Exploring the abundance, metabolic potential and gene expression of subseafloor Chloroflexi in million-year-old oxic and anoxic abyssal clay (original) (raw)
ABSTRACT
Chloroflexi are widespread in subsurface environments, and recent studies indicate that they represent a major fraction of the communities in subseafloor sediment. Here, we compare the abundance, diversity, metabolic potential and gene expression of Chloroflexi from three abyssal sediment cores from the western North Atlantic Gyre (water depth >5400 m) covering up to 15 million years of sediment deposition, where Chloroflexi were found to represent major components of the community at all sites. Chloroflexi communities die off in oxic red clay over 10–15 million years, and gene expression was below detection. In contrast, Chloroflexi abundance and gene expression at the anoxic abyssal clay site increase below the seafloor and peak in 2–3 million-year-old sediment, indicating a comparably higher activity. Metatranscriptomes from the anoxic site reveal increased expression of Chloroflexi genes involved in cell wall biogenesis, protein turnover, inorganic ion transport, defense mechanisms and prophages. Phylogenetic analysis shows that these Chloroflexi are closely related to homoacetogenic subseafloor clades and actively transcribe genes involved in sugar fermentations, gluconeogenesis and Wood–Ljungdahl pathway in the subseafloor. Concomitant expression of cell division genes indicates that these putative homoacetogenic Chloroflexi are actively growing in these million-year-old anoxic abyssal sediments.
Keywords: abyssal clay, Chloroflexi, Dehalococcoidia, SAR202 clade, western North Atlantic Gyre, homoacetogenesis, metagenomes, metatranscriptomes
Anaerobic Chloroflexi are transcriptionally active, grow and divide in anoxic abyssal clay that are millions of years old, whereas aerobic Chloroflexi die off in oxic abyssal red clay.
INTRODUCTION
Vast regions of the dark deep ocean have extremely slow sedimentation rates, ranging from 1 to 5 m of sediment deposition per million years in abyssal regions (D'Hondt et al. 2015) and most organic flux to the sediment is consumed before it can be buried (Røy et al. 2012). As a result, microbial communities in abyssal subseafloor sediment are characterized by very low cell densities (Kallmeyer et al. 2012) and experience extreme energy limitation over the long term (Hoehler and Jørgensen 2013), even over million year timescales under both oxic and anoxic conditions (D'Hondt et al. 2015).
Cells subsisting in abyssal clays may experience two types of energy limitation, limitation by electron donors and limitation by electron acceptors. In anoxic abyssal clay, high redox potential electron acceptors of oxygen and nitrate are consumed during organic matter oxidation at the sediment surface, and a main terminal electron acceptor is sulfate (Parkes et al. 2005; Jørgensen, Findlay and Pellerin 2019). In contrast, the cells subsisting in subseafloor oxic abyssal clay are constantly exposed to high redox potential electron acceptors of oxygen and nitrate throughout the entire sediment column (Røy et al. 2012), but are even more limited by the availability of electron donors in the form of organic matter (D'Hondt et al. 2015). This happens because the sedimentation rates are so slow in the oligotrophic ocean that what little organic matter makes it to the seafloor is exposed to oxic conditions for exceptionally long timescales before it is buried. This makes the total concentration of subseafloor organic matter in oxic abyssal red clay extremely low (Bradley, Amend and LaRowe 2019) compared to anoxic clays that have more organic matter but lower redox potential electron acceptors (Orsi 2018; D'Hondt et al. 2019). The composition of these abyssal communities is shaped by selective survival of taxa pre-adapted to these energy-limited conditions (Starnawski et al. 2017; Kirkpatrick, Walsh and D'Hondt 2019) and includes many poorly characterized clades of Chloroflexi (Inagaki et al. 2006; Nunoura et al. 2018). However, the structure of these communities, their relationship to redox stratification and their modes of selection as main constituents of the deep biosphere remain poorly constrained from the water-sediment interface down through the deep subseafloor sediment column (Durbin and Teske 2011; Walsh et al. 2016; Petro et al. 2019).
Chloroflexi are environmentally widespread. Their biogeography and modes of adaptation have been examined in various settings (Biddle et al. 2012) with variable salinities (Mehrshad et al. 2018), including shallow marine (Wilms et al. 2006; Petro et al. 2019), deep lacustrine (Vuillemin et al. 2018a; Kadnikov et al. 2019), aquifer sediments (Hug et al. 2013) and deep subseafloor sediments (Orsi 2018) where they have been found to be abundant and highly diverse. Considering the scale of the marine subsurface (Kallmeyer et al. 2012), subseafloor Chloroflexi may be one of the most abundant microbial components on Earth and may drive key sediment carbon cycling on a global scale (Zinger et al. 2011; Hug et al. 2013; Fincker et al. 2020). However, many groups of subsurface microorganisms, including the Chloroflexi, are still uncultivated (Biddle et al. 2012; Solden, Lloyd and Wrighton 2016). Thus, their metabolic properties remain poorly characterized, and the reasons for their broad ecological distribution and diversity have been elusive (Kaster et al. 2014; Wasmund et al. 2014; Sewell, Kaster and Spormann 2017).
Within representative isolates among classes of Anaerolineae and Dehalococcoidia, which are the two main Chloroflexi lineages found in marine sediments (Blazejak and Schippers 2010; Parkes et al. 2014), the main metabolic strategies identified include fermentation (Yamada et al. 2006 and 2017) and reductive dehalogenation (Duhamel and Edwards 2006; Futagami et al. 2009; Matturo et al. 2017). In terms of carbon cycling in marine sediments, the transformation of halogenated compounds remains barely understood (Kittelmann and Friedrich 2008; Kaster et al. 2014; Kawai et al. 2014; Atashgashi et al. 2018), while metagenome-assembled genomes (MAGs) and single-cell amplified genomes (SAGs) provide parallel evidence for respiration of sugars, fermentation, CO2 fixation via the Wood–Ljungdahl (W–L) pathway and acetogenesis with substrate-level phosphorylation (Hug et al. 2013; Sewell, Kaster and Spormann 2017), along with genes involved in environmental adaptation against oxygen and osmotic stress (Wasmund et al. 2014). The presence of genes encoding dissimilatory sulfite reductase (Dsr) and reversible adenylylsulfate reductase (apr) further imply roles in sulfur cycling and respiration (Wasmund, Mussmann and Loy 2017; Mehrshad et al. 2018; Vuillemin et al. 2018b). Altogether, these metagenomic data suggest versatile respiratory modes with intricate heterotrophic and lithoautotrophic metabolisms, which may support long-term survival in dormant states and dispersal in deep marine environments (Fullerton and Moyer 2016; Jørgensen and Marshall 2016).
In previous studies of slowly accumulating oxic and anoxic subseafloor clay from the abyssal North Atlantic covering up to 15 million years of depositional history (Vuillemin et al. 2019 and 2020), we found that Chloroflexi were a major component of the community in both settings. This abundance across multiple coring locations with varying redox states (oxic vs. anoxic) prompted a focused investigation of how the associated differences in long-term energy limitation select for metabolic features in Chloroflexi. In this study, we analyze 16S rRNA gene sequence, quantitative PCR (qPCR), metagenomic and metatranscriptomic data that identify metabolic features of the Chloroflexi associated with their selection and subsistence over million-year timescales in oxic and anoxic abyssal clay at multiple North Atlantic sampling locations. The data reveal differences in metabolism between aerobic and anaerobic Chloroflexi persisting under oxic and anoxic energy-limited conditions for millions of years below the abyssal seafloor.
METHODS
Sampling expedition
All samples were taken during Expedition KN223 of the R/V Knorr in the North Atlantic, from 26 October to 3 December 2014. At site 11 (22°47.0′ N, 56°31.0′ W, water depth ∼5600 m), site 12 (29°40.6′ N, 58°19.7′ W, water depth ∼5400 m) and site 15 (33°29.0’ N, 54°10.0’ W, water depth 5515 m), successively longer sediment cores were retrieved using a multicorer (∼0.4 m), gravity corer (∼3 m) and the Woods Hole Oceanographic Institution (WHOI) piston-coring device (∼29 m). Additional details of sampling are published elsewhere (D'Hondt et al. 2015 and 2019). Dissolved oxygen concentrations in the core sections were measured with optical O2 sensors as described previously (D'Hondt et al. 2015). Sediment subcores were retrieved on the ship aseptically using end-cut sterile syringes and kept frozen at -80°C without any RNA shield until extraction in spring 2018 in the home laboratory.
