Randomized Trial of a Vaccine Regimen to Prevent Chronic HCV Infection (original) (raw)
. Author manuscript; available in PMC: 2021 Aug 16.
Published in final edited form as: N Engl J Med. 2021 Feb 11;384(6):541–549. doi: 10.1056/NEJMoa2023345
Abstract
BACKGROUND
A safe and effective vaccine to prevent chronic hepatitis C virus (HCV) infection is a critical component of efforts to eliminate the disease.
METHODS
In this phase 1–2 randomized, double-blind, placebo-controlled trial, we evaluated a recombinant chimpanzee adenovirus 3 vector priming vaccination followed by a recombinant modified vaccinia Ankara boost; both vaccines encode HCV nonstructural proteins. Adults who were considered to be at risk for HCV infection on the basis of a history of recent injection drug use were randomly assigned (in a 1:1 ratio) to receive vaccine or placebo on days 0 and 56. Vaccine-related serious adverse events, severe local or systemic adverse events, and laboratory adverse events were the primary safety end points. The primary efficacy end point was chronic HCV infection, defined as persistent viremia for 6 months.
RESULTS
A total of 548 participants underwent randomization, with 274 assigned to each group. There was no significant difference in the incidence of chronic HCV infection between the groups. In the per-protocol population, chronic HCV infection developed in 14 participants in each group (hazard ratio [vaccine vs. placebo], 1.53; 95% confidence interval [CI], 0.66 to 3.55; vaccine efficacy, −53%; 95% CI, −255 to 34). In the modified intention-to-treat population, chronic HCV infection developed in 19 participants in the vaccine group and 17 in placebo group (hazard ratio, 1.66; 95% CI, 0.79 to 3.50; vaccine efficacy, −66%; 95% CI, −250 to 21). The geometric mean peak HCV RNA level after infection differed between the vaccine group and the placebo group (152.51×103 IU per milliliter and 1804.93×103 IU per milliliter, respectively). T-cell responses to HCV were detected in 78% of the participants in the vaccine group. The percentages of participants with serious adverse events were similar in the two groups.
CONCLUSIONS
In this trial, the HCV vaccine regimen did not cause serious adverse events, produced HCV-specific T-cell responses, and lowered the peak HCV RNA level, but it did not prevent chronic HCV infection. (Funded by the National Institute of Allergy and Infectious Diseases; ClinicalTrials.gov number, NCT01436357.)
Hepatitis C virus (HCV) infection remains one of the most prevalent blood-brone viral infections worldwide and is a leading cause of death from infectious disease globally.1–3 Despite high cure rates with direct-acting antiviral therapies, more than 71 million people live with chronic HCV infection, and an estimated 1.75 million new infections and approximately 400,000 deaths from HCV infection occur annually.1–3 From 2009 through 2018, the incidence of HCV infection tripled in the United States, fueled by increases in opioid injecting.4 Failure to prevent new HCV infections is the leading threat to the World Health Organization 2030 global elimination goal.2,5,6 A prophylactic HCV vaccine would provide an essential tool for achieving elimination goals by interrupting transmission.7,8
We assessed a heterologous prime–boost vaccination strategy with chimpanzee adenovirus 3 (ChAd3) and modified vaccinia Ankara (MVA) vectors encoding the nonstructural proteins (NS) of HCV genotype 1b (ChAd3-NSmut and MVA-NSmut, GlaxoSmithKline). In phase 1 testing, this vaccine regimen had a clinically acceptable safety profile and induced T-cell responses.9,10
The primary objectives of this trial were to assess the safety of ChAd3-NSmut and MVA-NSmut when administered to HCV-uninfected persons at high risk for infection and to determine whether the vaccine regimen would be more effective than placebo for the prevention of chronic HCV infection. The secondary objective of the trial was to evaluate the vaccine immunogenicity.
METHODS
TRIAL DESIGN AND PARTICIPANTS
We conducted this phase 1–2 double-blind, randomized, placebo-controlled trial between 2012 and 2018 at Johns Hopkins University; the University of California, San Francisco; and the University of New Mexico. Participants were healthy HCV-uninfected adults (18 to 45 years of age) who had injected drugs within 90 days before randomization. After 68 participants had been enrolled, the data and safety monitoring board recommended that phase 2 be initiated. Participants received risk-reduction counseling and referrals to substance-use treatment and syringe services at every study visit. All participants who acquired HCV infection were referred to independent physicians for clinical follow-up, including HCV treatment evaluation. The trial did not provide or pay for HCV treatment, which national guidelines did not uniformly recommend during acute infection at the time, or obtain treatment data after trial follow-up ended.
