Presence of Acetyl Coenzyme A (CoA) Carboxylase and Propionyl-CoA Carboxylase in Autotrophic Crenarchaeota and Indication for Operation of a 3-Hydroxypropionate Cycle in Autotrophic Carbon Fixation (original) (raw)
Abstract
The pathway of autotrophic CO2 fixation was studied in the phototrophic bacterium Chloroflexus aurantiacus and in the aerobic thermoacidophilic archaeon Metallosphaera sedula. In both organisms, none of the key enzymes of the reductive pentose phosphate cycle, the reductive citric acid cycle, and the reductive acetyl coenzyme A (acetyl-CoA) pathway were detectable. However, cells contained the biotin-dependent acetyl-CoA carboxylase and propionyl-CoA carboxylase as well as phosphoenolpyruvate carboxylase. The specific enzyme activities of the carboxylases were high enough to explain the autotrophic growth rate via the 3-hydroxypropionate cycle. Extracts catalyzed the CO2-, MgATP-, and NADPH-dependent conversion of acetyl-CoA to 3-hydroxypropionate via malonyl-CoA and the conversion of this intermediate to succinate via propionyl-CoA. The labelled intermediates were detected in vitro with either 14CO2 or [14C]acetyl-CoA as precursor. These reactions are part of the 3-hydroxypropionate cycle, the autotrophic pathway proposed for C. aurantiacus. The investigation was extended to the autotrophic archaea Sulfolobus metallicus and Acidianus infernus, which showed acetyl-CoA and propionyl-CoA carboxylase activities in extracts of autotrophically grown cells. Acetyl-CoA carboxylase activity is unexpected in archaea since they do not contain fatty acids in their membranes. These aerobic archaea, as well as C. aurantiacus, were screened for biotin-containing proteins by the avidin-peroxidase test. They contained large amounts of a small biotin-carrying protein, which is most likely part of the acetyl-CoA and propionyl-CoA carboxylases. Other archaea reported to use one of the other known autotrophic pathways lacked such small biotin-containing proteins. These findings suggest that the aerobic autotrophic archaea M. sedula, S. metallicus, and A. infernus use a yet-to-be-defined 3-hydroxypropionate cycle for their autotrophic growth. Acetyl-CoA carboxylase and propionyl-CoA carboxylase are proposed to be the main CO2 fixation enzymes, and phosphoenolpyruvate carboxylase may have an anaplerotic function. The results also provide further support for the occurrence of the 3-hydroxypropionate cycle in C. aurantiacus.
The capability to use carbon dioxide as the sole source of cell carbon (autotrophy) is found in almost all major groups of prokaryotes. The CO2 fixation pathways differ between groups, and there is no clear distribution pattern of the four presently known autotrophic pathways (8).
The reductive pentose phosphate cycle (Calvin-Bassham-Benson cycle) represents the CO2 fixation pathway in almost all aerobic autotrophic bacteria, for example, the cyanobacteria. This cycle became the autotrophic pathway of plants, and the CO2-fixing enzyme ribulose-1,5-bisphosphate carboxylase is one of the most abundant proteins in nature (for a recent review, see reference 43).
The reductive citric acid cycle was found in several strictly anaerobic bacteria, such as the phototrophic Chlorobium limicola (Chlorobiaceae [1, 6, 9, 21]) and the sulfate-reducing Desulfobacter hydrogenophilus (delta subgroup of Proteobacteria [33]), as well as in the microaerobic thermophilic hydrogen bacteria Hydrogenobacter thermophilus (36) and Aquifex pyrophilus (early branch-off of bacteria [2]), and in the sulfur-reducing crenarchaeon Thermoproteus neutrophilus (2). This pathway is characterized by the enzymes ATP citrate lyase, 2-oxoglutarate:acceptor oxidoreductase (2-oxoglutarate synthase), and pyruvate synthase.
The reductive acetyl coenzyme A (acetyl-CoA) pathway is confined to the strict anaerobic bacteria, such as gram-positive acetogenic bacteria (8, 45), the sulfate-reducing Desulfobacterium autotrophicum and relatives (delta subgroup group of Proteobacteria [32]), and the methanogenic bacteria (8). It is also found in euryarchaeota, e.g., in the denitrifying Ferroglobus placidus (40), and the sulfate-reducing Archaeoglobus lithotrophicus (41). This pathway is characterized by the CO2-fixing enzyme carbon monoxide dehydrogenase.
A new autotrophic pathway, the 3-hydroxypropionate cycle, has been discovered in Chloroflexus aurantiacus OK-70, a facultatively aerobic, phototrophic bacterium (5, 12, 13, 37, 38). The postulated outline of this pathway and the enzymes involved are shown in Fig. 1. Glyoxylate is formed from acetyl-CoA after the fixation of two molecules of CO2 by acetyl-CoA and propionyl-CoA carboxylases, while acetyl-CoA is regenerated. Phosphoenolpyruvate (PEP) carboxylase may have an anaplerotic function. The assimilation of the CO2 fixation product glyoxylate into cell material is at issue (20).
FIG. 1.

Proposed 3-hydroxypropionate cycle of autotrophic CO2 fixation in the phototrophic green nonsulfur bacterium C. aurantiacus (38). Enzymes: 1, acetyl-CoA carboxylase; 2, malonate-semialdehyde dehydrogenase; 3, 3-hydroxypropionate dehydrogenase; 4, 3-hydroxypropionate–CoA ligase; 5, 3-hydroxypropionyl–CoA dehydratase; 6, acrylyl-CoA reductase; 7, propionyl-CoA carboxylase; 8, methylmalonyl-CoA epimerase; 9, methylmalonyl-CoA mutase; 10, succinyl-CoA:malate-CoA transferase; 11, succinate dehydrogenase; 12, fumarase; 13, malyl-CoA lyase.
The pathways of autotrophic CO2 fixation in archaea have been studied in only a limited number of anaerobic species (see above), and the CO2 fixation pathways of aerobic autotrophic species are unknown. In the aerobic Haloferax mediterranei, ribulose-1,5-bisphosphate carboxylase was detected and purified from heterotrophically grown cells. Although the catalytic number of this key enzyme of the Calvin cycle was 100-fold lower than that in enzymes from other sources (30), this euryarchaeon may use the Calvin cycle for CO2 fixation under certain conditions, e.g., microaerobic conditions. Interestingly, the Methanococcus jannaschii, Archaeoglobus fulgidus, and Pyrococcus horikoshii genomes contain ribulose-1,5-bisphosphate carboxylase-like sequences (23, 24, 43). The situation is equally complex in the crenarchaeota. A reductive citric acid cycle has been reported for the strictly anaerobic sulfur-reducing T. neutrophilus (2, 37). Autotrophic members of the order Sulfolobales include Sulfolobus metallicus, Metallosphaera sedula, Metallosphaera prunae, Acidianus brierleyi, Acidianus infernus, Acidianus ambivalens, and Stygiolobus azoricus (34). Early studies, performed with aerobic or microaerophilic representatives of the Sulfolobales, indicated the presence of a nondefined carboxylic acid cycle in these organisms (22). Sulfolobus species grown aerobically under CO2 starvation showed an induced acetyl-CoA carboxylase activity (28). More recently, some enzymes of the proposed 3-hydroxypropionate cycle were detected in autotrophically grown A. brierleyi, including acetyl-CoA carboxylase, propionyl-CoA carboxylase, and malonate semialdehyde dehydrogenase (18). Furthermore, labelled products formed in vitro from 14CO2 in the presence of acetyl-CoA or propionyl-CoA included malate, fumarate, and succinate. 3-Hydroxypropionate was not found, and malyl-CoA lyase activity was undetectable. The authors concluded that a modified 3-hydroxypropionate cycle operates in A. brierleyi (18).
The aims of the present work were to study the pathway of autotrophic CO2 fixation in C. aurantiacus and in a representative of the aerobic crenarchaeota, M. sedula, and to screen other archaea for characteristic enzymes of the 3-hydroxypropionate cycle.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The following strains were used: T. neutrophilus V24Sta (DSM 2338), Thermoproteus tenax Kra1 (DSM 2078), M. sedula TH2 (DSM 5348), S. metallicus Kra23 (DSM 6482), A. infernus So4a (DSM 3191), Methanobacterium thermoautotrophicum YTB (DSM 1850), Methanosarcina barkeri UBS (DSM 1311), Methanogenium organophilum CV (DSM 3596), Methanococcus voltae PS (DSM 1537), Methanopyrus kandleri AV19 (DSM 6324), C. aurantiacus OK-70fl (DSM 636), A. pyrophilus KO15a (DSM 6858), D. autotrophicum HRM2 (DSM 3382), D. hydrogenophilus AcRS1 (DSM 3380), Alcaligenes eutrophus H16 (DSM 428), and Escherichia coli K-12 (DSM 423).
