc Natl Acad Sci U S A. 2005. October 18;102(42):15110–15115. PubMed PMID: 16219700; PubMed Central PMCID: PMCPMC1257700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fausch SC, Da Silva DM, Kast WM. Differential uptake and cross-presentation of human papillomavirus virus-like particles by dendritic cells and Langerhans cells. Cancer Res. 2003. July 01;63(13):3478–3482. PubMed PMID: 12839929. [PubMed] [Google Scholar]
- 74.McIntosh JD, Manning K, Chokshi S, et al. An engineered non-toxic superantigen increases cross presentation of hepatitis B virus nucleocapsids by human dendritic cells. PloS one. 2014;9(4):e93598 PubMed PMID: 24690680; PubMed Central PMCID: PMCPMC3972192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Banerjee D, Liu AP, Voss NR, et al. Multivalent display and receptor-mediated endocytosis of transferrin on virus-like particles. Chembiochem. 2010. June 14;11(9):1273–1279. PubMed PMID: 20455239; PubMed Central PMCID: PMCPMC4180096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mani B, Baltzer C, Valle N, et al. Low pH-dependent endosomal processing of the incoming parvovirus minute virus of mice virion leads to externalization of the VP1 N-terminal sequence (N-VP1), N-VP2 cleavage, and uncoating of the full-length genome. J Virol. 2006. January;80(2):1015–1024. PubMed PMID: 16379002; PubMed Central PMCID: PMCPMC1346861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen LS, Wang M, Ou WC, et al. Efficient gene transfer using the human JC virus-like particle that inhibits human colon adenocarcinoma growth in a nude mouse model. Gene Ther. 2010. August;17(8):1033–1041. PubMed PMID: 20410928. [DOI] [PubMed] [Google Scholar]
- 78.Ruedl C, Storni T, Lechner F, et al. Cross‐presentation of virus‐like particles by skin‐derived CD8–dendritic cells: a dispensable role for TAP. Eur J Immunol. 2002;32(3):818–825. [DOI] [PubMed] [Google Scholar]
- 79.Moffat JM, Cheong W-S, Villadangos JA, et al. Hepatitis B virus-like particles access major histocompatibility class I and II antigen presentation pathways in primary dendritic cells. Vaccine. 2013;31(18):2310–2316. [DOI] [PubMed] [Google Scholar]
- 80.Barth H, Ulsenheimer A, Pape GR, et al. Uptake and presentation of hepatitis C virus–like particles by human dendritic cells. Blood. 2005;105(9):3605–3614. [DOI] [PubMed] [Google Scholar]
- 81.Leclerc D, Beauseigle D, Denis J, et al. Proteasome-independent major histocompatibility complex class I cross-presentation mediated by papaya mosaic virus-like particles leads to expansion of specific human T cells. J Virol. 2007;81(3):1319–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morón VG, Rueda P, Sedlik C, et al. In vivo, dendritic cells can cross-present virus-like particles using an endosome-to-cytosol pathway. J Immunol. 2003;171(5):2242–2250. [DOI] [PubMed] [Google Scholar]
- 83.Haynes JR, Dokken L, Wiley JA, et al Influenza-pseudotyped Gag virus-like particle vaccines provide broad protection against highly pathogenic avian influenza challenge. Vaccine. 2009;27(4):530–541. [DOI] [PubMed] [Google Scholar]
- 84.Herzog C, Hartmann K, Künzi V, et al. Eleven years of Inflexal® V—a virosomal adjuvanted influenza vaccine. Vaccine. 2009;27(33):4381–4387. [DOI] [PubMed] [Google Scholar]
- 85.Mischler R, Metcalfe IC. Inflexal® V a trivalent virosome subunit influenza vaccine: production. Vaccine. 2002;20:B17–B23. [DOI] [PubMed] [Google Scholar]
- 86.Leser GP, Lamb RA. Influenza virus assembly and budding in raft-derived microdomains: a quantitative analysis of the surface distribution of HA, NA and M2 proteins. Virology. 2005;342(2):215–227. [DOI] [PubMed] [Google Scholar]
- 87.Rossman JS, Jing X, Leser GP, et al. Influenza virus m2 ion channel protein is necessary for filamentous virion formation. J Virol. 2010;84(10):5078–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Matlin KS, Reggio H, Helenius A, et al. Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol. 1981;91(3):601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Patterson S, Oxford J, Dourmashkin R. Studies on the mechanism of influenza virus entry into cells. J Gen Virol. 1979;43(1):223–229. [DOI] [PubMed] [Google Scholar]
