Tumor Mutational Burden (TMB) as a Predictive Biomarker in Solid Tumors (original) (raw)
. Author manuscript; available in PMC: 2021 Jun 1.
Abstract
TMB, defined as the number of somatic mutations per megabase of interrogated genomic sequence, varies across malignancies. Panel sequencing-based estimates of TMB have largely replaced whole exome sequencing-derived TMB in the clinic. Retrospective evidence suggests that TMB can predict the efficacy of immune checkpoint inhibitors, and data from KEYNOTE-158 led to the recent FDA approval of pembrolizumab for the TMB-high tumor subgroup. Unmet needs include prospective validation of TMB cutoffs in relationship to tumor type and patient outcomes. Furthermore, standardization and harmonization of TMB measurement across test platforms are important to the successful implementation of TMB in clinical practice.
Keywords: TMB, cancer, prediction, MSI, PD-L1, PD-1
Introduction
A major advance in cancer treatment is the development of immune checkpoint inhibitors (ICIs) that have produced durable responses and improved survival in multiple solid malignancies (1–8). However, a majority of patients treated with ICIs do not derive benefit and therefore, identification of predictive biomarkers of ICIs response are needed to enable more selective use of ICIs as well as to elucidate and overcome mechanisms of treatment resistance. Tumor mutational burden (TMB) is broadly defined as the number of somatic mutations per megabase of interrogated genomic sequence. TMB is believed to be a key driver in the generation of immunogenic neopeptides displayed on major histocompatibility complexes (MHC) on the tumor cell surface that influence patient response to ICIs. Tumor-specific neoantigens arise from somatic mutations (9,10) and can play a pivotal role in tumor-specific T cell-mediated, anti-tumor immunity after inhibition of checkpoint signals (11–14). In addition to neoantigen quantity, evidence suggests that quaIity may also be important in that high-quality neoantigens might include expressed clonal neoantigens in essential genes, which bind to multiple HLA alleles and cannot be repressed or deleted by virtue of their genomic position (15). Accumulating evidence suggests that TMB may be a predictive biomarker of tumor response to ICIs in several cancer types (10,16,17). The most robust initial responses to ICIs were observed in melanoma and non-small cell lung cancer (NSCLC) which typically have high mutation burden owing to the mutagenic effects of ultraviolet light and tobacco smoke, respectively (18). Subsequently, significant associations between high TMB and response to ICIs were reported in other solid tumor types (19–21). However, outliers have been observed that include renal cell carcinoma and Merkel cell carcinoma which responded better than expected on the basis of TMB alone, suggesting the importance of other as yet undefined factors (16,22,23). In a study in patients with previously treated, unresectable or metastatic solid tumors (KEYNOTE-158), TMB-high status (≥10 mut/Mb) was associated with a clinically meaningful improvement in efficacy of the anti-PD-1 antibody, pembrolizumab (24). Responses were observed across tumor types and MSI-H status did not account for all of the increased clinical benefit in the TMB-high subgroup (24). Based on these data, the U.S. Food and Drug Administration (FDA) approved pembrolizumab monotherapy for the subgroup of solid tumor patients with TMB ≥10 mut/Mb. While data from KEYNOTE-158 demonstrated a role for TMB in selection of patients for cancer treatment, important issues remain including the selection and implementation of a fixed TMB cutoff based on pan-cancer data. In this regard, observed cancer-type-related differences in TMB distributions are relevant to determining optimal TMB cutoffs to enable its use as a predictive biomarker for immunotherapy.
Tumor TMB is accurately measured by whole exome sequencing (WES), but this approach is impractical for use in the clinic. While panel-based sequencing of tumor tissue is commonplace in clinical practice, differences in panel size, mutation types, and bioinformatic platforms exist. TMB is associated with certain other biomarkers including microsatellite instability-high (MSI-H) that is detected in a subset of human cancers and is due to deficient DNA mismatch repair (dMMR). Tumors with MSI-H/dMMR typically display high TMB (25–27), and MSI-H/dMMR is an established predictive biomarker for the efficacy of ICIs. Expression of programmed death ligand-1 (PD-L1) on tumor cells and immune cells has become a widely used predictive biomarker for responsiveness to ICIs in several cancer types (6,28–31). However, TMB levels and response to immunotherapy in many cancers is independent of the level of PD-L1 expression (4,32,33) , suggesting a potential role of TMB to identify additional subgroups of patients who may benefit from ICIs (34). In this review, we will discuss the need for a consensus definition of TMB as well as the need to standardize TMB measurement among gene panels. Furthermore, we will review TMB variability across solid tumors with implication for cutoff selection, its association with benefit from ICIs, and discuss strategies to optimize TMB as a predictive biomarker for ICIs in clinical practice. We also discuss the current conundrum of supportive retrospective evidence, but the relative paucity of prospective data confirming the clinical utility of TMB.
Challenges in TMB definition and measurement
TMB can be assessed using a number of next-generation sequencing (NGS) platforms, including whole-genome sequencing (WGS), WES, or targeted panel sequencing. WES is the “gold standard” for measuring TMB, allowing for the detection of somatic coding mutations (non-synonymous) present within the entire exome. WES targets ~30 Mb of coding regions, covering all ~22 000 genes and making up ~1% of the genome. The definition of TMB varies by the measurement method utilized. While TMB has been accurately measured by WES in several studies, this is currently not feasible in clinical practice due to its high cost, relatively long turnaround time, and the need for sufficient tissue samples. Multiple commercially available gene panels designed for TMB estimation cover between approximately 0.80 and 2.40 Mb representing <5% of the total coding sequence (35–44). The number of genes in each of the gene panels ranges between 324 and 595 genes. Importantly, panels are not necessarily limited to the coding regions since many panels include intronic regions needed for gene fusion detection. Shown in Fig. 1 are confidence intervals relative to the level of TMB of four theoretical gene panels (size from 0.5 Mb to 4Mb) that vary markedly according to panel size. The coefficient of variation (CV) of TMB derived from panel sequencing decreases in a manner that is inversely proportional with both the square root of the panel size and the square root of the TMB level; for example, halving the CV requires a four-fold increase in panel size (45). Multiple NGS panels are commercially available that include Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) and FoundationOne CDx® (Foundation Medicine, Inc.), both of which are approved by the U.S. Food and Drug Administration (FDA), that cover ~1.14 Mb over 468 genes or ~0.8 Mb over 324 genes, respectively (Table 1). Each commercial laboratory uses their own bioinformatic algorithms and workflow that were optimized using sequencing methods, mutation types, and filters that best suit their own panel specifications.
Figure 1.
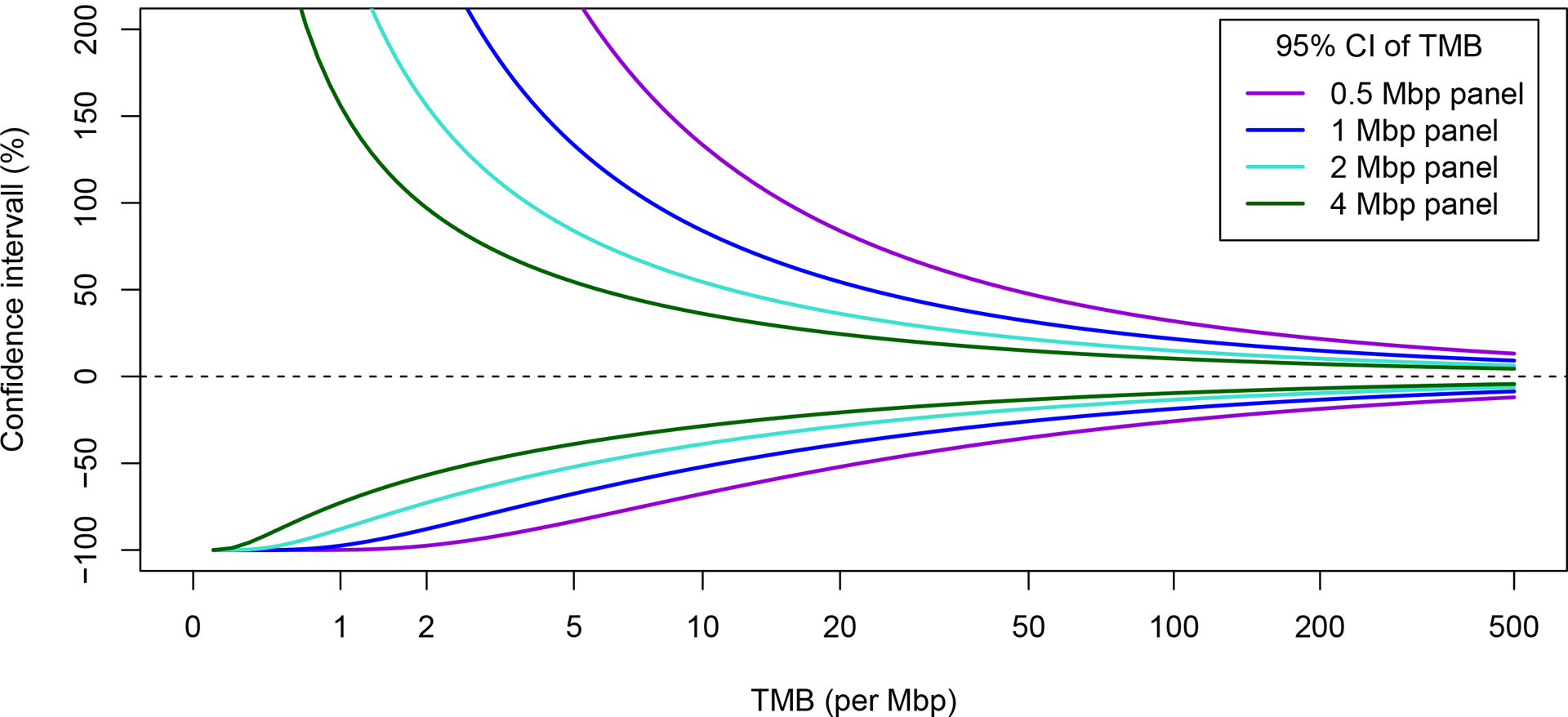
Panel-sequencing derived TMB. Confidence intervals (CIs) of panel sequencing-derived measurement of TMB showing the stochastic variability due to limited panel size. CIs can be reconstructed (TMB - lower limit in % × TMB, TMB + upper limit in % × TMB) from the upper and lower limits that are presented as percentages relative to the TMB. CIs were calculated using the Clopper-Pearson method (134).
Table 1.
Diagnostic Next Generation Sequencing (NGS) Panels Enable TMB Determination**
| Laboratory | Panel name | Number of genes | Total region covered (Mb) | TMB region covered* (Mb) | Type of exonic mutations included in TMB estimation | References |
|---|---|---|---|---|---|---|
| ACT Genomics | ACTOnco+ | 440 | 1.80 | 1.12 | Non-synonymous†, synonymous | NA |
| Caris | SureSelect XT | 592 | 1.60 | 1.40 | Non-synonymous† | (38) |
| Foundation Medicine | FoundationOne CDx®‡ | 324 | 2.20 | 0.80 | Non-synonymous, synonymous | (35–37) |
| Guardant Health | GuardantOMNI§ | 500 | 2.15 | 1.00 | Non-synonymous, synonymous | (39) |
| Illumina | TSO500 (TruSight Oncology 500) | 523 | 1.97 | 1.33 | Non-synonymous, synonymous | (47) |
| Memorial Sloan Kettering Cancer Center | MSK-IMPACT¶ | 468 | 1.53 | 1.14 | Non-synonymous | (40,41) |
| NeoGenomics | NeoTYPE Discovery Profile for Solid Tumors | 372 | 1.10 | 1.03 | Non-synonymous, synonymous | NA |
| Personal Genome Diagnostics | PGDx elio tissue complete | 507 | 2.20 | 1.33 | Non-synonymous, synonymous | (42) |
| QIAGEN | QIAseq TMB panel | 486 | 1.33 | 1.33 | Non-synonymous, synonymous | NA |
| Thermo Fisher Scientific | Oncomine Tumor Mutation Load Assay | 409 | 1.70 | 1.20 | Non-synonymous | (42,43) |
| TEMPUS | TEMPUS Xt | 595 | 2.40 | 2.40 | Non-synonymous | (44) |
TMB outputs from gene panel assays are usually normalized to mutations per Mb because they differ in the number of genes and target region size. Both MSK-IMPACT and FoundationOne CDx® panels detect somatic coding mutations (non-synonymous) per megabase of tumor genome examined, inclusive of frameshift, point mutations, and small insertions and deletions (indels) (see Supplementary Data). While synonymous mutations are detected by these panels (not reported by MSK-IMPACT), they are not involved in neoantigen production although their inclusion may reduce sampling noise and improve the approximation of TMB across the whole genome if tumor-normal pairs are sequenced (35). MSK-IMPACT and FoundationOne CDx® have been shown to be moderately concordant with WES in TMB assessment (35,46). Importantly, differences such as the location and size of the sequenced region, the number of sequenced genes, the mutation types detected, as well as differences in the definition of TMB among panels, create confusion in the interpretation of TMB and in comparing TMB values across test platforms. Genetic changes in cancer include non-synonymous (including missense, nonsense, frameshift and splice-site mutations) and synonymous mutations, insertion or deletion mutations (indels), and gene copy number alterations (CNAs). Indel calling can vary depending on the bioinformatic pipeline used, and whether indels generate a higher number of immunogenic neoantigens needs to be determined. Calculation of TMB from panel-based sequencing data has important limitations (35). There is a need to harmonize the types of mutations analyzed as missense mutations are included in all panels, but other types can vary. Panel sequencing-derived TMB measurement extrapolates the total number of mutations in the coding sequence by analysis of a limited panel of genes. For clinical purposes, evidence suggests that gene panels of at least 1 Mb are needed for TMB measurement (35,47,48). Even for large panels, the stochastic error related to panel size represents the largest of all contributions to total TMB variance (49). One can convert mutation number from WES data to mut/Mb that is reported in gene panel sequencing data (50); however, it is dependent on multiple sequencing-related parameters (e.g., sequencing methodology, WES enrichment kits, bioinformatics pipelines, etc.) such that a static conversion rate does not exist.