The Sulfolobales members were grown on Allen mineral medium as previously described (34). M. sedula was grown microaerobically with a gas phase of H2-CO2-O2 (78:19:3; 250 kPa), at 65°C and pH 2.0 (generation time, 10 h [15, 16]). As a control, cells grown aerobically with 0.05% yeast extract were used (generation time, 10 h [15]). A. infernus was grown at pH 2.5, aerobically with sulfur and anaerobically with sulfur and H2, both in the presence of 0.02% yeast extract at 80°C as reported in reference 34. S. metallicus was grown aerobically autotrophically on either metal ore or sulfur at 65°C (14). T. neutrophilus was grown anaerobically on mineral medium at 85°C under a gas phase of H2-CO2 (80:20) and elemental sulfur (37). T. tenax was also grown with CO2, H2, and thiosulfate but at 80°C (47). The methanogens M. voltae and M. kandleri were grown anaerobically with H2 and CO2 at 37 and 80°C, respectively (26, 42). M. barkeri was grown anaerobically on salt medium with acetate at 37°C, and M. organophilum was grown on salt medium with ethanol and CO2 at 30°C as reported in reference 44. A. pyrophilus was grown microaerobically on SME medium at 85°C with a gas phase of H2-CO2-O2 (78:19:3; 250 kPa), and thiosulfate (17). A. eutrophus was grown aerobically at 30°C and pH 7.0 under heterotrophic conditions with fructose (0.04% [wt/vol]) as organic substrate as reported in reference 7. D. hydrogenophilus was grown anaerobically at 30°C on a sulfide-reduced marine mineral medium under an atmosphere of H2-CO2 (80:20) and 6 g of sodium sulfate per liter as previously described (33). D. autotrophicum was similarly grown but at 28°C. C. aurantiacus was grown under phototrophic anaerobic conditions on a mineral salt medium supplemented with vitamins and gassed with a mixture of H2-CO2 (80:20), at 55°C as described elsewhere (38). E. coli was grown aerobically at 37°C with glucose in minimal salt medium. Cells were kept frozen in liquid nitrogen until use.
Cell extracts.
Cell extracts were prepared anaerobically. Cells were suspended in 2× 100 mM Tris-HCl (pH 7.8) buffer containing 2 mM dithioerythritol (DTE) and 1 mg of DNase I per 4 ml of cell suspension. The cell slurry was passed through a French pressure cell at 137 MPa, followed by centrifugation (100,000 × g) at 4°C for 1 h; the supernatant was recovered and used immediately or kept frozen at −70°C. In some experiments, the extracts were centrifuged at only 40,000 × g for 30 min. All tests were carried out under anaerobic conditions, unless insensitivity of enzyme reactions towards oxygen had been demonstrated. The protein content of the crude extracts was determined by the Lowry method (27) and usually was between 20 and 40 mg/ml.
Enzyme assays.
Enzyme assays with crude extracts of C. aurantiacus (20 mg of total protein/ml) and M. sedula (12 mg of total protein/ml) were performed at 55°C. ATP citrate lyase was measured by the citrate-, Mg2+-, ATP-, and CoA-dependent oxidation of NADH due to the reduction of the product oxaloacetate to malate (32) as modified in reference 2. Ribulose-1,5-bisphosphate carboxylase was tested based on the fixation of 14CO2 into acid-stable products, dependent on ribulose-1,5-bisphosphate (29). In controls, ribulose-1,5-bisphosphate was omitted. The pyruvate and 2-oxoglutarate dehydrogenase assay mixture (0.5 ml) contained 100 mM MOPS (morpholinopropanesulfonic acid)-KOH buffer (pH 7.0), 10 mM MgCl2, 5 mM DTE, 1 mM CoA, 1 mM NAD(P), 1 mM 2-oxoacid, and 10 μl of cell extract. Carbon monoxide dehydrogenase was tested as the CO-dependent reduction of methyl viologen (4). Pyruvate:viologen dye oxidoreductase and 2-oxoglutarate:viologen dye oxidoreductase activities were assayed by monitoring the 2-oxoacid- and CoA-dependent reduction of methyl or benzyl viologen dyes in the presence of thiamine diphosphate (46). The 14CO2 isotope exchange reaction with the C1-carboxyl group of the 2-oxoacid was tested as described in reference 33. Acetyl-CoA carboxylase and propionyl-CoA carboxylase were assayed as described in reference 38. Typically, the carboxylase assay (1 ml; 2-ml headspace) contained 100 mM Tris-HCl (pH 7.8), 0.4 mM substrate, 2 mM ATP, 4 mM MgCl2, 5 mM DTE, 10 mM NaHCO3, and 17 kBq of [14C]Na2CO3. The reaction mixture was preincubated at the reaction temperature, and the reaction was started by the addition of different volumes of cell extract (50 to 100 μl). When both carboxylating activities were measured simultaneously, 0.4 mM acetyl-CoA and 0.4 mM propionyl-CoA were added. The malonyl-CoA reduction to 3-hydroxypropionate was monitored spectrophotometrically by the oxidation of NADPH. The assay mixture (0.5 ml) contained 100 mM Tris-HCl (pH 7.8), 5 mM MgCl2, 5 mM DTE, 0.5 mM NADPH, 0.5 mM malonyl-CoA, and 0.2 mg of protein. The reaction was started by the addition of the substrate. The reductive conversion of 3-hydroxypropionate to propionyl-CoA was measured as the Mg2+-, ATP-, K+-, and CoA-dependent oxidation of NADPH. The reaction mixture contained 5 mM MgCl2, 3 mM ATP, 10 mM KCl, 0.5 mM CoASH, 0.5 mM NADPH, and different amounts of the cell extract, and the reaction was started by adding the substrate 3-hydroxypropionate to a 1 mM concentration. PEP carboxylase and pyruvate carboxylase activities were assayed as described in references 3 and 35 with minor modifications. The tests were coupled to a system in which the oxaloacetate formed was reduced to malate in the presence of endogenous malate dehydrogenase. Both tests were monitored spectrophotometrically at 365 nm. The PEP carboxylase assay mixture contained 100 mM Tris-HCl (pH 7.8), 5 mM DTE, 5 mM MgCl2, 0.5 mM NADH, 10 mM NaHCO3, and different amounts of cell extract, and the reaction was started by adding PEP at a 2 mM concentration. The pyruvate carboxylase assay mixture had a similar composition but with 3 mM ATP, and the reaction was started with 3 mM pyruvate. CO dehydrogenase and pyruvate:acceptor and 2-oxoglutarate:acceptor oxidoreductases were oxygen sensitive and therefore were assayed anaerobically. All other enzyme reactions were oxygen insensitive.
Synthesis of [14C]acetyl-CoA.
Radioactively labelled acetyl-CoA was enzymatically synthesized. The 1-ml reaction mixture contained 0.3 mM CoA, 0.2 mM [U-14C]acetate (54 μCi/μmol; Amersham, Braunschweig, Germany), 1 mM ATP, 0.4 U of acetyl-CoA synthetase (Boehringer Mannheim, Mannheim, Germany), and 1 mM MgCl2 in 50 mM Tris-HCl (pH 8.4). In addition, an ATP-regenerating system, consisting of 1 mM PEP, 0.4 mM NADH, and the enzymes myokinase (1 U), pyruvate kinase (1.5 U), and lactate dehydrogenase (2.8 U), was included. The reaction was carried out at 37°C, and NADH oxidation was monitored photometrically at 365 nm. The radioactive acetyl-CoA was purified by using an RP-C18 extraction minicolumn (ICT, Bad Homburg, Germany) following the protocols of the supplier.
Synthesis of CoA thioesters.
CoA thioesters of acetate, propionate, malonate, and succinate were synthesized according to the method described in reference 31, and the thioester of 3-hydroxypropionate was synthesized according to the method described in reference 11. 3-Hydroxypropionate was obtained by hydrolysis of 3-hydroxypropionitrile.
Detection of carboxylation products.
Radioactive products generated during the 14CO2 fixation tests were separated and identified by both high-pressure liquid chromatography (HPLC) and thin-layer chromatography (TLC). The reactions were stopped by adding 30 μl of 6 M H2SO4, and the reaction mixtures were incubated on ice for 10 min and centrifuged. The supernatant was retained and divided into two fractions. For scintillation counting, the free 14CO2 was removed from the sample by adding 100 μl of 6 M formic acid and gassing the samples for 15 min with a CO2 stream; later, 150 μl of 1 M KHCO3 was added and the gassing step was repeated for another 15 min. For HPLC, 60 μl of the centrifuged sample was injected onto a C18 RP HPLC column (LicroCART; Merck, Darmstadt, Germany) and chromatographed with a 20-min gradient of 1 to 8% acetonitrile in 50 mM phosphate buffer, pH 6.7. Simultaneous detection of standard compounds and reaction products was possible by using two detectors (UV and radioactivity) in series. The acyl-CoA esters present in 100 μl of the same nonvolatile sample were hydrolyzed at pH 12 for 30 min at 60°C. After cooling and neutralizing of the sample, 4 μl was loaded onto two TLC plates (Kieselgel 60; Merck) and chromatographed in two different solvent systems as mobile phase. One contained diisopropylether-formic acid-water (90:7:3), and the other contained butanol-acetic acid-water (12:3:5). The plates were exposed and developed with an imaging analyzer (Fujix BAS1000; Fuji Film, Tokyo, Japan). The radioactive products were identified by cochromatography of both radioactive and nonradioactive standard compounds. These nonradioactive standards were detected with a bromocresol green solution (Fluka Chemie, Buchs, Switzerland).
Detection of biotinylated proteins.
Biotinylated proteins in the bacterial cell extracts were detected with peroxidase-conjugated avidin. After one-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 15% gels, the proteins were electrotransferred to nitrocellulose membranes. The membranes were first blocked by shaking them for 2 h in a solution containing 5% skim milk powder in Tris-buffered saline (TBS) (20 mM Tris-HCl [pH 7.5], 500 mM NaCl). They were then washed three times with TBS and subsequently incubated for 1.5 h with 8.5 μg of avidin-peroxidase conjugate (Sigma, Deisenhofen, Germany) per ml in TBS. The membranes were washed again, and bound peroxidase was detected by developing the sheets with 4-chloronaphthol and hydrogen peroxide as described in reference 10.
RESULTS
Search for key enzymes of established autotrophic pathways in M. sedula.
M. sedula grows autotrophically at 65°C with CO2, S, and O2 as carbon, electron, and energy sources, respectively, with a generation time of 20 h. This corresponds to a specific carbon assimilation rate of 48 nmol of CO2 fixed min−1 mg of total cell protein−1. The calculation is based on an assumed carbon and protein content of 50% of cell dry matter each. Extracts of M. sedula, grown autotrophically under these conditions, were tested at 55 or 65°C for CO2 fixing and other key enzyme activities of known autotrophic pathways. As a control, extracts of other autotrophic bacteria, using different CO2 fixation pathways, were analyzed at their respective growth temperatures. Table 1 summarizes the results of these tests. Ribulose-1,5-bisphosphate carboxylase was below the limit of detection in M. sedula, whereas even heterotrophically grown A. eutrophus contained more than 180 nmol min−1 mg of protein−1. Carbon monoxide dehydrogenase was also not detectable in M. sedula, whereas D. autotrophicum contained large amounts of the enzyme activity. Similarly, ATP citrate lyase was below the detection limit, while a very high specific activity of the protein was measured in cell extracts of D. hydrogenophilus. Only very low, if any, activity of 2-oxoglutarate:acceptor oxidoreductase (2-oxoglutarate synthase) was detectable in M. sedula under anoxic conditions, both in the spectrophotometric and in the 14CO2 isotope exchange assay. As a control, cell extracts of A. pyrophilus, which efficiently catalyzed both reactions at 75°C, were used. Pyruvate synthase activity was detectable but was 140 times lower than that in the control reaction with D. autotrophicum. Furthermore, no NAD+-dependent pyruvate dehydrogenase or 2-oxoglutarate dehydrogenase activities were detectable.
TABLE 1.
Specific activity of key enzymes of different CO2 fixation pathways in M. sedulaa
| Enzyme tested | M. sedula | Control |
|---|---|---|
| Established CO2 fixation pathwaysb | ||
| Ribulose-1,5-bisphosphate carboxylase | <1 | >180 (Alcaligenes eutrophus) |
| Carbon monoxide dehydrogenase | <1 | 2,000 (Desulfobacterium autotrophicum) |
| Pyruvate:acceptor oxidoreductase (MV) | 3 | 560 (Desulfobacterium autotrophicum) |
| ATP citrate lyase | <1 | 1,800 (Desulfobacter hydrogenophilus) |
| Pyruvate dehydrogenase (NAD+) | <1 | |
| 2-Oxoglutarate dehydrogenase (NAD+) | <1 | |
| 2-Oxoglutarate:acceptor oxidoreductase (MV) 14C isotope exchange | 3c | 130 (Aquifex pyrophilus [BV reduction]) |
| <1 | 270 (Aquifex pyrophilus) | |
| 3-Hydroxypropionate cycled | ||
| Acetyl-CoA carboxylase | 13e | 15 (Chloroflexus aurantiacus) |
| Reduction of malonyl-CoA to 3-hydroxypropionic acid | 78e | 90 (Chloroflexus aurantiacus) |
| Reductive transformation of 3-hydroxypropionic acid to propionyl-CoA | 62e | 16 (Chloroflexus aurantiacus) |
| Propionyl-CoA carboxylase | 12e | 24 (Chloroflexus aurantiacus) |
| Acetyl-CoA plus propionyl-CoA carboxylase | 29e | 40 (Chloroflexus aurantiacus) |
| PEP carboxylase | 5e | 60 (Chloroflexus aurantiacus) |
C. aurantiacus grows autotrophically at 55°C under anaerobic conditions with light, H2, and CO2 as energy, electron, and carbon sources, respectively, with a generation time of approximately 20 h. This corresponds to a specific carbon assimilation rate of 48 nmol of CO2 fixed min−1 mg of total cell protein−1. Similarly to M. sedula, C. aurantiacus crude extracts showed a low activity of pyruvate:acceptor oxidoreductase at 55°C. With methyl viologen, the specific activity reached 15 nmol min−1 mg of cell protein−1, and in the isotope exchange reaction 10 nmol min−1 mg of cell protein−1 was measured. 2-Oxoglutarate:acceptor oxidoreductase activity was below the detection limit (<0.1 nmol min−1 mg of cell protein−1 measured with both methyl viologen and the isotope exchange reaction). No NAD+-dependent pyruvate dehydrogenase or 2-oxoglutarate dehydrogenase was detectable (<1 nmol min−1 mg of cell protein−1).
These results suggested that the Calvin cycle, the reductive acetyl-CoA pathway, and the reductive citric acid cycle do not function in M. sedula when grown with S and O2. Rather, the presence of a fourth autotrophic pathway has to be postulated.
Detection of CO2 fixation enzymes of the postulated 3-hydroxypropionate cycle in M. sedula.
It was tested whether M. sedula uses the postulated 3-hydroxypropionate cycle as an autotrophic pathway. This cycle uses acetyl-CoA carboxylase (reaction a) and propionyl-CoA carboxylase (reaction b) for the fixation of two molecules of CO2 per cycle. Since intermediates of the 3-hydroxypropionate cycle participate in the citric acid cycle, an active anaplerotic reaction is also required. Candidates for possible anaplerotic enzymes are pyruvate carboxylase (reaction c), PEP carboxylase (reaction d), and PEP carboxytransphosphorylase (reaction e).
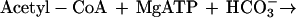 |
a |
|---|
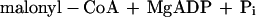
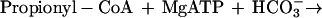 |
b |
|---|
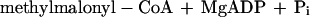
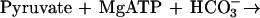 |
c |
|---|
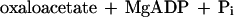
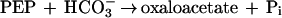 |
d |
|---|
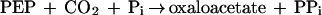 |
e |
|---|
Extracts of autotrophically grown M. sedula were tested at 55°C for the presence of these enzymes. Control extracts of autotrophically grown C. aurantiacus were tested in parallel at the same temperature. M. sedula contained both acetyl-CoA and propionyl-CoA carboxylase activities (Table 1). The specific enzyme activities were similar to those found in C. aurantiacus. When both substrates, acetyl-CoA (0.4 mM) and propionyl-CoA (0.4 mM), were simultaneously added to the assay, the initial CO2 fixation rates measured with M. sedula extracts were roughly additive (Table 1). This result suggests the presence of two different carboxylases, since both activities were measured near substrate saturation (Fig. 2). Both activities were completely abolished by low concentrations (1 nM) of avidin. The rate of 14CO2 fixation in these assays rapidly decreased for unknown reasons. The rates given refer to the amount of 14CO2 fixed after 2 min of incubation.
FIG. 2.

Carboxylation activity in M. sedula cell extracts at 55°C. Acetyl-CoA (●) and propionyl-CoA (○) were specifically carboxylated in vitro with 14CO2. CO2 fixation in the presence of both substrates is also shown (▿). The carboxylation reaction with both substrates was inhibited by 1 nM avidin (▾). For assay conditions, see Materials and Methods.
In addition to the two CO2-fixing enzyme activities, extracts of both organisms, M. sedula and C. aurantiacus, contained PEP carboxylase (Table 1), while pyruvate carboxylase and PEP carboxytransphosphorylase activities were not detectable (<1 nmol min−1 mg of cell protein−1).
Demonstration of enzyme activities converting acetyl-CoA via malonyl-CoA to 3-hydroxypropionate.
The acetyl-CoA carboxylase assay depends on the fixation of the radioactivity from 14CO2 into acid-stable products in the presence of MgATP and acetyl-CoA. Under these conditions, cell extracts of M. sedula catalyzed the MgATP- and CO2-dependent conversion of [14C]acetyl-CoA to [14C]malonyl-CoA (Fig. 3A). Formation of this intermediate was completely abolished by 1 nM avidin. When NADPH was included in this assay, 3-[14C]hydroxypropionate was formed in addition to [14C]malonyl-CoA (Fig. 3B). In this experiment, [14C]malonate was released from labelled malonyl-CoA by alkali treatment. Similar results were obtained when 14CO2 and acetyl-CoA were used (Fig. 4). This shows that acetyl-CoA is first carboxylated to malonyl-CoA by the avidin-sensitive acetyl-CoA carboxylase and then converted, by reduction, to 3-hydroxypropionate. The intermediate semialdehyde could not be identified, partly because it was not commercially available as a reference. The reduction of malonyl-CoA to 3-hydroxypropionate requires the oxidation of two molecules of NADPH per molecule of malonyl-CoA. This reaction was monitored in an aerobic spectrophotometric assay catalyzed by M. sedula cell extracts (Fig. 3C). The absorption decrease followed a biphasic curve, which may be due to different specific activities of the two sequential oxidoreductases, the first one catalyzing a fast reduction of malonyl-CoA to malonate semialdehyde, followed by a slower reduction of malonate semialdehyde to 3-hydroxypropionate (see also Fig. 1). The initial high rate corresponded to a specific NADPH oxidation rate of 156 nmol min−1 mg of cell protein−1, while the final lower rate was 78 nmol min−1 mg of cell protein−1. Both rates were linearly protein dependent in the range 0 to 0.15 mg of cell protein ml of assay mixture−1. No reaction occurred when NADH was used instead of NADPH in the assay (data not shown).
FIG. 3.

Carboxylation of acetyl-CoA to malonyl-CoA in the presence of ATP and conversion of malonyl-CoA to 3-hydroxypropionate in the presence of NADPH by cell extracts of M. sedula. (A) HPLC analysis of [14C]malonyl-CoA formed from [14C]acetyl-CoA and CO2. The chromatograms were recorded by using a radioactivity monitor calibrated for 14C detection. (I) Control reaction without cell extract after 30 min of incubation. (II) [14C]malonyl-CoA formation after a 30-min reaction. (III) Carboxylation reaction specifically inhibited with 20 μg of avidin. Retention times: [14C]acetyl-CoA, 13.3 min; [14C]malonyl-CoA, 7.5 min. Assay conditions (0.3-ml assay mixture, 1.2-ml headspace): 100 mM Tris-HCl (pH 7.8), 2 mM ATP, 2.5 mM MgCl2, 3 μmol of KHCO3, 9.3 nmol of [14C]acetyl-CoA (18 kBq), 1.5 mg of protein. (B) TLC detection of labelled products formed after carboxylation of [14C]acetyl-CoA. The solvent system used was butanol-acetic acid-water (12:3:5). The CoA esters present in the samples (4 μl) were hydrolyzed to the free acid form before being loaded onto the TLC plate. Note that acetic acid is volatile under these conditions. Lane 1, carboxylation reaction inhibited with 1 nM avidin. Lane 2, carboxylation of [14C]acetyl-CoA after a 30-min reaction. Lane 3, formation of 3-hydroxypropionate (3-OHP) after addition of NADPH (0.5 mM) to the reaction in lane 2 and further incubation for 30 min. Assay conditions were as described for panel A. (C) Spectrophotometric assay of the reduction of malonyl-CoA to 3-hydroxypropionate by NADPH. The reaction was started by adding the substrate malonyl-CoA (0.5 mM) as indicated. The dotted lines indicate the initial and final rates of NADPH oxidation.
FIG. 4.

TLC analysis of labelled products formed by cell extracts of M. sedula from 14CO2 and acetyl CoA. (A) Sample (4 μl) not hydrolyzed after 0 (lane 1), 10 (lane 2), and 30 (lane 3) min of incubation. (B) Samples (4 μl) as described for panel A but after alkaline hydrolysis of CoA thioesters. Assay conditions (85-μl assay mixture, 1.4-ml headspace): 100 mM Tris-HCl (pH 7.8), 1 mM ATP, 5 mM MgCl2, 0.3 mM NADPH, 10 mM KCl, 5 mM DTE, 3 mM acetyl-CoA, 90 nmol of [14C]Na2CO3 (180 kBq), 0.12 mg of protein. The solvent system was as described for Fig. 3. Note that acetic, acrylic, and propionic acids are volatile under these conditions. 3-OHP, 3-hydroxypropionate.
Control experiments were performed with C. aurantiacus cell extracts with acetyl-CoA and 14CO2 (Fig. 5) or with [14C]acetyl-CoA and CO2 (data not shown), in the presence of MgATP and NADPH. It is to be expected that malonyl-CoA is formed only transiently because this intermediate is rapidly reduced by NADPH to 3-hydroxypropionate. As expected, in both cases the early formation of 3-[14C]hydroxypropionate via [14C]malonyl-CoA was observed. According to the working hypothesis, 3-hydroxypropionate should subsequently be converted to succinyl-CoA via propionyl-CoA. As expected, after alkali treatment [14C]succinate and an unidentified labelled product (X) accumulated in time and were observed in two different TLC solvent systems (Fig. 5, lane 4).
FIG. 5.

TLC detection of labelled products formed by cell extracts of C. aurantiacus at 45°C from 14CO2 and acetyl-CoA in the presence of NADPH, after 0 (lane 1), 0.5 (lane 2), 2.5 (lane 3), and 10 (lane 4) min of incubation. The CoA esters in the samples (4 μl) were hydrolyzed by alkali treatment. The solvent system was as described for Fig. 3. Assay conditions (90-μl assay mixture, 1.4-ml headspace): 100 mM Tris-HCl (pH 7.8), 1 mM ATP, 5 mM MgCl2, 0.3 mM NADPH, 10 mM KCl, 5 mM DTE, 0.1 mM CoA, 90 nmol of [14C]Na2CO3 (180 kBq), 2.7 mM acetyl-CoA, 0.8 mg of protein. 3-OHP, 3-hydroxypropionate. X, unknown product.
These results showed that both C. aurantiacus and M. sedula convert acetyl-CoA to 3-hydroxypropionate via malonyl-CoA, as postulated for the 3-hydroxypropionate cycle.
Demonstration of enzyme activities converting 3-hydroxypropionate to propionyl-CoA.
The 3-hydroxypropionate cycle postulates the MgATP- and CoA-dependent activation, by a CoA ligase enzyme, of the characteristic intermediate of the cycle, 3-hydroxypropionate, to 3-hydroxypropionyl–CoA. This is followed by K+-dependent beta-elimination of water, generating acrylyl-CoA as the next intermediate. This intermediate is subsequently reduced to propionyl-CoA by NAD(P)H (Fig. 1). After sufficient formation of CoA thioesters, this sequence of reactions results in the oxidation of NADPH. This oxidation was the basis for an aerobic spectrophotometric assay of the reactions (Fig. 6A). NADPH oxidation required approximately 2 min to reach its maximal rate after the coupled spectrophotometric assay was started by addition of 3-hydroxypropionate. This is to be expected, because NADPH oxidation is initiated only after substantial amounts of the intermediates 3-hydroxypropionyl–CoA and acrylyl-CoA are formed. The final rate of NADPH oxidation with M. sedula cell extracts was even higher than that found in C. aurantiacus (Table 1). The final rate of NADPH oxidation observed after 3-hydroxypropionate addition was linearly dependent on the amount of cell protein in the range 0 to 0.15 mg ml of assay mixture−1. It has been shown before with C. aurantiacus that NADPH oxidation could also be started with acrylate instead of 3-hydroxypropionate (38).
FIG. 6.

Reductive conversion of 3-hydroxypropionate to propionyl-CoA via 3-hydroxypropionyl–CoA in the presence of CoA, ATP, and NADPH by cell extracts of M. sedula. (A) Spectrophotometric recording of NADPH oxidation during the formation of propionyl-CoA from 3-hydroxypropionate. The assay was started by the addition of the substrate as indicated. The dotted line indicates the point at which the reaction rates were determined. For assay conditions, see Materials and Methods. (B) HPLC chromatograms showing the transient formation of the intermediate 3-hydroxypropionyl–CoA (3-OHP-CoA) and the accumulation of the product propionyl-CoA during the reaction in panel A. The samples injected into the column were taken at different reaction times: 0 min, before addition of 3-hydroxypropionate; 5 min, after addition of 3-hydroxypropionate; 12 min, after addition of 3-hydroxypropionate. Control, assay without 3-hydroxypropionate after 12 min of incubation. The chromatograms were recorded with a UV monitor at 260 nm. Retention times: ATP, 3 min; NADP, 4 min; NADPH, 6.5 min; CoASH, 10.5 min; 3-hydroxypropionyl–CoA, 12.5 min; propionyl-CoA, 18.5 min. Note that CoASH and succinyl-CoA have identical retention times. Assay conditions (0.5-ml assay mixture): 100 mM Tris-HCl (pH 7.8), 1 mM 3-hydroxypropionate, 1 mM CoA, 5 mM ATP, 5 mM MgCl2, 0.5 mM NADPH, 10 mM KCl, 0.5 mg of protein.
HPLC analysis of the samples taken at different reaction times (Fig. 6B) showed that cell extracts of M. sedula produced 3-hydroxypropionyl–CoA and subsequently reduced it to propionyl-CoA in the presence of the corresponding cofactors and cosubstrates. In control experiments, without 3-hydroxypropionate, none of these products was detected. Under the chromatographic conditions used, the substrate, 3-hydroxypropionic acid, does not separate and elutes together with other polar substances during the first 2 min. The chromatograms also show the consumption of the corresponding cofactor CoASH and cosubstrate NADPH and the production of NADP.
Evidently, 3-hydroxypropionate is not a metabolic dead-end product but can be reduced, in vitro, to propionyl-CoA, the substrate of the second postulated carboxylating enzyme.
Demonstration of enzyme activities converting propionyl-CoA to succinate via methylmalonyl-CoA.
The second CO2 fixation step proposed in the 3-hydroxypropionate cycle involves the carboxylation of propionyl-CoA to methylmalonyl-CoA. This reaction is catalyzed by the biotin-dependent enzyme propionyl-CoA carboxylase and requires ATP and Mg2+. The product of this reaction, methylmalonyl-CoA, is further isomerized to succinyl-CoA and later de- or transesterified to succinate (Fig. 1).
Cell extracts of M. sedula catalyzed the MgATP-dependent carboxylation of propionyl-CoA, measured as the incorporation of 14CO2 into acid-stable products. HPLC analysis of the reaction time course showed the formation of [14C]methylmalonyl-CoA, in initial stages of the reaction (Fig. 7A). Later, [14C]methylmalonyl-CoA was consumed in favor of [14C]succinate production. The identity of these intermediates was confirmed by performing a second HPLC analysis after alkaline hydrolysis of the samples. The radioactive peaks coeluted with the free acids [14C]methylmalonate and [14C]succinate (Fig. 7B). The [14C]methylmalonate and [14C]succinate formed during the reaction were also identified by TLC with two different solvent systems (not shown).
FIG. 7.

Time course of the carboxylation of propionyl-CoA to methylmalonyl-CoA with 14CO2 and subsequent conversion to succinate catalyzed by cell extracts of M. sedula at 55°C. (A) HPLC chromatograms showing the incorporation of 14CO2 into propionyl-CoA, generating [14C]methylmalonyl-CoA as transient intermediate. This intermediate is subsequently converted to [14C]succinate. (B) HPLC chromatograms after alkaline hydrolysis of the CoA esters present in the samples. The labelled compounds in both panels were preliminarily identified by cochromatography with authentic compounds. Assay conditions (1-ml assay mixture, 2-ml headspace): 100 mM Tris-HCl (pH 7.8), 1 mM ATP, 5 mM MgCl2, 3 mM DTE, 0.5 mM propionyl-CoA, 5 μmol of [14C]Na2CO3 (10 kBq), 0.4 mg of protein.
The presence of the two enzymes methylmalonyl-CoA epimerase and methylmalonyl-CoA mutase can be inferred from the detection of the reaction product [14C]succinate. These enzymes catalyze reactions 8 and 9 of the 3-hydroxypropionate cycle (Fig. 1).
Detection of carboxylase activities and biotin-containing peptides in M. sedula, S. metallicus, and A. infernus.
Since archaea do not contain substantial amounts of fatty acids—if any—neither acetyl-CoA carboxylase nor propionyl-CoA carboxylase plays a significant role in their central carbon metabolism. Therefore, it was not expected that these carboxylases would be found in archaea. All bacterial acetyl-CoA carboxylases studied so far contain a small and abundantly synthesized biotin-carboxy-carrier protein subunit (BCCP; 16 to 24 kDa) (reviewed in reference 39). Biotin-containing proteins can be specifically detected by a reaction with peroxidase-conjugated avidin. By this technique, several autotrophic archaeal species were screened for biotin-containing peptides. Cell extracts of the bacteria C. aurantiacus (Fig. 8A, lane 1) and E. coli (Fig. 8A, lane 4) served as positive controls. These two bacteria contained substantial amounts of a small biotinylated peptide. Autotrophically grown C. aurantiacus was shown to contain some larger biotinylated proteins in addition to the putative BCCP peptide (Fig. 8A, lane 1).
FIG. 8.

Detection of biotin-containing proteins in cell extracts (80 μg of protein) from various microorganisms by avidin-staining technique. (A) Bacteria and Crenarchaeota. Lane 1, C. aurantiacus, grown autotrophically; lane 2, M. sedula, grown autotrophically; lane 3, M. sedula, grown heterotrophically; lane 4, E. coli; lanes 5 and 6, S. metallicus, grown autotrophically on metal ore; lanes 7 and 8, S. metallicus grown autotrophically on sulfur; lanes 9 and 10, T. tenax grown autotrophically; lanes 11 and 12, A. infernus grown autotrophically aerobically; lanes 13 and 14, A. infernus grown autotrophically anaerobically. (B) Euryarchaeota. Lanes 1 and 2, M. voltae; M, molecular mass marker (schematic drawing of the positions of 67-, 29-, and 14-kDa marker proteins); lanes 3 and 4, M. barkeri; lanes 5 and 6, M. organophilum; lanes 7 and 8, M. kandleri; lanes 9 and 10, M. thermoautotrophicum. For growth conditions, refer to Materials and Methods.
M. sedula as well as other autotrophic representatives of the Sulfolobales order, namely, S. metallicus and A. infernus, contained significant amounts of a small biotinylated protein (Fig. 8A). In M. sedula, it corresponded to the most abundant biotin protein detected, while in S. metallicus and A. infernus, this was the only biotin-containing protein detected. Additionally, in M. sedula there was no difference in the contents of the likely BCCP between cells grown autotrophically and those grown heterotrophically (Fig. 8A, lanes 2 and 3). In the case of S. metallicus, no difference in the amount of potential BCCP was observed between cells grown autotrophically on sulfidic ore (Fig. 8A, lanes 5 and 6) and those grown with sulfur (Fig. 8A, lanes 7 and 8). In A. infernus, the BCCP-like protein was easily detected in anaerobically grown cells (Fig. 8, lanes 13 and 14), while aerobic cells showed a similar but much weaker band (Fig. 8A, lanes 11 and 12).
The occurrence of a small biotinylated peptide was also investigated in several archaea with known autotrophy pathways. T. tenax, a strictly anaerobic autotrophic member of the Crenarchaeota, which probably uses the reductive citric acid cycle for CO2 fixation (as shown for T. neutrophilus), did not contain detectable amounts of biotin-carrying peptides (Fig. 8A, lanes 9 and 10). A small BCCP-like protein was also absent in autotrophic Euryarchaeota with a reductive acetyl-CoA pathway for CO2 fixation. These bacteria either contained virtually no biotin enzymes, as in the case of M. organophilum (Fig. 8B, lanes 5 and 6), M. kandleri (Fig. 7B, lanes 7 and 8), and M. thermoautotrophicum (Fig. 8B, lanes 9 and 10), or contained various biotinylated proteins of higher molecular mass, as found in M. voltae (Fig. 8B, lanes 1 and 2) and M. barkeri (Fig. 8B, lanes 3 and 4).
Acetyl-CoA and propionyl-CoA carboxylase activities were tested with extracts of various archaea. Acetyl-CoA-plus propionyl-CoA-dependent fixation of 14CO2 was detected, albeit at a low rate, in S. metallicus (2.5 nmol min−1 mg of total cell protein−1 at 60°C) and A. infernus (0.18 nmol min−1 mg of total cell protein−1 at 60°C). Note, however, that A. infernus was grown at 80°C in the presence of 0.02% yeast extract. In T. tenax (grown at 80°C, measured at 60°C) and M. voltae (grown and measured at 37°C), no activity was observed (<0.01 nmol min−1 mg of total cell protein−1).
These results indicate that acetyl-CoA and propionyl-CoA carboxylase are actively synthesized not only in M. sedula but also in S. metallicus and A. infernus, whereas these enzymes appear to be lacking in T. tenax and in the methanogens tested. The optimal conditions for autotrophic growth and enzyme assay in these bacteria are yet to be established.
DISCUSSION
In vitro evidence for the 3-hydroxypropionate cycle of CO2 fixation in Chloroflexus and in archaea.
The 3-hydroxypropionate cycle of CO2 fixation (5, 13, 38) has been proposed as the fourth, alternative pathway for autotrophic growth in nature. It was first described in the early-branching phototrophic thermophilic bacterium C. aurantiacus. We have demonstrated in vitro the essential partial reactions of this cycle from acetyl-CoA via 3-hydroxypropionate and propionyl-CoA to succinate. The radioactive intermediates were formed irrespective of which carbon precursor, acetyl-CoA or bicarbonate, was 14C labelled. At a generation time of 20 h, the specific CO2 fixation rate reaches 48 nmol min−1 mg of protein−1. Since two molecules of CO2 are fixed per turn of the postulated CO2 fixation cycle, a minimal specific activity of the enzymes of the cycle of 24 nmol min−1 mg of protein−1 is required to explain the rate of autotrophic growth. The reactions rates measured approximated these postulated in vivo rates of autotrophically growing cells.
C. aurantiacus remains, so far, the only representative of the bacteria where strong evidence for this cycle has been found. K+, MgATP, CoA, and NADPH were required for reaction. We could not confirm the operation of an alternative CO2 fixation cycle as proposed elsewhere (19, 25), since pyruvate:acceptor oxidoreductase activity was low and the labelling pattern of alanine precluded synthesis of pyruvate from acetyl-CoA and CO2 (5). We assume that the low levels of pyruvate:acceptor oxidoreductase function in acetyl-CoA synthesis. PEP carboxylase activity is required as an anaplerotic enzyme of the 3-hydroxypropionic acid cycle.
Recent evidence has indicated the presence of a similar pathway operating in A. brierleyi (18). This organism belongs to the Sulfolobales, a crenarchaeal order consisting of lithoautotrophic aerobes, facultative anaerobes, and obligate anaerobes; characteristically, these organisms thrive by aerobic sulfur oxidation or anaerobic sulfur reduction (34). We have extended these studies and have examined M. sedula, another autotrophic member of the Sulfolobales (15), for the presence of the 3-hydroxypropionate cycle. This organism does not express key enzymes of the reductive pentosephosphate cycle (ribulose-1,5-bisphosphate carboxylase), the reductive acetyl-CoA pathway (carbon monoxide dehydrogenase), or the reductive citric acid cycle (ATP citrate lyase). In contrast, the two carboxylating enzymes of the 3-hydroxypropionate cycle, acetyl-CoA carboxylase and propionyl-CoA carboxylase, were found. Additionally, the enzyme activities and intermediates involved in the CO2 fixation following the 3-hydroxypropionate cycle up to succinate have been verified. The specific activities of the enzymes of this cycle measured in cells grown on O2, S, and CO2 (generation time, 20 h) were close to the calculated minimal enzyme activities required for growth. It should be noted that the actual specific activity of the carboxylating enzymes at growth temperature (65°C) should be twice as high as that measured with the assay at 55°C.
Acetyl-CoA and propionyl-CoA carboxylase activities and small biotin-carrying proteins in archaea.
A fast, specific, and sensitive test for the presence of biotin-containing proteins in cell extracts, with particular focus on the BCCP fragment of the acetyl-CoA and propionyl-CoA carboxylases, was used to screen autotrophic archaea. The rationale behind the test was that lipids of archaea do not contain fatty acids, since their membranes are formed by isoprenol glycerol ether lipids. It is therefore assumed that archaea normally do not require, and therefore do not synthesize, acetyl-CoA and propionyl-CoA carboxylases. The presence of significant amounts of a small BCCP-like protein and acetyl-CoA plus propionyl-CoA carboxylase activities in cell extracts of archaea would indicate that these carboxylases are being synthesized and involved in a process other than lipid biosynthesis.
A clear correlation was found between the presence of a small biotin-containing protein and the detection of acetyl-CoA- and propionyl-CoA-dependent carboxylation activity in cell extracts. This positive correlation was found only in members of the Sulfolobales (M. sedula, S. metallicus, and A. infernus). In all other autotrophic archaea tested, which use one of the other pathways for CO2 fixation, neither an abundant BCCP-like fragment nor acetyl-CoA and propionyl-CoA carboxylase activities were detected.
Occurrence of acetyl-CoA carboxylase in archaea.
The acetyl-CoA carboxylase protein is well conserved in nature; it has been purified and characterized not only in bacteria but also in yeasts, plants, and animals. Two basic types of this enzyme are found. The “bacterial type” contains four different subunits organized in three functional domains. Four different genes (accABCD) encode the peptides (AccABCD) of approximately 35, 17, 49, and 33 kDa, respectively. The “eukaryotic type” is composed of a single large peptide of approximately 280 kDa (39). The subunit composition of the acetyl-CoA carboxylase in archaea requires further studies. The recent sequencing of the complete genomes of several archaea allows a preliminary picture of the distribution of putative acetyl-CoA carboxylase subunits to be drawn (Table 2). The genomes of the Euryarchaeota M. jannaschii, M. thermoautotrophicum, A. fulgidus, and P. horikoshii contain DNA sequences coding for a protein domain similar to the bacterial BCCP peptide as part of a larger open reading frame, which possibly encodes a biotin-dependent decarboxylase. No candidate genes encoding the carboxytransferase chains (accA and accD) of the acetyl-CoA carboxylase were found in the genomes available to date. Putative genes coding for the acetyl-CoA carboxylase of S. metallicus are deposited in the sequence database. The derived gene products show a high similarity to acetyl-CoA carboxylases from organisms belonging to very distant evolutionary branches. These genes of S. metallicus correspond to accB, accC, and accD, genes found in the bacterial-type acetyl-CoA carboxylases, with the accA gene missing. The predicted mass of the S. metallicus AccB peptide would be in close agreement with that of the bacterial-type BCCP (18.6 kDa) and with that of the fragment detected in our tests (Fig. 8A). The other two polypeptides of the S. metallicus acetyl-CoA carboxylase reported have predicted masses of 56 and 57 kDa, respectively, significantly larger than those of similar polypeptides of the bacterial-type acetyl-CoA carboxylase (Table 2). The subunit composition of this protein therefore would be unique, though it would still resemble the bacterial-type acetyl-CoA carboxylases.
TABLE 2.
Presence of DNA sequences with similarity to E. coli acetyl-CoA carboxylase genes accABCD in archaeal genomes and their putative productsa
| Organism | BCCP (accB; 17 kDa) | Biotin carboxylase (accC; 49 kDa) | Carboxytransferase α chain (accA; 35 kDa) | Carboxytransferase β chain (accD; 33 kDa) |
|---|---|---|---|---|
| Sulfolobus metallicus | accB, 18.6 kDa (AF042099) | accC, 57 kDa (AF042099) | Not found | accD, 56 kDa (AF042099) |
| Archaeoglobus fulgidus | accB, 15.6 kDa, subunit of putative oxaloacetate decarboxylase (AE000952); | accC, 57.4 kDa (AE001090) | Putative α subunit of methylmalonyl-CoA decarboxylase (AE000952) | Same as accA |
| accB, 15.7 kDa, subunit of putative methylmalonyl-CoA decarboxylase (AE000960) | ||||
| Methanococcus jannaschii | BCCP domain at the C terminus of putative oxaloacetate decarboxylase, α chain, 64 kDa (U67563) | accC, 55 kDa (Q58626) | Not found | Not found |
| Pyrococcus horikoshii | BCCP domain at the C terminus of a putative transcarboxylase which is similar to oxaloacetate decarboxylase, α chain, 64 kDa (AP000003 and AP000005) | Not found | Putative α subunit of methylmalonyl-CoA decarboxylase (AB009510) | Same as accA |
| Methanobacterium thermoautotrophicum | BCCP domain at the C terminus of a putative oxaloacetate decarboxylase, α chain, 64 kDa (AF039105) | accC, 54.6 kDa (AE000942) | Not found | Not found |
A more extensive screening, with the biotin test presented, would help to establish the possible occurrence of the 3-hydroxypropionate cycle in autotrophic organisms of other orders, with still-uncharacterized CO2 fixation pathways.
Further reactions of the 3-hydroxypropionate cycle and possible role in acetyl-CoA assimilation.
At the present stage of our research with M. sedula, the fate of the CO2 fixed after the formation of succinate remains an open question. However, it can be assumed that the rest of the reactions postulated in the 3-hydroxypropionate cycle involving the regeneration of the initial substrate, acetyl-CoA, are in operation. Tests aimed at clarifying this issue and the fate of glyoxylate are under way.
Furthermore, the assimilation of acetyl-CoA into C4 compounds via the initial reactions of the 3-hydroxypropionate cycle should be considered as a possible pathway in those microorganisms that lack one, or both, of the characteristic enzymes of the glyoxylate cycle (isocitrate lyase and malate synthase). The new pathway of succinate synthesis from acetyl-CoA and 2CO2 would allow growth on ethanol, acetate, or fatty acids and may be responsible for the assimilation of these substrates in the microorganisms studied here.
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie.
Thanks are due to Monika Beh for contributing to the initial stages of this work; to Rainer Hedderich, Marburg, for a gift of several methanogenic bacteria; to Kerstin Roth, Regensburg, for technical assistance; and to Johann Heider, Freiburg, for reading the manuscript.
Footnotes
†
Dedicated to Volkmar Braun, Tübingen, on the occasion of his 60th birthday.
REFERENCES
- 1.Antranikian G, Herzberg C, Gottschalk G. Characterization of ATP citrate lyase from Chlorobium limicola. J Bacteriol. 1982;152:1284–1287. doi: 10.1128/jb.152.3.1284-1287.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beh M, Strauß G, Huber R, Stetter K O, Fuchs G. Enzymes of the reductive citric acid cycle in the autotrophic eubacterium Aquifex pyrophilus and in the archaebacterium Thermoproteus neutrophilus. Arch Microbiol. 1993;160:306–311. [Google Scholar]
- 3.Cánovas J L, Kornberg H L. Phosphoenolpyruvate carboxylase from Escherichia coli. Methods Enzymol. 1969;XIII:288–292. [Google Scholar]
- 4.Daniels L, Fuchs G, Thauer R K, Zeikus G. Carbon monooxide oxidation by methanogenic bacteria. J Bacteriol. 1977;132:118–126. doi: 10.1128/jb.132.1.118-126.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisenreich W, Strauss G, Werz U, Fuchs G, Bacher A. Retrobiosynthesis analysis of carbon fixation in the phototrophic eubacterium Chloroflexus aurantiacus. Eur J Biochem. 1993;215:619–632. doi: 10.1111/j.1432-1033.1993.tb18073.x. [DOI] [PubMed] [Google Scholar]
- 6.Evans M C W, Buchanan B B, Arnon D I. A new ferrodoxin-dependent carbon reduction cycle in a photosynthetic bacterium. Proc Natl Acad Sci USA. 1966;55:928–934. doi: 10.1073/pnas.55.4.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedrich C G, Bowien G, Friedrich B. Formate and oxalate metabolism in Alcaligenes eutrophus. J Gen Microbiol. 1979;115:185–192. [Google Scholar]
- 8.Fuchs G. Alternative pathways of autotrophic CO2 fixation. In: Schlegel H G, Bowien B, editors. Autotrophic bacteria. Berlin, Germany: Springer-Verlag; 1989. pp. 365–382. [Google Scholar]
- 9.Fuchs G, Stupperich E, Eden G. Autotrophic CO2 fixation in Chlorobium limicola. Evidence for the operation of a reductive tricarboxylic acid cycle in growing cells. Arch Microbiol. 1980;128:64–71. [Google Scholar]
- 10.Gallagher S, Winston S E, Fuller S A, Hurrell J G R. Immunoblotting and immunodetection. In: Ausubel F M, et al., editors. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons, Inc.; 1997. pp. 10.8.1–10.8.21. [Google Scholar]
- 11.Gross G G, Zenk M H. Darstellung und Eigenschaften von Coenzym A-Thioestern substituierter Zimtsäuren. Z Naturforsch. 1966;21b:683–690. [Google Scholar]
- 12.Holo H. Chloroflexus aurantiacus secretes 3-hydroxypropionate, a possible intermediate in the assimilation of CO2 and acetate. Arch Microbiol. 1989;151:252–256. [Google Scholar]
- 13.Holo H, Sirevåg R. Autotrophic growth and CO2 fixation in Chloroflexus aurantiacus. Arch Microbiol. 1986;145:173–180. [Google Scholar]
- 14.Huber G, Stetter K O. Sulfolobus metallicus, sp. nov., a novel strictly chemolithoautotrophic thermophilic archaeal species of metal-mobilizers. Syst Appl Microbiol. 1991;14:372–378. [Google Scholar]
- 15.Huber G, Spinnler C, Gambacorta A, Stetter K O. Metallosphaera sedula gen. and sp. nov. represents a new genus of aerobic, metal-mobilizing, thermoacidophilic archaebacteria. Syst Appl Microbiol. 1989;12:38–47. [Google Scholar]
- 16.Huber G, Drobner E, Huber H, Stetter K O. Growth by aerobic oxidation of molecular hydrogen in archaea—a metabolic property so far unknown for this domain. Syst Appl Microbiol. 1992;15:502–504. [Google Scholar]
- 17.Huber R, Wilharm T, Huber D, Tincone A, Burggraf S, König H, Rachel R, Rockinger I, Fricke H, Stetter K O. Aquifex pyrophilus gen. nov., sp. nov., represents a novel group of marine hyperthermophilic hydrogen-oxidizing bacteria. Syst Appl Microbiol. 1992;15:340–351. [Google Scholar]
- 18.Ishii M, Miyake T, Satoh T, Sugiyama H, Oshima Y, Kodama T, Igarashi Y. Autotrophic carbon dioxide fixation in Acidianus brierleyi. Arch Microbiol. 1996;166:368–371. doi: 10.1007/BF01682981. [DOI] [PubMed] [Google Scholar]
- 19.Ivanovsky R N, Krasilnikova E N, Fal Y I. A pathway of the autotrophic CO2 fixation in Chloroflexus aurantiacus. Arch Microbiol. 1993;159:257–264. [Google Scholar]
- 20.Ivanovsky R N, Krasilnikova E N. Glyoxylate metabolism in Chloroflexus aurantiacus. Microbiology. 1995;64:257–261. [Google Scholar]
- 21.Ivanovsky R N, Sintsov N V, Kondratieva E N. ATP-linked citrate lyase activity in the green sulfur bacterium Chlorobium limicola forma thiosulfatophilum. Arch Microbiol. 1980;128:239–241. [Google Scholar]
- 22.Kandler O, Stetter K O. Evidence for autotrophic CO2 assimilation in Sulfolobus brierleyi via a reductive carboxylic acid pathway. Zentbl Bakteriol Hyg I Abt Orig C. 1981;2:111–121. [Google Scholar]
- 23.Kawarabayasi Y, Sawada M, Horikawa H, Haikawa Y, Hino Y, Yamamoto S, Sekine M, Baba S, Kosugi H, Hosoyama A, Nagai Y, Sakai M, Ogura K, Otuka R, Nakazawa H, Takamiya M, Ohfuku Y, Funahashi T, Tanaka T, Kudoh Y, Yamazaki J, Kushida N, Oguchi A, Aoki K, Nakamura Y, Robb T F, Horikoshi K, Masuchi Y, Shizuya H, Kikuchi H. Complete sequence and gene organization of the genome of a hyperthermophilic archaebacterium, Pyrococcus horikoshii OT3. DNA Res. 1998;5:55–76. doi: 10.1093/dnares/5.2.55. [DOI] [PubMed] [Google Scholar]
- 24.Klenk H P, Clayton R A, Tomb J F, White O, Nelson K E, Ketchum K A, Dodson R J, Gwinn M, Hickey E K, Peterson J D, Richardson D L, Kerlavage A R, Graham D E, Kyrpides N C, Fleischmann R D, Quackenbush J, Lee N H, Sutton G G, Gill S, Kirkness E F, Dougherty B A, McKenney K, Adams M D, Loftus B, Venter J C. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- 25.Kondratieva E N, Ivanovsky R N, Krasilnikova E N. Carbon metabolism in Chloroflexus aurantiacus. FEMS Microbiol Lett. 1992;100:269–272. [Google Scholar]
- 26.Kurr M, Huber R, König H, Jannasch H W, Fricke H, Trincone A, Kristjansson J K, Stetter K O. Methanopyrus kandleri, gen. and sp. nov., represents a novel group of hyperthermophilic methanogens, growing at 110°C. Arch Microbiol. 1991;156:239–247. [Google Scholar]
- 27.Lowry O H, Rosebrough N J, Farr A L, Randall R J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 28.Norris P, Nixon A, Hart A. Acidophilic, mineral-oxidizing bacteria: the utilization of carbon dioxide with particular reference to autotrophy in Sulfolobus. In: Da Costa M S, Duarte J C, Williams R A D, editors. Microbiology of extreme environments and its potential for biotechnology. London, United Kingdom: Elsevier; 1989. pp. 24–43. [Google Scholar]
- 29.Quayle J R, Keech D B. Carbon assimilation by Pseudomonas oxalaticus (OX 1). 2. Formate and carbon dioxide utilization by cell-free extracts of the organism grown on formate. Biochem J. 1959;72:631–637. doi: 10.1042/bj0720631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rajagopalan K, Altekar W. Characterisation and purification of ribulose-bisphosphate carboxylase from heterotrophically grown halophilic archaebacterium, Haloferax mediterranei. Eur J Biochem. 1994;221:863–869. doi: 10.1111/j.1432-1033.1994.tb18801.x. [DOI] [PubMed] [Google Scholar]
- 31.Schachter D, Taggart J V. Benzoyl coenzyme A and hippurate synthesis. J Biol Chem. 1976;203:925–933. [PubMed] [Google Scholar]
- 32.Schauder R, Preuß A, Jetten M, Fuchs G. Oxidative and reductive acetyl-CoA/carbon monoxide dehydrogenase pathway in Desulfobacterium autotrophicum. 2. Demonstration of the enzymes of the pathway and comparison of CO dehydrogenase. Arch Microbiol. 1989;151:84–89. [Google Scholar]
- 33.Schauder R, Widdel F, Fuchs G. Carbon assimilation pathways in sulfate-reducing bacteria. II. Enzymes of a reductive citric acid cycle in the autotrophic Desulfobacter hydrogenophilus. Arch Microbiol. 1987;148:218–225. [Google Scholar]
- 34.Segerer A H, Stetter K O. The order Sulfolobales. In: Balows A, Trüper H G, Dworkin M, Harder W, Schleifer K H, editors. The prokaryotes. Berlin, Germany: Springer-Verlag; 1992. pp. 684–701. [Google Scholar]
- 35.Seubert W, Weicker H. Pyruvate carboxylase from Pseudomonas. Methods Enzymol. 1969;XIII:258–262. [Google Scholar]
- 36.Shiba H, Kawasumi T, Igarashi Y, Kodama T, Minoda Y. The CO2 assimilation via the reductive tricarboxylic acid cycle in an obligately autotrophic, aerobic hydrogen-oxidizing bacterium, Hydrogenobacter thermophilus. Arch Microbiol. 1985;141:198–203. [Google Scholar]
- 37.Strauß G, Eisenreich W, Bacher A, Fuchs G. 13C-NMR study of autotrophic CO2 fixation pathways in the sulfur-reducing archaebacterium Thermoproteus neutrophilus and in the phototrophic eubacterium Chloroflexus aurantiacus. Eur J Biochem. 1992;205:853–866. doi: 10.1111/j.1432-1033.1992.tb16850.x. [DOI] [PubMed] [Google Scholar]
- 38.Strauß G, Fuchs G. Enzymes of a novel autotrophic CO2 fixation pathway in the phototrophic bacterium Chloroflexus aurantiacus, the 3-hydroxypropionate cycle. Eur J Biochem. 1993;215:633–643. doi: 10.1111/j.1432-1033.1993.tb18074.x. [DOI] [PubMed] [Google Scholar]
- 39.Toh H, Kondo H, Tanabe T. Molecular evolution of biotin-dependent carboxylases. Eur J Biochem. 1993;215:687–696. doi: 10.1111/j.1432-1033.1993.tb18080.x. [DOI] [PubMed] [Google Scholar]
- 40.Vorholt J A, Hafenbradl D, Stetter K O, Thauer R K. Pathways of autotrophic CO2 fixation and of dissimilatory nitrate reduction to N2O in Ferroglobus placidus. Arch Microbiol. 1997;167:19–23. doi: 10.1007/s002030050411. [DOI] [PubMed] [Google Scholar]
- 41.Vorholt J, Kunow J, Stetter K O, Thauer R K. Enzymes and coenzymes of the carbon monoxide dehydrogenase pathway for autotrophic CO2 fixation in Archaeoglobus lithotrophicus and the lack of carbon monoxide dehydrogenase in the heterotrophic A. profundus. Arch Microbiol. 1995;163:112–118. [Google Scholar]
- 42.Ward J M, Smith P H, Boone D R. Emended description of strain PS (= OGC 70 = ATCC 33273 = DSM 1537), the type strain of Methanococcus voltae. Int J Syst Bacteriol. 1989;39:493–494. [Google Scholar]
- 43.Watson G M, Tabita F R. Microbial ribulose 1,5-bisphosphate carboxylase/oxygenase: a molecule for phylogenetic and enzymological investigation. FEMS Microbiol Lett. 1997;146:13–22. doi: 10.1111/j.1574-6968.1997.tb10165.x. [DOI] [PubMed] [Google Scholar]
- 44.Widdel F, Rouviere P E, Wolfe R S. Classification of secondary alcohol-utilizing methanogens including a new thermophilic isolate. Arch Microbiol. 1988;150:477–481. [Google Scholar]
- 45.Wood H G, Ljungdahl L G. Autotrophic character of the acetogenic bacteria. In: Shively J M, Barton L L, editors. Variations in autotrophic life. San Diego, Calif: Academic Press, Inc.; 1991. pp. 201–250. [Google Scholar]
- 46.Zeikus J G, Fuchs G, Kenealy W, Thauer R K. Oxidoreductases involved in cell carbon synthesis of Methanobacterium thermoautotrophicum. J Bacteriol. 1977;132:604–613. doi: 10.1128/jb.132.2.604-613.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zillig W, Stetter K O, Schäfer W, Janekovic D, Wunderl S, Holz I, Palm P. Thermoproteales: a novel type of extremely thermoacidophilic anaerobic archaebacteria isolated from Icelandic solfatares. Zentbl Bakteriol Hyg Abt Orig C. 1981;2:205–227. [Google Scholar]