- 90.Chen C, Zhuang X. Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc Natl Acad Sci. 2008;105(33):11790–11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Vries E, Tscherne DM, Wienholts MJ, et al. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 2011;7(3):e1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lakadamyali M, Rust MJ, Zhuang X. Endocytosis of influenza viruses. Microb Infect. 2004;6(10):929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sieczkarski SB, Whittaker GR. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol. 2002;76(20):10455–10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chao C-N, Lin M-C, Fang C-Y, et al. Gene therapy for human lung adenocarcinoma using a suicide gene driven by a lung-specific promoter delivered by JC virus-like particles. PloS one. 2016;11(6):e0157865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hoffmann DB, Böker KO, Schneider S, et al. In vivo siRNA delivery using JC virus-like particles decreases the expression of RANKL in rats. Mol Ther Nucleic Acids. 2016;5:e298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Elphick GF, Querbes W, Jordan JA, et al. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306(5700):1380–1383. [DOI] [PubMed] [Google Scholar]
- 97.Pho M, Ashok A, Atwood WJ. JC virus enters human glial cells by clathrin-dependent receptor-mediated endocytosis. J Virol. 2000;74(5):2288–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ashok A, Atwood WJ. Contrasting roles of endosomal pH and the cytoskeleton in infection of human glial cells by JC virus and simian virus 40. J Virol. 2003;77(2):1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.An K, Gillock ET, Sweat JA, et al. Use of the baculovirus system to assemble polyomavirus capsid-like particles with different polyomavirus structural proteins: analysis of the recombinant assembled capsid-like particles. J Gen Virol. 1999;80(4):1009–1016. [DOI] [PubMed] [Google Scholar]
- 100.Chang C-F, Wang M, Ou W-C, et al. Human JC virus-like particles as a gene delivery vector. Expert Opin Biol Ther. 2011;11(9):1169–1175. [DOI] [PubMed] [Google Scholar]
- 101.Jariyapong P, Chotwiwatthanakun C, Somrit M, et al. Encapsulation and delivery of plasmid DNA by virus-like nanoparticles engineered from Macrobrachium rosenbergii nodavirus. Virus Res. 2014;179:140–146. [DOI] [PubMed] [Google Scholar]
- 102.Shao W, Paul A, Abbasi S, et al. A novel polyethyleneimine-coated adeno-associated virus-like particle formulation for efficient siRNA delivery in breast cancer therapy: preparation and in vitro analysis. Int J Nanomedicine. 2012;7:1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.El Mehdaoui S, Touzé A, Laurent S, et al. Gene transfer using recombinant rabbit hemorrhagic disease virus capsids with genetically modified DNA encapsidation capacity by addition of packaging sequences from the L1 or L2 protein of human papillomavirus type 16. J Virol. 2000;74(22):10332–10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang G, Jia T, Xu X, et al. Novel miR-122 delivery system based on MS2 virus like particle surface displaying cell-penetrating peptide TAT for hepatocellular carcinoma. Oncotarget. 2016;7(37):59402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bachmann MF, Zinkernagel RM. Neutralizing antiviral B cell responses. Annu Rev Immunol. 1997;15(1):235–270. [DOI] [PubMed] [Google Scholar]
- 106.Chackerian B, Durfee MR, Schiller JT. Virus-like display of a neo-self antigen reverses B cell anergy in a B cell receptor transgenic mouse model [Research Support, N.I.H., Extramural]. J Immunology. 2008. May 1;180(9):5816–5825. PubMed PMID: 18424700; PubMed Central PMCID: PMC3493123. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bessa J, Zabel F, Link A, et al. Low-affinity B cells transport viral particles from the lung to the spleen to initiate antibody responses. Proc Natl Acad Sci. 2012;109(50):20566–20571. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This research article investigates the transportation of VLP following intranasal delivery, mediated by low-affinity B cells. Intranasal vaccination and the induction of mucosal IgA anti-VLP production is an important vaccination outcome for VLP vaccines against viruses that infect through mucosal surfaces.