Other relevant factors include the bioinformatic protocols used to calculate TMB and methods of filtering germline mutations (43,47,48,51,52). Germline mutation filtering is an important step in panel-based TMB measurement since only somatic mutations in the tumor can be recognized by the immune system. Germline mutation filtering can be performed in silico using bioinformatic pipelines or alternatively, paired normal tissue or blood samples can be sequenced and used as a filtering tool. In panel-based sequencing, fewer total mutations were called when a patient-matched normal tissue was used in variant calling because germline SNVs were appropriately filtered out (53). TMB variability introduced by errors in somatic mutation detection is only moderate compared to stochastic error related to panel size and other confounders (54). Panel size is the most important contributor to TMB variance since panels represent only a very small proportion of the exome, especially in tumors of small to medium TMB (up to 10 mut/Mb) (54). With regard to gene panel composition and its contribution to TMB variability, an in silico study found that TMB variability increased by 8% when using 1 MB panels composed of oncogenes and tumors suppressor genes (TSGs) compared to 1 MB panels composed of random genes (45). Thus, including oncogenes and TSGs only slightly increases TMB variability. Bioinformatics pipelines usually include negative filtering for cancer hotspot mutations further mitigating the influence of the panel composition.
Variability in TMB across solid tumors
TMB is a continuous variable and variability of TMB (ranging from 0.001/Mb to more than 1000/Mb) has been observed across and within cancer types (18,35,41). Studies indicate that some cancer types have less variability in TMB such as lung and head and neck cancers, and some having greater variability such as colon, bladder, and uterine cancers (55). Cancers related to chronic mutagenic exposures such as lung (tobacco) and melanoma (UV light), exhibit the highest TMB whereas leukemia and certain childhood cancers have the lowest TMB (18). In an analysis of 24 cancer types using the TCGA database, only three cancer types, adenocarcinoma of the colorectum, stomach and uterus, harbor bi- or multimodal TMB distribution (56) (Fig. 2A). Also shown are the percentage of tumors with TMB above 10 mut/MB and the contribution of MSI-H. In these cancers, TMB distribution is shaped by the occurrence of hypermutation in MMR deficient and/or POLE/POLD1 mutated tumors and permits relatively clean dichotomization. For most other cancer types including adenocarcinoma or squamous cell carcinoma of the lung or cutaneous melanoma, TMB is unimodally distributed with a dense point cloud of TMB scores scattered around the cutoff (56) (Fig. 2A). Eleven cancer types include a high or at least moderate percentage of tumors above the cutoff of 10 mut/Mb: melanoma (71%), lung squamous cell carcinoma (50%), lung adenocarcinoma (44%), uterus adenocarcinoma (39%), transitional cell carcinoma (36%), stomach adenocarcinoma (29%), head and neck SCC (19%), colorectal carcinoma (18%), cervix carcinoma (15%), esophagus adenocarcinoma (9%) and sarcoma (6%) (Fig. 2A). The percentage of tumors above the cutoff for the remaining 13 cancer types was low (<5%). For uterus, stomach and colorectal carcinomas, the majority of tumors above the cutoff were MSI-H (77%, 69% and 78%). Accordingly, 9% of adenocarcinomas of the uterus and stomach and 4% of colorectal carcinomas were above the cutoff and MSS/MSI-L. Additional data for TMB distributions is provided in sections for individual tumor types.
Figure 2.
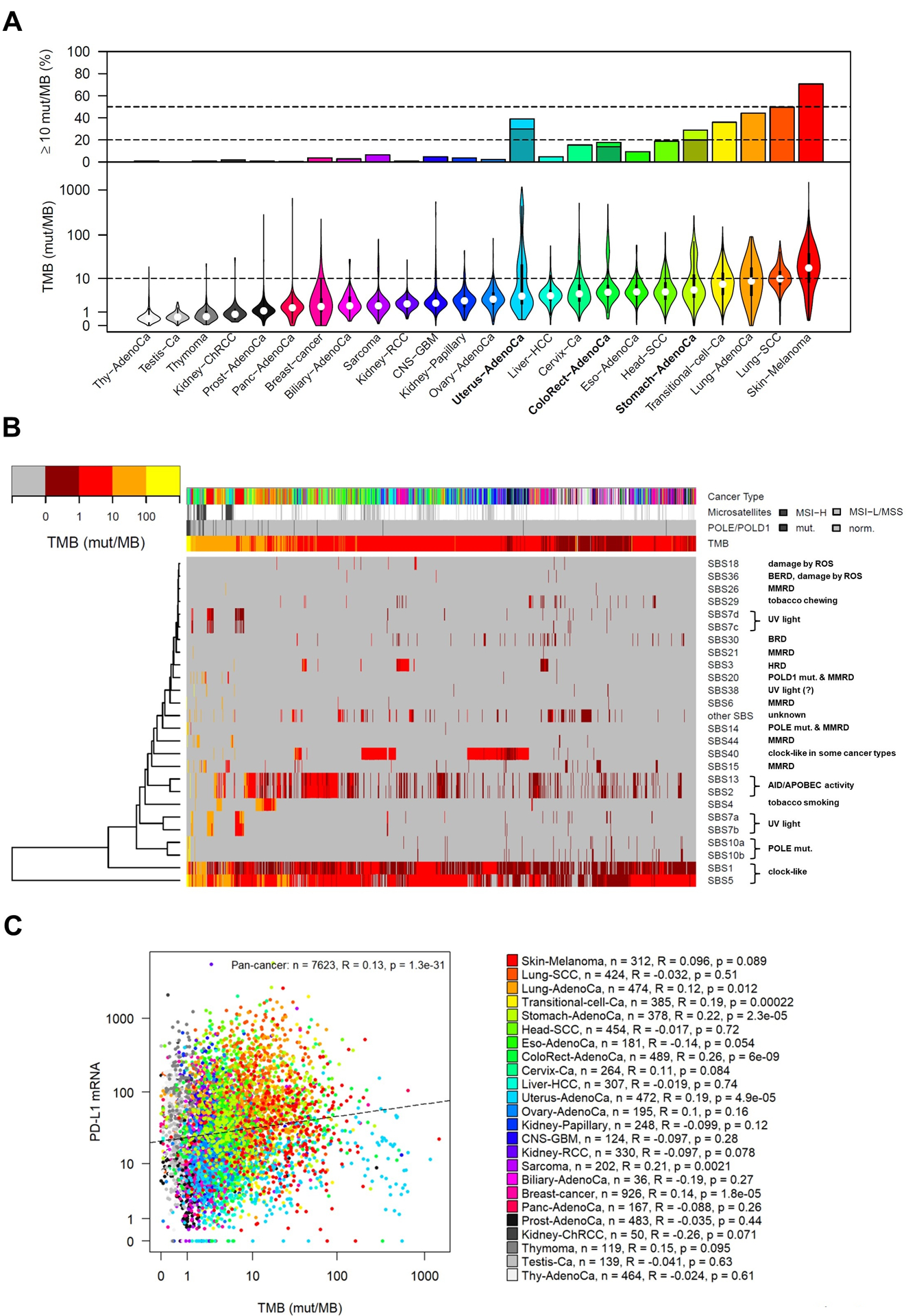
Analysis of TMB and mutational signatures in 24 cancer types (8273 tumors, TCGA pan-cancer atlas). Numbers of missense mutations detected by WES were converted to TMB per Mb using the correspondence of 199 mutations to 10 mut/Mb (50). A, Violin plots show markedly different median TMB levels and TMB variability in different cancer types. For most cancer types, unimodal distributions of TMB were observed, but bimodal distributions were observed in adenocarcinomas of the colorectum and stomach, and a multimodal distribution was observed in uterine adenocarcinoma. Upper panel: Percentage of tumors with TMB above 10 mut/Mb including MSI-H adenocarcinoma of uterus, colorectum and stomach (dark bars). B, Heatmap and hierarchical clustering of 26 single base substitution (SBS) signatures using the Manhattan distance and the average linkage method (135). SBS signatures are annotated by the known or putative underlying mutational processes. SBS signatures that are not linked to an underlying mutational process (other SBS) were pooled. Mutational processes that can cause a very high TMB and hypermutation include POLE/POLD1 mutations (SBS10a, 10b, 14 and 20), DNA mismatch repair deficiency (SBS6, SBS15, SBS21, SBS15 and SBS44), UV light (SBS7a and 7b), tobacco smoking (SBS4), AID/APOBEC activation (SBS2 and SBS13) and the three clock-like processes (SBS1 and SBS5). MSI = microsatellite instability, MMRD = mismatch repair deficiency, HRD = homologous recombination deficiency, BERD = base excision repair deficiency. A detailed description of the methods for analysis of mutational signatures can be found at (https://www.nature.com/articles/nature12477). C, Correlation analysis of PD-L1 mRNA expression and TMB level. Significant positive correlations were observed in 5 cancer types, while correlations were not significant in the remaining 19 cancer types. R = Spearman correlation, * = significant after Bonferroni correction (p < 0.05/24). Somatic mutation and mRNA expression data were obtained from [https://gdc.cancer.gov/about-data/publications/pancanatlas], MSI data from [https://gdac.broadinstitute.org] and the levels of the SBS mutational signatures from [https://www.synapse.org/#!Synapse:syn11804040].
Thousands of somatic mutations can now be identified in single cancer samples offering the possibility of deciphering various mutational signatures even when they are caused by several mutational processes (18,57) that can differ by cancer types and between individual tumors. Using the TCGA pan-cancer cohort, analysis of TMB and mutational signatures was performed in 24 cancer types (8,273 tumors), and a new type of heatmap was generated to analyze the contribution of single base substitution-derived mutational signatures to TMB and in particular, to hypermutation and ultra-hypermutation (Fig. 2B). To gain insight in the biological processes underlying high TMB in specific tumors, we took advantage of a method of cancer genome analysis recently developed by Alexandrov and Stratton (18). Ultra-hypermutation (>100 mut/MB) or hypermutation (>10 mut/MB) can either result from the activity of a single mutational process or by accumulation over multiple such processes. Mutational processes that can cause a very high TMB and hypermutation include: POLE/POLD1 mutation, mismatch repair deficiency, UV light, tobacco smoking, AID/APOBEC activation and the three clock-like mutational processes (SBS1, SBS5) (Fig. 2B). TMB is to a large extent independent of PD-L1 status in most cancers (34) as shown in an analysis of 24 cancer types included in the TCGA database (Fig. 2C).
A TMB cutoff is a function of the gene panel (genomic footprint and bioinformatics platform) that is used in a given study. Since data obtained from a given gene panel cannot be directly applied to another panel without a conversion algorithm, direct comparisons of results between panels can be very problematic. As indicated, there are cancer-type related biological TMB distributions and accordingly, these distributions may be relevant to evaluation of the clinical utility of TMB and for determining optimal TMB cutoffs. Due to diverse TMB distributions, optimal TMB cutoff values to discriminate potential responder’s vs non-responders to ICIs may vary significantly among cancer types (35,58). Cohort-specific TMB cutoffs have been defined differently across studies, tumor types, testing platforms and using variable bioinformatics methods. Importantly, cancer type-related biological TMB distributions are distinct from prognostic cutoffs whereby the former may not predict the latter.