HCV-uninfected persons who inject drugs were randomly assigned to receive intramuscular injections of ChAd3-NSmut vaccine (2.5×1010 viral particles) on day 0 and MVA-NSmut vaccine (1.8×108 plaque-forming units) on day 56 (vaccine group) or saline placebo on days 0 and 56 (placebo group). Randomization was performed in a 1:1 ratio and was stratified according to sex and IFNL3 genotype, because both factors alter the likelihood of progression to chronic HCV infection.11,12 Both ChAd3-NSmut and MVA-NSmut encoded NS3, NS4, NS5A, and NS5B from the HCV 1b genotype with an inactivating mutation introduced in the catalytic site of the HCV polymerase.9
Inclusion and exclusion criteria are provided in Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org. The data and safety monitoring board reviewed interim analyses with a focus on safety, immunogenicity, and recruitment milestones for power and sample-size requirements. The trial investigators were unaware of the randomization assignments and results throughout the trial. Participants were followed monthly for HCV infection for 20 months after enrollment and for 9 months after HCV detection.
OVERSIGHT
The trial was performed in accordance with federal and local ethical standards under an Investigational New Drug protocol. The trial protocol (available at NEJM.org) and trial documents were reviewed and approved by human subjects review committees at Johns Hopkins University; the University of California, San Francisco; the University of New Mexico; the National Institute of Allergy and Infectious Diseases (NIAID); and the Food and Drug Administration. A certificate of confidentiality was obtained to further protect sensitive participant data. Written informed consent was obtained from all participants after a comprehensive explanation of the nature and risks of the trial and successful completion of a comprehension assessment.
Okairos, a company acquired by GlaxoSmithKline, provided the prime and boost vaccines and saline placebo, as well as consultation for the trial. Representatives from GlaxoSmithKline reviewed the protocol and contributed to the writing of the manuscript that was submitted, including reviewing before submission. Data analyses were conducted by the Emmes Company. The trial sponsor (the NIAID) and the trial principal investigators (the first and last authors) vouch for the accuracy and completeness of the data and for the fidelity of the trial to the protocol, and the principal investigators and lead statistician (the sixth-to-last author) vouch for the data analyses.
EVALUATIONS OF SAFETY
Safety analyses included all participants who received the first injection of vaccine or placebo. The primary safety end points were vaccine-related serious adverse events occurring at any time during the trial period, severe local or systemic solicited adverse events occurring during the 8 days after each injection, and laboratory adverse events assessed at baseline and 1 month after each injection. Participants recorded their body temperature and the presence and intensity of postinjection adverse events daily for 8 days after injection of vaccine or placebo (see Section 2 in the Supplementary Appendix). Laboratory evaluations included monthly white-cell and platelet counts and hemoglobin, alanine aminotransferase (ALT), and creatinine levels; laboratory adverse events are defined in Table S2. All women underwent urine pregnancy testing at screening and before each injection of vaccine or placebo. Pregnant participants were not given vaccine or placebo.
EVALUATIONS OF EFFICACY
Participants underwent monthly qualitative HCV RNA testing by means of transcription-mediated amplification (Gen-Probe). Positive results were confirmed by quantitative HCV RNA and genotype testing in the Johns Hopkins University laboratory13 or at Quest Diagnostics. Incident HCV infection was defined as a confirmed positive HCV RNA test after a previous negative HCV RNA test (or tests). The date of HCV infection was defined as the midpoint between the last negative and first positive HCV RNA tests.
The primary efficacy end point was chronic HCV infection, defined as persistent viremia for 6 months. Chronicity is established in most infected persons by then.14 Persistent viremia was defined as the presence of the same virus (confirmed by sequencing of the HCV core–E1 region and phylogenetic analysis) in blood obtained at the first visit in which HCV RNA was detected and at month 6 after incident infection, with a third HCV RNA–positive sample identified between the two visits. An independent expert in HCV sequence analysis compared sequences at the time of incident infection and at later time points to confirm infection with the same virus.13 An end-point review committee, the members of which were unaware of the randomization assignments, reviewed all cases to confirm the infection end point.
The date of viral clearance was defined as the midpoint of the interval between the last test with detectable HCV RNA and the first of two consecutive tests with undetectable HCV RNA. Exploratory efficacy analyses included assessments of whether the vaccines had an effect on the incidence of (primary) HCV infection, the incidence of chronic HCV infection for 9 months, the geometric mean peak HCV RNA level after infection, the duration of HCV viremia in participants in whom incident HCV infection was cleared, and the incidence of chronic infection with HCV of genotype 1 as compared with non–genotype 1 infection.
MEASUREMENT OF VACCINE IMMUNOGENICITY
T-cell responses were measured by interferon-γ enzyme-linked immunosorbent spot (ELISpot) assays at baseline (before any injection was received) and at 30 and 56 days after the first injection (ChAd3-NSmut or placebo), as well as at 7 and 34 days after the second injection (MVA-NSmut or placebo). Assays were performed with thawed peripheral blood mononuclear cells (PBMCs) and peptide pools derived from the HCV virus used in the vaccine.9 A positive immune response was defined as more than 48 spot-forming cells per million PBMCs and at least three times the mean background number of spots per million PBMCs. Participants were defined as having had a response to the vaccine or placebo if they had tested negative for HCV-specific immune responses at baseline and had a positive immune response to at least one peptide pool detected after either injection.