DNA extraction, quantitative PCR, 16S rRNA genes
For sites 11 and 12, total DNA was extracted from 10 g of sediment per sample and concentrated to a volume of 100 μL using 50-kDa Amicon centrifugal filters (Millipore, Burlington, United-States) as previously described (Vuillemin et al. 2019). At sites 11 and 12, we obtained DNA extracts from a total of 17 and 12 sediment depths, respectively. For site 15, we used 0.7 g of sediment per sample from 20 different sediment depths and diluted the final extracts 10 times in ultrapure PCR water (Roche, Basel, Switzerland). DNA concentrations, quantified with a Qubit 3.0 and dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, United-States), were systematically below detection. DNA templates were used in quantitative PCR (qPCR) amplifications with updated 16S rRNA gene primer pair 515F (5′- GTG YCA GCM GCC GCG GTA A -3′) with 806R (5′- GGA CTA CNV GGG TWT CTA AT -3′) to increase our coverage of Archaea and marine clades and run as previously described (Pichler et al. 2018). All qPCR reactions were set up in 20 µL volumes with 4 µL of DNA template and performed as previously described (Coskun et al. 2019). Reaction efficiency values in all qPCR assays were between 90 and 110% with R2 values >0.95% for the standards. For 16S rRNA gene library preparation, PCR runs were performed with barcoded primer pair 515F and 806R. All 16S rRNA gene amplicons were purified from 1.5% agarose gels, normalized to 1 nM solutions and pooled. Library preparation was carried out according to the MiniSeq System Denature and Dilute Libraries Guide (Protocol A, Illumina b). We combined 500 µL of the denatured and diluted 16S rRNA library (1.8 pM) with 8 µL of denatured and diluted Illumina generated PhiX control library (1.8 pM) to assess sequencing error rates. For each run, we used four custom sequencing primers Read 1, Index 1, Index 2 and Read 2, which were diluted and loaded into the correct position of the reagent cartridge. An additional Index 2 sequencing primer was designed to enable the dual-index barcoding method on the MiniSeq (Pichler et al. 2018). Pooled libraries were sequenced on the Illumina MiniSeq platform at the GeoBio-Center LMU.
Demultiplexing and base calling were both performed using bcl2fastq Conversion Software v. 2.18 (Illumina, Inc., San Diego, United-States). We used USEARCH (Edgar 2010 and 2013) and QIIME version 1.9.1 (Caporaso et al. 2010 and 2012) for MiniSeq read trimming and assembly, OTU picking and clustering at 97% sequence identity, which we previously tested with mock communities sequenced on the same platform (Pichler et al. 2018). The initial step was to assemble paired-end reads using the fastq_merge pairs command with default parameters allowing for a maximum of five mismatches in the overlapping region. Stringent quality filtering was carried out using the fastq_filter command. We discarded low quality reads by setting the maximum expected error threshold (E_max), which is the sum of the error probability provided by the Q score for each base, to 1. Reads were de-replicated and singletons discarded. OTU representative sequences were identified by BLASTn searches against the SILVA 16S rRNA SSU NR99 reference database release 132 (Quast et al. 2013). All operational taxonomic units (OTUs) assigned to Chloroflexi were aligned with SINA online v.1.2.11 (Pruesse et al. 2007) and inserted in a Maximum Likelihood RAxML phylogenetic tree selecting the best tree among 100 replicates, using ARB (Ludwig et al. 2004). Partial OTU sequences were added to the tree using the maximum parsimony algorithm without allowing changes of tree typology.
Metagenomes and metatranscriptomes
Whole genome amplifications were performed on DNA extracts diluted 10-fold through a multiple displacement amplification (MDA) step of 6–7 h using the REPLI-g Midi Kit (QIAGEN, Hilden, Germany) and following the manufacturer's instructions. MDA-amplified PCR products were then diluted to DNA concentrations of 0.2 ng/μL and used in metagenomic library preparations with the Nextera XT DNA Library Prep Kit (Illumina, San Diego, CA), then quantified on an Agilent 2100 Bioanalyzer System (Agilent Genomics, Santa Clara, CA) and normalized with the Select-a-Size DNA Clean and Concentrator MagBead Kit (Zymo Research, Irvine, CA) as previously described (Vuillemin et al. 2019), diluted to 1 nM and pooled for further sequencing on the MiniSeq platform (Illumina). We acknowledge that the use of a MDA step produces many short fragments potentially biased towards GC-rich sequences that do not allow high-quality binning and full genome completion.
For site 15, total RNA extractions were obtained from 3.5 g of wet sediments using the FastRNA Pro Soil-Direct Kit (MP Biomedicals, Irvine, CA) following the manufacturer's instructions, with the addition of 4 μL glycogen (0.1 g/mL) to increase yield during precipitation of the RNA pellet and final elution in 40 µL PCR-grade water (Roche). Extraction blanks were processed alongside to assess laboratory contamination. RNA extracts and extraction blanks, quantified with the QuBit RNA HS Assay Kit (Thermo Fisher Scientific), were all below detection. DNAse treatment, synthesis of complementary DNA and library construction were processed on the same day from 10 µL of RNA templates, without any prior MDA step, using the Trio RNA-Seq kit protocol (NuGEN Technologies, Redwood City, CA). The Trio RNA-Seq kit allows RNA amplification from initial concentrations as low as 50 pg/μL. All libraries were quantified as described above, diluted to 1 nM and pooled for further sequencing on the MiniSeq platform (Illumina). For sites 11 and 12, RNA yields were too low to achieve amplification and sequencing.
Because assigning taxonomic affiliation to metagenomic and transcriptomic data is challenging (Breitwieser, Lu and Salzberg 2019), we applied our previously published bioinformatics pipeline (Orsi, Richards and Francis 2018; Ortega-Arbulù et al. 2019; Vuillemin et al. 2019) that involves a large aggregated genome database of predicted proteins including the SEED (www.theseed.org) and NCBI RefSeq databases updated with all predicted proteins from recently described high-quality draft subsurface MAGs and SAGs from the NCBI protein database and all fungal genomes from the NCBI RefSeq database. The total number of predicted proteins in the updated database was 37.8 millions. This approach, which assigns ORFs to higher-level taxonomic groups (Orsi et al. 2020a,b), does not allow us to draw conclusions about specific populations within those groups, as would be possible with MAGs or SAGs, but does allow us to draw conclusions about metabolic traits derived specifically from higher-level taxonomic groups, such as Chloroflexi-related microorganisms given the ORF annotations provided. All scripts and code used to produce the following analysis have been posted on GitHub (github.com/williamorsi/MetaProt-database), and we provide a link to the MetaProt on the GitHub page, as well as instructions within the scripts regarding how to conduct the workflows that we used.
In brief, the MiniSeq reads were trimmed and paired-end reads assembled into contigs, using CLC Genomics Workbench 9.5.4 (https://www.qiagenbioinformatics.com/), using a word size of 20, bubble size of 50 and a minimum contig length of 300 nucleotides. Reads were then mapped to the contigs using the following parameters (mismatch penalty = 3, insertion penalty = 3, deletion penalty = 3, minimum alignment length = 50% of read length, minimum % identity = 95%). Coverage values were obtained from the number of reads mapped to a contig divided by its length (i.e. average coverage). Only contigs with an average coverage >5 were selected for open reading frame (ORF) searches, and downstream analysis. This protocol does not assemble ribosomal RNA (rRNA), and thus transcript results are only discussed in terms of messenger RNA (mRNA). Protein encoding genes and ORFs were extracted using FragGeneScan v. 1.30 (Rho, Tang and Ye 2010). Taxonomic identifications were integrated with the functional annotations, performing BLASTp searches of ORFs against our large aggregated genome database of predicted proteins described above. We used the DIAMOND protein aligner version 0.9.24 (Buchfink, Xie and Huson 2015). Cut-off values for assigning the best hit to specific taxa were performed at a minimum bit score of 50, minimum amino acid similarity of 60 and an alignment length of 50 residues. We chose to focus on the coverage of total annotated protein-encoding ORFs detected, as opposed to the number of reads mapping per kilobase per ORF (for example, RPKM), to reduce potential bias from small numbers of ‘housekeeping’ genes with potentially higher expression levels (Orsi, Richards and Francis 2018; Orsi et al.2020a,b). In addition, COG categories were assigned by comparing the metatranscriptome and metagenome annotated protein-encoding ORFs against the COG database (Galperin et al. 2015). Statistical analyses of beta-diversity were performed using RStudio v. 3.3.3 with the Bioconductor package (Huber et al. 2015). For both sites 11 and 12, the metagenome was sequenced for samples from four different depths to an average depth of 15 million reads, and de novo assembly resulted in a total of 177 498 contigs across all samples sequenced (Table S1, Supporting Information). For site 15, metagenomes were sequenced for samples from five different depths at an average depth of 8.4 million reads (± 2.5 millions), assembled into 70 157 contigs. The metatranscriptomes from each sample at site 15 were produced in biological replicates and sequenced at an average depth of 4.0 million reads (± 1.5 million). De novo assembly resulted in a total of 91 199 contigs across all samples sequenced (Table S1, Supporting Information). For all metagenomes and metatranscriptomes, the read coverage of annotated protein-encoding ORFs assigned to Chloroflexi was normalized to the coverage of all transcripts and results shown as % of read coverage. Data are publicly available through NCBI BioProject PRJNA473406 and PRJNA590088. Metagenomes from sites 11 and 12 have Short Read Archive (SRA) BioSample accession numbers SAMN10924458 and SAMN10924459, corresponding to run accession numbers SRX5372537–SRX5372545. Metagenomes and metatranscriptomes from site 15 have accession number SAMN13317858–SAMN13317870, corresponding to run accession numbers SRR10481880–SRR1048192. The 16S rRNA gene data are available in SRA BioSample accessions SAMN10929403–SAMN10929517 and SAMN13324854–SAMN13324920.