TMB as a predictive biomarker for cancer immunotherapy
The rationale for the association between TMB and benefit from immunotherapy is based on the hypothesis that tumor mutation-specific neoantigens can be displayed on major histocompatibility complexes (MHC) on the tumor cell surface, and then recognized by tumor infiltrating T-cells. Accordingly, a higher TMB will generate more neoantigens that can then trigger intratumoral T-cells whose ability to attack and destroy tumor cells is enabled by ICIs. (10,17,59). The first evidence to support this hypothesis came from studies of melanoma and NSCLC treated with anti-CTLA-4 and anti-PD-1 antibodies, respectively, whereby a higher nonsynonymous mutation burden was associated with improved objective response rate (ORR) and progression-free survival (PFS) (10,17). Using an optimized receiver operating characteristic (ROC) in patients with melanoma, TMB levels that were divided into high (>23.1 mut/Mb), intermediate (3.3–23.1 mut/Mb), and low (<3.3 mut/Mb) groups showed superior prediction of ICI efficacy compared with a binary classification (60). Evaluation of TMB in a NSCLC (CheckMate 568) study using the FoundationOne Cdx® assay utilized ROC curves to determine an optimal TMB cutoff in patients receiving first-line therapy with nivolumab plus ipilimumab. The ORR was increased in patients with higher TMB, and the benefit was observed to plateau with a TMB threshold of ≥10 mut/Mb. This TMB cutoff of ≥10 mut/Mb was subsequently evaluated in the first prospective phase III trial known as CheckMate 227 in patients with NSCLC where TMB served as the co-primary efficacy endpoint (61–63). Patients whose tumors had a prespecified TMB cutoff of ≥10 mut/Mb had significantly prolonged PFS, but not OS (independent of PD-L1 expression) with a combination of nivolumab + ipilimumab vs. chemotherapy (63). Despite the negative data for OS, PFS is considered to be more informative for predictive biomarker evaluation since OS is influenced by therapies given after progression on ICIs.
Multiple studies have shown an association between TMB level and the efficacy of immunotherapy in melanoma, lung cancer and urothelial cancer, although TMB only weakly discriminated responders from nonresponders (AUC 0.6 to 0.7) among these tumor types (6,10,17,59,61,64). In a pan cancer analysis of 151 patients treated with anti-PD-1/PD-L1 monotherapy, tumor response rate and TMB level were linearly associated and there was also a significant association with the dichotomized TMB level (≥20 mut/Mb) which was consistent across tumor types (65). A nonrandomized and open label phase II study of pembrolizumab monotherapy was conducted in patients with multiple advanced solid tumors who progressed on or were intolerant to one or more lines of prior therapy (KEYNOTE-158). In this study, 751 patients had evaluable TMB data and of these, 99 (13.2%) were TMB-high (≥10 mut/Mb per FoundationOne CDx®). The most common tumor types that showed TMB-high included SCLC (34.3%) and carcinomas of the uterine cervix (16.2%), endometrium (15.2%), and anus (14.1%). Most common non-TMB high tumors were mesothelioma (12.7%) and carcinomas of neuroendocrine origin (12.3%), salivary gland (12.0%) and endometrium (10.3%). The ORR and PFS were superior in patients with TMB-high vs. low tumors (ORR: 30.3% vs 6.8%; PFS at 12 months: 26.4% vs 14.1%). Among the TMB-high group, 85/99 (85.9%) tumors were microsatellite stable (MSS) indicating that MSI-H status did not account for the predictive utility of TMB-high (24). Based on these impressive data, the FDA approved pembrolizumab monotherapy for the treatment of patients with solid tumors showing TMB-high status (≥10 mut/Mb), as determined by an FDA-approved test, who had progressed following prior treatment and lack alternative treatment options. In this report, we analyzed 24 cancer types (8,273 tumors) from the TCGA database whereby TMB-high (≥10 mut/Mb) was found in more than 20% of cutaneous melanomas, NSCLC (squamous carcinoma and adenocarcinoma), transitional cell carcinoma, and adenocarcinomas of stomach and uterus (of which most are MSI-H) (Fig. 2A). In addition, more than 10% of squamous carcinomas of the head/neck and cervix and colorectal adenocarcinomas (most MSI-H) were TMB-high. Using this cutoff for TMB, many solid tumors are eligible for treatment with ICIs with the potential for clinical benefit. While the KEYNOTE-158 data and related FDA approval are practice changing, a pan-cancer and prespecificed TMB-high cutoff may not be an optimal approach for individual tumor types.
We acknowledge that cutoff values for TMB are critical for use in clinical decision-making and propose three potential approaches to determine cutoff values of TMB as a predictive biomarker: (1) a single cutoff for all cancer types, i.e., one-size fits all that is ideally determined in a pan-cancer trial; (2) cancer-specific cutoffs which increase complexity and cost, and (3) a variable cutoff that relates to some upper percentiles for each individual cancer type (58). A pan-cancer TMB cutoff, as approved by FDA for use of pembrolizumab, is intended to enrich for responders to ICIs and thus, enables patient selection for such therapy. However, a static cutoff for TMB may not be an optimal approach to identify tumor type-specific responders to ICIs and thus, fine tuning of TMB cutoffs by tumor type is an area of future research. A further limitation of a pan-cancer TMB cutoff approach is illustrated using a dataset where genomic correlates of response to ICIs were examined in MSS solid tumors (66). Using this dataset of MSS solid tumors, we analyzed the impact of TMB cutoff selection on sensitivity and specificity of ICI response prediction. As shown in Fig. 3A–D, a cutoff of 10 mut/Mb corresponded to a sensitivity of approximately 75% for melanoma, lung and bladder cancers although specificity varied widely and was much lower for melanoma (44%) than for lung and bladder cancers (89% and 70%). Using this cutoff, 20–25% of responders would be missed. Decreasing the cutoff to 5 mut/Mb would increase sensitivity, but at the expense of decreasing specificity to 29%, 68% and 20% for the cancer types under consideration. This analysis raises concern for use of a tumor type–agnostic designation for high TMB to predict ICI response. If a cutoff is selected for each specific cancer type, then complexity is increased in that prospective clinical trials are needed to validate TMB cutoffs in individual tumor types. The third approach requires a comparator set/benchmark against which percentiles are chosen. A universal standard would be needed (how many cases, which tumor types, which spectrum of TMB). Regardless, all three approaches and their determined cutoffs are a function of the gene panels used such that comparability is ideally achieved only if the same panel is used. Other potential options in need of further research are probabilistic approaches of low-intermediate-high cutoffs. A current limitation for in-depth comparative study of the three approaches as well as for TMB cutoff optimization is the limited number of patients with high quality outcome data and available TMB data. While statistical methods such as Subpopulation Treatment Effect Pattern Plot (STEPP) and Cutoff Finder for cutoff optimization are available (67,68), clinical studies are typically well-powered for comparison of immunotherapy with a reference therapy, but underpowered for biomarker analysis with a fixed cutoff and even more underpowered for cutoff optimization.
Figure 3.
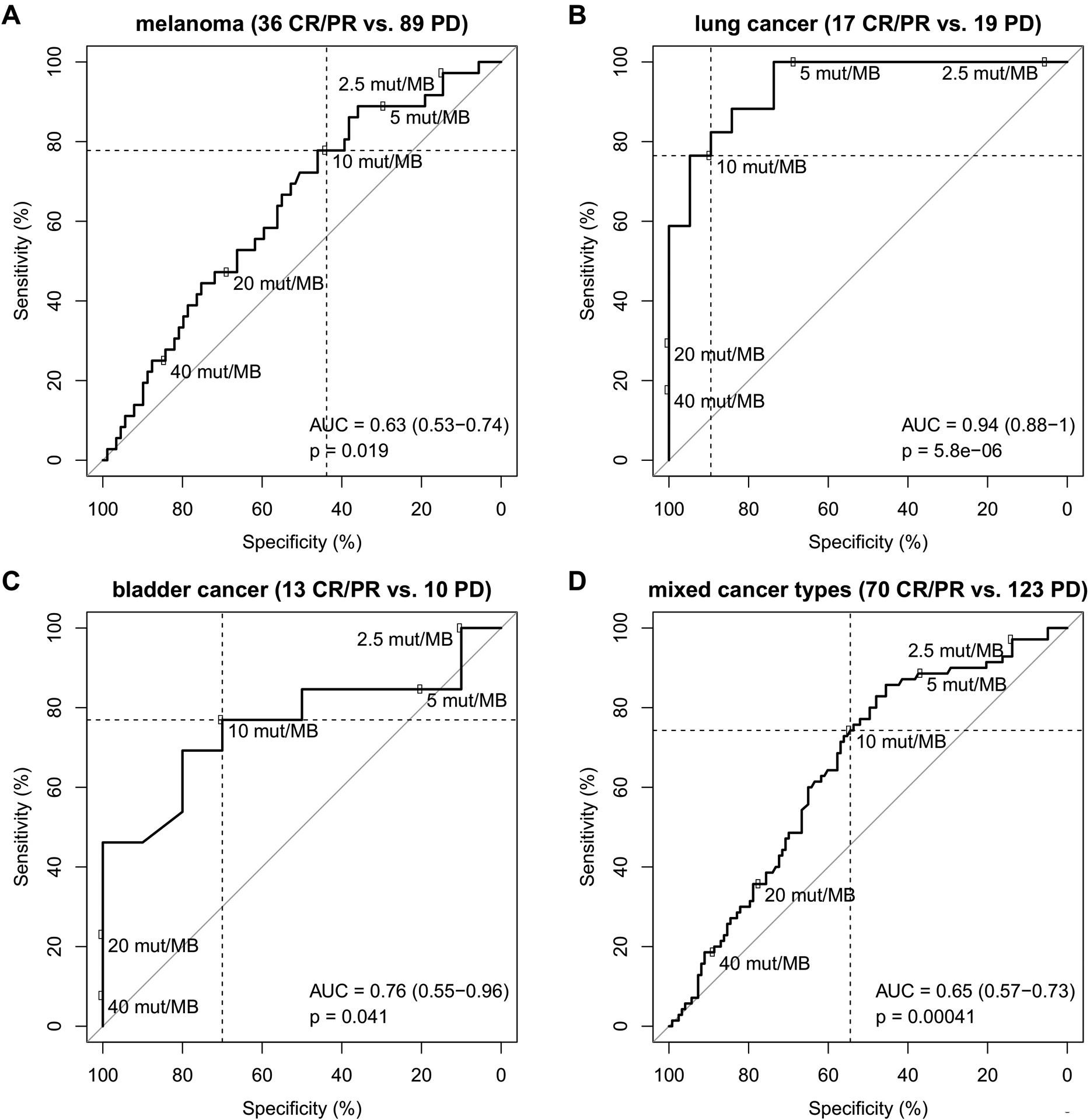
Impact of TMB cutoff choice on sensitivity and specificity of response prediction. Analysis of the Miao et al. cohort (66) of ICB-treated patients with microsatellite-stable solid tumors and clinical annotation of complete response, partial response (CR/PR) or progressive disease (PD). Numbers of missense mutations detected by WES were converted to TMB per Mb using the correspondence of 199 mutations and 10 mut/Mb (50). A, melanoma subcohort (n=125). B, lung cancer subcohort (n=36). C, bladder cancer subcohort (n=23). D, entire cohort of mixed cancer types (n=193).
Melanoma
Consistent with the known high TMB in cutaneous melanoma, metastatic melanoma also showed an increase in median non-synonymous mutation burden assessed by WES (18,64). Importantly, TMB differed significantly among melanomas harboring mutations in BRAF, NRAS, _NF_1 or triple wild-type (WT) tumors with median TMB values of 12.0, 17.6, 62.7, and 2.2 mut/Mb, respectively (p< 0.001) (60). Melanomas with _NF_1 mutations are associated with chronic UV damage and thus, have a high TMB. Of note, TMB differed among melanoma subtypes with cutaneous and occult melanomas having higher TMB than did acral and mucosal subtypes, as well as higher response rates to anti-PD 1 blockade (69).