- 108.MacLennan I, Toellner KM, Cunningham AF, et al. Extrafollicular antibody responses. Immunol Rev. 2003;194(1):8–18. [DOI] [PubMed] [Google Scholar]
- 109.Nutt SL, Tarlinton DM. Germinal center B and follicular helper T cells: siblings, cousins or just good friends [quest]. Nat Immunol. 2011;12(6):472–477. [DOI] [PubMed] [Google Scholar]
- 110.Radbruch A, Muehlinghaus G, Luger EO, et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;6(10):741–750. [DOI] [PubMed] [Google Scholar]
- 111.Fairfax KA, Kallies A, Nutt SL, et al, editors. Plasma cell development: from B-cell subsets to long-term survival niches. Seminars in immunology; 2008; Elsevier. [DOI] [PubMed] [Google Scholar]
- 112.Yang R, Murillo FM, Delannoy MJ, et al. B lymphocyte activation by human papillomavirus-like particles directly induces Ig class switch recombination via TLR4-MyD88. J Immunol. 2005;174(12):7912–7919. [DOI] [PubMed] [Google Scholar]
- 113.Bessa J, Bachmann MF. T cell-dependent and-independent IgA responses: role of TLR signalling. Immunol Invest. 2010;39(4–5):407–428. [DOI] [PubMed] [Google Scholar]
- 114.Bessa J, Jegerlehner A, Hinton HJ, et al. Alveolar macrophages and lung dendritic cells sense RNA and drive mucosal IgA responses. J Immunol. 2009;183(6):3788–3799. [DOI] [PubMed] [Google Scholar]
- 115.Bessa J, Schmitz N, Hinton HJ, et al. Efficient induction of mucosal and systemic immune responses by virus‐like particles administered intranasally: implications for vaccine design. Eur J Immunol. 2008;38(1):114–126. [DOI] [PubMed] [Google Scholar]
- 116.Brandtzaeg P. Mucosal immunity in the female genital tract. J Reprod Immunol. 1997;36(1):23–50. [DOI] [PubMed] [Google Scholar]
- 117.Mestecky J, Kutteh W, Jackson S. Mucosal immunity in the female genital tract: relevance to vaccination efforts against the human immunodeficiency virus. AIDS Res Hum Retroviruses. 1994;10:S11–20. [PubMed] [Google Scholar]
- 118.Parr E, Parr M. A comparison of antibody titres in mouse uterine fluid after immunization by several routes, and the effect of the uterus on antibody titres in vaginal fluid. J Reprod Fertil. 1990;89(2):619–625. [DOI] [PubMed] [Google Scholar]
- 119.Hocini H, Barra A, Belec L, et al. Systemic and secretory humoral immunity in the normal human vaginal tract. Scand J Immunol. 1995;42(2):269–274. [DOI] [PubMed] [Google Scholar]
- 120.Kozlowski PA, Cu-Uvin S, Neutra MR, et al. Comparison of the oral, rectal, and vaginal immunization routes for induction of antibodies in rectal and genital tract secretions of women. Infect Immun. 1997;65(4):1387–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nardelli-Haefliger D, Wirthner D, Schiller JT, et al. Specific antibody levels at the cervix during the menstrual cycle of women vaccinated with human papillomavirus 16 virus–like particles. J Natl Cancer Inst. 2003;95(15):1128–1137. [DOI] [PubMed] [Google Scholar]
- 122.Olsson SE, Villa LL, Costa RL, et al. Induction of immune memory following administration of a prophylactic quadrivalent human papillomavirus (HPV) types 6/11/16/18 L1 virus-like particle (VLP) vaccine. Vaccine. 2007. June 21;25(26):4931–4939. PubMed PMID: 17499406. [DOI] [PubMed] [Google Scholar]
- 123.Hillaire ML, Osterhaus AD, Rimmelzwaan GF. Induction of virus-specific cytotoxic T lymphocytes as a basis for the development of broadly protective influenza vaccines. J Biomed Biotechnol. 2011;2011:939860 PubMed PMID: 22007149; PubMed Central PMCID: PMCPMC3189652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schotsaert M, Saelens X, Leroux-Roels G. Influenza vaccines: T-cell responses deserve more attention. Expert Rev Vaccines. 2012. August;11(8):949–962. PubMed PMID: 23002976. [DOI] [PubMed] [Google Scholar]; •• This review article discusses the importance of inducing a potent cytotoxic immune response for vaccination against select virus infections.