As is the case in all metagenomic studies, the incomplete nature of genomes in databases, together with the lower representation of sequenced genomes from candidate clades compared to cultured ones, make it likely that our pipeline misses annotation of ORFs that are derived from as-of-yet unsequenced Chloroflexi genomes that are not yet in databases. We acknowledge that some genes in databases annotated as being present in Chloroflexi MAGs might not actually derive from Chloroflexi chromosomes, but have been assigned to bins according to criteria that differ from study to study. Thus, we confirmed the accuracy of annotating ORFs to Phyla based on ‘best BLAST’ by an in silico test for true and false positive annotations based on 151 randomly selected peptide fragments extracted from a Chloroflexi MAG predicted proteome (Anantharaman et al. 2016), as well as other bacterial and archaeal genomes (Figure S1, Supporting Information). In this analysis, 50 randomly selected predicted proteins were randomly cut into peptide fragments ranging from 20 to 140 amino acid residues in length, in order to replicate partial ORFs typically recovered in metagenomes. The random peptide fragments were then searched against our large aggregate database for their top hits with BLASTp.
Taxonomic assignment of protein-encoding genes to Chloroflexi among the SAR202 and Dehalococcoidia clades was further confirmed in our metagenomes by phylogenetic analysis of the RNA polymerase sigma factor (RpoD) gene proteins annotated as Chloroflexi, using alignments of 619 amino acid residues. We chose this gene for three reasons: (1) it is a universal single copy gene (Kawai et al. 2014); (2) the RpoD gene has a faster evolutionary rate compared to 16S rRNA genes and can be used to distinguish taxa at a relatively finer taxonomic level in phylogenetic analyses (Ghyselinck et al. 2013; Gupta, Chander and George 2013) and (3) the RpoD gene was detected in Chloroflexi annotated contigs from metagenomes created at both oxic and anoxic sites, allowing for a comparative phylogenetic analysis of Chloroflexi in the metagenomes across sites.
Phylogenetic analyses of the predicted alpha and beta subunits of the Dsr and apr gene proteins were performed for all the corresponding annotated taxa in our metagenomes and metatranscriptomes, using 466, 433, 754 and 157 aligned amino acid sites respectively (Orsi, Jørgensen and Biddle 2016). For each of the five marker gene phylogenies (RpoD, DsrA, DsrB, aprA and_aprB_), all ORFs annotated to those genes from our bioinformatics pipeline were aligned against their top two BLASTp hits in the NCBI-nr and SEED databases using MUSCLE (Edgar 2004). Conserved regions of the alignments were selected in SeaView version 4.7 (Gouy, Guindon and Gascuel 2010), using Gblocks with the following settings: allowing for smaller final blocks, gap positions within the final blocks and less strict flanking positions. Phylogenetic analysis of the resulting amino acid alignments of the predicted proteins were conducted in SeaView version 4.7 (Gouy, Guindon and Gascuel 2010), using RAxML (Stamatakis et al. 2012, 2014) with BLOSUM62 as the evolutionary model and 100 bootstrap replicates. All alignments are publicly available through the LMU Open Data website (https://doi.org/10.5282/ubm/data.190).
RESULTS
Sedimentation rate and pore water geochemistry
The three long (∼30 m) sediment cores from three abyssal (>5000 m water depth) coring locations sites from R/V Knorr expedition KN223 in the Western North Atlantic are comprised of abyssal clay (Fig. 1A), with the exception of sandy layers occurring around 15, 16, 18, 21 and 23 mbsf at site 15. Dissolved oxygen is present throughout the clay at sites 11 and 12, with concentrations at the water-sediment interface just below that of the overlying water (approximately 300 μM) and gradually decreasing with sediment depth (Figure S2, Supporting Information). Drawdown of O2 with sediment depth at these sites reflects oxidation of organic matter by aerobic microorganisms (Vuillemin et al. 2019). Sedimentation rate at both oxic sites sampled is estimated to be 1 m per million years (Kallmeyer et al. 2012; D'Hondt et al. 2015). Thus, the clay sampled at the deepest portion of the cores analyzed from both oxic sites (sites 11 and 12) is about 10–15 million years old (Vuillemin et al. 2019). In contrast, dissolved oxygen at site 15 is restricted to the top mm of sediment. Compared to the two oxic sites, site 15 is characterized by a mean sedimentation rate of about 3 m per million years (Fig. 1A). Given this rate, the deepest sediment sampled from the anoxic site 15 at 29 meters below seafloor (mbsf) is between 8 and 9 million years old.
Figure 1.

Map of sampling sites with bathymetry and sedimentation rate, and characterization of Chloroflexi in terms of beta diversity, density and taxonomy at the three sites. (A) Map of subseafloor sedimentation rate (modified after D'Hondt et al. 2015) with location of the three sampling sites. (B) Non-metrical dimensional scaling (NMDS) plot based on all OTUs assigned to Chloroflexi across the three sites. (C) Quantitative polymerase chain reaction (qPCR) of total (black dots) and qPCR- normalized to Chloroflexi (colored dots) 16S rRNA genes [gene copies/g sed], and Chloroflexi taxonomic assemblages [relative %].
Diversity, abundance and taxonomy of 16S rRNA genes
A total of four biological replicates of 16S rRNA genes (e.g. from separate DNA extractions) were sequenced at each sediment horizon from the anoxic sediment at site 15, which were compared to 16S rRNA genes from the two separate coring locations (sites 11 and 12) that contain oxic clay. Based on 3711 OTUs obtained across all samples, non-metric multidimensional scaling (NMDS) analysis groups samples from the oxic sites separately from those of the anoxic site (Figure S3, Supporting Information), demonstrating statistical significance on the community structure (ANOSIM: P 0.001, R = 0.897). All three sites exhibit similar values of total OTU richness (approximately 1500 at each site), Chloroflexi being the richest phylum (Figure S4, Supporting Information). Taxonomic assignment of 16S rRNA gene amplicons resulted in 791 Chloroflexi OTUs. Of these 791 OTUs, 555 are assigned to the class Dehalococcoidia, 136 are assigned to Anaerolineae and the remaining 100 OTUs are in nine different classes (Fig. 2). Based on NMDS analysis of these 791 OTUs, differences in Chloroflexi distribution in oxic and anoxic sediments are statistically significant (ANOSIM: P 0.001, R = 0.897; Fig. 1B).
Figure 2.
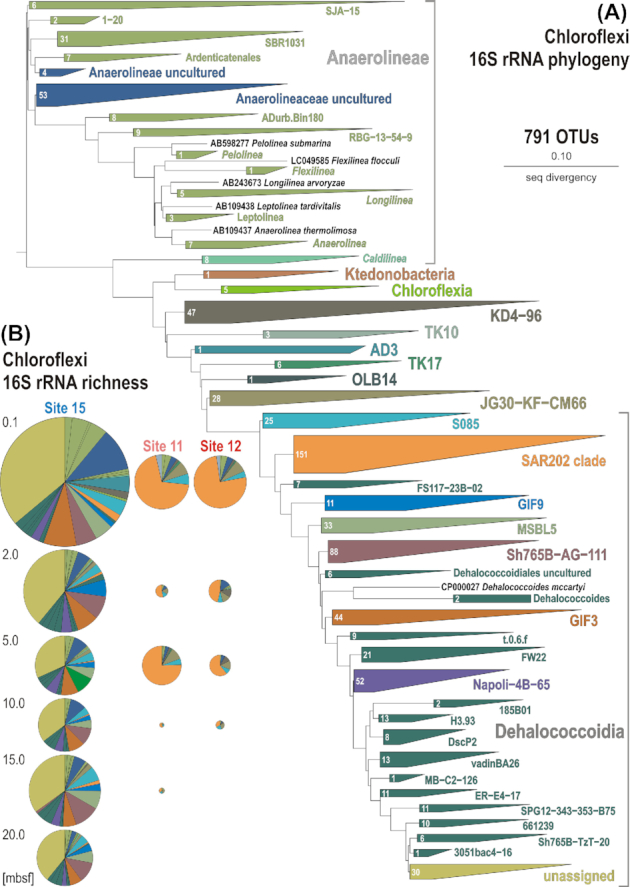
Phylogenetic analysis of 16S rRNA genes (V4 hypervariable region). (A) Phylogenetic tree obtained for all operational taxonomic units (OTUs) assigned to Chloroflexi in this study. (B) Pie charts displaying 16S rRNA gene richness of the whole assemblage of Chloroflexi for each of the three sites and at each sediment depth. The radii of pie charts are proportional to the number of OTUs. The clockwise color coding in the pie charts matches that of the phylogenetic tree from top to bottom, and bar charts from left to right (Fig. 1).