STATISTICAL ANALYSIS
Sample-size calculations were based on the detection of a 60% lower incidence of 6-month chronic HCV infection among vaccine recipients than among placebo recipients in the per-protocol population. We calculated that 43 chronic infection events would provide 85% power to detect such a difference with a two-sided logrank test conducted at an alpha level of 0.05. The incidence of chronic infection among placebo recipients was assumed to be 14% annually; thus, a total of 292.5 participants in the per-protocol population followed for 1.5 years would provide, on average, 43 events. Under the assumption that 65% of enrolled participants would be retained in the per-protocol population, the target enrollment was originally estimated as 450 participants. Subsequently, a protocol-specified blinded interim analysis for the reestimation of sample size was reviewed by the data and safety monitoring board, and because of a low incidence of chronic infection, the enrollment target was increased to 540 participants.
Efficacy analyses were performed in modified intention-to-treat and per-protocol populations. The primary efficacy analysis was performed in the per-protocol population; a secondary efficacy analysis of chronic HCV infection was performed in the modified intention-to-treat population. The modified intention-to-treat population included all participants who received the first injection, were HCV negative at the time of the first injection, and had sufficient follow-up data (at least three clinic visits after the second injection). The per-protocol population included participants who met the criteria for the modified intention-to-treat population, received both injections, and had no major protocol deviations that would compromise the assessment of vaccine efficacy. Because the efficacy analyses were time-to-event analyses, data from participants who underwent randomization were included at the point at which the protocol definition of the efficacy end point was met or were censored at the point at which at least one of the analysis population criteria was no longer met, whichever came first. Data from participants who discontinued participation in the trial early or who completed the trial without an observed end-point event were censored at the final visit.
The between-group difference in the incidence of 6-month chronic HCV infection (primary end point) was calculated from the hazard ratio of Cox proportional hazards models, stratified according to sex and IFNL3 status. Vaccine efficacy was calculated as 100×(1-hazard ratio). The methods used to assess primary and exploratory end points are described in Section 3 in the Supplementary Appendix. The immunogenicity analysis population included participants who received any vaccine or placebo and for whom immunogenicity end-point data were available.
Safety data were coded according to Medical Dictionary for Regulatory Activities preferred term and system organ class and were summarized on both on the participant level and the event level.
Because the statistical analysis plan did not include a provision for correcting for multiplicity for the secondary or other end points, the results are reported as point estimates and 95% confidence intervals. The widths of the confidence intervals were not adjusted for multiplicity, so they should not be used to infer definitive treatment effects for the secondary or other end points.
RESULTS
TRIAL POPULATION
A total of 991 persons were screened, and 548 were enrolled. Of the enrolled participants, 78% were male, 61% were White, 21% were Black or African American, and 14% were Hispanic. Sex, race, ethnic group, IFNL3 status, age, and body-mass index did not differ substantially between the groups (Table 1). The most common reason for not passing screening was not being deemed in good health by a trial physician and having clinical laboratory values outside the acceptable range (103 participants). Among the 548 enrolled participants, 274 (50%) were randomly assigned to the vaccine group and 274 (50%) to the placebo group; 1 participant who had been randomly assigned to the placebo group was erroneously given vaccine for both doses and is included in the vaccine group in all summaries and analyses. A total of 546 participants received the first injection of vaccine or placebo, and 455 participants received both injections (228 in the vaccine group and 227 in the placebo group). Table S3 shows the reasons for discontinuation of receipt of injections and early discontinuation of participation in the trial.
Table 1.
Baseline Characteristics of the Participants.
| Characteristic | Vaccine Group (N = 275)* | Placebo Group (N = 273)* | All Participants (N = 548) |
|---|---|---|---|
| Age — yr | |||
| Median | 30.0 | 29.0 | 29.0 |
| Range | 18–45 | 18–45 | 18–45 |
| Sex — % | |||
| Female | 22 | 23 | 22 |
| Male | 78 | 77 | 78 |
| Body-mass index† | |||
| Median | 24.4 | 24.3 | 24.3 |
| Range | 17.2–55.5 | 16.8–53.4 | 16.8–55.5 |
| Hispanic ethnic group — %‡ | 15 | 14 | 14 |
| Race or ethnic group — %‡ | |||
| American Indian or Alaska Native | 3 | <1 | 2 |
| Asian, Native Hawaiian, or Pacific Islander | 1 | 1 | 1 |
| Black or African American | 23 | 19 | 21 |
| White | 58 | 64 | 61 |
| Multiracial | 12 | 11 | 11 |
| Not reported | 3 | 4 | 4 |
| IFNL3 CC genotype — %§ | 41 | 41 | 41 |
In total, 75 participants became HCV-infected during follow-up (37 participants [13%] in the vaccine group and 38 [14%] in the placebo group) and 2 participants in the vaccine group received treatment for acute HCV infection outside the protocol. A total of 36 participants met the definition of having 6-month chronic infection — 19 (7%) in the vaccine group and 17 (6%) in the placebo group — and the infection was cleared in 9 participants (5 in the vaccine group and 4 in the placebo group), with no evidence of viremia at 6 months (Fig. S1). The numbers of the remaining participants who became infected (including the number who discontinued participation early or completed the trial without an observed end-point event) and the timing of the initial and chronic infections and infection genotypes are provided in Table S4.