- 125.Sommerfelt MA. T-cell-mediated and humoral approaches to universal influenza vaccines. Expert Rev Vaccines. 2011. October;10(10):1359–1361. PubMed PMID: 21988298. [DOI] [PubMed] [Google Scholar]
- 126.McElhaney JE, Dutz JP. Better influenza vaccines for older people: what will it take? J Infect Dis. 2008. September 01;198(5):632–634. PubMed PMID: 18652548. [DOI] [PubMed] [Google Scholar]
- 127.Ohmit SE, Petrie JG, Cross RT, et al. Influenza hemagglutination-inhibition antibody titer as a correlate of vaccine-induced protection. J Infect Dis. 2011. December 15;204(12):1879–1885. PubMed PMID: 21998477. [DOI] [PubMed] [Google Scholar]
- 128.Schwarz K, Meijerink E, Speiser DE, et al. Efficient homologous prime‐boost strategies for T cell vaccination based on virus‐like particles. Eur J Immunol. 2005;35(3):816–821. [DOI] [PubMed] [Google Scholar]
- 129.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001. March 15;344(11):783–792. PubMed PMID: 11248153. [DOI] [PubMed] [Google Scholar]
- 130.Li K, Peers-Adams A, Win SJ, et al. Antigen incorporated in virus-like particles is delivered to specific dendritic cell subsets that induce an effective antitumor immune response in vivo [Research Support, Non-U.S. Gov’t]. J Immunotherapy. 2013. January;36(1):11–19. PubMed PMID: 23211625; eng. [DOI] [PubMed] [Google Scholar]
- 131.Li J, Sun Y, Jia T, et al. Messenger RNA vaccine based on recombinant MS2 virus‐like particles against prostate cancer. Int J Cancer. 2014;134(7):1683–1694. [DOI] [PubMed] [Google Scholar]
- 132.Riabov V, Tretyakova I, Alexander RB, et al. Anti-tumor effect of the alphavirus-based virus-like particle vector expressing prostate-specific antigen in a HLA-DR transgenic mouse model of prostate cancer. Vaccine. 2015. October 5;33(41):5386–5395. PubMed PMID: 26319744; PubMed Central PMCID: PMCPMC4581984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.De Filette M, Martens W, Smet A, et al. Universal influenza A M2e-HBc vaccine protects against disease even in the presence of pre-existing anti-HBc antibodies. Vaccine. 2008. December 2;26(51):6503–6507. PubMed PMID: 18835315. [DOI] [PubMed] [Google Scholar]
- 134.Gedvilaite A, Zvirbliene A, Staniulis J, et al. Segments of puumala hantavirus nucleocapsid protein inserted into chimeric polyomavirus-derived virus-like particles induce a strong immune response in mice. Viral Immunol. 2004;17(1):51–68. PubMed PMID: 15018662. [DOI] [PubMed] [Google Scholar]
- 135.Jegerlehner A, Wiesel M, Dietmeier K, et al. Carrier induced epitopic suppression of antibody responses induced by virus-like particles is a dynamic phenomenon caused by carrier-specific antibodies. Vaccine. 2010. July 26;28(33):5503–5512. PubMed PMID: 20307591. [DOI] [PubMed] [Google Scholar]
- 136.Link A, Zabel F, Schnetzler Y, et al. Innate immunity mediates follicular transport of particulate but not soluble protein antigen. J Immunology. 2012. April 15;188(8):3724–3733. PubMed PMID: 22427639. [DOI] [PubMed] [Google Scholar]; •• This research article investigates the transportation and delivery of Qβ VLP to follicular dendritic cells within the lymph node, and the impairment of this delivery in the presence of anti-Qβ VLP antibodies.