At both oxic sites, the total 16S rRNA gene densities, inclusive of both Bacteria and Archaea, are respectively 107 and 106 copies/g wet sediment in the shallow subsurface and decrease by three orders of magnitude within the underlying 0.5 mbsf (Fig. 1C). At oxic site 12, our limit of detection (102 × 16S rRNA gene copies/g wet sediment) was reached at ∼8 mbsf, whereas at oxic site 11, the density of 16S rRNA genes increases by two orders of magnitude at ∼6 mbsf before reaching the detection limit at 15 mbsf. Similar to the procedure described by Lloyd et al. (2020), we normalized the fractional abundance of the Chloroflexi 16S rRNA abundance to quantitative values using the qPCR determined 16S rRNA gene concentrations as described previously (Vuillemin et al. 2019). The 16S rRNA gene density normalized to Chloroflexi decreases with depth at both oxic sites, reaching the limit of detection at 15 mbsf at site 11 and at 10 mbsf at site 12. In contrast, the relative abundance of 16S rRNA genes assigned to Chloroflexi increases gradually at both oxic sites from <10% at the surface to about 40% in the deepest samples (Fig. 1C). The corresponding taxonomic assemblage is mainly composed of candidate clade S085 and SAR202 among Dehalococcoidia and uncultured Anaerolineales (Fig. 1C).
As implied by the NMDS analysis (Fig. 1B), the community composition at the anoxic site was significantly different by comparison. In contrast to the declining abundance of subseafloor Chloroflexi at oxic sites 11 and 12, at the anoxic site 15 the 16S rRNA gene density normalized to Chloroflexi increases from 105 copies at the seafloor surface by more than one order of magnitude in the deeper subsurface samples down to 2 mbsf, spanning ∼1 million years of depositional history (Fig. 1C). Below this depth, the 16S rRNA gene density of Chloroflexi decreases by an order of magnitude and remains constant down to the deepest sampled interval at 29 mbsf. Throughout the anoxic sediment column, Chloroflexi account for about 20% of the complete taxonomic assemblage (Fig. 1C). Within the uppermost 0.3 mbsf at the anoxic site 15, Chloroflexi communities are more similar to those of the oxic sites, namely candidate clades S085 and SAR202 and diverse uncultured Anaerolineales. However, below this depth at the anoxic site 15, the assemblages shift to presumed anaerobic candidates (e.g. GIF9, MSBL5 and Sh7765B-AG-111) and uncultivated clades related to Dehalococcoides (Fig. 1C and Figure S4, Supporting Information). We acknowledge that primers targeting the V4–V6 regions as well as the average number of 16S rRNA gene copies in genomes of Chloroflexi (2.2 ± 1.2 copies) can potentially result in quantitative biases between the clades mentioned (Větrovský and Badrian 2013).
Assessing the detection limit in low biomass metatranscriptomes
At the anoxic site, the number of unique annotated protein-encoding ORFs assigned to Chloroflexi increases by more than one order of magnitude from the seafloor surface to 5 mbsf (Fig. 3). Below this depth, the number of unique ORFs assigned to Chloroflexi steadily decreases with depth until 15.9 mbsf. Below this depth, the only ORFs annotated corresponded to bacterial groups other than Chloroflexi that are known contaminants from molecular kits including those from human skin and soil (Salter et al. 2014). Many of these same groups are common laboratory contaminants found in dust samples from our lab in 16S rRNA gene surveys (Pichler et al. 2018), and include Pseudomonas, Rhizobium, Acinetobacter and Staphylococcus. We interpreted the sudden dominance of ORFs with similarity these common contaminants below 15.9 mbsf to be indicative of our limit of RNA detection, whereby a lower amount of extracted RNA from the in situ active community becomes overprinted by background ‘noise’ from contaminating DNA.
Figure 3.

Open reading frames (ORFs) assigned to Chloroflexi, their COG categories and metabolic potential obtained from metagenomes and metatranscriptomes. (A) Number of annotated protein-encoding ORFs obtained from metagenomes and attributed to Chloroflexi with sediment depth at the three sites; relative abundances of different COG categories and metabolic functions obtained from metagenomes at the three sites. (B) Number of annotated protein-encoding ORFs obtained from the metatranscriptomes and attributed to Chloroflexi with sediment depth at the anoxic site; relative abundances of different COG categories and metabolic functions that are expressed at the anoxic site in [read % coverage].
From the oxic site samples, no metatranscriptomes of sufficient quality could be obtained. We attempted to extract RNA and make metatranscriptome libraries from the samples, but in every sample the only recovered ORFs annotated were those assigned to groups of known contaminants from molecular kits (Salter et al. 2014) and dust samples from our lab (Pichler et al. 2018). We interpreted the dominance of ORFs with similarity to these common contaminants to be a clear indication of contamination, since the metagenomes and 16S rRNA gene datasets showed a completely different community structure that was dominated by the Chloroflexi (Figs 1–3). In none of the metatranscriptomes from the oxic site could we retrieve any ORFs annotated to Chloroflexi genomes, indicating that the gene expression and overall activity of Chloroflexi at the oxic sites were very low, compared to the anoxic site.
Assessing the accuracy of Chloroflexi ORF assignments
The results of our in silico experiment to determine how accurate ORFs annotations are using ‘best BLAST’, showed that 100% of all randomly cut peptide fragments from a Chloroflexi predicted proteome were true positives (Figure S1, Supporting Information). Namely, all ORFs that were randomly extracted from a Chloroflexi MAG predicted proteome had a predicted protein from the same Chloroflexi MAG as a top BLASTp hit. This trend held true for other bacteria and archaea tested in the in silico experiment, in all cases the rate of true positives using our approach was >90%. This shows that our use of this previously published similarity-based approach using ‘best BLAST’ (Orsi, Richards and Francis 2018; Orsi et al. 2020a,b) is adequate for assigning ORFs encoded on de novo assembled contigs to groups at high taxonomic levels (e.g. bacterial Phyla) including those derived from uncultivated Chloroflexi.
In addition to this ‘best BLAST’ approach, we furthermore identified the specific clades of Chloroflexi in our sampled communities using phylogenetic analyses of several marker proteins (Fig. 4). Phylogenetic analysis of the RpoD gene revealed the presence of several Chloroflexi clades among the SAR202 cluster in the oxic site metagenomes, and anaerobic clades of Dehalococcoidia in the anoxic site metagenomes (Fig. 4A). The RpoD genes detected had close affiliation to the recently published Chloroflexi genomes from Mid-Atlantic hydrothermal vent and deep-sea sediments (Fincker et al. 2020; Zhou et al. 2020), indicating that closely related Chloroflexi from these groups are also present in our sampled communities. RpoD encoding genes from Chloroflexi were only detected in the metagenomes, no RpoD encoding transcripts from Chloroflexi were detected in the metatranscriptomes.
Figure 4.

Phylogenetic analysis of predicted proteins encoded by selected marker genes in the metagenomes and metatranscriptomes from oxic and anoxic sites, based on RAxML using BLOSUM62 as the evolutionary model. (A) Phylogeny of Chloroflexi RNA polymerase sigma factor (RpoD) ORFs (619 aligned amino acid sites). (B) Phylogeny of all DsrA and rDsrA ORFs detected (466 aligned amino acid sites). (C) Phylogeny of all DsrB and rDsrB ORFs detected (433 aligned amino acid sites). (D) and (E) Phylogenies of all aprA and aprB ORFs detected (754 and 157 aligned amino acid sites, respectively). All phylogenies were conducted with 100 bootstrap replicates, circles on the nodes indicate support values (black > 90%, gray > 70%, white >50%). Dsr: dissimilatory sulfite reductase; rDsr: reverse dissimilatory sulfite reductase; apr: adenylylsulfate reductase; A and B: alpha and beta subunits.
Phylogenetic analysis of the alpha and beta subunits of the apr and Dsr genes revealed detection of these genes from Chloroflexi between 2 and 5 mbsf in the metagenomes (Fig. 4B–E). These apr and Dsr genes are affiliated to Chloroflexi known from previous surveys of anoxic marine sediments (Leloup et al. 2009; Wasmund et al. 2016; Dong et al. 2019). However, no aprAB or DsrAB encoding transcripts from Chloroflexi were detected in the metatranscriptomes. Instead, the detected aprAB or DsrAB encoding transcripts in the metatranscriptomes were derived mostly from groups of sulfate-reducing Deltaproteobacteria (Müller et al. 2015). This points to Deltaproteobacteria as the main active sulfate-reducing bacteria subsisting in the anoxic subseafloor clay at site 15, whereas a smaller number of expressed ORFs were affiliated with Gammaproteobacteria reverse DsrAB (rDsr) involved in sulfide oxidation (Loy et al. 2009). At the oxic site 11, at a depth of 0.1 mbsf, aprA and aprB subunits were found to be encoded in metagenomes with affiliation to Chloroflexi ORFs (Fig. 2D–E). Five ORFs assigned to aprAB genes in metagenomes from oxic site 11 and anoxic site 15 were affiliated to Chloroflexi MAGs among the SAR202 cluster (Mehrshad et al. 2018; Fig. 4). However, mRNA was below detection at the oxic sites, and thus the expression of these genes could not be found. Thus, the genomic potential for sulfur cycling in Chloroflexi is present at all sites, but the expression of the associated genes was below detection in all cases.