Among 32/110 (29%) patients with metastatic melanoma treated with anti-PD-1/PD-L1 antibodies, median TMB values were significantly higher in responders vs non-responders (45.6 vs 3.9 mut/Mb, p=0.003). Furthermore, the ORR to ICI treatment was significantly increased in tumors with high TMB (>23.1 mut/Mb) vs intermediate (3.3–23.1 mut/Mb) vs low (< 3.3 mut/Mb) groups (ORR: 82% vs 36% vs 10%, p=0.003) (60) (Table 2). Results were confirmed in an independent validation cohort (n=33) [p=0.002] (60). Certain genomic alterations such as mutations in _NF_1 or _BRCA_2 were more common in responders to ICI treatment compared to triple WT tumors (60). In a pooled dataset of 300 patients with cutaneous metastatic melanoma, TMB was categorized as high (7.1 mut/Mb) in 21% of tumors and was associated with a higher ORR to anti-PD-1/PD-L1 treatment compared to lower TMB (33% vs 21%) (70) (Table 2). Responders with mucosal or acral melanomas had a lower TMB than did progressors with cutaneous or occult melanoma, suggesting that melanoma subtype may confound the association between TMB and response to anti-PD1 therapy (69). In this regard, TMB failed to predict benefit from ICIs in a multivariable model adjusting for melanoma subtype (cutaneous, occult, acral and mucosal). Among aggregate data in melanoma patients, only a modest difference in clinical benefit by TMB level has been observed. Further data in molecular subgroups remain of interest with the caveat that large sample sizes are needed for such comparisons.
Table 2.
Overview of published studies assessing TMB across cancer types
| Cancer type /reference | Trial/Drug | Definition of TMB | TMB detection method | Cutoff of TMB | Type of benefit |
|---|---|---|---|---|---|
| multiple cancer types (n=151) / (65) | anti-PD-1/PD-L1, anti-CTLA4, or combination | number of somatic mutations by NGS | 182, 236, or 315 genes, panels of 1.2 Mb of genome | ≥ 20mut/Mb | ORR, PFS, OS |
| multiple cancer types (n=751) / (24) | KEYNOTE-158/ Pembrolizumab | nonsynonymous coding mutations in a tumor | FM | ≥ 10mut/Mb | ORR, PFS |
| Metastatic melanoma (n=65) / (60) | nivolumab or pembrolizumab or atezolizumab | Number of somatic mutations | 0.91 and 1.25 MB for the 236 gene and 315 gene versions (FM) | >23.1 mut /Mb | ORR, PFS, OS |
| Metastatic melanoma (n=300) / (70) | ipilimumab | Somatic nonsynonymous mutations | 409 cancer-related genes | >7.1 mut/Mb | ORR |
| NSCLC (n=312) / (77) | CheckMate-026/ nivolumab or platinum-doublet chemotherapy | total number of somatic missense mutations | WES | ≥243 mutations | ORR, PFS No benefit of OS |
| NSCLC (n=299) / (63) | CheckMate-227/ nivolumab + ipilimumab, or nivolumab alone, or chemotherapy | NA | NA | ≥10 mut/Mb | ORR, PFS No benefit of OS |
| SCLC (n=401) / (59) | CheckMate-032/ nivolumab alone (n=245) vs. nivolumab +ipilimumab (n=156) | number of somatic missense mutations | WES | ≥ 248 mutations | ORR, PFS, OS |
| advanced urothelial carcinoma (n=139) (19) | CheckMate-275/ nivolumab | the total number of missense somatic mutations per tumor | WES | ≥13 mut/Mb | ORR, PFS, OS |
| advanced urothelial carcinoma (n=150) (6) | atezolizumab | the number of somatic base substitutions or indels per megabase | 315 cancer-related genes (FM) | NA | ORR |
| SCCHN (n=81) /(20) | anti–PD-1/PD-L1 immunotherapy | the number of nonsynonymous somatic mutations per megabase | gene panel | >10 mut/Mb | ORR, OS |
| stage IV CRC (n=22) / (25) | anti-PD-1/PD-L1 immunotherapy | number of synonymous and non-synonymous mutations | mutations across a 0.8–1.2 Mb region(FM) | 37–41 mut/Mb | ORR, PFS, OS |
| advanced or metastatic CRC (n=843) / (27) | CALGB/SWOG80405/ chemotherapy +cetuximab /bevacizumab or cetuximab +bevacizumab | NA | 395 cancer-related genes and of 31 genes often rearranged or altered in cancer (FM) | >8 mut/Mb | NA |
| metastatic TNBC (n=62) / (93) | anti-PD-1/L1 therapy or in combination with chemotherapy or targeted therapy | nonsynonymous mutations | gene panel targeting full coding regions or selected intronic regions of 305–335 genes | ≥10 mut/Mb | ORR |
| metastatic breast cancer (n=28) / (94) | TAPUR study/ pembrolizumab | NA | FM | ≥9 mut/Mb | No benefit of PFS, OS |
Lung cancer
Lung cancers have the highest somatic mutation burden among solid tumors that is believed to be due to direct exposure to mutagens in tobacco smoking (18,71–73). Lung cancer in smokers was associated with significantly higher median TMB (10.5 mut/Mb) compared to never-smokers with lung cancer (0.6 mut/Mb) (74). Of note, smoking status is inversely related to prevalence of targetable oncogenic driver mutations in EGFR, ALK and ROS1 genes in lung adenocarcinoma (75). Among lung cancers [squamous carcinoma and small cell lung cancers (SCLC)] associated with cigarette smoking, mutations in BRAF, KRAS, PTEN and PIK3CA are most common (76). In a large lung cancer database, TMB was found to be relatively similar across lung cancer histologies, although squamous cell carcinomas had a slightly higher mean TMB (n=1,324, 11.3 mut/Mb) compared with adenocarcinomas (n=7,925, 9.1 mut/Mb) and SCLC (n=640, 10.3 mut/Mb) (75). Variability of TMB has been identified among molecular subgroups of lung cancer. In this regard, tumors with ALK/_ROS_1 (n=489), EGFR (n=1,775), and MET exon 14 (n=286) had mean TMB levels (mut/Mb) of 3.1, 4.5 and 6.2, respectively (75).
The ability of TMB to predict efficacy of the anti-PD-1 antibody pembrolizumab was first shown in patients with advanced NSCLC from two independent cohorts (10), and led to multiple clinical trials. In patients with metastatic or recurrent NSCLC, the CheckMate 026 study compared nivolumab vs. platinum-doublet chemotherapy as first-line treatment. Patients whose tumors had a higher TMB experienced an improved ORR (47% vs. 28%) and longer PFS (median (m) PFS: 9.7 vs. 5.8 months, HR 0.62; 95% CI, 0.38 to 1.00) in the nivolumab study arm (77) (Table 2). In the subsequent CheckMate 568 study of nivolumab plus ipilimumab in treatment naive patients with metastatic NSCLC, TMB data were available in 98 patients whereby ORRs (44% vs 12.0% ) and mPFS (7.1 months vs. 2.6 months) were improved in those whose tumors had high vs low TMB ( ≥10 mut/Mb) (61). In the phase 3 MYSTIC trial, 1,118 patients with treatment-naïve, metastatic NSCLC (without EGFR or ALK mutations) were randomized to durvalumab (PD-L1 inhibitor) alone or combined with tremelimumab (CTLA-4 inhibitor), or platinum-based doublet chemotherapy (78). Among 488 patients with tumor cell PD-L1 expression ≥25%, the primary endpoint was not met in that neither study arm showed a survival benefit compared to the chemotherapy arm. Among 460 patients with tumor TMB data, high TMB (≥ 10 mut/Mb) was associated with longer median OS that did not achieve statistical significance (78). Importantly, TMB levels from 352 matched tumor and blood samples tested were correlated (Spearman p = 0.6; Pearson r = 0.7), indicating that TMB testing in blood may be a useful adjunct when tissue testing is not feasible (78). In the first prospective trial (CheckMate 227) that examined TMB as a co-primary endpoint biomarker, nivolumab plus ipilimumab was compared to platinum-doublet chemotherapy in patients with advanced NSCLC in the first-line setting. Among patients with TMB data (n=1,004), tumors with high vs. low TMB (prespecified cutoff of ≥10 mut/Mb) demonstrated an ORR of 45.3% vs 26.9% and superior PFS of 7.2 vs 5.5 months (HR 0.58; 95% CI: 0.41 to 0.81, P <0.001) for nivolumab plus ipilimumab vs. chemotherapy, respectively, that was independent of PD-L1 expression (63) (Table 2). No difference was observed for OS in this study, yet PFS is a preferred endpoint for predictive biomarker evaluation since OS includes outcome of all subsequent treatments. In the CheckMate 568 study (61), TMB ≥10 mut/Mb was a cutpoint for ORR and was validated as a predictive biomarker when prospectively applied to CheckMate 227 (63) where it showed improved PFS for nivolumab and ipilimumab compared to standard chemotherapy. In these studies, PD-L1 and TMB were independent predictive biomarkers.
While a modest association of TMB with efficacy of dual checkpoint blockade was demonstrated in the Checkmate 568 and 227 studies, inconsistent data have been reported for the role of TMB as a predictor of response and survival to dual checkpoint blockade across clinical trials. Among 17 patients from a pan-cancer cohort treated with a combination of anti-PD-1 and anti-CTLA4 antibodies, TMB levels were not associated with treatment efficacy (65). In treatment naïve patients with metastatic NSCLC (N=955) randomized to tremelimumab plus durvalumab vs. platinum-based chemotherapy (NEPTUNE trial), TMB level (TMB high ≥20 mut/Mb) determined in blood samples was not shown to predict patient survival (https://www.astrazeneca.com/media-centre/press-releases/2019/update-on-the-phase-iii-neptune-trial-of-imfinzi-plus-tremelimumab-in-stage-iv-non-small-cell-lung-cancer-21082019.html). The explanation for the more modest association of TMB with clinical outcome among patients treated with dual checkpoint inhibitors is unclear, but relevant factors include retrospective design, relatively small numbers of cases with TMB data especially the TMB-high population that can limit statistical power, varied DNA sources, i.e., blood vs. tissue, and multiple platforms for TMB measurement as well as variable TMB cutoffs for correlation with outcomes.
The role of TMB as a predictive biomarker in extensive stage SCLC was studied in patients who had failed at least one prior chemotherapy regimen. In patients treated with nivolumab alone or combined with ipilimumab (CheckMate 032), the ORR by treatment arm increased stepwise in patients whose tumors showed TMB high vs. medium vs. low levels (59) (Table 2). Importantly, dual ICIs treatment was associated with an impressive ORR of 46.2% and an estimated 1-year OS rate of 62.4% in these previously treated patients with high TMB tumors (59).
Urothelial carcinoma
Cigarette smoking is the most common risk factor and is estimated to be responsible for approximately 50% of all urothelial carcinomas (79). Urothelial carcinoma carry the third highest mutation rate among solid tumors (80). In an analysis of 472 urothelial carcinomas using a panel of 237 cancer genes, a median TMB of 10.9 mut/Mb was found. TMB was increased in high vs low grade cancers with or without muscle invasion (81). Mutation in the apolipoprotein B editing enzyme (APOBEC) was common in all stages and locations of urothelial carcinoma, was strongly associated with TMB, and was more frequent in muscle invasive (MIBC) and high grade non muscle invasive bladder cancers (NMIBC) (81).
In the CheckMate 275 study, patients with unresectable locally advanced or metastatic urothelial carcinoma who failed at least one platinum-based regimen received nivolumab monotherapy. Of 270 patients, 139 (51%) had evaluable TMB. High TMB (≥ 13 mut/Mb) was associated with higher ORR, longer PFS, and longer OS in patients treated with nivolumab (19)[Table 2]. In a phase II single-arm trial I of atezolizumab monotherapy in this same study population, median TMB (measured in 150 patients by a 315-gene panel) was significantly increased in responders vs. non-responders (12.4 mut/Mb vs. 6.4 mut/Mb, p<0.0001) (6)[Table 2]. The role of TMB as a predictor for immunotherapy outcome was also explored in neoadjuvant setting. In the PURE-01 study of neoadjuvant pembrolizumab in MIBC patients, median TMB was higher in patients who achieved a pathological complete response (18.4 mut/Mb vs. 8.4 mut/Mb (82). These data suggest that TMB may enrich for responders to ICIs in patients with urothelial carcinomas.