VACCINE EFFICACY
No evidence of vaccine efficacy was detected in the primary per-protocol analysis of 6-month chronic infection, in which 202 participants (73%) in the vaccine group and 199 participants (73%) in the placebo group were followed to chronic infection or trial completion and data from the remaining participants were censored before trial completion. A total of 14 participants in the vaccine group and 14 participants in the placebo group were chronically infected at 6 months (Table 2). There was no evidence of vaccine efficacy in the analysis of the incidence of chronic HCV infection in the vaccine group and the placebo group (hazard ratio [vaccine vs. placebo], 1.53; 95% confidence interval [CI], 0.66 to 3.55; vaccine efficacy, −53%; 95% CI, −255 to 34) (Fig. S2). No evidence of vaccine efficacy was detected in the modified intention-to-treat analysis, with 19 participants in the vaccine group and 17 participants in the placebo group chronically infected at 6 months (hazard ratio, 1.66; 95% CI, 0.79 to 3.50; vaccine efficacy, −66%; 95% CI, −250 to 21).
Table 2.
Vaccine Efficacy against Chronic HCV Infection at 6 Months.*
| Analysis and Population† | Vaccine (N = 275) | Placebo (N =273) | Vaccine Efficacy (95% CI)‡ | Hazard Ratio (95% CI)§ | P Value¶ | ||
|---|---|---|---|---|---|---|---|
| Censored Data | Chronic Infection | Censored Data | Chronic Infection | ||||
| number of participants | percent | ||||||
| Primary efficacy analysis, per-protocol population‖ | 261 | 14 | 259 | 14 | −53 (−255 to 34) | 1.53 (0.66–3.55) | 0.31 |
| Secondary efficacy analysis, modified intention-to-treat population | 256 | 19 | 257 | 17 | −66 (−250 to 21) | 1.66 (0.79–3.50) | 0.18 |
EXPLORATORY EFFICACY ANALYSES
Results of the exploratory analyses of efficacy end points are provided in Table S6. There was no evidence of a vaccine effect on the incidence of chronic HCV infection at 9 months, the incidence of primary HCV infection (14.1 cases per 100 person-years of observation in the overall trial population), the time from incident infection to spontaneous viral clearance, or the incidence of chronic HCV infection with genotype 1 as compared with non–genotype 1 virus. In the placebo group, the geometric mean HCV RNA level increased after incident infection, peaking 1 month after incident infection (Table S7). In the vaccine group (both the per-protocol and modified intention-to-treat populations), the geometric mean peak HCV RNA level occurred at the time of incident infection; the ramp-up of viremia that follows incident infection in natural infection was not observed.15,16 In the modified intention-to-treat population, the geometric mean peak HCV RNA level was lower in the vaccine group than in the placebo group (152.51×103 IU per milliliter [95% CI, 33.5×103 to 686×103] vs. 1804.93×103 IU per milliliter [95% CI, 565×103 to 5764×103]). Similar results were observed in the per-protocol population.
SAFETY
There were no vaccine-related serious adverse events (Table 3). Serious adverse events were of similar type and incidence in the two groups, with most attributed to injection drug use. Severe solicited adverse events in the 8 days after injection were rare (Table 3). Solicited systemic adverse events that occurred in at least 10% of participants in either group occurred in similar percentages of vaccine and placebo recipients (Table S8). The most frequent laboratory adverse event was an elevation in ALT level, a finding known to be associated with substance use17 and with HCV infection. Other than ALT elevation, grade 3 or 4 laboratory adverse events occurred in less than 1% of participants (Table 3). Laboratory events of all grades are shown in Table S9.
Table 3.