Metabolic potential of subseafloor Chloroflexi in metagenomes and metatranscriptomes
For both oxic sites, the number of unique ORFs in metagenomes assigned to Chloroflexi is highest in surface sediment and decreases exponentially with increasing sediment depth (Fig. 3A). This mirrors the trend of declining Chloroflexi abundance and diversity with depth based on qPCR (Fig. 1C) and decreasing number of 16S rRNA gene OTUs with depth (Fig. 2B). In metagenomes from the shallow oxic sediment, Chloroflexi-derived ORFs could be assigned to clusters of orthologous genes (COGs) involved in energy conversion, transport and metabolism of amino acids, carbohydrates and coenzyme metabolism (Fig. 3A). Potential for aerobic respiration via cytochrome C oxidase and the tricarboxylic acid (TCA) cycle were also identified in Chlorolexi assigned ORFs (Fig. 5). The potential mechanisms of energy production could be identified by ORFs annotated as ATP synthase, NADH-quinone and -flavin reductase based on similarity to predicted proteins in previously sequenced Chloroflexi genomes. ORFs with highest similarity to dehalogenases, peptidases, lipases, glycosidases, acyl-CoA dehydrogenase and acetate kinase from annotated predicted proteins in previously sequenced Chloroflexi genomes were all detected in the metagenomes (Figure S5, Supporting Information).
Figure 5.

Presence and absence of Chloroflexi assigned predicted proteins as detected in the present metagenomes and metatranscriptomes. Presence (colored) or absence (white) of metabolic functions (circles) and expressed genes (squares) identified in the metagenomes and metatranscriptomes at the oxic (red) and anoxic (blue) sites across all depths sampled. Presence/absence of substrate transporters, catabolytic enzymes, apr and Dsr genes are shown on the left hand side. Metabolic pathways listed correspond to sugar metabolism, pyruvate fermentation, aerobic respiration, Wood-Ljungdahl pathway, electron transport, tricarboxylic acid (TCA) cycle, hydrogen metabolism and oxidative and non-oxidative pentose phosphate pathway. The complete list of ORFs with assignment and coverage is available as Supplementary Data.
At the anoxic site, the main COG categories detected in the metagenomes with highest similarity to previously sequenced Chloroflexi genomes are energy production, transport and metabolism of amino acids, carbohydrates, coenzymes and lipids (Fig. 3A). There were several COG categories detected in the metatranscriptomes with highest similarity to previously sequenced Chloroflexi genomes that were not detected in the metagenomes (Fig. 3B). The ORFs assigned to Chloroflexi also had similarity to genes involved in glycolysis/gluconeogenesis (Seshadri et al. 2005; Say and Fuchs 2010), the pentose phosphate pathway, TCA cycle and the W–L pathway. ORFs annotated as energy metabolism (electron carrier) genes from the Chloroflexi included pyruvate and aldehyde ferredoxin oxidoreductase, as well as NADH-quinone and -flavin oxidoreductase (Fig. 5). At the anoxic site, Chloroflexi assigned ORFs corresponded to several COG categories were detected exclusively in the metatranscriptomes, which were not detected in the metagenomes from the same depths. The relative abundance of these categories peaked between 2 and 5 mbsf (Fig. 3B). At 0.1 mbsf at the anoxic site, expressed Chloroflexi COG categories detected exclusively in the metatranscriptomes included ORFs with similarity to predicted proteins involved in cellular motility (Fig. 3B). Interestingly below 0.1 mbsf, there was no longer any gene expression of Chloroflexi assigned ORFs encoding proteins involved in motility. Rather, in the deeper depths several Chloroflexi-assigned COG categories increased in relative abundance in the metatranscriptomes, which included cellular defense, prophages, cell wall biogenesis, protein turnover and secondary metabolite biosynthesis (Fig. 3B). Because these COG categories were expressed in the metatranscriptomes but were not detected in the metagenomes, they likely correspond to activities that increase over time since burial.
An NMDS analysis based on all annotated protein-encoding ORFs obtained from metagenomes and metatranscriptomes (ANOSIM: P 0.001, R = 0.56), as well as those annotated specifically to Chloroflexi (ANOSIM: P 0.005, R = 0.34), clearly separates all of the metagenome and metatranscriptome samples from both oxic sites from those recovered from the anoxic site (Fig. 6). This demonstrates that the metabolic potential is significantly different, not only within the entire microbial community, but between aerobic and anaerobic Chloroflexi inhabiting the ancient oxic and anoxic abyssal clays as well.
Figure 6.

Non-metrical dimensional scaling (NMDS) plot for all three sites based on metagenomes and metatranscriptomes. (Left) NMDS plot for all three sites based on all ORFs from the metagenomes (circles) and metatranscriptomes (squares) for the entire microbial communities. (Right) NMDS plot for all three sites based on annotated protein-encoding ORFs from the metagenomes (circles) and metatranscriptomes (squares) assigned to Chloroflexi.
DISCUSSION
We present new data from sediments that are millions of year old below the North Atlantic oceanic gyres that provide new insights into how life survives under energy-limited sediment conditions, in both oxic and anoxic sediments, over million-year timescales. Since Chloroflexi constitute a substantial part of the composition of subseafloor communities that were studied under both settings (Vuillemin et al. 2019 and 2020), we investigated metabolic potentials and strategies that may allow them to actively survive under extreme energy limitation and persist in subseafloor sediment for millions of years after burial.
Chloroflexi in oxic abyssal clay
At both oxic sites, the Chloroflexi were dominated by the SAR202 clade, and the 16S rRNA gene density of Chloroflexi decreased exponentially with depth at site 12 (Fig. 1C), indicating that Chloroflexi die off over this interval of burial, whereas at site 11 this exponential decrease occurs below 5 mbsf, potentially due to variability in sedimentation rate and delivery of organic matter to the sediment. The clade SAR202 dominates communities in global ocean waters (Mehrshad et al. 2018) and abyssal oxic sediment in many locations (Durbin and Teske 2011), indicating it is relatively well-suited to cope with the otherwise unfavorable (e.g. energy-limited) conditions for life. Phylogenetic analysis of the Chloroflexi RpoD gene proteins confirmed taxonomic affiliation of protein-encoding ORFs to the SAR202 clade in metagenomes from the oxic sites (Fig. 4A).
Genomic evidences indicate that the SAR202 clade lives off of refractory organic matter, such as hydrocarbons (Wang et al. 2020), via cyclic alkane monooxygenases (Landry et al. 2017; Saw et al. 2020), peptidases (Mehrshad et al. 2018) and potentially haloalkane and haloacetate dehalogenases (Wasmund et al. 2016; Yang et al. 2020). We detected ORFs with similarity to all these genes at the oxic site metagenomes (Figure S5, Supporting Information), which indicates potential for the SAR202 and other clades of Dehalococcoidia to consume refractory organic matter in the million year old clay. The organic matter in deep-sea clay has a low reactivity (Estes et al. 2019) which probably makes it difficult to access by many microbes and the ability of SAR202 to access refractory organic matter may explain its abundance in the setting seen here. However, the fact that the extractable RNA was below detection in the oxic red clay suggests that the aerobic Chloroflexi have a relatively lower level of activity, compared to the anaerobic Chloroflexi that were subsisting at the anoxic site that had detectable levels of transcriptional activity that relatively increase with depth (Fig. 3A and B). According to Mehrshad et al. (2018), some SAR202 Chloroflexi may use the apr gene to oxidize sulfite, thiosulfate and organic sulfur molecules. The SAR202 detected in the subseafloor oxic clay metagenomes here also encode ORFs with high similarity to the apr gene (Fig. 4D), which is closest related to Euryarchaeota (Mehrshad et al. 2018). Because mRNA was below detection in the oxic red clays, it remains unknown whether the subseafloor SAR202 Chloroflexi expressed this apr gene or not, and whether they are engaged in sulfite or thiosulfate oxidation. Nevertheless, the presence of ORFs with similarity to the apr gene in the metagenomes indicates the metabolic potential by SAR202 Chloroflexi in the oxic subseafloor red clay.
Chloroflexi in anoxic abyssal clay
The metabolic activity and biomass of the anaerobic Chloroflexi apparently increases with sediment depth at the anoxic site based on: (1) increasing density of Chloroflexi 16S rRNA genes with depth (Fig. 1C); (2) increasing number of Chloroflexi annotated protein-encoding ORFs in the metatranscriptomes with depth (Fig. 3B); and (3) the increasing number of different COG categories in the metatranscriptomes assigned to Chloroflexi with depth (Fig. 3B). All of these results suggest that Chloroflexi have increased in activity after burial, actively grew and divided over millions of years since deposition below the seafloor. The Chloroflexi 16S rRNA genes below 0.5 mbsf is dominated by anaerobic clades of Dehalococcoides (Figs 1C and 2B), which was confirmed by the phylogenetic analysis of RpoD encoded proteins with similarity to Dehaloccoidia clades that are common from subsurface habitats (Fig. 4A). These metagenomes indicate that the actively growing Chloroflexi at the anoxic site have the potential to conserve energy via sugar fermentation/gluconeogenesis (Seshadri et al. 2005), the potentially reversible W-L pathway, pyruvate ferredoxin oxidoreductase, ATP synthase and NADH-quinone oxidoreductase (Fig. 5 and Figure S5, Supporting Information).