Head and neck cancer
Etiologic factors associated with squamous cell carcinoma of the head and neck (SCCHN) include tobacco and alcohol consumption and virus infection (human papillomavirus, HPV and Epstein-Barr virus, EBV) (16). Of 126 patients with SCCHN who received anti-PD-1/PD-L1 treatment, 64% had evaluable TMB that ranged from 1.5 to 76.0 mut/Mb (median 7.6 mut/Mb), and 13% of tumors showing TMB values greater than 20 mut/Mb (20). The median TMB level was significantly higher among responders vs. nonresponders (17.7 vs. 7.1, P < 0.01) to ICIs. Interestingly, patients with non-virus vs. virus-mediated tumors had a higher median TMB (8.2 vs. 4.7, P < 0.01) which is consistent with prior studies (83,84). TMB predicted anti-PD-1/PD-L1 response among HPV- and EBV-negative tumors, but not among HPV- or EBV-positive tumors. Moreover, TMB correlated with longer OS among virus-negative patients, but not among their virus-positive counterparts. Among responders to anti-PD-1/PD-L1 treatment, the most commonly mutations were in NOTCH1, TP53, KMT2D, and SMARCA4 genes (20)[Table 2]. These data support TMB as a promising biomarker for ICI efficacy in SCCHN.
Gastrointestinal (GI) cancers
Among various GI cancer types (4,125 patients), mean level of TMB ranged from 5.1 to 13.0 mut/Mb (592 gene panel) (Table 2) (85). Adenocarcinomas of the right colon and small-bowel exhibited the highest mean TMB (13 and 10.2 mut/Mb, respectively) while pancreatic adenocarcinoma, pancreatic neuroendocrine tumors (NET) and GIST had the lowest TMB levels (6.1 mut/Mb, 5.8 mut/Mb and 5.1 mut/Mb, respectively). Cancers with the highest known TMB levels are ultramutated and are caused by mutations in polymerase epsilon (POLE) that impair DNA proofreading (86). Such tumors are nearly exclusively microsatellite stable (MSS), have TMB values ranging from 122 mut/Mb to 303 mut/Mb, and comprise approximately 1–2% of all MSS CRCs (26,43,87). Also hypermutated are GI cancers with deficient DNA mismatch repair (dMMR) resulting in microsatellite instability-high (MSI-H). MSI-H have a significantly higher median TMB level compared to MSS tumors (25–27), yet lower than those with POLE. Analysis of MSI-H cancers by WES showed a mean of 1,782 somatic mutations per tumor compared with 73 mutations per tumor in patients with MSS cancers (P = 0.007) (5).
To date, results for TMB and outcome from ICIs in GI cancers is limited with the exception of the subset of tumors with MSI-H, especially CRC. Among MSI-H cancers, high ORRs and prolonged PFS have been observed and are discussed below under “TMB and Tumor MSI Status.” To date, only limited data exist among GI cancers with MSS treated with ICIs (5,21,26,88,89). In a study of 54 advanced gastric cancers, median TMB for MSS cases was 6.6 mut/Mb (range 0– 30.0) and there was one MSI-H tumor. Evaluation of the predictive utility of TMB for the anti-PD-1 antibody, toripalimab, revealed that patients with TMB-high (≥ 12 mut/Mb) vs. low tumors had better ORR (33.3% vs. 7.1%, P=0.017), similar PFS (2.5 vs. 1.9 months, P=0.055), but significantly improved OS (14.6 vs 4.0 months, P=0.038) (21). Among 17 patients with MSS advanced hepatocellular carcinoma treated with an anti-PD-1 antibody, one patient (TMB 15 mut/Mb) had a sustained complete response to nivolumab lasting >2 years. However, TMB did not segregate patients by response criteria (89). Future studies are needed to evaluate ICI efficacy in GI cancer patients with MSS cancers and high TMB, which will also enable comparison of results with MSI-H cancers.
Breast cancer
Evidence indicates that breast cancers typically have a lower TMB compared to NSCLC and melanoma, although TMB has been shown to vary both within and across breast cancer subtypes (90–92). Using WES and panel-based sequencing data from 3,969 patients from 6 cohorts, median TMB was 2.63 mut/Mb with 5% of patients showing high TMB (≥10 mut/Mb). Among breast cancer subtypes, median TMB was significantly higher in triple-negative breast cancer (TNBC) (1.8 mut/Mb) compared to hormone receptor-positive (1.1 mut/Mb, P < 0.001) or Her2-positive cancers (1.3 mut/Mb, P = 0.003) (91). While these differences in TMB were statistically significant, they are within the error range of large gene panels.
To date, only limited data are available for the evaluation of TMB as a predictive biomarker in patients with breast carcinoma treated with ICIs. In a single center cohort of patients with metastatic TNBC (N=62) treated with an anti-PD-1/PD-L1 antibody, patients whose tumors had high TMB (≥10 mut/Mb) had a 2-fold increase in likelihood of response compared to those with lower TMB (93) (Table 2). In a prospective clinical trial known as TAPUR, patients with heavily pre-treated metastatic breast cancer were treated with pembrolizumab which was associated with a modest benefit in patients with high TMB (≥ 9 mut/Mb) tumors (94) (Table 2). Based on available data, TMB appears to be less relevant to ICI treatment outcome in breast cancer given that only 5% of these tumors have high TMB (≥10 mut/Mb). This TMB cutoff was associated with pembrolizumab benefit in a recent pan-cancer study (KEYNOTE-158) that led to FDA approval of this antibody for this high TMB subgroup.
TMB and other biomarkers
TMB and PD-L1
Studies have shown that TMB is to a large extent independent of PD-L1 status in most cancers (34) and might therefore, identify additional subgroups of patients who may benefit from ICIs (46,59,65,77). Among 24 cancer types included in the TCGA database, TMB and PD-L1 mRNA expression were examined and significant correlations were observed in only 5 of 24 cancer types (Fig. 2C). These comprised adenocarcinoma of the colorectum, stomach and uterus (R=0.26, R=0.22 and R=0.19) as well as transitional cell carcinoma (R=0.19) and breast cancer (R=0.14). Across cancer types we observed a weak, but highly significant positive correlation (R=0.13). In a retrospective analysis of 11,348 patients across 26 cancer types, 7.7% of tumors were found to be TMB-high (≥17 mut/Mb) and of these, 44% were also PD-L1 immunopositive (≥ 1%; PD-L1 antibody: SP142) (38). Among 4,125 GI cancers examined, squamous cell carcinomas of the esophagus and anus showed high PD-L1 expression yet low or negative TMB or infrequent MSI-H. Other tumor types such as right-sided colon cancers and small-bowel adenocarcinomas showed high TMB or MSI-H, yet low PD-L1 expression (85).
Multiple studies have analyzed PD-L1 expression as a potential predictive biomarker for response to ICIs targeting PD-1 or its ligand, PD-L1. Determination of PD-L1 expression is approved by the FDA as a companion diagnostic test for pembrolizumab treatment in patients with NSCLC, gastroesophageal junction and gastric cancer, cervical cancer, and urothelial cancer (28,29,95–98). However, this is not the case in other tumor types (99,100) and therefore, the predictive utility of PD-L1 remains limited (101). Whereas TMB and MSI-H describe features of the tumor, PD-L1 expression depends on the specific cell types examined and the score that is applied (tumor only, immune cell only, or their combinations), while quantification of lymphocyte densities or specific immune signatures, e.g., IFNgamma or T-cell clonality, highlight a specific state of the local immune environment and effector compartment. Both quantitative and qualitative measurements of the tumor compartment and the effector compartment are important for understanding outcomes of immunotherapy, and information on both compartments is likely needed for arriving at meaningful conclusions in a clinical setting.
Evidence suggests that complimentary utilization of both TMB and PD-L1 may predict responsiveness to ICIs better than either alone (see Supplementary Data) (46,61). In patients with NSCLC, the CheckMate 568 study demonstrated superior ORR for nivolumab plus ipilimumab independent of PD-L1 status in NSCLC patients with TMB high (≥10 mut/Mb) vs. TMB low tumors (ORR: PD-L1≥1%, 48% vs. 18%; PD-L1<1%, 47% vs. 5%). Importantly, patients with tumors that had PD-L1 expression < 1% and TMB < 10 mut/Mb had an ORR of only 5%, suggesting that using the combination of PD-L1 and TMB testing can identify a subgroup of patients who have a low likelihood of benefit from the combination of PD-1 and CTLA-4 checkpoint blockade (61).
TMB and Tumor MSI status
MSI-H tumors show hypermutation including frameshift mutations that generate numerous neopeptides (71). Tumors with MSI-H due to dMMR typically have high TMB levels. WES revealed a mean of 1,782 somatic mutations per MSI-H tumors as compared with 73 in MSS tumors (P=0.007), suggesting that a markedly increased number of mutation-associated neoantigens is responsible for enhanced anti-PD-1 response (5). Among 4,125 GI cancers of 14 different types, TMB-high (≥ 17 mut/Mb; 592 gene panel) was strongly correlated with MSI-H status in most cancer types indicating that high TMB and MSI-H are inextricably linked. Exceptions among TMB-high tumors included squamous cell carcinomas of the anus and esophagus that were generally MSS (85). A majority of MSI-H/dMMR cancers are CRC or endometrial cancers (102–105).
MSI-H/dMMR has been shown to be a predictive biomarker for treatment with ICIs (106). In this regard, pembrolizumab was approved by the FDA for the treatment of MSI-H cancers agnostic of primary tumor type. Pembrolizumab is standard of care for treatment refractory, MSI-H solid tumors and more recently, pembrolizumab was shown to be superior to and less toxic than chemotherapy in MSI-H CRC in the first-line setting (107). Among 124 patients with MSI-H/dMMR CRC treated with pembrolizumab (KEYNOTE-164), the ORR was 33% overall and median PFS was 2.3 months (≥ 2 prior lines of therapy) or 4.1 months (≥ 1 prior therapy) (108). In another phase II study, nivolumab (anti-PD-1) provided a response rate of 31% and a 12-month OS rate of 73% in heavily pretreated patients with metastatic MSI-H/dMMR CRC, while its combination with ipilimumab demonstrated a response rate of 55% and 12-month OS rate of 85% (8). In a separate study, 22 patients with MSI-H metastatic CRCs were treated with an anti-PD-1 or PD-L1 antibody. Median TMB level in responders was significantly greater than in non-responders (54 mut/Mb vs 29 mut/Mb, p<0.001). Of the 13 patients with TMB high tumors, define as ≥ 37–41 mut/Mb, an objective response was observed in all while only 3 of 9 (33%) patients with low TMB tumors had disease control (25) (Table 2). These data suggest that high TMB in MSI-H/dMMR tumors is associated with increased and durable responses to ICIs (5,109,110), and that TMB may further identify responders to ICIs within MSI-H cancers (25). However, TMB can occur due to multiple and distinct mutational processes, and the relative contribution of mutation load vs. mutational process has yet to be clarified. In addition to variability in TMB observed in MSI-H cancers (25), heterogeneity in densities of tumor infiltrating lymphocytes (TILs) are also found in this tumor subgroup that can prognostically stratify these tumors (111,112). Further study is needed to examine the relationships of TMB with neoantigen load and TIL density.
Evidence indicates that a subset of MSS cancers have high TMB (85,88,113). In the study of 4,125 patients with GI cancers, anal squamous cell carcinomas exhibited the highest prevalence (8.3%) of MSS/TMB-high (≥17 mut/Mb) followed by esophageal squamous cancer (3.5%) (85). In a study of MSS cancers of 14 different histologies, treatment with an ICI produced longer median PFS (26.8 vs 4.3 mo., p=0.0173) in TMB-high (≥20 mut/Mb) vs TMB-low tumors (113). In other studies, the prevalence of TMB-high (≥ 11.7 mut/Mb) in MSS CRCs was 2.9% (164/5,702)(26) and in a study of multiple cancer types, TMB-high (≥ 20 mut/Mb) was identified in 5.4% (7,972/148,803) of cancers (113). Importantly, the prevalence of MSI-H was only 1.5% (2,179/148,803) in this study so it was significantly exceeded by the TMB-high subgroup(113). These data suggest that mechanisms beside DNA repair defects, such as DNA replication mutations (POLD1 and POLE) or TP53 mutations (35,114,115), may underlie their increased TMB. Furthermore, evidence suggests that hypermutation with high mutation-specific neoantigenic load is a critical factor responsible for anti-tumor efficacy of ICIs since both MSI-H and MSS POLE tumors show relatively high response rates to ICIs. Of clinical relevance is that patient selection for ICIs based on TMB status may potentially expand the candidate pool for cancer immunotherapy. In this regard, data from the pan-cancer cohort of predominantly MSS solid tumors (KEYNOTE-158) found an ORR of 27.1% for patients with MSS tumors and TMB ≥ 10 mut/Mb (24).