- 137.Chuan YP, Rivera-Hernandez T, Wibowo N, et al. Effects of pre-existing anti-carrier immunity and antigenic element multiplicity on efficacy of a modular virus-like particle vaccine. Biotechnol Bioeng. 2013. September;110(9):2343–2351. PubMed PMID: 23532896. [DOI] [PubMed] [Google Scholar]
- 138.Donaldson B. Immunomodulation and vaccination with RHDV VLP: a thesis submitted for the degree of Doctor of Philosophy, University of Otago, Dunedin, New Zealand. Dunedin: University of Otago; 2017. [Google Scholar]
- 139.Zhou H, Guo L, Wang M, et al. Prime immunization with rotavirus VLP 2/6 followed by boosting with an adenovirus expressing VP6 induces protective immunization against rotavirus in mice. Virol J. 2011;8(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.McClintock SL. Can different virus-like particles be used for prime-boost vaccination? Dunedin: University of Otago; 2011. [Google Scholar]
- 141.Jariyapong P, Xing L, van Houten NE, et al. Chimeric hepatitis E virus-like particle as a carrier for oral-delivery. Vaccine. 2013. January 2;31(2):417–424. PubMed PMID: 23107594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lin S-Y, Chung Y-C, Chiu H-Y, et al. Evaluation of the stability of enterovirus 71 virus-like particle. J Biosci Bioeng. 2014;117(3):366–371. [DOI] [PubMed] [Google Scholar]
- 143.Kissmann J, Ausar SF, Foubert TR, et al. Physical stabilization of Norwalk virus-like particles. J Pharm Sci. 2008. October;97(10):4208–4218. PubMed PMID: 18300304. [DOI] [PubMed] [Google Scholar]
- 144.Peixoto C, Sousa MF, Silva AC, et al. Downstream processing of triple layered rotavirus like particles. J Biotechnol. 2007. January 10;127(3):452–461. PubMed PMID: 16959354. [DOI] [PubMed] [Google Scholar]
- 145.Kramer RM, Zeng Y, Sahni N, et al. Development of a stable virus-like particle vaccine formulation against Chikungunya virus and investigation of the effects of polyanions. J Pharm Sci. 2013. December;102(12):4305–4314. PubMed PMID: 24129946; PubMed Central PMCID: PMCPMC3869236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lynch A, Meyers AE, Williamson AL, et al. Stability studies of HIV-1 Pr55gag virus-like particles made in insect cells after storage in various formulation media. Virol J. 2012. September 18;9:210 PubMed PMID: 22988963; PubMed Central PMCID: PMCPMC3502365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lang R, Winter G, Vogt L, et al. Rational design of a stable, freeze-dried virus-like particle-based vaccine formulation. Drug Dev Ind Pharm. 2009;35(1):83–97. [DOI] [PubMed] [Google Scholar]
- 148.Lan NT, Kim HJ, Han HJ, et al. Stability of virus-like particles of red-spotted grouper nervous necrosis virus in the aqueous state, and the vaccine potential of lyophilized particles. Biologicals: Journal Int Assoc Biol Standardization. 2018. January;51:25–31. PubMed PMID: 29174141. [DOI] [PubMed] [Google Scholar]
- 149.Seth A, Kong IG, Lee SH, et al. Modular virus-like particles for sublingual vaccination against group A streptococcus. Vaccine. 2016. December 12;34(51):6472–6480. PubMed PMID: 27866769. [DOI] [PubMed] [Google Scholar]
- 150.Mohr J, Chuan YP, Wu Y, et al. Virus-like particle formulation optimization by miniaturized high-throughput screening. Methods. 2013. May 1;60(3):248–256. PubMed PMID: 23639868. [DOI] [PubMed] [Google Scholar]