Because gene expression of Chloroflexi assigned ORFs encoding proteins involved in motility was no longer detectable below 1 mbsf (Fig. 3B), the lack of detection below this depth indicates that motility of Chloroflexi was possibly reduced in the deeper sediment layers (Zhu and Gau2020). The detection of COG categories exclusively in the metatranscriptomes indicate that cellular functions involving cell wall biogenesis, cellular defense, prophages and production of secondary metabolites had a relatively higher expression in the deeper depths compared to the surface sediment (Fig. 3B). In the deepest metatranscriptome at 15.9 mbsf, the only annotated protein-encoding ORFs that were assigned to Chloroflexi in the metatranscriptomes were involved in DNA repair, nucleotide transport, metabolism and protein turnover, which are indicative of cellular maintenance processes in sediments that have an estimated age of 5 million years (Fig. 3B).
The metatranscriptomes from the anoxic site indicate fermentation and acetogenesis as potential metabolisms (Fig. 5), which has been previously predicted from genome analysis of subseafloor Chloroflexi (Sewell, Kaster and Spormann 2017; Fincker et al. 2020). The expression of ORFs assigned to Chloroflexi with similarity to the methyl-viologen Fe–S reducing and F420 non-reducing Ni–Fe hydrogenases (MVHs) indicates the potential for production of molecular hydrogen (Vignais, Billoud and Meyer 2001; Shafaat et al. 2013) (Fig. 5). A partial WLP is used by the Dehalococcoides mccartyi, whereby CO2 and/or acetate are assimilated (Löffler et al. 2013; Zhuang et al. 2014; Sewell, Kaster and Spormann 2017). We detected expression of ORFs assigned to Chloroflexi with similarity to W–L pathway including formate dehydrogenase (fdh), methylenetetrahydrofolate reductase (MTHFR), carbon monoxide dehydrogenase (CODH) and acetyl-CoA synthase (cdhA; Figs 3 and 5). Furthermore, expression of ORFs with highest similarity to annotated pyruvate ferredoxin oxidoreductase from previously sequenced Chloroflexi genomes were detected (Fig. 3B). As suggested previously (Wasmund et al. 2016), pyruvate ferredoxin oxidoreductase may provide a link between the W–L pathway and other anabolic pathways in some Chloroflexi. Many acetogenic bacteria use the Rnf complex to generate a chemiosmotic gradient of Na+ ions in order to synthesize ATP at the membrane (Peters et al. 2016; Schuchmann and Müller 2019). However, Dehalococcoidia appear to use the NADH-quinone oxidoreductase (Nuo) to create a proton gradient in order to generate a chemiosmotic potential for ATP synthesis (Wasmund et al. 2016). Nuo yields reduced energy compared to the flavin-dependent oxidoreductase (Buckel and Thauer 2018). ORFs with similarity to Nuo and ATP synthases were expressed by the Chloroflexi at depths down to 10 mbsf (Fig. 3B). This could be potentially related to active ATP production via chemiosmotic proton pumping with Nuo. ORFs encoding proteins with similarity to ferredoxin and flavodoxin (Figs 3 and 4) were expressed by the Chloroflexi at these same depths and are likely involved as electron carriers in cellular redox reactions (Peters et al. 2016; Buckel and Thauer 2018).
The detection of acyl-CoA dehydrogenase (ACAD) also indicates that some of the anaerobic Chloroflexi in the anoxic subseafloor clay have metabolic potential for β-oxidation of volatile fatty acids (Wasmund et al. 2014; Sewell, Kaster and Spormann 2017; Supplementary Data). Chloroflexi ORFs annotated as acetate kinase, which is the enzyme that yields one ATP during substrate level phosphorylation (SLP) in (homo)acetogenic bacteria (Schuchmann and Müller 2019), was detected in the metagenomes, but not in the metatranscriptomes (Supplementary Data). This indicates the genomic potential for ATP production via SLP, albeit its expression remained undetected.
Potential mechanisms of long term survival
The organic matter present at very low concentrations in abyssal clay is dominated by amide, carboxylic carbon and oxidized glycoproteins, and thus is mainly proteinaceous (Estes et al. 2019). Most of cellular material is protein by weight (Orsi et al. 2020c) and, although necromass is considered a minor substrate compared to allochthonous organic material, amino acids are possibly the most important of all fermentation substrates in the subseafloor for cells that subsist in dormancy (Bradley, Amend and LaRowe 2019). Based on the relatively high expression levels of peptide transporters and peptidases (Fig. 3B), we suggest that Chloroflexi actively utilize this organic matter in the anoxic subseafloor clays.
The genomic potential for dissimilatory sulfate reduction in uncultivated clades of Chloroflexi from marine sediments has been identified previously (Wasmund et al. 2016 and Wasmund, Mussmann and Loy 2017). However, expression of ORFs encoding the aprA and DsrAB proteins assigned to Chloroflexi were not detected in the metatranscriptomes from the same samples (Figs 2 and 4), which suggests that dissimilatory sulfate reduction is not a major energy yielding activity under these conditions compared to the Deltaproteobacteria that, in contrast, expressed many ORFs of DsrAB and aprAB genes (Fig. 2B–E). The most genes expressed by the anaerobic Chloroflexi at the anoxic site appear to be rather involved in a homoacetogenic lifestyle, and have rpoD gene phylogenies (Fig. 4A) that place them in close affiliation to known homoacetogenic subsurface Chloroflexi (Finker et al. 2020). The lack of expression of Chloroflexi DsrAB genes suggests the possibility that some Chloroflexi avoid competing for sulfate with sulfate-reducing bacteria, by applying a homoacetogenic lifestyle (Lever 2012; Wiechmann et al. 2020).
Our metatranscriptomes show that Deltaproteobacteria are the main sulfate-reducing bacteria that are presently active in the anoxic subseafloor (Fig. 4B–E). However, the phylogenetic analysis of expressed ORFs identified some with high similarity to the reverse dissimilatory sulfite reductase rDsrAB (Loy et al. 2009; Müller et al. 2015) of Gammaproteobacteria known to be involved in sulfide oxidation (Fig. 4). If these rDsr expressing bacteria are indeed performing H2S oxidation, it is unclear what would be used as a terminal electron acceptor in anoxic sediment. Potentially, Fe3+ or Mn4+ could be used for iron or manganese reduction from sulfide oxidation. The rDsrABL complex from Allochromatium vinosum is reversible in vitro and can catalyze NADH-dependent sulfite reduction (Löffler et al. 2020), indicating the potential for some rDsr containing bacteria to function as sulfate reducers. An expressed ORF with >90% amino acid similarity to the rDsr from A. vinosum was detected in the metatranscriptomes at 2 mbsf (Fig. 4C), supporting this possibility. More experiments with pure cultures of subseafloor rDsr containing bacteria are needed to confirm the physiological capabilities of subseafloor rDsr containing Gammaproteobacteria, and whether they are able to function as sulfate reducers
CONCLUSIONS
Our findings demonstrate that transcriptionally active anaerobic, potentially homoacetogenic Chloroflexi exhibit higher activity in anoxic abyssal clay compared to aerobic Chloroflexi from oxic abyssal red clay, in subseafloor sediments that are up to 3 million years old. The close affiliation of RpoD encoding Chloroflexi ORFs from the anoxic site with a clade from deep-sea Chloroflexi genomes with a proposed homoacetogenic metabolism indicates that many of the transcriptionally active Chloroflexi at the anoxic site have the potential to survive via homoacetogenesis. This is consistent with the expression of Chloroflexi assigned ORFs with similarity to proteins of the W–L pathway, sugar fermentations and gluconeogenesis, which is apparently a fitness advantage for anaerobic Chloroflexi surviving in the energy-starved anoxic abyssal subseafloor over millions of years.
Supplementary Material
fiaa223_Supplemental_File
ACKNOWLEDGEMENTS
We thank Sergio Vargas, Ömer K. Coskun and Tobias Magritsch (LMU München) for assistance with laboratory procedures and sequencing on the Illumina MiniSeq. We also thank Robert Pockalny, David C. Smith (University of Rhode Island) and Richard W. Murray (Boston University) for collection of the sediment samples during Expedition KN223 of the R/V Knorr in the North Atlantic. Two anonymous reviewers are acknowledged for their comments on the manuscript. This work was supported by the LMU Junior Researcher Fund (to W.D.O.) and LMU Mentoring Program (to A.V.) of the LMU Munich's Institutional Strategy LMUexcellent within the framework of the German Excellence Initiative.
Contributor Information
Aurèle Vuillemin, Department of Earth and Environmental Sciences, Paleontology & Geobiology, Ludwig-Maximilians-Universität München, Richard-Wagner-Strasse 10, 80333 Munich, Germany.
Zak Kerrigan, Graduate School of Oceanography, University of Rhode Island, Narragansett Bay Campus, 215 South Ferry Road, Narragansett, RI 02882, USA.
Steven D'Hondt, Graduate School of Oceanography, University of Rhode Island, Narragansett Bay Campus, 215 South Ferry Road, Narragansett, RI 02882, USA.
William D Orsi, Department of Earth and Environmental Sciences, Paleontology & Geobiology, Ludwig-Maximilians-Universität München, Richard-Wagner-Strasse 10, 80333 Munich, Germany; GeoBio-CenterLMU, Ludwig-Maximilians-Universität München, Richard-Wagner-Strasse 10, 80333 Munich, Germany.
FUNDING
This work was supported primarily by the Deutsche Forschungsgemeinschaft (DFG) project OR 417/1-1 granted to W.D.O. The expedition was funded by the US National Science Foundation through grant NSF-OCE-1433150. Shipboard microbiology efforts were supported by the Center for Dark Energy Biosphere Investigations (C-DEBI grant NSF-OCE-0939564). This is C-DEBI publication 551. This is a contribution of the Deep Carbon Observatory (DCO).
Conflicts of Interest
None declared.
REFERENCES
- Anantharaman K, Brown CT, Hug LA et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Comm. 2016;7:13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atashgahi S, Häggblom MM, Smidt H. Organohalide respiration in pristine environments: implications for the natural halogen cycle. Environ Microbiol. 2018;20:934–48. [DOI] [PubMed] [Google Scholar]
- Biddle JF, Sylvan JB, Brazeltin WJ et al. Prospects for the study of evolution in the deep biosphere. Front Microbiol. 2012;2:e285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazejak A, Schippers A. High abundance of JS-1 and _Chloroflexi_-related Bacteria in deeply buried marine sediments revealed by quantitative, real-time PCR. FEMS Microbiol Ecol. 2010;72:198–207. [DOI] [PubMed] [Google Scholar]
- Bradley JA, Amend JP, LaRowe DE. Survival of the fewest: microbial dormancy and maintenance in marine sediments through deep time. Geobiology. 2019;17:43‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitwieser FP, Lu J, Salzberg SL. A review of methods and databases for metagenomic classification and assembly. Brief Bioinform. 2019;20:1125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60. [DOI] [PubMed] [Google Scholar]
- Buckel W, Thauer RK. Flavin-based electron bifurcation, a new mechanism of biological energy coupling. Chem Rev. 2018;118:3862–86. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun OK, Ozen V, Wankel SD et al. Quantifying population-specific growth in benthic bacterial communities under low oxygen using H2(18)O. ISME J. 2019;5:1546–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hondt S, Inagaki F, Zarikian CA et al. Presence of oxygen and aerobic communities from sea floor to basement in deep-sea sediments. Nat Geosci. 2015;8:299–304. [Google Scholar]
- D'Hondt S, Pockalny R, Fulfer VM et al. Subseafloor life and its biogeochemical impacts. Nat Commun. 2019;10:3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Greening C, Rattray JE et al. Metabolic potential of uncultured bacteria and archaea associated with petroleum seepage in deep-sea sediments. Nat Commun. 2019;10:1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhamel M, Edwards EA. Microbial composition of chlorinated ethene-degrading cultures dominated by Dehalococcoides. FEMS Microbiol Ecol. 2006;58:538–49. [DOI] [PubMed] [Google Scholar]
- Durbin AM, Teske A. Microbial diversity and stratification of South Pacific abyssal marine sediments. Environ Microbiol. 2011;13:3219–34. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792‐1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–61. [DOI] [PubMed] [Google Scholar]
- Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–8. [DOI] [PubMed] [Google Scholar]
- Estes ER, Pockalny R, D'Hondt S et al. Persistent organic matter in oxic subseafloor sediment. Nat Geosci. 2019;12:126–31. [Google Scholar]
- Fincker M, Huber JA, Orphan VJ et al. Metabolic strategies of marine subseafloor Chloroflexi inferred from genome reconstructions. Environ Microbiol. 2020;22:3188–204. [DOI] [PubMed] [Google Scholar]
- Fullerton H, Moyer CL. Comparative single-cell genomics of Chloroflexi from the Okinawa Trough deep-subsurface biosphere. Appl Environ Microb. 2016;82:3000–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futagami T, Morono Y, Terada T et al. Dehalogenation activities and distribution of reductive dehalogenase homologous genes in marine subsurface sediments. Appl Environ Microb. 2009;75:6905–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin MY, Makarova KS, Wolf YI et al. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015;43:261–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghyselinck J, Coorevits A, Van Landschoot A et al. An rpoD gene sequence based evaluation of cultured Pseudomonas diversity on different growth media. Microbiology. 2013;159:2097–108. [DOI] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–4. [DOI] [PubMed] [Google Scholar]
- Gupta RS, Chander P, George S. Phylogenetic framework and molecular signatures for the class Chloroflexi and its different clades; proposal for division of the class Chloroflexi class. nov. into the suborder Chloroflexineae subord. nov., consisting of the emended family Oscillochloridaceae and the family Chloroflexaceae fam. nov., and the suborder Roseiflexineae subord. nov., containing the family Roseiflexaceae fam. nov. Anton van Leeuw J Microb. 2013;103:99–119. [DOI] [PubMed] [Google Scholar]
- Hoehler TM, Jørgensen BB. Microbial life under extreme energy limitation. Nat Rev Microbiol. 2013;11:83–94. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 2015;12:115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug LA, Castelle CJ, Wrighton KC et al. Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome. 2013;1:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki F, Nunoura T, Nakagawa S et al. Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc Natl Acad Sci USA. 2006;103:2815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen BB, Findlay AJ, Pellerin A. The biogeochemical sulfur cycle of marine sediments. Front Microbiol. 2019;10:e849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen BB, Marshall I. Slow microbial life in the seabed. Annu Rev Mar Sci. 2016;8:311–32. [DOI] [PubMed] [Google Scholar]
- Kadnikov VV, Savvichev AS, Mardanov AV et al. Microbial communities involved in the methane cycle in the near-bottom water layer and sediments of the meromictic subarctic Lake Svetloe. Anton van Leeuw J Microb. 2019;112:1801–14. [DOI] [PubMed] [Google Scholar]
- Kallmeyer J, Pockalny R, Adhikari R et al. Global distribution of microbial abundance and biomass in subseafloor sediment. Proc Natl Acad Sci USA. 2012;109:16213–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaster A, Blackwell-Mayer K, Pasarell B et al. Single cell study of Dehalococcoidetes species from deep-sea sediments of the Peruvian Margin. ISME J. 2014;8:1831–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai M, Futagami T, Toyoda A et al. High frequency of phylogenetically diverse reductive dehalogenase-homologous genes in deep subseafloor sedimentary metagenomes. Front Microbiol. 2014;5:e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick JB, Walsh EA, D'Hondt S. Microbial selection and survival in subseafloor sediment. Front Microbiol. 2019;10:e956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittelmann S, Friedrich MW. Novel uncultured Chloroflexi dechlorinate perchloroethene to _trans_-dichloroethene in tidal flat sediments. Environ Microbiol. 2008;10:1557–70. [DOI] [PubMed] [Google Scholar]
- Landry Z, Swan BK, Herndl GJ et al. SAR202 genomes from the dark ocean predict pathways for the oxidation of recalcitrant dissolved organic matter. mBio. 2017;8:e00413–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leloup J, Fossing H, Kohls K et al. Sulfate-reducing bacteria in marine sediment (Aarhus Bay, Denmark): abundance and diversity related to geochemical zonation. Environ Microbiol. 2009;11:1278–91. [DOI] [PubMed] [Google Scholar]
- Lever MA. Acetogenesis in the energy-starved deep biosphere - a paradox? Front Microbiol. 2012;2:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd KG, Bird JT, Buongiorno J et al. Evidence for a growth zone for deep subsurface microbial clades in near-surface anoxic sediments. Appl Environ Microb. 2020;86:e02224–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loy A, Duller S, Baranyi C et al. Reverse dissimilatory sulfite reductase as phylogenetic marker for a subgroup of sulfur-oxidizing prokaryotes. Environ Microbiol. 2009;11:289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig W, Strunk O, Westram R et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löffler FE, Yan J, Ritalahti KM et al. Dehalococcoides mccartyi gen. nov., sp. nov., obligately organohalide respiring anaerobic bacteria relevant to halogen cycling and bioremediation, belong to a novel bacterial class, Dehalococcoidia classis nov., order Dehalococcoidales ord. nov. and family Dehalococcoidaceae fam. nov., within the phylum Chloroflexi. Int J Syst Evol Microbiol. 2013;63:625–35. [DOI] [PubMed] [Google Scholar]
- Löffler M, Feldhues J, Venceslau SS et al. DsrL mediates electron transfer between NADH and rDsrAB in Allochromatium vinosum. Environ Microbiol. 2020;22:783–95. [DOI] [PubMed] [Google Scholar]
- Matturro B, Frascadore E, Rossetti S. High-throughput sequencing revealed novel Dehalococcoidia in dechlorinating microbial enrichments from PCB-contaminated marine sediments. FEMS Microbiol Ecol. 2017;93:fix134. [DOI] [PubMed] [Google Scholar]
- Mehrshad M, Rodriguez-Valera F, Amoozegar MA et al. The enigmatic SAR202 cluster up close: shedding light on a globally distributed dark ocean lineage involved in sulfur cycling. ISME J. 2018;12:655–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller AL, Kjeldsen KU, Rattei T et al. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 2015;9:1152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunoura T, Nishizawa M, Hirai M et al. Microbial diversity in sediments from the bottom of the Challenger Deep, the Mariana Trench. Microbes Environ. 2018;33:186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi WD, Jørgensen BB, Biddle JF. Transcriptional analysis of sulfate reducing and chemolithoautotrophic sulfur oxidizing bacteria in the deep subseafloor. Environ Microbiol Rep. 2016;8:452–60. [DOI] [PubMed] [Google Scholar]
- Orsi WD, Morard R, Vuillemin A et al. Anaerobic metabolism of Foraminifera thriving below the seafloor. ISME J. 2020b;14:2580–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi WD, Richards TA, Francis WR. Predicted microbial secretomes and their target substrates in marine sediment. Nat Microbiol. 2018;3:32–37. [DOI] [PubMed] [Google Scholar]
- Orsi WD, Schink B, Buckel W et al. Physiological limits to life in anoxic subseafloor sediment. FEMS Microbiol Rev. 2020c;44;fuaa004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi WD, Vuillemin A, Rodriguez P et al. Metabolic activity analyses demonstrate that Lokiarchaeon exhibits homoacteogenesis in sulfidic marine sediments. Nat Microbiol. 2020a;5:248–55. [DOI] [PubMed] [Google Scholar]
- Orsi WD. Ecology and evolution of seafloor and subseafloor microbial communities. Nat Rev Microbiol. 2018;16:671–83. [DOI] [PubMed] [Google Scholar]
- Ortega-Arbulù A-S, Pichler M, Vuillemin A et al. Effect of organic matter and low oxygen on the mycobenthos in a coastal lagoon. Environ Microbiol. 2019;21:374–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes RJ, Cragg B, Roussel E et al. A review of prokaryotic populations and processes in sub-seafloor sediments, including biosphere:geosphere interactions. Mar Geol. 2014;352:409–25. [Google Scholar]
- Parkes RJ, Webster G, Cragg B et al. Deep sub-seafloor prokaryotes stimulated at interfaces over geological time. Nature. 2005;436:390–4. [DOI] [PubMed] [Google Scholar]
- Peters JW, Miller A-F, Jones AK et al. Electron bifurcation. Curr Opin Chem Biol. 2016;31:146–52. [DOI] [PubMed] [Google Scholar]
- Petro C, Zäncker B, Starnawski P et al. Marine deep biosphere microbial communities assemble in near-surface sediments in Aarhus Bay. Front Microbiol. 2019;10:e758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichler M, Coskun ÖK, Ortega AS et al. A 16S rRNA gene sequencing and analysis protocol for the Illumina MiniSeq platform. MicrobiologyOpen. 2018;7:e00611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho M, Tang H, Ye Y. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acid Res. 2010;38:e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Røy H, Kallmeyer J, Adhikari RR et al. Aerobic microbial respiration in 86-million-year-old deep-sea red clay. Science. 2012;336:922–5. [DOI] [PubMed] [Google Scholar]
- Salter SJ, Cox MJ, Turek EM et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saw J, Nunoura T, Hirai M et al. Pangenomics analysis reveals diversification of enzyme families and niche specialization in globally abundant SAR202 Bacteria. mBio. 2020;11:e02975–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Say RS, Fuchs G. Fructose 1,6-biphosphate aldolase/phosphatase may be an ancestral gluconeogenic enzyme. Nature. 2010;464:1077–81. [DOI] [PubMed] [Google Scholar]
- Schuchmann K, Müller V. Energetics and application of heterotrophy in acetogenic bacteria. Appl Environ Microb. 2019;82:4056–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri R, Adrian L, Fouts DE et al. Genome sequence of the PCE-dechlorinating Bacterium Dehalococcoidetes ethenogenes. Science. 2005;307:105–8. [DOI] [PubMed] [Google Scholar]
- Sewell HL, Kaster A-K, Spormann AM. Homoacetogenesis in deep-sea Chloroflexi, as inferred by single-cell genomics, provides a link to reductive dehalogenation in terrestrial Dehalococcoidetes. mBio. 2017;8:e02022–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafaat HS, Rüdiger O, Ogata H et al. [NiFe] hydrogenases: a common active site for hydrogen metabolism under diverse conditions. Geochim Cosmochim Ac. 2013;1827:986–1002. [DOI] [PubMed] [Google Scholar]
- Solden L, Lloyd K, Wrighton K. The bright side of microbial dark matter: lessons learnt from the uncultivated majority. Curr Opin Microbiol. 2016;31:217–26. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Aberer AJ, Goll C et al. RAxML-Light: a tool for computing terabyte phylogenies. Bioinformatics. 2012;28:2064‐2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312‐1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starnawski P, Bataillon T, Ettema TJG et al. Microbial community assembly and evolution in subseafloor sediment. Proc Natl Acad Sci USA. 2017;114:2940–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignais PM, Billoud B, Meyer J. Classification and phylogeny of hydrogenases. FEMS Microbiol Rev. 2001;25:455–501. [DOI] [PubMed] [Google Scholar]
- Vuillemin A, Ariztegui D, Horn F et al. Microbial community composition along a 50,000-year lacustrine sediment sequence. FEMS Microbiol Ecol. 2018a;94:fiy029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillemin A, Horn F, Friese A et al. Metabolic potential of microbial communities from ferruginous sediments. Environ Microbiol. 2018b;20:4297–313. [DOI] [PubMed] [Google Scholar]
- Vuillemin A, Vargas S, Coskun ÖK et al. Atribacteria reproducing over millions of years in the Atlantic abyssal subseafloor. mBio. 2020;11:e01937–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillemin A, Wankel SD, Coskun ÖK et al. Archaea dominate oxic subseafloor communities over multimillion-year time scales. Sci Adv. 2019;5:eaaw4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Větrovský T, Badrian P. The variability of 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One. 2013;8:e57923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh EA, Kirkpatrick JB, Rutherford SD et al. Bacterial diversity and community from seasurface to subseafloor. ISME J. 2016;10:979–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li Z, Zeng L et al. The oxidation of hydrocarbons by diverse heterotrophic and mixotrophic bacteria that inhabit deep-sea hydrothermal ecosystems. ISME J. 2020;14:1994–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmund K, Cooper M, Schreiber L et al. Single-cell genome and group-specific dsrAB sequencing implicate marine members of the Class Dehalococcoidia (Phylum Chloroflexi) in sulfur cycling. mBio. 2016;7:e00266–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmund K, Mussmann M, Loy A. The life sulfuric: microbial ecology of sulfur cycling in marine sediments. Environ Microbiol Rep. 2017;9:323–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmund K, Schreiber L, Lloyd KG et al. Genome sequencing of a single cell of the widely distributed marine subsurface Dehalococcoidia, phylum Chloroflexi. ISME J. 2014;8:383–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiechmann A, Ciurus S, Oswald F et al. It does not always take two to tango: “Syntrophy” via hydrogen cycling in one bacterial cell. ISME J. 2020;14:1561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilms R, Köpke B, Sass H et al. Deep biosphere-related bacteria within the subsurface of tidal flat sediments. Environ Microbiol. 2006;8:709–19. [DOI] [PubMed] [Google Scholar]
- Yamada T, Imachi H, Ohashi A et al. Bellilinea caldifistulae gen. nov., sp. nov. and Longilinea arvoryzae gen. nov., sp. nov., strictly anaerobic, filamentous bacteria of the phylum Chloroflexi isolated from methanogenic propionate-degrading consortia. Int J Sys Evol Microb. 2017;57:2299–306. [DOI] [PubMed] [Google Scholar]
- Yamada T, Sekiguchi Y, Hanada S et al. Anaerolinea thermolimosa sp. nov., Levilinea saccharolytica gen. nov., sp. nov. and Leptolinea tardivitalis gen. nov., sp. nov., novel filamentous anaerobes, and description of the new classes Anaerolineae classis nov. and Caldilineae classis nov. in the bacterial phylum Chloroflexi. Int J Sys Evol Microb. 2006;56:1331–40. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zhang Y, Cápiro NL et al. Genomic characteristics distinguish geographically distributed Dehalococcoidia. Front Microbiol. 2020;11:e546063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Liu Y, Xu W et al. Genome- and community-level interaction insights into carbon utilization and element cycling functions of Hydrothermarchaeota in hydrothermal sediment. mSystems. 2020;5:e00795–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang W-Q, Yi S, Bill M et al. Incomplete Wood–Ljungdahl pathway facilitates one-carbon metabolism in organohalide-respiring Dehalococcoides mccartyi. Proc Natl Acad Sci USA. 2014;111:6419–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Gao B. Bacterial flagella loss under starvation. Trends Microbiol. 2020; 28785–788. [DOI] [PubMed] [Google Scholar]
- Zinger L, Amaral-Zettler LA, Fuhrman JA et al. Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PlosONE. 2011;6:e24570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
fiaa223_Supplemental_File