TMB and DNA Damage Response and Repair
Alterations in DNA Damage Response and Repair (DDR) genes are associated with genomic instability and increased somatic tumor mutational burden, which may enhance immunogenicity through increased tumor-specific neoantigen load (116,117). The relationship between TMB and DDR genes has been explored in various cancer types including NSCLC, urothelial cancer and GI cancers (81,118–120). Recent evidence revealed deleterious somatic DDR mutations in approximately 50% of patients with NSCLC or urothelial carcinomas (81,120). Patients with DDR mutations had significantly increased tumor TMB levels (81,120) and longer PFS and OS independent of covariates (120). The prevalence of DDR alterations was 17% among 17,486 GI carcinomas (119), of which ARID1A (9.2%) and ATM (4.7%) were most common followed by BRCA2 (2.3%), BRCA1 (1.1%) and CHEK2 (1.0%). DDR mutations were associated with increased TMB and of DDR-altered/TMB-high cases, 87% were also MSI-H. Of note, MSI-H and high TMB (≥20 mut/Mb) were found in 19% and 21% of DDR-mutated cases, respectively. Even among MSS tumors, TMB-high was significantly more frequent in DDR-mutated vs. non-mutated cases (119). An important caveat is that tumors with high TMB and/or MSI-H are more likely to harbor DDR mutations which suggest the potential for confounding.
Strategies to Optimize TMB as a Predictive Biomarker
Observed cancer-type related biological TMB distributions offer the potential to determine tumor-specific and optimal TMB cutoffs. Various strategies to optimize TMB as a predictive biomarker for ICIs are being explored (Table 3). A novel three-tier (high, intermediate and low) TMB classification scheme was introduced to reduce the possibility of misclassification by the current two-tier (high, low) TMB classification scheme. A so-called “gray zone” for TMB was identified to potentially aid future panel designs, trial design, and clinical decision making (45). Transforming unadjusted TMB values into standardized z scores (converts the right-skewed TMB distributions to normal distributions) has been proposed to standardize and compare TMB across panels from different platforms (121). However, this would require similar TMB mean and standard deviation from datasets of comparable cohorts. Other strategies include screening for actionable mutations or biomarkers, refining immunotherapy response prediction (such as negative predictors of response and variants predisposing to toxic effects), align panel-based TMB values to a WES-based TMB reference to ensure consistency across assays, standardize bioinformatic algorithms used for mutation calling and filtering, use variant allele frequency (VAF) as proxy for clonality to further refine TMB quantification and allow calibration of results from different studies. Friends of Cancer Research (Friends) and Quality Assurance Initiative Pathology (QuIP) are two international organizations that have proposed approaches to standardize and harmonize TMB assessment across assays and centers globally (54,55,122,123). The Friends of Cancer Research TMB Harmonization Consortium made the following recommendations: 1) reporting of TMB in mutations/megabase (mut/Mb) to keep these values consistent and comparable among different studies; 2) validation studies for TMB estimation should be standardized to include assessment of analytical accuracy, precision and sensitivity, and 3) ensure consistency across panels through alignment of panel TMB values to WES-derived universal reference standards (55).
Table 3.
Key parameters for the standardization and harmonization of TMB analysis and workflow
| Parameter | Principles | |
|---|---|---|
| Pre--analytical | standardize sample processing protocols minimize inter-laboratory variability | |
| Gene panel specifications | genome coverage | > 1 Mb |
| composition | screen for actionable mutations or biomarkers refine immunotherapy response prediction by including:- negative predictors of response- variants predisposing to toxic effects- other potential immunotherapy biomarkers (MSI/MMR, PD-L1) | |
| TMB definition | non-synonymous mutations | involved in creating neoantigens |
| synonymous mutations | indirectly involved in creating neoantigens reduce sampling noise and improve approximation of TMB across the whole genome | |
| indels (insertion+deletion) | generate a higher number of immunogenic neoantigens than non-synonymous SNVs; more strongly associated with response to ICIs than non-synonymous SNV load | |
| TMB cutoff | method | use method such as ROC curves instead of percentiles which are affected by outliers |
| type | cancer-specific cutoffs (diverse TMB distribution across different cancer types) | |
| classification scheme | novel three-tier (high, intermediate and low) TMB classification to reduce the possibility of misclassification by the two-tier (high, low) | |
| Bioinformatics | standardization of workflow | align panel-based TMB values to a WES-based TMB reference to ensure consistency across assays |
| standardize bioinformatic algorithms used for mutation calling and filtering | ||
| filter germline variants with matched normal samples | ||
| use non-tumor samples to establish the limit of blank for TMB, yielding results close to, but not always equal to 0 mut/Mb | ||
| enhance TMB predictive power | use variant allele frequency (VAF) as proxy for clonality to further refine TMB quantification | |
| combine TMB with potential immunotherapy biomarkers (CNA, T-cell-inflamed GEP or PD-L1, tumor burden) | ||
| addition of paired normal tissue to the sequencing panel to get individualized germline data | ||
| Comparison of results | calibration of outputs | ensure report consistency (eg. TMB should be reported in mutations /megabase) |
| allow calibration of results from different studies |
Determination of TMB in commercially available gene panels relies mainly on missense mutations. While gene panel sequencing platforms generally detect and report indels (Table 1), indel calling can vary based on the bioinformatics pipeline (124). Data in CRC and melanoma suggest that frameshift indels generate a higher number of immunogenic neoantigens than do non-synonymous single-nucleotide variant (SNV) mutations (47,125,126) which awaits confirmation. Indel load was strongly associated with ICI response although controversy exists (126,127). Furthermore, the proportion of indels in conjunction with TMB values can identify different tumor types and genetic subgroups, including MSI-H cases (127) (Table 3). Evidence suggests that underlying biology is likely driving and shaping TMB with prime examples of hypermutation being due to MSI-H (due to defective DNA mismatch repair) or MSS POLE (exonuclease domain mutations). Both MSI-H and POLE tumors show relatively high response rates to ICIs, suggesting that TMB is a proxy or parameter governed by tumor biology. The relative contribution of mutation load vs mutational process to TMB has yet to be clarified. Considering mutational signatures in the context of TMB might help to decipher the biology that creates TMB. Furthermore, high TMB in MSI-H tumors can be identified and was associated with increased and durable responses to ICIs (5,109,110), suggesting that stratification of MSI-H tumor using TMB may distinguish responders vs nonresponders to ICIs (25). Data suggests that complimentary utilization of both TMB and PD-L1 may predict responsiveness to ICIs better than either alone (46,61), and further evaluation of this approach in selected tumor types is warranted. Other studies indicate that tumors that have high levels of TMB and inflammatory markers [T-cell-inflamed gene-expression profile (GEP) or PD-L1] represent a population with the highest likelihood of response to ICIs (128,129). In this regard, TMB and GEP exhibited joint predictive utility in identifying responders and nonresponders to pembrolizumab. TMB and GEP were independently predictive of response and demonstrated a low correlation, suggesting that they capture distinct features of neoantigenicity and T cell activation (128,129). Pretreatment tumor burden may also influence the efficacy of ICIs. A hypothesis has been proposed that evaluates the ratio of TMB to tumor burden that could provide a more effective prediction of ICI efficacy, and warrants testing in clinical trials (130).
Evidence indicates that relative TMB can be determined through sequencing analysis of cell-free DNA (cfDNA). Measurement of TMB in blood plasma has been developed and in limited studies, has been shown to provide predictive information for immunotherapy response in patients with NSCLC (131,132). The number of mutations detected in cfDNA was positively correlated with ICIs efficacy and OS across various cancer types (n=69) (133). Furthermore, retrospective analysis of cfDNA from two randomized trials (n=211 and 583) demonstrated that cfDNA-derived TMB is associated with improved survival in patients with NSCLC treated with an anti-PD-L1 antibody (132). While a blood-based assay has clear advantages for clinical application, further development and evaluation of this assay technology is awaited in addition to its comparison to tissue-based approaches.
Conclusions and Future Perspectives
Accumulating evidence suggests that TMB may serve as a predictive biomarker for immunotherapy in multiple solid tumors, although further prospective validation is needed. Use of TMB is now a component of routine oncologic practice based on recent FDA approval of pembrolizumab for TMB-high solid tumors. The TMB-high subgroup was identified using a prespecified cutoff of at least 10 mut/MB, and issues remain regarding optimal cutoffs per tumor type based, in part, on baseline TMB distributions. Furthermore, prospective studies are needed for further evaluation and validation of the predictive utility of TMB in clinical practice. TMB has been shown to be a predictive biomarker for ICIs in both MSI-H and MSS cancers, and patient selection based on TMB levels may better select patients for ICIs or potentially expand the candidate pool for immunotherapy. Although current evidence indicates that TMB is associated with ICIs efficacy, the mechanism(s) underlying the association between TMB and benefit from immunotherapy is incompletely understood. TMB is independent of PD-L1 status in most cancer types, although the complementary utilization of TMB, PD-L1 and MSI-H has the potential to predict ICIs responsiveness better than each alone. As a novel biomarker, there is an urgent need to harmonize and standardize TMB measurement, testing platforms and reporting of TMB. The Friends of Cancer Research TMB Harmonization Consortium is working to establish guidelines to harmonize TMB across diagnostic platforms and results of this effort are eagerly awaited. Larger datasets for TMB and clinical outcome of ICI-treated patients will facilitate the optimization of TMB cutoffs within specific cancer types and potentially extend the approval of immune therapies to larger patient populations. Furthermore, prospective and randomized studies are needed to validate a TMB-high cutoff and to explore optimal TMB cutoffs in specific tumor types. TMB combined with other potential biomarkers and computational assistance is paving the way towards a precision immunotherapy approach.
Supplementary Material
1
Statement of Significance.
Evaluation of TMB as a predictive biomarker creates the need to harmonize panel-based TMB estimation and standardize its reporting. TMB can improve the predictive accuracy for immunotherapy outcomes, and has the potential to expand the candidate pool of patients for treatment with immune checkpoint inhibitors.
Financial Support:
The study was also supported, in part, by an NCI grant [grant number R01 CA210509-01A1] (to FAS). D. Sha was supported by the China Scholarship Council and Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, P.R. China.
Footnotes
Conflict of Interest: The authors report no conflicts of interest related to the content of this manuscript.