- 151.Czyz M, Dembczynski R, Marecik R, et al. Freeze-drying of plant tissue containing HBV surface antigen for the oral vaccine against hepatitis B. Biomed Res Int. 2014;2014:485689 PubMed PMID: 25371900; PubMed Central PMCID: PMCPMC4209752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kanojia G, Have RT, Soema PC, et al. Developments in the formulation and delivery of spray dried vaccines. Hum Vaccin Immunother. 2017. October 3;13(10):2364–2378. PubMed PMID: 28925794; PubMed Central PMCID: PMCPMC5647985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tumban E, Muttil P, Escobar CA, et al. Preclinical refinements of a broadly protective VLP-based HPV vaccine targeting the minor capsid protein, L2. Vaccine. 2015. June 26;33(29):3346–3353. PubMed PMID: 26003490; PubMed Central PMCID: PMCPMC4468037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Saboo S, Tumban E, Peabody J, et al. Optimized formulation of a thermostable spray-dried virus-like particle vaccine against human papillomavirus. Mol Pharm. 2016. May 2;13(5):1646–1655. PubMed PMID: 27019231; PubMed Central PMCID: PMCPMC4853272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Peabody J, Muttil P, Chackerian B, et al. Characterization of a spray-dried candidate HPV L2-VLP vaccine stored for multiple years at room temperature. Papillomavirus Res. 2017. June;3:116–120. PubMed PMID: 28720444; PubMed Central PMCID: PMCPMC5604873. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This research article demonstrates the compatibility of a recombinant MS2-16L2 VLP vaccine with spray-drying, and the retention of vaccine immunogenicity after prolonged storage at room temperature.
- 156.Zhu Q, Zarnitsyn VG, Ye L, et al. Immunization by vaccine-coated microneedle arrays protects against lethal influenza virus challenge. Proc Natl Acad Sci U S A. 2009. May 12;106(19):7968–7973. PubMed PMID: 19416832; PubMed Central PMCID: PMCPMC2683119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Koutsonanos DG, Del Pilar Martin M, Zarnitsyn VG, et al. Transdermal influenza immunization with vaccine-coated microneedle arrays. PloS one. 2009;4(3):e4773 PubMed PMID: 19274084; PubMed Central PMCID: PMCPMC2651574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pasquale AD, Preiss S, Silva FTD, et al. Vaccine adjuvants: from 1920 to 2015 and beyond. Vaccines. 2015;3(2):320–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Temizoz B, Kuroda E, Ishii KJ. Vaccine adjuvants as potential cancer immunotherapeutics. Int Immunol. 2016. July;28(7):329–338. PubMed PMID: 27006304; PubMed Central PMCID: PMCPMC4922024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gorski KS, Waller EL, Bjornton-Severson J, et al. Distinct indirect pathways govern human NK-cell activation by TLR-7 and TLR-8 agonists. Int Immunol. 2006. July;18(7):1115–1126. PubMed PMID: 16728430. [DOI] [PubMed] [Google Scholar]
- 161.Hart OM, Athie-Morales V, O’Connor GM, et al. TLR7/8-mediated activation of human NK cells results in accessory cell-dependent IFN-gamma production. J Immunology. 2005. August 1;175(3):1636–1642. PubMed PMID: 16034103. [DOI] [PubMed] [Google Scholar]