Safety End Points.*
| Event | Participants with an Event after Dose 1 | Participants with an Event after Dose 2 | Participants with an Eventafter Either Dose | |||
|---|---|---|---|---|---|---|
| no./total no. | % (95% CI) | no./total no. | % (95% CI) | no./total no. | % (95% CI) | |
| Vaccine- or placebo-related serious adverse event | ||||||
| Vaccine | 0/274 | 0 (0–1) | 0/228 | 0 (0–2) | 0/274 | 0 (0–1) |
| Placebo | 0/272 | 0 (0–1) | 0/227 | 0 (0–2) | 0/272 | 0 (0–1) |
| Severe solicited local adverse event†‡ | ||||||
| Vaccine | 0/274 | 0 (0–1) | 1/228 | <1 (0–2) | 1/274 | <1 (0–2) |
| Placebo | 0/272 | 0 (0–1) | 0/227 | 0 (0–2) | 0/272 | 0 (0–1) |
| Severe solicited systemic adverse event†§ | ||||||
| Vaccine | 0/274 | 0 (0–1) | 1/228 | <1 (0–2) | 1/274 | <1 (0–2) |
| Placebo | 0/272 | 0 (0–1) | 0/227 | 0 (0–2) | 0/272 | 0 (0–1) |
| Any laboratory adverse event | ||||||
| Vaccine | 70/258 | 27 (22–33) | 73/220 | 33 (27–40) | 102/262 | 39 (33–45) |
| Placebo | 48/259 | 19 (14–24) | 49/224 | 22 (17–28) | 76/261 | 29 (24–35) |
| Grade 3 or 4 laboratory adverse events | ||||||
| Increase in ALT level¶ | ||||||
| HCV infected | ||||||
| Vaccine | 1/4 | 25 (1–75) | 4/8 | 50 (19–81) | 4/8 | 50 (19–81) |
| Placebo | 0/5 | 0 (50–100) | 3/9 | 33 (10–68) | 3/10 | 30 (9–62) |
| HCV uninfected | ||||||
| Vaccine | 1/258 | <1 (0–2) | 0/213 | 0 (0–2) | 1/262 | <1 (0–2) |
| Placebo | 0/258 | 0 (0–1) | 0/217 | <1 (0–2) | 1/260 | <1 (0–2) |
| Increase in creatinine level | ||||||
| Vaccine | 0/258 | 0 (0–1) | 0/220 | 0 (0–2) | 0/262 | 0 (0–1) |
| Placebo | 0/259 | 0 (0–1) | 0/224 | 0 (0–2) | 0/261 | 0 (0–1) |
| Decrease in hemoglobin level | ||||||
| Vaccine | 0/258 | 0 (0–1) | 0/220 | 0 (0–2) | 0/262 | 0 (0–1) |
| Placebo | 0/259 | 0 (0–1) | 0/224 | 0 (0–2) | 0/261 | 0 (0–1) |
| Increase in white-cell count | ||||||
| Vaccine | 0/258 | 0 (0–1) | 0/220 | 0 (0–2) | 0/262 | 0 (0–1) |
| Placebo | 0/259 | 0 (0–1) | 0/224 | 0 (0–2) | 0/261 | 0 (0–1) |
| Decrease in platelet count | ||||||
| Vaccine | 1/258 | <1 (0–2) | 0/220 | 0 (0–2) | 1/262 | <1 (0–2) |
| Placebo | 0/259 | 0 (0–1) | 0/224 | 0 (0–2) | 0/261 | 0 (0–1) |
IMMUNOGENICITY
Because HCV infection induces HCV-specific T-cell responses, immunogenicity was assessed before HCV infection. Immunogenicity data were available for 145 vaccine recipients (53%) and 149 placebo participants (54%). T-cell responses to HCV were detected in 78% of vaccine recipients and 3% of placebo recipients. The geometric mean ELISpot responses in each group over time are shown in Table S10. Among the placebo recipients, ELISpot responses did not change substantially over the course of the trial. The peak interferon-γ ELISpot responses across each vaccine antigen pool among vaccine recipients are shown in Figure 1. The median of maximum interferon-γ ELISpot results summed for all pools for all vaccine recipients was 428.3 spot-forming cells per million PBMCs (range, 0 to 3443).
Figure 1. Peak Vaccine-Induced T-Cell Responses in the Vaccine Group.
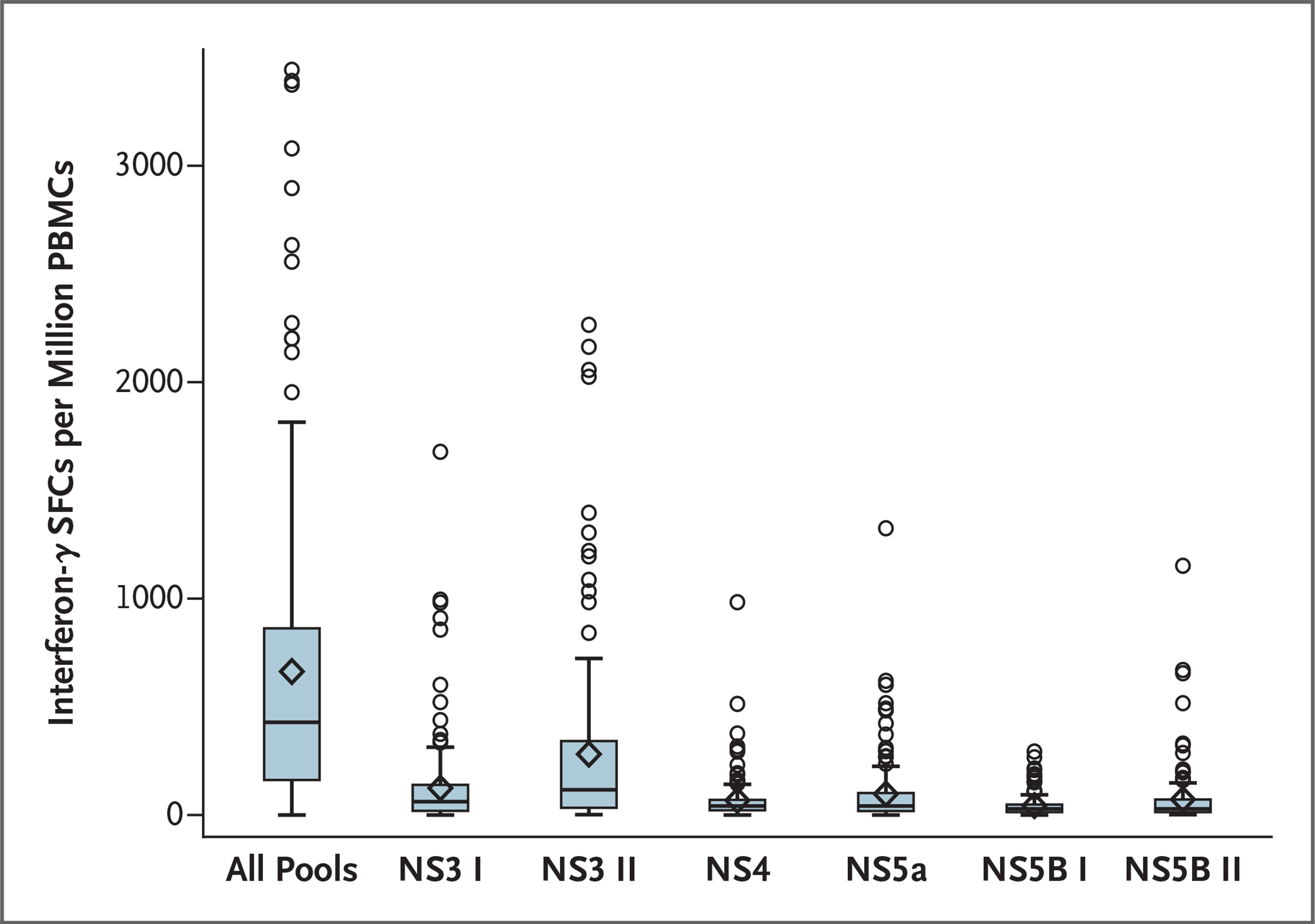
Peak responses (at 1 week after the MVA-NSmut injection) were assessed by inteferon-γ enzyme-linked immunosorbent spot assay according to nonstructural (NS) protein pool. In the box-and-whisker plots, the horizontal line indicates the median, the top and bottom of the box the interquartile range, the diamond the mean, and the whiskers the 95% confidence interval. PBMC denotes peripheral blood mononuclear cell, and SFC spot-forming cell.