References
- 1.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363(8):711–23 doi 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369(2):122–33 doi 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med 2015;373(17):1627–39 doi 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015;373(19):1803–13 doi 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015;372(26):2509–20 doi 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016;387(10031):1909–20 doi 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med 2018;378(14):1277–90 doi 10.1056/NEJMoa1712126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol 2018;36(8):773–9 doi 10.1200/JCO.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 9.Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015;350(6266):1387–90 doi 10.1126/science.aad1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348(6230):124–8 doi 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desrichard A, Snyder A, Chan TA. Cancer Neoantigens and Applications for Immunotherapy. Clin Cancer Res 2016;22(4):807–12 doi 10.1158/1078-0432.CCR-14-3175. [DOI] [PubMed] [Google Scholar]
- 12.Efremova M, Finotello F, Rieder D, Trajanoski Z. Neoantigens Generated by Individual Mutations and Their Role in Cancer Immunity and Immunotherapy. Front Immunol 2017;8:1679 doi 10.3389/fimmu.2017.01679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348(6230):69–74 doi 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 14.Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nature reviews Cancer 2017;17(4):209–22 doi 10.1038/nrc.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGranahan N, Swanton C. Neoantigen quality, not quantity. Sci Transl Med 2019;11(506) doi 10.1126/scitranslmed.aax7918. [DOI] [PubMed] [Google Scholar]
- 16.Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 2017;377(25):2500–1 doi 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371(23):2189–99 doi 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500(7463):415–21 doi 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galsky MD, Saci A, Szabo PM, Han GC, Grossfeld G, Collette S, et al. Nivolumab in Patients with Advanced Platinum-resistant Urothelial Carcinoma: Efficacy, Safety, and Biomarker Analyses with Extended Follow-up from CheckMate 275. Clin Cancer Res 2020. doi 10.1158/1078-0432.CCR-19-4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanna GJ, Lizotte P, Cavanaugh M, Kuo FC, Shivdasani P, Frieden A, et al. Frameshift events predict anti-PD-1/L1 response in head and neck cancer. JCI Insight 2018;3(4) doi 10.1172/jci.insight.98811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang F, Wei XL, Wang FH, Xu N, Shen L, Dai GH, et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemo-refractory gastric cancer treated with toripalimab, a PD-1 antibody in phase Ib/II clinical trial NCT02915432. Ann Oncol 2019;30(9):1479–86 doi 10.1093/annonc/mdz197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gallo M, Guarnotta V, De Cicco F, Rubino M, Faggiano A, Colao A. Immune checkpoint blockade for Merkel cell carcinoma: actual findings and unanswered questions. J Cancer Res Clin Oncol 2019;145(2):429–43 doi 10.1007/s00432-019-02839-w. [DOI] [PubMed] [Google Scholar]
- 23.Harms PW, Harms KL, Moore PS, DeCaprio JA, Nghiem P, Wong MKK, et al. The biology and treatment of Merkel cell carcinoma: current understanding and research priorities. Nat Rev Clin Oncol 2018;15(12):763–76 doi 10.1038/s41571-018-0103-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marabelle AFM, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of Tumor Mutational Burden with Outcomes in Patients with Select Advanced Solid Tumors Treated with Pembrolizumab in KEYNOTE-158. Ann Oncol 2019;30 (suppl_5):v475–v532 doi 10.1093/annonc/mdz253. [DOI] [Google Scholar]
- 25.Schrock AB, Ouyang C, Sandhu J, Sokol E, Jin D, Ross JS, et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann Oncol 2019;30(7):1096–103 doi 10.1093/annonc/mdz134. [DOI] [PubMed] [Google Scholar]
- 26.Fabrizio DA, George TJ Jr., Dunne RF, Frampton G, Sun J, Gowen K, et al. Beyond microsatellite testing: assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J Gastrointest Oncol 2018;9(4):610–7 doi 10.21037/jgo.2018.05.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Innocenti F, Ou FS, Qu X, Zemla TJ, Niedzwiecki D, Tam R, et al. Mutational Analysis of Patients With Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient Outcome. J Clin Oncol 2019;37(14):1217–27 doi 10.1200/JCO.18.01798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med 2016;375(19):1823–33 doi 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 29.Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M, et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol 2018;4(5):e180013 doi 10.1001/jamaoncol.2018.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 2014;20(19):5064–74 doi 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 2018;379(22):2108–21 doi 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 32.Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol 2018;19(11):1480–92 doi 10.1016/S1470-2045(18)30700-9. [DOI] [PubMed] [Google Scholar]
- 33.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372(4):320–30 doi 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 34.Yarchoan M, Albacker LA, Hopkins AC, Montesion M, Murugesan K, Vithayathil TT, et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019;4(6) doi 10.1172/jci.insight.126908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9(1):34 doi 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31(11):1023–31 doi 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fabrizio D, Milbury C, Yip WK, Ramamurthy L, Bai X, Pattani V, et al. 56PDAnalytic validation of tumor mutational burden as a companion diagnostic for combination immunotherapy in non-small cell lung cancer. Annals of Oncology 2018;29 doi 10.1093/annonc/mdy269.054. [DOI] [Google Scholar]
- 38.Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med 2018;7(3):746–56 doi 10.1002/cam4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quinn K, Helman E, Nance T, Artieri C, Yen J, Zhao J, et al. Development and analytical validation of a plasma-based tumor mutational burden (TMB) score from next-generation sequencing panels. Annals of Oncology 2018;29:viii41 doi 10.1093/annonc/mdy269.129. [DOI] [Google Scholar]
- 40.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015;17(3):251–64 doi 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23(6):703–13 doi 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wood DE, White JR, Georgiadis A, Van Emburgh B, Parpart-Li S, Mitchell J, et al. A machine learning approach for somatic mutation discovery. Sci Transl Med 2018;10(457) doi 10.1126/scitranslmed.aar7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Endris V, Buchhalter I, Allgauer M, Rempel E, Lier A, Volckmar AL, et al. Measurement of tumor mutational burden (TMB) in routine molecular diagnostics: in silico and real-life analysis of three larger gene panels. Int J Cancer 2019;144(9):2303–12 doi 10.1002/ijc.32002. [DOI] [PubMed] [Google Scholar]
- 44.Beaubier N, Tell R, Lau D, Parsons JR, Bush S, Perera J, et al. Clinical validation of the tempus xT next-generation targeted oncology sequencing assay. Oncotarget 2019;10(24):2384–96 doi 10.18632/oncotarget.26797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Budczies J, Allgauer M, Litchfield K, Rempel E, Christopoulos P, Kazdal D, et al. Optimizing panel-based tumor mutational burden (TMB) measurement. Ann Oncol 2019;30(9):1496–506 doi 10.1093/annonc/mdz205. [DOI] [PubMed] [Google Scholar]
- 46.Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J Clin Oncol 2018;36(7):633–41 doi 10.1200/JCO.2017.75.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buchhalter I, Rempel E, Endris V, Allgauer M, Neumann O, Volckmar AL, et al. Size matters: Dissecting key parameters for panel-based tumor mutational burden analysis. Int J Cancer 2019;144(4):848–58 doi 10.1002/ijc.31878. [DOI] [PubMed] [Google Scholar]
- 48.Garofalo A, Sholl L, Reardon B, Taylor-Weiner A, Amin-Mansour A, Miao D, et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med 2016;8(1):79 doi 10.1186/s13073-016-0333-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Budczies J, Kazdal D, Allgauer M, Christopoulos P, Rempel E, Pfarr N, et al. Quantifying potential confounders of panel-based tumor mutational burden (TMB) measurement. Lung Cancer 2020;142:114–9 doi 10.1016/j.lungcan.2020.01.019. [DOI] [PubMed] [Google Scholar]
- 50.Chang H, Sasson A, Srinivasan S, Golhar R, Greenawalt DM, Geese WJ, et al. Bioinformatic Methods and Bridging of Assay Results for Reliable Tumor Mutational Burden Assessment in Non-Small-Cell Lung Cancer. Mol Diagn Ther 2019;23(4):507–20 doi 10.1007/s40291-019-00408-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol 2019;30(1):44–56 doi 10.1093/annonc/mdy495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Allgauer M, Budczies J, Christopoulos P, Endris V, Lier A, Rempel E, et al. Implementing tumor mutational burden (TMB) analysis in routine diagnostics-a primer for molecular pathologists and clinicians. Transl Lung Cancer Res 2018;7(6):703–15 doi 10.21037/tlcr.2018.08.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parikh K, Huether R, White K, Hoskinson D, Beaubier N, Dong H, et al. Tumor Mutational Burden From Tumor-Only Sequencing Compared With Germline Subtraction From Paired Tumor and Normal Specimens. JAMA Netw Open 2020;3(2):e200202 doi 10.1001/jamanetworkopen.2020.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stenzinger A, Endris V, Budczies J, Merkelbach-Bruse S, Kazdal D, Dietmaier W, et al. Harmonization and Standardization of Panel-Based Tumor Mutational Burden Measurement: Real-World Results and Recommendations of the Quality in Pathology Study. J Thorac Oncol 2020;15(7):1177–89 doi 10.1016/j.jtho.2020.01.023. [DOI] [PubMed] [Google Scholar]
- 55.Merino DM, McShane LM, Fabrizio D, Funari V, Chen SJ, White JR, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother Cancer 2020;8(1) doi 10.1136/jitc-2019-000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Budczies J, Seidel A, Christopoulos P, Endris V, Kloor M, Gyorffy B, et al. Integrated analysis of the immunological and genetic status in and across cancer types: impact of mutational signatures beyond tumor mutational burden. Oncoimmunology 2018;7(12):e1526613 doi 10.1080/2162402X.2018.1526613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature 2020;578(7793):94–101 doi 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 2019;51(2):202–6 doi 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018;33(5):853–61 e4 doi 10.1016/j.ccell.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson DB, Frampton GM, Rioth MJ, Yusko E, Xu Y, Guo X, et al. Targeted Next Generation Sequencing Identifies Markers of Response to PD-1 Blockade. Cancer Immunol Res 2016;4(11):959–67 doi 10.1158/2326-6066.CIR-16-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ready N, Hellmann MD, Awad MM, Otterson GA, Gutierrez M, Gainor JF, et al. First-Line Nivolumab Plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer (CheckMate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J Clin Oncol 2019;37(12):992–1000 doi 10.1200/JCO.18.01042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramalingam SS, Hellmann MD, Awad MM, Borghaei H, Gainor J, Brahmer J, et al. Abstract CT078: Tumor mutational burden (TMB) as a biomarker for clinical benefit from dual immune checkpoint blockade with nivolumab (nivo) + ipilimumab (ipi) in first-line (1L) non-small cell lung cancer (NSCLC): identification of TMB cutoff from CheckMate 568. Cancer Research 2018;78(13 Supplement):CT078–CT doi 10.1158/1538-7445.Am2018-ct078. [DOI] [Google Scholar]
- 63.Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N Engl J Med 2018;378(22):2093–104 doi 10.1056/NEJMoa1801946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350(6257):207–11 doi 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther 2017;16(11):2598–608 doi 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miao D, Margolis CA, Vokes NI, Liu D, Taylor-Weiner A, Wankowicz SM, et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet 2018;50(9):1271–81 doi 10.1038/s41588-018-0200-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lazar AA, Cole BF, Bonetti M, Gelber RD. Evaluation of treatment-effect heterogeneity using biomarkers measured on a continuous scale: subpopulation treatment effect pattern plot. J Clin Oncol 2010;28(29):4539–44 doi 10.1200/JCO.2009.27.9182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Budczies J, Klauschen F, Sinn BV, Gyorffy B, Schmitt WD, Darb-Esfahani S, et al. Cutoff Finder: a comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS One 2012;7(12):e51862 doi 10.1371/journal.pone.0051862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Amon L, et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med 2019;25(12):1916–27 doi 10.1038/s41591-019-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morrison C, Pabla S, Conroy JM, Nesline MK, Glenn ST, Dressman D, et al. Predicting response to checkpoint inhibitors in melanoma beyond PD-L1 and mutational burden. J Immunother Cancer 2018;6(1):32 doi 10.1186/s40425-018-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017;171(5):1042–56 e10 doi 10.1016/j.cell.2017.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pfeifer GP. Environmental exposures and mutational patterns of cancer genomes. Genome Med 2010;2(8):54 doi 10.1186/gm175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davis AA, Chae YK, Agte S, Pan A, Mohindra NA, Villaflor VM, et al. Association of tumor mutational burden with smoking and mutation status in non-small cell lung cancer (NSCLC). Journal of Clinical Oncology 2017;35(7_suppl):24- doi 10.1200/JCO.2017.35.7_suppl.24.28034071 [DOI] [Google Scholar]
- 74.Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012;150(6):1121–34 doi 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spigel DR, Schrock AB, Fabrizio D, Frampton GM, Sun J, He J, et al. Total mutation burden (TMB) in lung cancer (LC) and relationship with response to PD-1/PD-L1 targeted therapies. Journal of Clinical Oncology 2016;34(15_suppl):9017- doi 10.1200/JCO.2016.34.15_suppl.9017. [DOI] [Google Scholar]