- 162.Ma F, Zhang J, Zhang J, et al. The TLR7 agonists imiquimod and gardiquimod improve DC-based immunotherapy for melanoma in mice. Cell Mol Immunol. 2010. September;7(5):381–388. PubMed PMID: 20543857; PubMed Central PMCID: PMCPMC4002679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhou Z, Yu X, Zhang J, et al. TLR7/8 agonists promote NK–DC cross-talk to enhance NK cell anti-tumor effects in hepatocellular carcinoma. Cancer Lett. 2015;369(2):298–306. [DOI] [PubMed] [Google Scholar]
- 164.Zou BB, Wang F, Li L, et al. Activation of Toll-like receptor 7 inhibits the proliferation and migration, and induces the apoptosis of pancreatic cancer cells. Mol Med Rep. 2015;12(4):6079–6085. [DOI] [PubMed] [Google Scholar]
- 165.Klimek L, Bachmann MF, Senti G, et al. Immunotherapy of type-1 allergies with virus-like particles and CpG-motifs. Expert Rev Clin Immunol. 2014;10(8):1059–1067. [DOI] [PubMed] [Google Scholar]
- 166.Tumban E, Peabody J, Peabody DS, et al. A universal virus-like particle-based vaccine for human papillomavirus: longevity of protection and role of endogenous and exogenous adjuvants. Vaccine. 2013. September 23;31(41):4647–4654. PubMed PMID: 23933337; PubMed Central PMCID: PMCPMC3785330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Martins KA, Cooper CL, Stronsky SM, et al. Adjuvant-enhanced CD4 T cell responses are critical to durable vaccine immunity. EBioMedicine. 2016;3:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Herzog C. Influence of parenteral administration routes and additional factors on vaccine safety and immunogenicity: a review of recent literature. Expert Rev Vaccines. 2014;13(3):399–415. [DOI] [PubMed] [Google Scholar]
- 169.Gillet Y, Steri G, Behre U, et al. Immunogenicity and safety of measles-mumps-rubella-varicella (MMRV) vaccine followed by one dose of varicella vaccine in children aged 15 months–2 years or 2–6 years primed with measles-mumps-rubella (MMR) vaccine. Vaccine. 2009;27(3):446–453. [DOI] [PubMed] [Google Scholar]
- 170.Quan F-S, Kim Y-C, Song J-M, et al. Long-term protective immunity from an influenza virus-like particle vaccine administered with a microneedle patch. Clin Vaccine Immunol. 2013;20(9):1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tamminen K, Malm M, Vesikari T, et al. Mucosal antibodies induced by intranasal but not intramuscular immunization block norovirus GII.4 virus-like particle receptor binding. Viral Immunol. 2016. June;29(5):315–319. PubMed PMID: 27135874. [DOI] [PubMed] [Google Scholar]
- 172.Jiao YY, Fu YH, Yan YF, et al. A single intranasal administration of virus-like particle vaccine induces an efficient protection for mice against human respiratory syncytial virus. Antiviral Res. 2017. August;144:57–69. PubMed PMID: 28529001. [DOI] [PubMed] [Google Scholar]
- 173.Huang Z, Elkin G, Maloney BJ, et al. Virus-like particle expression and assembly in plants: hepatitis B and Norwalk viruses. Vaccine. 2005;23(15):1851–1858. [DOI] [PubMed] [Google Scholar]
- 174.Mason HS, Ball JM, Shi -J-J, et al. Expression of Norwalk virus capsid protein in transgenic tobacco and potato and its oral immunogenicity in mice. Proc Natl Acad Sci. 1996;93(11):5335–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Berardi A, Lomonossoff GP, Evans DJ, et al. Plant-expressed Hepatitis B core antigen virus-like particles: characterization and investigation of their stability in simulated and pig gastro-intestinal fluids. Int J Pharm. 2017;522(1–2):147–156. [DOI] [PubMed] [Google Scholar]