DISCUSSION
The data from this randomized, double-blind, placebo-controlled trial of ChAd3-NSmut and MVA-NSmut show that the vaccine regimen did not cause serious adverse events and did elicit T-cell responses against HCV proteins; however, it was not associated with a lower incidence of chronic HCV infection than placebo.
Because persons who inject drugs have the highest incidence of HCV infection, targeting this population for testing and implementing preventive vaccines is critical18; however, it is also challenging. Scheduling interim analyses to assess retention was important, and ongoing outreach was essential to maximize participant engagement. The trial required significant resources and expertise to engage persons who inject drugs; however, it showed the feasibility of conducting rigorous vaccine research involving this population.
Some studies have shown lower vaccine immunogenicity in persons who inject drugs.19,20 Consistent with these observations, peak immune responses were lower in our trial than in the trials of this vaccine regimen that have involved healthy volunteers.9 Responses in healthy volunteers may better represent vaccine immunogenicity if the vaccine is used universally as a way to prevent transmission. Nevertheless, an effective vaccine against HCV will ideally have sufficient immunogenicity to provide protection if it is administered to persons who inject drugs.
Randomization was stratified according to sex because vaccines can be less immunogenic in men and because women have higher rates of spontaneous HCV clearance.11,21,22 Because persons who inject drugs are predominantly male and because screening failure due to anemia was more common among women, men were disproportionately enrolled in the trial, which limited our ability to examine sex-associated outcomes. Strengths of this trial include the racial and ethnic diversity of the study population and the use of placebo to provide data on background levels of adverse events related to drug use and HCV infection.
The reasons for the lack of a vaccine effect on the incidence of chronic infection are unknown. Adenoviral vectors can be less immunogenic in persons with vector cross-reactive antibodies, which may be more common in persons who inject drugs.23,24 Alternatively, previous exposure to trace amounts of HCV that are insufficient to induce seroconversion could reduce immune responses during subsequent infection, as shown previously in nonhuman primates.25 Finally, the vaccine did not contain HCV envelope proteins, the target of neutralizing antibodies that could reduce incident infection or enhance clearance.26,27 Persistence in the face of vaccine-induced T-cell responses could be due to viral escape from immune pressure, as occurs in persistent natural HCV infection, or to the limited cross-reactivity of vaccine-induced T cells to infecting HCV strains.28–30
Studies showing that persons with HCV infection who inject drugs rarely seek HCV treatment, in spite of the safety and efficacy of current treatments, underscore the importance of vaccination to prevent infection.31–33 In addition to other strategies for HCV infection prevention, screening, and treatment, a prophylactic HCV vaccine will be needed for successful global control of HCV infection.