- 76.Gou LY, Niu FY, Wu YL, Zhong WZ. Differences in driver genes between smoking-related and non-smoking-related lung cancer in the Chinese population. Cancer 2015;121 Suppl 17:3069–79 doi 10.1002/cncr.29531. [DOI] [PubMed] [Google Scholar]
- 77.Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N Engl J Med 2017;376(25):2415–26 doi 10.1056/NEJMoa1613493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Waqar SN, Govindan R. The Mystic Role of Tumor Mutational Burden in Selecting Patients With Lung Cancer for First-Line Immunotherapy. JAMA Oncology 2020;6(5):674–5 doi 10.1001/jamaoncol.2020.0264 %JJAMAOncology. [DOI] [PubMed] [Google Scholar]
- 79.Freedman ND, Silverman DT, Hollenbeck AR, Schatzkin A, Abnet CC. Association between smoking and risk of bladder cancer among men and women. Jama 2011;306(7):737–45 doi 10.1001/jama.2011.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014;507(7492):315–22 doi 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nassar AH, Umeton R, Kim J, Lundgren K, Harshman L, Van Allen EM, et al. Mutational Analysis of 472 Urothelial Carcinoma Across Grades and Anatomic Sites. Clin Cancer Res 2019;25(8):2458–70 doi 10.1158/1078-0432.CCR-18-3147. [DOI] [PubMed] [Google Scholar]
- 82.Necchi A, Raggi D, Gallina A, Madison R, Colecchia M, Lucianò R, et al. Updated Results of PURE-01 with Preliminary Activity of Neoadjuvant Pembrolizumab in Patients with Muscle-invasive Bladder Carcinoma with Variant Histologies. European Urology 2020;77(4):439–46 doi 10.1016/j.eururo.2019.10.026. [DOI] [PubMed] [Google Scholar]
- 83.Goh G, Walradt T, Markarov V, Blom A, Riaz N, Doumani R, et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2016;7(3):3403–15 doi 10.18632/oncotarget.6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Swanton C, McGranahan N, Starrett GJ, Harris RS. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov 2015;5(7):704–12 doi 10.1158/2159-8290.Cd-15-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Salem ME, Puccini A, Grothey A, Raghavan D, Goldberg RM, Xiu J, et al. Landscape of Tumor Mutation Load, Mismatch Repair Deficiency, and PD-L1 Expression in a Large Patient Cohort of Gastrointestinal Cancers. Mol Cancer Res 2018;16(5):805–12 doi 10.1158/1541-7786.MCR-17-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Castellucci E, He T, Goldstein DY, Halmos B, Chuy J. DNA Polymerase ɛ Deficiency Leading to an Ultramutator Phenotype: A Novel Clinically Relevant Entity. The oncologist 2017;22(5):497–502 doi 10.1634/theoncologist.2017-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gong J, Wang C, Lee PP, Chu P, Fakih M. Response to PD-1 Blockade in Microsatellite Stable Metastatic Colorectal Cancer Harboring a POLE Mutation. J Natl Compr Canc Netw 2017;15(2):142–7. [DOI] [PubMed] [Google Scholar]
- 88.Gong J, Robertson MD, Kim E, Fakih M, Schrock AB, Tam KW, et al. Efficacy of PD-1 Blockade in Refractory Microsatellite-Stable Colorectal Cancer With High Tumor Mutation Burden. Clin Colorectal Cancer 2019;18(4):307–9 doi 10.1016/j.clcc.2019.08.001. [DOI] [PubMed] [Google Scholar]
- 89.Ang C, Klempner SJ, Ali SM, Madison R, Ross JS, Severson EA, et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget 2019;10(40):4018–25 doi 10.18632/oncotarget.26998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013;502(7471):333–9 doi 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barroso-Sousa R, Jain E, Cohen O, Kim D, Buendia-Buendia J, Winer E, et al. Prevalence and mutational determinants of high tumor mutation burden in breast cancer. Ann Oncol 2020;31(3):387–94 doi 10.1016/j.annonc.2019.11.010. [DOI] [PubMed] [Google Scholar]
- 92.Budczies J, Bockmayr M, Denkert C, Klauschen F, Lennerz JK, Györffy B, et al. Classical pathology and mutational load of breast cancer - integration of two worlds. The journal of pathology Clinical research 2015;1(4):225–38 doi 10.1002/cjp2.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barroso-Sousa R, Keenan TE, Pernas S, Exman P, Jain E, Garrido-Castro AC, et al. Tumor Mutational Burden and PTEN Alterations as Molecular Correlates of Response to PD-1/L1 Blockade in Metastatic Triple-Negative Breast Cancer. Clin Cancer Res 2020;26(11):2565–72 doi 10.1158/1078-0432.CCR-19-3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alva AS, Mangat PK, Garrett-Mayer E, Halabi S, Alvarez RH, Calfa CJ, et al. Pembrolizumab (P) in patients (pts) with metastatic breast cancer (MBC) with high tumor mutational burden (HTMB): Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Journal of Clinical Oncology 2019;37(15_suppl):1014- doi 10.1200/JCO.2019.37.15_suppl.1014. [DOI] [Google Scholar]
- 95.Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 2016;387(10027):1540–50 doi 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 96.Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S, et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol 2016;17(6):717–26 doi 10.1016/S1470-2045(16)00175-3. [DOI] [PubMed] [Google Scholar]
- 97.Bellmunt J, Bajorin DF. Pembrolizumab for Advanced Urothelial Carcinoma. N Engl J Med 2017;376(23):2304 doi 10.1056/NEJMc1704612. [DOI] [PubMed] [Google Scholar]
- 98.Kojima T, Muro K, Francois E, Hsu C-H, Moriwaki T, Kim S-B, et al. Pembrolizumab versus chemotherapy as second-line therapy for advanced esophageal cancer: Phase III KEYNOTE-181 study. Journal of Clinical Oncology 2019;37(4_suppl):2- doi 10.1200/JCO.2019.37.4_suppl.2. [DOI] [PubMed] [Google Scholar]
- 99.O’Neil BH, Wallmark JM, Lorente D, Elez E, Raimbourg J, Gomez-Roca C, et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced colorectal carcinoma. PLoS One 2017;12(12):e0189848 doi 10.1371/journal.pone.0189848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389(10088):2492–502 doi 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kerr KM, Tsao MS, Nicholson AG, Yatabe Y, Wistuba II, Hirsch FR, et al. Programmed Death-Ligand 1 Immunohistochemistry in Lung Cancer: In what state is this art? J Thorac Oncol 2015;10(7):985–9 doi 10.1097/JTO.0000000000000526. [DOI] [PubMed] [Google Scholar]
- 102.Koopman M, Kortman GA, Mekenkamp L, Ligtenberg MJ, Hoogerbrugge N, Antonini NF, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer 2009;100(2):266–73 doi 10.1038/sj.bjc.6604867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moreira L, Balaguer F, Lindor N, de la Chapelle A, Hampel H, Aaltonen LA, et al. Identification of Lynch syndrome among patients with colorectal cancer. Jama 2012;308(15):1555–65 doi 10.1001/jama.2012.13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shih KK, Garg K, Levine DA, Kauff ND, Abu-Rustum NR, Soslow RA, et al. Clinicopathologic significance of DNA mismatch repair protein defects and endometrial cancer in women 40years of age and younger. Gynecol Oncol 2011;123(1):88–94 doi 10.1016/j.ygyno.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 105.Goodfellow PJ, Buttin BM, Herzog TJ, Rader JS, Gibb RK, Swisher E, et al. Prevalence of defective DNA mismatch repair and MSH6 mutation in an unselected series of endometrial cancers. Proc Natl Acad Sci U S A 2003;100(10):5908–13 doi 10.1073/pnas.1030231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010;28(19):3167–75 doi 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Andre T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt CJA, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high/mismatch repair deficient metastatic colorectal cancer: The phase 3 KEYNOTE-177 Study. J Clin Oncol 2020;38(18_suppl):LBA4–LBA doi 10.1200/JCO.2020.38.18_suppl.LBA4. [DOI] [Google Scholar]
- 108.Le DT, Kim TW, Van Cutsem E, Geva R, Jäger D, Hara H, et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J Clin Oncol 2020;38(1):11–9 doi 10.1200/jco.19.02107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357(6349):409–13 doi 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017;552(7683):116–20 doi 10.1038/nature24673. [DOI] [PubMed] [Google Scholar]
- 111.Lee H, Sha D, Foster NR, Shi Q, Alberts SR, Smyrk TC, et al. Analysis of tumor microenvironmental features to refine prognosis by T, N risk group in patients with stage III colon cancer (NCCTG N0147) (Alliance). Ann Oncol 2020;31(4):487–94 doi 10.1016/j.annonc.2020.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yoon HH, Shi Q, Heying EN, Muranyi A, Bredno J, Ough F, et al. Intertumoral Heterogeneity of CD3(+) and CD8(+) T-Cell Densities in the Microenvironment of DNA Mismatch-Repair-Deficient Colon Cancers: Implications for Prognosis. Clin Cancer Res 2019;25(1):125–33 doi 10.1158/1078-0432.CCR-18-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Goodman AM, Sokol ES, Frampton GM, Lippman SM, Kurzrock R. Microsatellite-Stable Tumors with High Mutational Burden Benefit from Immunotherapy. Cancer Immunol Res 2019;7(10):1570–3 doi 10.1158/2326-6066.CIR-19-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez-Vega F, Ahuja A, et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018;33(5):843–52.e4 doi 10.1016/j.ccell.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 2007;28(6):622–9 doi 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 116.Mouw KW, Goldberg MS, Konstantinopoulos PA, D’Andrea AD. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov 2017;7(7):675–93 doi 10.1158/2159-8290.CD-17-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chae YK, Davis AA, Raparia K, Agte S, Pan A, Mohindra N, et al. Association of Tumor Mutational Burden With DNA Repair Mutations and Response to Anti-PD-1/PD-L1 Therapy in Non-Small-Cell Lung Cancer. Clin Lung Cancer 2019;20(2):88–96 e6 doi 10.1016/j.cllc.2018.09.008. [DOI] [PubMed] [Google Scholar]
- 118.Teo MY, Seier K, Ostrovnaya I, Regazzi AM, Kania BE, Moran MM, et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit From PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. 2018;36(17):1685–94 doi 10.1200/jco.2017.75.7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Parikh AR, He Y, Hong TS, Corcoran RB, Clark JW, Ryan DP, et al. Analysis of DNA Damage Response Gene Alterations and Tumor Mutational Burden Across 17,486 Tubular Gastrointestinal Carcinomas: Implications for Therapy. Oncologist 2019;24(10):1340–7 doi 10.1634/theoncologist.2019-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ricciuti B, Recondo G, Spurr LF, Li YY, Lamberti G, Venkatraman D, et al. Impact of DNA Damage Response and Repair (DDR) Gene Mutations on Efficacy of PD-(L)1 Immune Checkpoint Inhibition in Non-Small Cell Lung Cancer. Clin Cancer Res 2020;26(15):4135–42 doi 10.1158/1078-0432.CCR-19-3529. [DOI] [PubMed] [Google Scholar]
- 121.Vokes NI, Liu D, Ricciuti B, Jimenez-Aguilar E, Rizvi H, Dietlein F, et al. Harmonization of Tumor Mutational Burden Quantification and Association With Response to Immune Checkpoint Blockade in Non–Small-Cell Lung Cancer. JCO Precision Oncology 2019(3):1–12 doi 10.1200/PO.19.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stenzinger A, Allen JD, Maas J, Stewart MD, Merino DM, Wempe MM, et al. Tumor mutational burden standardization initiatives: Recommendations for consistent tumor mutational burden assessment in clinical samples to guide immunotherapy treatment decisions. Genes Chromosomes Cancer 2019;58(8):578–88 doi 10.1002/gcc.22733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fancello L, Gandini S, Pelicci PG, Mazzarella L. Tumor mutational burden quantification from targeted gene panels: major advancements and challenges. J Immunother Cancer 2019;7(1):183 doi 10.1186/s40425-019-0647-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sandmann S, Karimi M, de Graaf AO, Rohde C, Gollner S, Varghese J, et al. appreci8: a pipeline for precise variant calling integrating 8 tools. Bioinformatics 2018;34(24):4205–12 doi 10.1093/bioinformatics/bty518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep 2016;17(4):1206 doi 10.1016/j.celrep.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol 2017;18(8):1009–21 doi 10.1016/S1470-2045(17)30516-8. [DOI] [PubMed] [Google Scholar]
- 127.Mandal R, Samstein RM, Lee KW, Havel JJ, Wang H, Krishna C, et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019;364(6439):485–91 doi 10.1126/science.aau0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ott PA, Bang YJ, Piha-Paul SA, Razak ARA, Bennouna J, Soria JC, et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated With Pembrolizumab Across 20 Cancers: KEYNOTE-028. J Clin Oncol 2019;37(4):318–27 doi 10.1200/JCO.2018.78.2276. [DOI] [PubMed] [Google Scholar]
- 129.Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018;362(6411) doi 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Qin BD, Jiao XD, Zang YS. Tumor mutation burden to tumor burden ratio and prediction of clinical benefit of anti-PD-1/PD-L1 immunotherapy. Med Hypotheses 2018;116:111–3 doi 10.1016/j.mehy.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 131.Rizvi NA, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn M-J, et al. Blood tumor mutational burden (bTMB) and tumor PD-L1 as predictive biomarkers of survival in MYSTIC: First-line durvalumab (D) ± tremelimumab (T) versus chemotherapy (CT) in metastatic (m) NSCLC. Journal of Clinical Oncology 2019;37(15_suppl):9016- doi 10.1200/JCO.2019.37.15_suppl.9016. [DOI] [Google Scholar]
- 132.Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med 2018;24(9):1441–8 doi 10.1038/s41591-018-0134-3. [DOI] [PubMed] [Google Scholar]
- 133.Khagi Y, Goodman AM, Daniels GA, Patel SP, Sacco AG, Randall JM, et al. Hypermutated Circulating Tumor DNA: Correlation with Response to Checkpoint Inhibitor-Based Immunotherapy. Clin Cancer Res 2017;23(19):5729–36 doi 10.1158/1078-0432.CCR-17-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Clopper CPES. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika 1934;26(4):404–13 doi 10.1093/biomet/26.4.404. [DOI] [Google Scholar]
- 135.Venables W, Ripley BD. Modern Applied Statistics with S. 2002, Fourth edition New York: Springer-Verlag. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1