Supplementary Material
Supplement1
Acknowledgments
Supported by the National Institute of Allergy and Infectious Diseases, NIH, Department of Health and Human Services (contract no. HHSN266200400074C). The Switzerland-based company Okairos, which was acquired by GlaxoSmithKline in May 2013, provided the vaccines and consultation for the trial.
Dr. Page reports receiving grant support, paid to the University of New Mexico, from Gilead Sciences and grant support, paid to Greenville Health System, from the Patient-Centered Outcomes Research Institute (PCORI); Dr. Melia, receiving grant support, paid to Johns Hopkins University, from Gilead Sciences; Dr. Vassilev, being employed by GlaxoSmithKline; and Dr. Lin, being employed by and owning stock options in GlaxoSmithKline. No other potential conflict of interest relevant to this article was reported.
Footnotes
The contents of this report are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health (NIH).
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
A data sharing statement provided by the authors is available with the full text of this article at NEJM.org.
Contributor Information
Kimberly Page, University of New Mexico, Albuquerque, Maryland
Michael T. Melia, Johns Hopkins University, Baltimore, Maryland
Rebecca T. Veenhuis, Johns Hopkins University, Baltimore, Maryland
Matthew Winter, Johns Hopkins University, Baltimore, Maryland
Kimberly E. Rousseau, Johns Hopkins University, Baltimore, Maryland
Guido Massaccesi, Johns Hopkins University, Baltimore, Maryland
William O. Osburn, Johns Hopkins University, Baltimore, Maryland
Michael Forman, Johns Hopkins University, Baltimore, Maryland
Elaine Thomas, University of New Mexico, Albuquerque, Maryland
Karla Thornton, University of New Mexico, Albuquerque, Maryland
Katherine Wagner, University of New Mexico, Albuquerque, Maryland
Ventzislav Vassilev, GSK Vaccines, Rixensart, Belgium
Lan Lin, GSK Vaccines, Rixensart, Belgium
Paula J. Lum, University of California, San Francisco, San Francisco
Linda C. Giudice, University of California, San Francisco, San Francisco
Ellen Stein, University of California, San Francisco, San Francisco
Alice Asher, University of California, San Francisco, San Francisco Centers for Disease Control and Prevention, Office of Policy, Planning, and Partnerships, Atlanta.
Soju Chang, Division of Microbiology and Infectious Diseases, National Institute of Allergy and Infectious Diseases, Maryland
Richard Gorman, Division of Microbiology and Infectious Diseases, National Institute of Allergy and Infectious Diseases, Maryland
Marc G. Ghany, Liver Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland
T. Jake Liang, Liver Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland
Michael R. Wierzbicki, Emmes Company, Rockville, Maryland
Elisa Scarselli, ReiThera, Rome, Italy
Alfredo Nicosia, CEINGE, Naples, Italy
Antonella Folgori, ReiThera, Rome, Italy
Stefania Capone, ReiThera, Rome, Italy
Andrea L. Cox, Johns Hopkins University, Baltimore, Maryland
REFERENCES
- 1.Jefferies M, Rauff B, Rashid H, Lam T, Rafiq S. Update on global epidemiology of viral hepatitis and preventive strategies. World J Clin Cases 2018;6:589–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hill AM, Nath S, Simmons B. The road to elimination of hepatitis C: analysis of cures versus new infections in 91 countries. J Virus Erad 2017;3:117–23. [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization. Global hepatitis report, 2017. (https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/).
- 4.Ryerson AB, Schillie S, Barker LK, Kupronis BA, Wester C. Vital signs: newly reported acute and chronic hepatitis C cases — United States, 2009–2018. MMWR Morb Mortal Wkly Rep 2020;69:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Razavi H, Waked I, Sarrazin C, et al. The present and future disease burden of hepatitis C virus (HCV) infection with today’s treatment paradigm. J Viral Hepat 2014;21:Suppl 1:34–59. [DOI] [PubMed] [Google Scholar]
- 6.Maticic M, Lombardi A, Mondelli MU, Colombo M. Elimination of hepatitis C in Europe: can WHO targets be achieved? Clin Microbiol Infect 2020;26:818–23. [DOI] [PubMed] [Google Scholar]
- 7.Bartenschlager R, Baumert TF, Bukh J, et al. Critical challenges and emerging opportunities in hepatitis C virus research in an era of potent antiviral therapy: considerations for scientists and funding agencies. Virus Res 2018;248:53–62. [DOI] [PubMed] [Google Scholar]
- 8.Roingeard P, Beaumont E. Hepatitis C vaccine: 10 good reasons for continuing. Hepatology 2020;71:1845–50. [DOI] [PubMed] [Google Scholar]
- 9.Swadling L, Capone S, Antrobus RD, et al. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci Transl Med 2014;6:261ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartnell F, Brown A, Capone S, et al. A novel vaccine strategy employing serologically different chimpanzee adenoviral vectors for the prevention of HIV-1 and HCV coinfection. Front Immunol 2019;9: 3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grebely J, Page K, Sacks-Davis R, et al. The effects of female sex, viral genotype, and IL28B genotype on spontaneous clearance of acute hepatitis C virus infection. Hepatology 2014;59:109–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009;461:798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osburn WO, Fisher BE, Dowd KA, et al. Spontaneous control of primary hepatitis C virus infection and immunity against persistent reinfection. Gastroenterology 2010;138:315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Page K, Hahn JA, Evans J, et al. Acute of hepatitis C virus infection in young adult injection drug users: a prospective study of incident infection, resolution, and reinfection. J Infect Dis 2009;200:1216–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glynn SA, Wright DJ, Kleinman SH, et al. Dynamics of viremia in early hepatitis C virus infection. Transfusion 2005;45: 994–1002. [DOI] [PubMed] [Google Scholar]
- 16.Hajarizadeh B, Grady B, Page K, et al. Patterns of hepatitis C virus RNA levels during acute infection: the InC3 study. PLoS One 2015;10(4):e0122232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larrey D, Ripault MP. Illegal and recreational compounds: hepatotoxicity of psychotropic drugs and drugs of abuse. In: Kaplowitz N, DeLeve LD, eds. Drug-induced liver disease. 3rd ed. Philadelphia:Elsevier, 2013:456–7. [Google Scholar]
- 18.Page K, Cox A, Lum PJ. Opioids, hepatitis C virus infection, and the missing vaccine. Am J Public Health 2018;108:156–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baral S, Sherman SG, Millson P, Beyrer C. Vaccine immunogenicity in injecting drug users: a systematic review. Lancet Infect Dis 2007;7:667–74. [DOI] [PubMed] [Google Scholar]
- 20.McElrath MJ, Corey L, Montefiori D, et al. A phase II study of two HIV type 1 envelope vaccines, comparing their immunogenicity in populations at risk for acquiring HIV type 1 infection. AIDS Res Hum Retroviruses 2000;16:907–19. [DOI] [PubMed] [Google Scholar]
- 21.Esmaeili A, Mirzazadeh A, Morris MD, et al. The effect of female sex on hepatitis C incidence among people who inject drugs: results from the International Multicohort InC3 Collaborative. Clin Infect Dis 2018;66:20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flanagan KL, Fink AL, Plebanski M, Klein SL. Sex and gender differences in the outcomes of vaccination over the life course. Annu Rev Cell Dev Biol 2017;33:577–99. [DOI] [PubMed] [Google Scholar]
- 23.Pine SO, Kublin JG, Hammer SM, et al. Pre-existing adenovirus immunity modifies fies a complex mixed Th1 and Th2 cytokine response to an Ad5/HIV-1 vaccine candidate in humans. PLoS One 2011; 6(4):e18526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frahm N, DeCamp AC, Friedrich DP, et al. Human adenovirus-specific T cells modulate HIV-specific T cell responses to an Ad5-vectored HIV-1 vaccine. J Clin Invest 2012;122:359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park SH, Veerapu NS, Shin EC, et al. Subinfectious hepatitis C virus exposures suppress T cell responses against subsequent acute infection. Nat Med 2013;19: 1638–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinchen VJ, Cox AL, Bailey JR. Can broadly neutralizing monoclonal antibodies lead to a hepatitis C virus vaccine? Trends Microbiol 2018;26:854–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choo Q-L, Kuo G, Ralston R, et al. Vaccination of chimpanzees against infection by the hepatitis C virus. Proc Natl Acad Sci U S A 1994;91:1294–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erickson AL, Kimura Y, Igarashi S, et al. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity 2001;15:883–95. [DOI] [PubMed] [Google Scholar]
- 29.Cox AL, Mosbruger T, Mao Q, et al. Cellular immune selection with hepatitis C virus persistence in humans. J Exp Med 2005;201:1741–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelly C, Swadling L, Brown A, et al. Cross-reactivity of hepatitis C virus specific vaccine-induced T cells at immunodominant epitopes. Eur J Immunol 2015; 45:309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehta SH, Genberg BL, Astemborski J, et al. Limited uptake of hepatitis C treatment among injection drug users. J Community Health 2008;33:126–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris MD, Mirzazadeh A, Evans JL, et al. Treatment cascade for hepatitis C virus in young adult people who inject drugs in San Francisco: low number treated. Drug Alcohol Depend 2019;198:133–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.ksher AK, Portillo CJ, Cooper BA, Dawson-Rose C, Vlahov D, Page KA. Clinicians’ views of hepatitis C virus treatment candidacy with direct-acting anti? viral regimens for people who inject drugs. Subst Use Misuse 2016;51:1218–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement1