Cell Density and Phosphorylation Control the Subcellular Localization of Adenomatous Polyposis Coli Protein (original) (raw)
Abstract
Loss of functional adenomatous polyposis coli protein (APC) leads to uncontrolled proliferation of colonic epithelial cells, as evidenced by polyp formation, a prelude to carcinogenesis. As a tumor suppressor, APC targets the oncogene β-catenin for proteasome-mediated cytoplasmic degradation. Recently, it was demonstrated that APC also interacts with nuclear β-catenin, thereby reducing β-catenin's activity as a transcription cofactor and enhancing its nuclear export. The first objective of this study was to analyze how cellular context affected APC distribution. We determined that cell density but not cell cycle influenced APC's subcellular distribution, with predominantly nuclear APC found in subconfluent MDCK and intestinal epithelial cells but both cytoplasmic and nuclear APC in superconfluent cells. Redistribution of APC protein did not depend on continual nuclear export. Focusing on the two defined nuclear localization signals in the C-terminal third of APC (NLS1APC and NLS2APC), we found that phosphorylation at the CK2 site increased and phosphorylation at the PKA site decreased NLS2APC-mediated nuclear translocation. Cell density-mediated redistribution of β-galactosidase was achieved by fusion to NLS2APC but not to NLS1APC. Both the CK2 and PKA sites were important for this density-mediated redistribution, and pharmacological agents that target CK2 and PKA instigated relocalization of endogenous APC. Our data provide evidence that physiological signals such as cell density regulate APC's nuclear distribution, with phosphorylation sites near NLS2APC being critical for this regulation.
The tumor suppressor gene adenomatous polyposis coli (APC) was identified a decade ago as mutated in the inherited colon cancer syndrome, familial adenomatous polyposis (8, 20, 23, 30). Further research revealed that mutation of APC is an early step in the progression of most sporadic colon cancers as well (35). How do mutations in the APC gene initiate unregulated cell growth in the colon, manifested by polyp formation and colorectal carcinogenesis? Colonocytes originate from stem cells located approximately 30 cells below the luminal surface of the colon (21). In the course of its short life, a colonocyte moving toward the luminal surface will divide a few times, differentiate, undergo apoptosis, and ultimately be shed into the lumen (34). Identification of proteins that interact with APC has implicated this tumor suppressor in cell division, migration, and apoptosis (25, 26).
β-Catenin was the first APC binding partner identified (39, 44). In addition to its participation in E-cadherin-mediated epithelial cell adhesion, β-catenin is a key player in the Wnt signaling pathway. In the absence of a Wnt signal, APC promotes the degradation of cytoplasmic β-catenin. This proteolysis requires several additional proteins, including axin, glycogen synthase kinase 3β (GSK3β), and components of the proteasome (3, 9, 13, 15, 40). In the presence of a Wnt signal, β-catenin accumulates in the cytoplasm, translocates to the nucleus, and coordinates with T-cell factor/lymphoid-enhancer factor to activate gene transcription. Some of the genes transcriptionally activated by β-catenin, such as cyclin D1 and c-myc, are important for control of cell cycle progression and proliferation (10, 42, 45). If APC fails to down-regulate β-catenin, as is the case in many colon cancers, then constitutive expression of cyclin D1 and myc can drive cell proliferation.
Discovery of APC protein in the nuclei of human epithelial cells (28) provided the potential for APC to directly influence not only cytoplasmic β-catenin but also nuclear β-catenin levels. Recently we demonstrated that APC contains two functional nuclear import signals (NLS1APC and NLS2APC), which are necessary for optimal nuclear import of full-length APC (47). We classified NLS1APC and NLS2APC as bona fide nuclear localization signals based on the following two criteria (5). Mutation of both NLS1APC and NLS2APC leads to cytoplasmic localization of full-length APC protein (29), and each NLSAPC is sufficient to direct the nonnuclear protein β-galactosidase (β-Gal) to the nucleus (47). Because truncated forms of APC, lacking both NLS1APC and NLS2APC, are still able to enter the nucleus (11, 38), it is likely that yet a third NLS exists in the N-terminal half of APC. Although this putative third NLSAPC appears sufficient to target considerably truncated versions of APC protein to the nucleus, there is no indication that it functions in the context of full-length APC protein. APC also contains at least two endogenous nuclear export signals (NESs) and shuttles between the nucleus and the cytoplasm (11, 27, 38). Nuclear APC can sequester nuclear β-catenin and facilitate its nuclear export (11, 29). Thus, nuclear-cytoplasmic shuttling of APC can influence β-catenin nuclear levels and activity.
Since proliferation of epithelial cells lining the colon must normally be under exquisite control, we were interested in defining the signals in normal epithelial cells that dictate APC's localization and ultimately its function. We previously demonstrated that phosphorylation of Ser2054 at the PKA site adjacent to NLS2APC negatively regulated the NLS2APC-mediated nuclear import of a chimeric β-Gal fusion protein as well as the nuclear import of full-length APC (47). Therefore, phosphorylation is implicated in the regulation of APC's nuclear import.
The classic example of NLS-mediated nuclear import modulated by phosphorylation comes from the simian virus 40 (SV40) T-ag protein. The SV40 T-ag regulatory domain consists of a casein kinase 2 (CK2) site, a cyclin-dependent kinase 2 (cdc2/cdk2) site and a classical monopartite NLS (17, 19, 36) (see NLSSV40 T-ag, Fig. 5A). The mechanism of SV40 T-ag nuclear import has been studied extensively. The crystal structure of nuclear import receptor importin α in the presence of NLSSV40 T-ag revealed a combination of electrostatic and hydrophobic features, which necessitate that the NLSSV40 T-ag be composed of positively charged lysines and arginines (4, 7). Phosphorylation at the CK2 site regulates the nuclear import rate of SV40 T-ag, presumably by increasing the affinity and rate of association with importin α; phosphorylation at the cdc2/cdk2 site inhibits the maximal level of nuclear accumulation, apparently through cytoplasmic retention (18). The NLS2APC region is similar to this regulatory motif of SV40 T-ag protein, suggesting possible conservation of function.
FIG. 5.

The CK2APC site upstream of NLS2APC modifies nuclear import of a β-Gal-NLS2APC chimera. (A) Alignment of NLS2APC with NLSSV40 T-ag reveals similarity of NLS sequence as well as the adjacent phosphorylation sites. β-Gal chimeras were expressed in L cells (B) or MDCK cells (C) and stained as described for Fig. 4. β-Gal-NLS2APC and β-Gal-NLS2APCmCK2S/A localized to both the cytoplasm and nucleus. β-Gal-NLS2APCmCK2S/D was predominantly nuclear. Bars, 10 μm (B) and 20 μm (C). A negative charge, which mimics phosphorylation of Ser2034 in the potential CK2APC site increases NLS2APC-mediated nuclear import in both L cells and MDCK cells. The results of three independent experiments are presented as the incidence of MDCK cells with predominantly cytoplasmic β-Gal staining as follows: β-Gal-NLS2APC, 8%; β-Gal-NLS2APCmCK2S/D, 2%; β-Gal-NLS2APCmCK2S/A, 22%.
In the present study, we examined the effects of various physiological signals on the subcellular distribution of APC. We found that cell density, but not cell cycle, influenced the subcellular distribution of APC in epithelial cells. NLS2APC was sufficient to confer cell density-dependent nuclear localization when fused to the cytoplasmic protein β-Gal and both a CK2 and a PKA site influenced this NLS2APC-mediated nuclear translocation. Moreover, manipulating CK2 and PKA activities with pharmacological agents altered the localization pattern of endogenous APC in proliferating and quiescent epithelial cells. The results suggest that dynamic APC localization achieved through differential phosphorylation of amino acid residues near NLS2APC might affect β-catenin activity, with global implications for normal colonocyte proliferation and homeostasis.
MATERIALS AND METHODS
Cell culture.
L cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. MDCK cells were maintained in minimal Eagle's medium supplemented with 10% fetal bovine serum. IEC-6 cells were maintained in Dulbecco's modified Eagle's medium (high glucose) supplemented with 0.1 U of insulin/liter and 5% fetal bovine serum. MDCK and IEC-6 cells seeded at ≈10% confluence on glass chamber slides were allowed to grow to 50% confluence (subconfluent cells) or for an additional 36 h after reaching 100% confluence (superconfluent cells).
Expression vector construction.
A kozak sequence (GCC GCC ACC) and a start codon were inserted near the 5′ end of the MCS region in pCMV-βFUSa (16). Synthetic oligonucleotides coding for NLS1APC or NLS2APC were inserted into the pCMV-βFUSa plasmid immediately following the kozak sequence. All β-Gal expression constructs were generated using the same strategy. The β-Gal-NLS1APC, β-Gal-NLS2APC, and β-Gal-NLSSV40 T-ag expression constructs encode residues QLDGKKKKPTSPVKPIPQ (amino acids 1764 to 1781), SSLSIDSEDDLLQECISSAMPKKKKPSRLKGD (amino acids 2028 to 2058), and LFCSEEMPSSDDEATADSQHSTPPKKKRKVEDP (amino acids 103 to 135), respectively. In β-Gal-NLS1APCmCDK2S/A and β-Gal-NLS1APCmCDK2S/D, the serine at the potential CDK2 site (Ser1774) was changed to alanine and aspartic acid, respectively. In β-Gal-NLS2APCmCK2S/A and β-Gal-NLS2APCmCK2S/D, the serine at the potential CK2APC site (Ser2034) was changed to alanine and aspartic acid, respectively. In β-Gal-NLS2APCmPKAS/A and β-Gal-NLS2APCmPKAS/D, the serine at the potential PKAAPC site (Ser2054) was changed to alanine and aspartic acid, respectively. β-Gal-NLS2APCmCK2S/DmPKAS/A contains aspartic acid at the potential CK2APC site (Ser 2034) and alanine at the potential PKAAPC site (Ser2054).
Cell synchronization and BrdU labeling.
MDCK or IEC-6 cells were seeded at a density of 4 × 105 cells/10-cm-diameter dish. Nocodazole (1 μM final concentration; Sigma) was added directly into the medium for 12 h to arrest cells in M phase. Alternatively, mimosine (1 mM final concentration; Sigma) was added directly into the medium for 18 h to block cells in late G1. Following removal of mimosine, cells were cultured for an additional 0, 3, 6, or 9 h in regular medium before analysis. Synchronized cells were trypsinized, fixed with methanol, and analyzed by flow cytometry using a Becton Dickinson FACScan as described previously (46). Bromodeoxyuridine (BrdU) was added directly into the culture medium (10 μM) for 30 min before fixation.
Transfection and immunofluorescence.
Cos7 and L cells plated on glass chamber slides were transfected using Fugene 6 (Boehringer Mannheim) and Superfect (Qiagen), respectively, following manufacturers' instructions. APC immunostaining was performed on MDCK or L cells grown on glass chambers as described previously (28) with the following exceptions. The Na2BH3 incubation step was eliminated, and 0.1% Triton X-100 was included in the fixation and antibody incubation steps. Antibodies used were as follows: APC Ab-4 and Ab-1 (mouse immunoglobulin G [IgG]; Oncogene Sciences), 1:100 and 1:50; goat anti-mouse IgG-fluorescein isothiocyanate (FITC) (Southern Biotechnology Associates), 1:200; APC C-20 and N-15 (rabbit; Santa Cruz), 1:70; goat anti-rabbit IgG-FITC (Southern Biotechnology Associates), 1:200; RanBP-1 (goat, catalog no. sc1159; Santa Cruz) 1:100; rabbit anti-goat IgG-FITC (Sigma), 1:200; BrdU (Becton Dickinson), 1:100; goat anti-mouse IgG-Texas red (Southern Biotechnology Associates), 1:200; β-Gal antibody (mouse IgG; Promega), 1:1,000. For RanBP-1 staining the normal goat serum in the antibody dilution buffer was replaced with normal rabbit serum. MDCK and mouse L cells were fixed 24 or 36 h after transfection. Nuclei were counterstained with 4, 6-diamidino-2-phenylindole (DAPI) or TO-PRO-3 (Molecular Probes) following the secondary antibody incubation. For BrdU staining, cells were incubated in 0.5% Triton X-100–2 M HCl in phosphate-buffered saline for 30 min following fixation to denature the DNA. Stained cells were examined using an Axioplan fluorescence microscope (Zeiss) with a 63× objective. Alternatively, cells were examined using a Fluoview 200 laser scanning confocal microscope (Olympus). For protein localization scoring, at least 40 transfected cells for each condition were scored with results presented as means for three independent experiments.
Drug treatments.
Cells were incubated with the following drug combinations for 30 min (or 8 h) prior to fixation and staining for APC (or cyclin D1). For superconfluent cells, PKA inhibitors (100 μM Rp diastereomer of cyclic AMP [cAMP] and 100 μM 4-cyano-3-methylisoquinoline) were mixed with CKII agonists (10 mM insulin and 100 ng of epidermal growth factor/ml). For subconfluent cells**,** PKA agonists (1 mM 8-bromo-cAMP and 1 mM dibutylyl-cAMP) were mixed with CKII inhibitor (1 mM 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole [DRB]; Calbiochem). Dimethyl sulfoxide (DMSO), used to dissolve some of the drugs, was added to cells at comparable concentrations to control the effects of the dissolving reagent. To block nuclear export, cells were treated with leptomycin B (LMB; a generous gift from Minoru Yoshida) 8 h prior to fixation.
RESULTS
Subcellular distribution of APC does not change during the cell cycle.
Proteins that control cell cycle progression are often differentially expressed or localized at various stages of the cell cycle. To test if APC fit that description, we examined the subcellular distribution of APC at various stages of the MDCK cell cycle. MDCK cells are derived from normal canine kidney epithelia. In culture, confluent MDCK cells polarize and form junctions, thus serving as a popular model system for investigation of epithelial cell dynamics. MDCK cells were synchronized by treatment with mimosine or nocodazole. Mimosine is a natural plant drug that reversibly arrests cells at the late G1 phase without affecting cytoskeletal structures (12, 32, 46). When mimosine is removed, cells progress into S phase and eventually G2/M. Nocodazole treatment causes destabilization of microtubules, thereby inhibiting formation of the mitotic spindle which leads to arrest in M.
Using populations that represented various stages of the cell cycle as revealed by fluorescence-activated cell sorting (FACS) analysis, we determined the subcellular localization of APC by immunofluorescence microscopy. In mimosine-treated MDCK cells arrested in G1, APC was located in the nucleus and the cytoplasm, showing a predominantly nuclear staining and a bright edge staining (Fig. 1A, mimosine). Following removal of mimosine from the medium, cells progressed through S (Fig. 1A, 3 and 6 h) and into G2/M (9 h postmimosine) with no associated changes in the subcellular distribution of APC. A slight accumulation of APC near the nuclear membrane as cells progressed from G0/G1 to G2 was occasionally observed, but not with all APC antibodies used. In mitotic cells, enriched by nocodazole treatment, APC was excluded from the chromatin (Fig. 1A, nocodazole). This chromatin exclusion during M was also observed in asynchronous populations of MDCK cells (data not shown). APC's constant localization throughout the cell cycle was observed in cells stained using any one of three different APC antibodies.
FIG. 1.
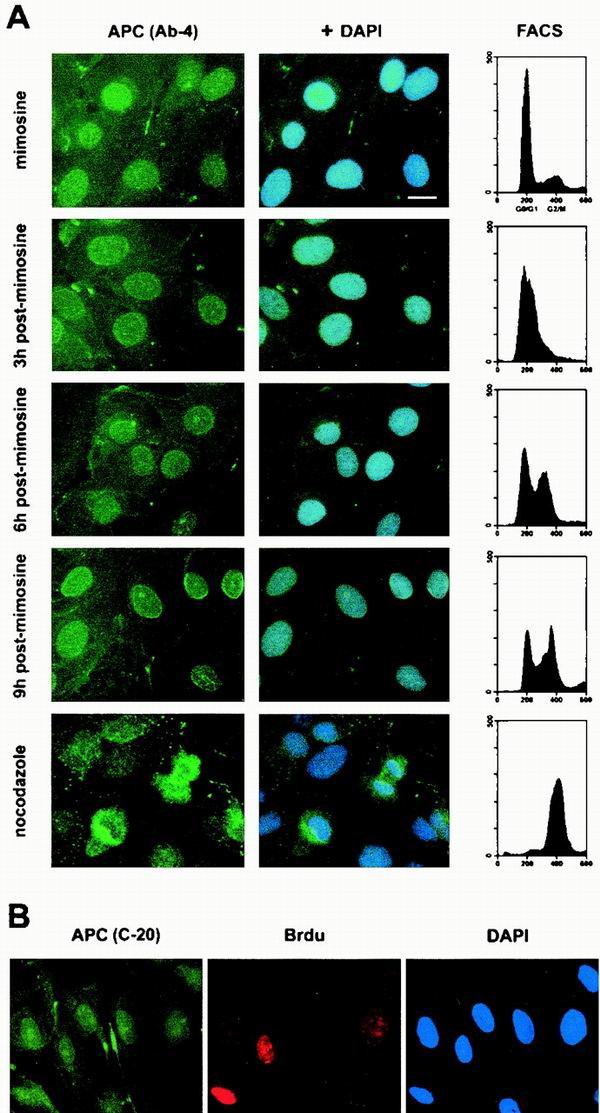
APC is predominantly nuclear throughout the MDCK cell cycle. (A) FACS analysis revealed that most MDCK cells were arrested in G0/G1 after an 18-h incubation with mimosine. Many cells harvested 3 h after mimosine removal had entered S phase. By 6 h following mimosine removal, the majority of cells were in S phase. After 9 h, most cells had a 4c DNA content, suggesting that they were in G2/M. The majority of cells showed a 4c DNA content after nocodazole treatment, suggesting arrest in M. Using immunofluorescence microscopy and a monoclonal antibody against APC (Ab-4), APC was located in the cytoplasm and predominantly in the nucleus throughout the cell cycle (left panels, green). Nuclei were visualized by DAPI staining (blue). Bar, 20 μm. (B) Asynchronous MDCK cells were pulse-labeled with BrdU prior to fixation. Cells in S phase were identified using an anti-BrdU antibody (red). APC was identified using a polyclonal antibody raised against APC (C-20, green). Nuclei were visualized with DAPI (blue).
FIG. 3.

Redistribution of APC does not depend on sustained nuclear export. Super- and subconfluent MDCK cells were treated with LMB prior to fixation and staining with αAPC, Ab-1 (left panel) and RanBP-1 (right panel). LMB treatment resulted in an increase in nuclear APC in subconfluent but not superconfluent cells. RanBP-1 relocated from the cytoplasm to the nucleus in both super- and subconfluent cells following LMB treatment. Bar, 20 μm.
FIG. 6.

Cell density influences NLS2APC- but not NLS1APC-mediated nuclear translocation of β-Gal in MDCK cells. β-Gal fusion proteins expressed in MDCK cells grown to different densities were localized as previously described. β-Gal was predominantly cytoplasmic in both superconfluent and subconfluent MDCK cells. β-Gal-NLSSV40 T-ag was predominantly nuclear under both conditions. β-Gal-NLS1APC was evenly distributed in the nucleus and cytoplasm of over half of the super-confluent and subconfluent MDCK cells, with slightly more nuclear localization in superconfluent cells. β-Gal-NLS2APC was predominantly nuclear in subconfluent cells, but was more evenly distributed between the nucleus and cytoplasm in superconfluent cells. MDCK cells from at least three independent experiments, scored for protein localization, were placed in the following categories: cytoplasmic > nuclear (white bar), cytoplasmic = nuclear (grey bar), and cytoplasmic < nuclear (black bar).
To further distinguish MDCK cells in S phase from those in G1 and G2, S-phase cells were identified in asynchronous populations by briefly labeling with BrdU. The APC staining pattern did not vary in BrdU-positive (S-phase) cells of asynchronous MDCK populations (Fig. 1B). In summary, APC localized to both the cytoplasm and the nucleus in subconfluent MDCK cells, with a predominantly nuclear distribution. Cytoplasmic APC was dispersed throughout the cell, with concentration near a single edge. The subcellular distribution of APC did not change during the G1, S, and G2 phases of the cell cycle and was comparable using any one of three different APC antibodies for the immunofluorescence analysis. In mitotic cells, APC was excluded from chromatin.
Cell density influences the subcellular distribution of APC in kidney and intestinal epithelial cells.
Reasoning that APC might function in the context of the colonic crypt to restrict cellular proliferation, we tested whether APC location differed in subconfluent (proliferating) cells versus superconfluent (G0) cells. In contrast to subconfluent cells, in which APC was located predominantly in the nucleus and somewhat in the cytoplasm, in G0 cells, obtained by maintaining the MDCK cells for several days after they reached confluence, APC was more evenly distributed between the cytoplasm and nucleus (Fig. 2). This striking redistribution was observed in cells stained using any one of a panel of APC antibodies including two monoclonal antibodies (Ab-1 and Ab-4) and two polyclonals (N-15 and C-20). These antibodies were raised against the N terminus (Ab-1 and N-15) and C terminus (Ab-4 and C-20) of APC and have been used routinely for APC recognition. Use of the nuclear marker TO-PRO-3 and confocal microscopy to collect images insured proper classification of the cytoplasmic and nuclear compartments. APC signal was fully attenuated with APC peptides containing the appropriate epitope (data not shown). Additionally, intestinal epithelial cells displayed a similar density-mediated redistribution of endogenous APC, showing predominantly nuclear APC in subconfluent cells and predominantly cytoplasmic APC in superconfluent cells (data not shown). These data indicate that the subcellular distribution of APC is influenced by cell density.
FIG. 2.

The subcellular distribution of APC in MDCK cells is influenced by cell density. The distribution of APC in subconfluent and superconfluent MDCK cells was determined using immunofluorescence confocal microscopy and four different antibodies raised against APC. Nuclei were visualized with TO-PRO-3. Bar, 20 μm. FACS analysis confirmed that subconfluent cells were distributed throughout the cell cycle whereas superconfluent cells were mostly in G0/G1.
The cytoplasmic redistribution of APC in quiescent MDCK cells is not dependent on continuous Crm-1-mediated nuclear export.
Since APC is a nuclear-cytoplasmic shuttling protein, with both intrinsic nuclear export and nuclear import signals, it is possible that the cell density-related accumulation of cytoplasmic APC depends on sustained nuclear export. To test this hypothesis directly, super- and subconfluent MDCK cells were treated with LMB to block Crm-1-mediated nuclear export (Fig. 3). Subconfluent MDCK cells treated with LMB displayed slightly more nuclear APC than did untreated cells, as noted in previous reports of other cell lines (11, 27). In contrast, treating superconfluent MDCK cells with LMB had no apparent effect on APC distribution (Fig. 3). To ensure that superconfluent MDCK cells were capable of Crm-1-dependent nuclear export and therefore sensitive to LMB, cells were stained for the nuclear-cytoplasmic shuttling protein, RanBP-1 (33). Treatment with LMB of MDCK cells grown at either density resulted in a shift of RanBP-1 from the cytoplasm to the nucleus (Fig. 3, right panels). These observations suggest that the density-mediated redistribution of APC is not dependent on continuous nuclear export, implicating the NLSAPC in this relocalization.
NLS1APC-mediated nuclear translocation of a β-Gal chimera is not regulated by the adjacent CDK2 site.
Our previous work suggested that phosphorylation near NLS2APC could impact APC's subcellular distribution by modulating NLS2APC-mediated nuclear localization. This observation led us to investigate other potential phosphorylation sites close to NLS1APC and NLS2APC for modulating effects on the nuclear localization of APC. To test if a potential CDK2 site just downstream of NLS1APC (Ser1774) regulates NLS1APC-mediated nuclear localization, we expressed four different β-Gal-NLS1APC fusion proteins in mouse L cells (Fig. 4). β-Gal protein has no inherent NLS, forms a large homotetramer (≈480 kDa) and is thus predominantly cytoplasmic when expressed in L cells. β-Gal-NLS1APC contains wild-type APC sequence, including the potential CDK2 site Ser1774 and, as shown previously (47), is significantly more nuclear than β-Gal alone. In contrast, β-Gal-mNLS1APC, carrying substitution of alanine for the four lysines of NLS1APC (Lys1768-1771), was predominantly cytoplasmic. Substitution of alanine (β-Gal-NLS1APCmCDK2S/A) or aspartic acid (β-Gal-NLS1APCmCDK2S/D) for Ser1774 did not alter the expression pattern significantly from that of β-Gal-NLS1APC. The alanine substitution mimics a dephosphorylated form of NLS1APC, whereas the aspartic acid substitution mimics the negative charge of phosphorylation. When 100 cells were scored, the increased incidence of nuclear β-Gal staining observed for β-Gal-NLS1APCmCDKS/D (83%) compared to that observed for β-Gal-NLS1APCmCDKS/A (58%) and β-Gal-NLS1APC (73%) suggested that phosphorylation of Ser1774 might promote nuclear localization, although these differences were only slight. The relatively similar staining patterns of β-Gal-NLS1APCmCDK2S/A and β-Gal-NLS1APCmCDK2S/D suggested that NLS1APC-mediated nuclear translocation of the β-Gal chimera is not significantly regulated by the adjacent CDK2 site.
FIG. 4.
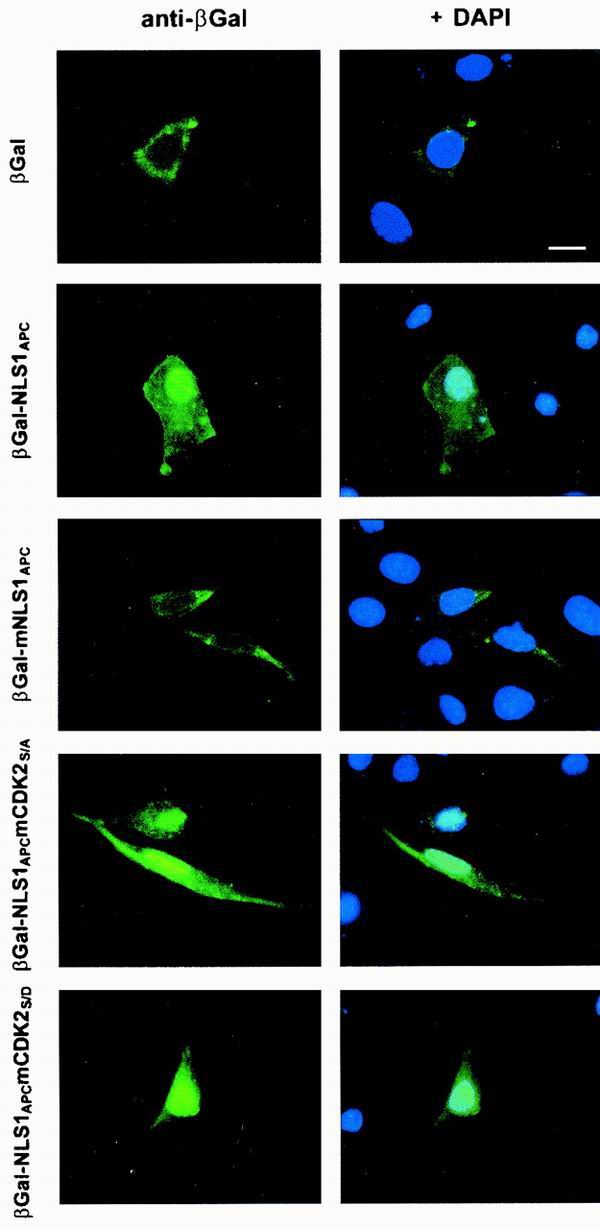
The adjacent CDK2 site does not regulate the NLS1APC-mediated nuclear translocation of a β-Gal chimera. β-Gal fusion proteins were expressed in mouse L cells and detected using immunofluorescence microscopy. Nuclei were visualized with DAPI. Areas of overlap between the β-Gal fusion protein (green) and the nuclei (blue) appear in aqua. Bar, 10 μm. For each construct, 100 cells were scored for β-Gal fusion protein localization. The results of three independent experiments are presented as the incidence of nuclear β-Gal staining ± standard deviations as follows: β-Gal, 27% ± 6%; β-Gal-NLS1APC, 73% ± 11%; β-Gal-mNLS1APC, 30% ± 13%; β-Gal-NLS1APCmCDKS/A, 58% ± 10%; β-Gal-NLS1APCmCDKS/D, 83% ± 6%.
A negative charge upstream of NLS2APC increases the relative nuclear level of β-Gal-NLS2APC.
We noticed that the similarity between NLS2APC and NLSSV40 T-ag extended beyond the NLS to the CK2 phosphorylation sites ≈14 amino acids upstream of each NLS as well as to potential phosphorylation sites immediately adjacent to each NLS (Fig. 5A). Both the CK2APC and the PKAAPC sites are evolutionarily conserved among human, mouse, rat, and frog APC proteins, suggesting the potential for functional significance. Using site-directed mutagenesis, we showed that a negative charge, which mimics phosphorylation of Ser2054, inhibited NLS2APC-mediated nuclear localization (47). To analyze how phosphorylation at the CK2APC site upstream of NLS2APC affects nuclear localization we used the same approach. The β-Gal-NLS2APC chimera was distributed rather evenly between the cytoplasm and the nucleus in transfected L cells (Fig. 5B). Substitution of aspartic acid for the potential CK2 site serine (Ser2034) mimics the negative charge resulting from phosphorylation. As predicted from the NLSSV40 T-ag analogy, the chimeric protein with this substitution (β-Gal-NLS2APCmCK2S/D) showed a predominantly nuclear staining pattern (Fig. 5B). This suggests that phosphorylation of Ser2034 promotes nuclear localization. In contrast, the β-Gal-NLS2APCmCK2S/A protein showed a more cytoplasmic distribution than either β-Gal-NLS2APC or β-Gal-NLS2APCmCK2S/D (Fig. 5B). Similar results were obtained with MDCK cells (Fig. 5C).
Cell density-mediated redistribution of β-Gal is achieved by fusion to NLS2APC.
The observation that phosphorylation regulates NLS2APC activity in both L cells and MDCK cells compelled us to integrate phosphorylation with the cell density-dependent redistribution of APC observed in MDCK cells. We hypothesized that cell density influences the subcellular distribution of APC by modulating its nuclear import. Since both NLS1APC and NLS2APC are necessary for optimal nuclear translocation of full-length APC, we initially tested if nuclear translocation mediated by either NLS1APC or NLS2APC is influenced by cell density in MDCK cells. Various β-Gal chimeras were expressed in superconfluent or subconfluent MDCK cells and localized using immunofluorescence microscopy (Fig. 6). The control β-Gal protein was seen predominantly in the cytoplasm of all superconfluent and subconfluent MDCK cells scored. On the other hand, β-Gal fused with the NLSSV40 T-ag was seen predominantly in the nuclei of all superconfluent and subconfluent MDCK cells. β-Gal-NLS1APC was evenly distributed in the nuclei and cytoplasms of over half of the superconfluent and subconfluent MDCK cells, with slightly more nuclear localization in superconfluent cells. In contrast, β-Gal-NLS2APC was predominantly nuclear in subconfluent cells but was more evenly distributed between the nuclei and cytoplasms of superconfluent cells. The decrease in nuclear β-Gal-NLS2APC in superconfluent MDCK cells indicated that cell density negatively impacts the NLS2APC-mediated nuclear localization of the β-Gal chimera. More importantly, this redistribution was strikingly similar to that of endogenous APC protein in MDCK cells (Fig. 2), suggesting that NLS2APC is sufficient to impart this property to a heterologous protein.
CK2 and PKA sites are both required for the cell density-influenced nuclear localization of β-Gal-NLS2APC.
Ideally, we wanted to fully integrate the observation that phosphorylation regulates the nuclear localization of APC with the finding that APC localization is dependent on cell density. To that end, we hypothesized that the phosphorylation status of the potential PKAAPC and CK2APC sites near NLS2APC is critical for density-mediated regulation of nuclear import. Our data suggested that phosphorylation at multiple sites controlled nuclear import. At a given site, one phosphorylation state promoted nuclear import and the opposite state impeded nuclear import. We already showed that phosphorylation at the PKAAPC2054 and the CK2APC2034 sites had opposite effects on nuclear localization mediated by NLS2APC. To simplify the explanation of the next set of experiments, we will describe each potential phosphorylation site as a “switch” and refer to the amino acid substitutions as “on” (promoting nuclear localization) or “off” (blocking nuclear localization) based on our previous results. Specifically, the CK2APC switch is on when the serine is mutated to aspartic acid and off when mutated to alanine. In contrast, the PKAAPC switch is on when the serine is mutated to alanine and off when it is mutated to aspartic acid.
To test if the phosphorylation status of the PKAAPC2054 and CK2APC2034 sites affected the density-mediated regulation of nuclear localization, various β-Gal-NLS2APC expression constructs with amino acid substitutions at either the CK2APC2034 or the PKAAPC2054 site were transfected into MDCK cells grown to different cellular densities (Fig. 7). β-Gal-NLS2APCmCK2S/A, with the CK2APC switch turned off, was located predominantly in the cytoplasms of both superconfluent and subconfluent MDCK cells, with somewhat higher levels in the nuclei of subconfluent cells (Fig. 7A, left panel). β-Gal-NLS2APCmPKAS/D, with the PKAAPC switch turned off, had a similar distribution (Fig. 7A, right panel). These results indicate that turning off either the CK2APC or the PKAAPC switch impairs nuclear translocation of β-Gal-NLS2APC yet still allows some differential localization in subconfluent versus superconfluent cells. To explore this further, we expressed β-Gal-NLS2APC with either the CK2APC or the PKAAPC switch constitutively turned on. When β-Gal-NLS2APCmCK2S/D was expressed in superconfluent cells, it distributed evenly between the cytoplasm and nucleus in 44% of the cells and was predominantly cytoplasmic in 40% of the cells. In subconfluent cells, β-Gal-NLS2APCmCK2S/D localization was distinctly more nuclear. Similarly, β-Gal-NLS2APCmPKAS/A was evenly distributed between the nucleus and cytoplasm in about half of the superconfluent cells but was predominantly nuclear in nearly all subconfluent cells (Fig. 7B). This localization pattern was similar to that of β-Gal-NLS2APC and, more importantly, to endogenous APC. Therefore, if either the CK2APC or the PKAAPC switch is constitutively turned on, then the cell density-dependent regulation of β-Gal-NLS2APC localization is preserved. These data suggest that nuclear localization of β-Gal-NLS2APC in subconfluent MDCK cells is enhanced if both CK2APC and PKAAPC are on. However, one phosphorylation site in the off position is sufficient to promote cytoplasmic accumulation of β-Gal-NLS2APC in superconfluent MDCK cells.
FIG. 7.

The potential CK2 and PKA sites control β-Gal-NLS2APC distribution in a manner influenced by cell density. (A) If either the CK2APC or the PKAAPC site is turned off, then nuclear translocation of β-Gal-NLS2APC is impaired. β-Gal fusion proteins were expressed in MDCK cells grown to different cell densities and were located as previously described. β-Gal-NLS2APCmCK2S/A and β-Gal-NLS2APCmPKAS/D were both predominantly cytoplasmic in superconfluent MDCK cells with a slight increase in nuclear accumulation when cells were subconfluent. (B) Mutation of either the CK2APC or the PKAAPC site to the on position did not abolish the cell density-influenced β-Gal-NLS2APC redistribution. β-Gal-NLS2APCmCK2S/D and β-Gal-NLS2APCmPKAS/A were both nuclear in subconfluent cells but had more cytoplasmic localizations in superconfluent cells (see quantification in graph). Bar, 20 μm.
Mutation of both CK2 and PKA sites abolished the cell density-regulated β-Gal-NLS2APC localization.
Having shown that both CK2APC2034 and PKAAPC2054 sites were important for the cell density-dependent regulation of β-Gal-NLS2APC distribution, there was still the possibility that another element in the NLS2APC region also participated in modification of nuclear localization. To exclude the possibility that amino acids other than the serines at potential CK2APC and PKAAPC sites were sufficient to drive the cytoplasmic accumulation of NLS2APC in superconfluent cells, we expressed β-Gal-NLS2APC containing substitutions in both the CK2APC and PKAAPC sites (β-Gal-NLS2APCmCK2S/DmPKAS/A). Each single amino acid substitution should promote nuclear localization (phosphorylation site switches turned on). If other elements in the NLS2APC region can confer cell density-dependent cytoplasmic accumulation of β-Gal-NLS2APC, then fixing both phosphorylation sites in the on position would still result in increased cytoplasmic β-Gal-NLS2APC in superconfluent cells. On the contrary, β-Gal-NLS2APCmCK2S/D mPKAS/A was predominantly nuclear in both subconfluent and superconfluent MDCK cells (Fig. 8). These data demonstrate that the CK2APC and PKAAPC phosphorylation sites are necessary for the cell density-dependent redistribution of β-Gal-NLS2APC in MDCK cells.
FIG. 8.
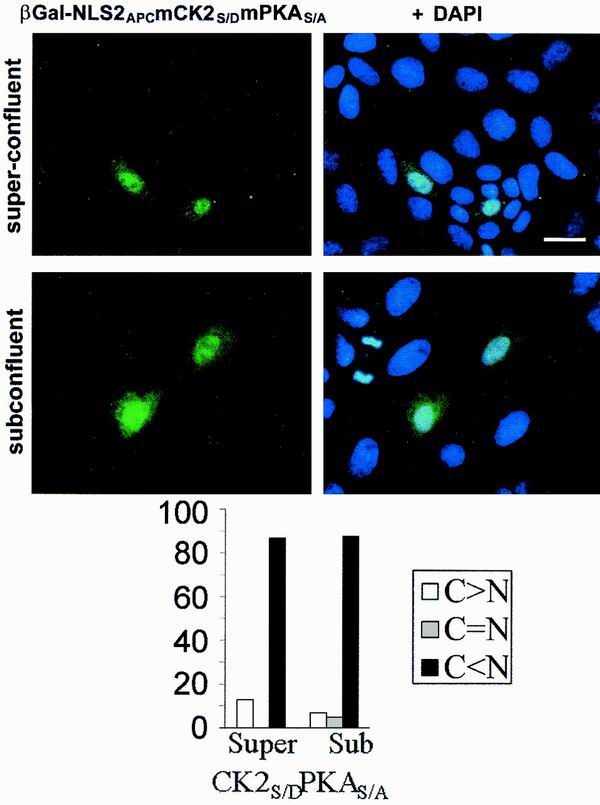
If both CK2APC and PKAAPC switches are constitutively turned on, then the cell density-influenced redistribution of β-Gal-NLS2APC is abolished. β-Gal-NLS2APCmCK2S/DmPKAS/A was expressed in MDCK cells grown to different cell densities and was located as described previously. β-Gal-NLS2APCmCK2S/DmPKAS/A was predominantly nuclear in both superconfluent and subconfluent cells (see results displayed in graphic form below images). Bar, 20 μm.
PKA and CK2 activities affect endogenous APC localization in kidney and intestinal epithelial cells.
Our results with exogenous NLS2APC imply that both phosphorylation sites mediate the cell density-dependent redistribution of full-length APC as well. If true, then manipulation of PKA and CK2 activities would result in altered localization of endogenous APC. To test this directly, superconfluent MDCK cells were treated briefly with a combination of CK2 agonists and PKA inhibitors. While cells treated with DMSO (used to solubilize the drugs) displayed APC in both the cytoplasm and the nucleus, drug treatment resulted in a significant shift toward a more nuclear APC distribution (Fig. 9A, left panel). This increase in nuclear APC accompanying CK2 activation and PKA inhibition was predicted from the β-Gal-NLS2APC data and indicates that localization of endogenous APC is controlled by CK2 and PKA. As further evidence, subconfluent MDCK cells were treated with a combination of CK2 inhibitors and PKA agonists. Whereas DMSO-treated subconfluent cells displayed predominant nuclear APC, as did untreated cells (Fig. 2), simultaneous inhibition of CK2 and activation of PKA resulted in a more cytoplasmic distribution (Fig. 9A, right panel). Similar results were obtained using normal intestinal epithelial cells (Fig. 9B). Thus, we were able to instigate the redistribution of endogenous APC protein in both subconfluent and superconfluent cells using pharmacological agents. Furthermore, the redistributions that were observed with endogenous APC in two different epithelial cell lines confirmed our results from the analysis of β-Gal-NLS2APC.
FIG. 9.

PKA and CK2 activities affect endogenous APC in MDCK and intestinal epithelial cells. MDCK (A) or IEC-6 (B) cells were treated for 30 min with various drug combinations prior to fixation and immunofluorescence microscopy with αAPC Ab-1 or C-20, respectively. Superconfluent cells (left panels) were treated with CK2 agonist (insulin and epidermal growth factor) and PKA inhibitors (Rp diastereomer of cAMP and 4-cyano-3-methylisoquinoline). Subconfluent cells were treated with CK2 inhibitor (DRB) and PKA agonists (8-bromo-cAMP and dibutylyl-cAMP). Both treatments altered localization of endogenous APC. Bar, 20 μm.
DISCUSSION
A healthy human colon is in a continuous state of cellular renewal, and consequently, epithelial cells lining the colon undergo a highly regulated series of cell divisions. Since loss of APC function initiates colorectal tumor formation, characterized by unregulated cellular proliferation, we hypothesized that APC normally functions to maintain normal colon cell proliferation. Furthermore, in serving this function, APC might display a distinct subcellular localization in proliferating cells compared to quiescent cells. We found that the subcellular localization of APC was stable as MDCK epithelial cells passed through the cell cycle (Fig. 1). APC was predominantly nuclear, with some cytoplasmic concentration near the cell's edge, consistent with previous reports using asynchronous MDCK cells (6). However, when epithelial cells from kidney or intestine became superconfluent, presumably on entering G0, APC was evenly distributed between the cytoplasm and nucleus (Fig. 2). This is the first demonstration that cellular context influences the subcellular localization of APC.
Our observation that blocking Crm-1-mediated nuclear export with LMB did not result in a nuclear accumulation of APC in superconfluent epithelial cells (Fig. 3) suggests that only a small fraction of the total APC population shuttles between the nucleus and cytoplasm in quiescent cells. Alternatively, it is possible that most APC molecules shuttle between the nucleus and cytoplasm in quiescent cells, but only rarely. After showing that the increased cytoplasmic APC in quiescent cells was not dependent on continual nuclear export, we focused on the role of nuclear import signals. We demonstrated that a stretch of 32 amino acids containing NLS2APC was sufficient to confer nuclear localization to the otherwise cytoplasmic protein β-Gal, in a manner dependent on cell density (Fig. 5). NLS1APC, although adequate to drive the nuclear localization of β-Gal, was not markedly regulated by phosphorylation (Fig. 4) and was not influenced by cell density (Fig. 6). Additionally, we showed that two intrinsic phosphorylation sites modulated nuclear localization driven by NLS2APC and were necessary for the differential localization of β-Gal-NLS2APC in quiescent versus proliferating cells (Fig. 5 and 7).
We hypothesize, based on the similarity between NLS2APC and the NLS of SV40 T-ag, that NLS2APC mediates nuclear import of APC. Demonstration that fusion of either NLS1APC or NLS2APC with the large cytoplasmic protein β-Gal resulted in its nuclear localization further implicated NLS1APC and NLS2APC as mediators of nuclear import. The steady-state localization of APC might also be influenced by nuclear and/or cytoplasmic retention. Since the APC sequence analyzed in this study extends beyond the monopartite basic stretch of amino acids that we classified as NLS2APC, it is possible that other elements in this region affect nuclear or cytoplasmic retention and ultimately, APC's subcellular localization.
Apart from containing two potential phosphorylation sites, the NLS2APC peptide used in our experiments encompasses the third axin-binding motif of APC, SAMP#3 (3). It is possible that binding of the NLS2APC region to axin participates in the differential localization of NLS2APC-β-Gal in quiescent cells. Altering both phosphorylation sites in NLS2APC so as to promote nuclear import resulted in the loss of the cytoplasmic β-Gal in quiescent cells (Fig. 8). Therefore, if axin-APC interactions are responsible for cytoplasmic retention of APC, the single amino acid substitutions that promoted nuclear localization must have disrupted axin and APC binding. Conversely, if axin-APC interactions are responsible for nuclear retention of APC, the single amino acid substitutions that promoted nuclear localization of β-Gal-NLS2APC in quiescent cells must have enhanced axin and APC binding. The structure of a portion of axin, crystallized with APC-SAMP#3, predicted contact between the potential CK2 site, Ser2034, and axin, with no contact at the potential PKA site, Ser2054 (43). Then again, the equivalent position of Ser2034 is an aspartic acid in human, murine, and Xenopus SAMP#1 and an alanine in Drosophila melanogaster SAMP#2, suggesting tolerance for both amino acid substitutions used in this study. Detailed analyses of axin's subcellular localization as well as the binding kinetics of endogenous axin with mutant APC are necessary to further examine this intriguing possibility.
Using various β-Gal chimeras, we demonstrated that potential phosphorylation sites near NLS2APC are critical modulators of nuclear localization, responsible for differences in β-Gal-NLS2APC localization in proliferating versus quiescent cells. We predicted that phosphorylated serine residues, Ser2034APC and Ser2054APC, function in a similar manner in full-length APC in vivo. Indeed, a brief activation of CK2 and inhibition of PKA in quiescent MDCK or IEC-6 cells resulted in a dramatic relocalization of endogenous APC from the cytoplasm to the nucleus (Fig. 9). Similarly, simultaneous inhibition of CK2 and activation of PKA in proliferating MDCK or IEC-6 cells resulted in a shift of endogenous APC from the nucleus to the cytoplasm. In the future it will be interesting to determine whether Ser2034 and Ser2054 are differentially phosphorylated within the context of endogenous APC protein in cells grown under different conditions. So far, our attempts to use matrix-assisted laser desorption ionization–time of flight analysis of purified APC protein digested with trypsin to determine the phosphorylation status of specific APC residues have been impeded by the relatively low abundance and large size of APC protein (K. Neufeld, personal communication).
Our data support the proposal that APC localization is controlled by a balance of nuclear import, mediated by NLSAPC, and nuclear export, mediated by the intrinsic NESAPC. A schematic of NLS2 modulation within the APC protein is depicted in Fig. 10. In proliferating cells, where APC is primarily nuclear, CK2APC2034 is phosphorylated but PKAAPC2054 is not (Fig. 10A). As cells become superconfluent and cease to divide, APC is more evenly distributed between the cytoplasm and the nucleus, indicating a shift in the balance of APC phosphorylation (Fig. 10B). Some of the cellular APC is dephosphorylated at the CK2APC2034 site, phosphorylated at the PKAAPC2054 site or both, thereby inhibiting nuclear import mediated by NLS2APC. Consequently, we suggest that CK2APC and PKAAPC, together with NLS2APC, constitute a phosphorylation-regulated module, providing a mechanism for regulated nuclear localization of APC.
FIG. 10.

Model for the cell density-influenced regulation of APC's subcellular distribution. The CK2APC and PKAAPC sites function as switches promoting nuclear localization (on) or impeding nuclear localization (off). The CK2APC switch is on when the CK2APC site serine is phosphorylated. The PKAAPC switch is on when the PKAAPC site is dephosphorylated. (A) In subconfluent and/or proliferating cells, APC primarily exists in the form with both switches on and is therefore localized predominantly to the nucleus. (B) Superconfluent and/or quiescent cells contain APC with either or both switches turned off, resulting in an increased cytoplasmic pool of APC.
Our mutagenesis strategy clearly indicated that Ser2034APC and Ser2054APC were critical mediators of NLS2APC activity. Within APC, these serine residues are surrounded by amino acids that fit the consensus pattern for phosphorylation by CK2 and PKA, respectively, and thus we have referred to them as potential CK2 and PKA sites (22). Additionally, pharmacological agents that specifically target PKA and CK2 influenced the localization of endogenous APC (Fig. 9). CK2 inhibitors changed the predominantly nuclear APC distribution in proliferating cells to one that was more cytoplasmic, suggesting that phosphorylation of NLS2APC by CK2 is important for nuclear APC localization. Conversely, CK2 activation in quiescent cells resulted in increased nuclear APC. Our observations correlate with reported CK2 activities. Elevated CK2 levels and activity have been documented in actively proliferating cells, including those from human tumors and normal tissue (reviewed in reference 14). Here we report that CK2 and PKA have opposing effects on APC localization, suggesting greater PKA activity in quiescent than in proliferating cells. A correlation between PKA activity and cell quiescence is not well established; however, activity of one PKA isozyme has been associated with reduced colonic proliferation (1). It remains possible that APC is also phosphorylated at these serines by alternative kinases. Precise identification of the kinase responsible for phosphorylation of a given APC residue in vivo will be challenging but ultimately will greatly increase our understanding of how APC regulation is affected by cellular context.
What are the consequences of APC redistribution to the cytoplasm of quiescent epithelial cells? Our results are compatible with the observation that overexpression of APC in mouse fibroblast cells blocks cell cycle progression from G0/G1 to S (2). APC-mediated β-catenin degradation appears to occur exclusively in the cytoplasm and not in the nucleus (11, 27). Thus, our observation of more cytoplasmic APC in quiescent cells than in proliferating cells suggests that cytoplasmic APC controls β-catenin levels in quiescent cells. Consistent with this theory and not surprising given its well-documented role in cell-cycle progression (41), cyclin D1, a gene transcriptionally activated by β-catenin, was expressed in five times more proliferating than quiescent cells (F. Zhang, personal communication). Furthermore, pharmacological agents that caused APC redistribution (Fig. 9) affected subsequent changes in cyclin D1 levels. As a general rule, higher levels of cytoplasmic APC correlated with less cyclin D1 expression; lower levels of cytoplasmic APC correlated with increased cyclin D1 expression (data not shown). We have previously observed that nuclear APC can bind to and inactivate nuclear β-catenin (27). Whereas the present results appear to contradict this previous finding, β-catenin binding by nuclear APC is likely regulated, occurring only under specific conditions. As such, we envision the nuclear pool of APC acting as a sentry, ready to quickly dampen the β-catenin/LEF-1 signal under particular circumstances. In this capacity, nuclear APC would be crucial in proliferating cells, where β-catenin activity might be tightly regulated. Additionally, we predict that nuclear APC has functions distinct from its role in β-catenin regulation.
Several other tumor suppressor proteins, such as p53, BRCA1, and von Hippel-Lindau, shuttle between the nucleus and the cytoplasm (24, 31, 37). Regulated protein movement in and out of the nucleus provides a simple, reversible, and rapid means to control nuclear activity and coordinate nuclear and cytoplasmic events. Regulated nuclear localization of tumor suppressor proteins likely serves as a general mechanism to control responses to different environmental conditions and stimuli in normal cells; therefore, the control of nuclear localization is critical for tumor suppressor function. Moreover, regulated nuclear localization of APC, through differential phosphorylation, is expected to affect β-catenin target genes and may prove critical for normal colonocytes to respond rapidly and precisely to different physiological conditions and extracellular signals.
ACKNOWLEDGMENTS
We thank C. Anderson for confocal microscopy assistance, Z. Izsvak for the CMV-βFUSa expression construct, M. Yoshida for leptomycin C, E. Meenan and R. Schackmann for oligonucleotide synthesis, and M. Robertson for DNA sequencing.
Grant 5PO1 CA73992-02 and the Huntsman Cancer Institute supported this work.
REFERENCES
- 1.Aukema H M, Davidson L A, Pence B C, Jiang Y H, Lupton J R, Chapkin R S. Butyrate alters activity of specific cAMP-receptor proteins in a transgenic mouse colonic cell line. J Nutr. 1997;127:18–24. doi: 10.1093/jn/127.1.18. [DOI] [PubMed] [Google Scholar]
- 2.Baeg G, Matsumine A, Kuroda T, Bhattacharjee R N, Miyashiro I, Toyoshima K, Akiyama T. The tumour suppressor gene product APC blocks cell cycle progression from G0/G1 to S phase. EMBO J. 1995;14:5618–5625. doi: 10.1002/j.1460-2075.1995.tb00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behrens J, Jerchow B A, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with β-catenin, APC, and GSK3β. Science. 1998;280:596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- 4.Conti E, Uy M, Leighton L, Blobel G, Kuriyan J. Crystallographic analysis of the recognition of a nuclear localization signal by the nuclear import factor karyopherin-α. Cell. 1998;94:193–204. doi: 10.1016/s0092-8674(00)81419-1. [DOI] [PubMed] [Google Scholar]
- 5.Dingwall C, Laskey R A. Nuclear targeting sequences–a consensus? Trends Biochem Sci. 1991;16:478–481. doi: 10.1016/0968-0004(91)90184-w. [DOI] [PubMed] [Google Scholar]
- 6.Erdmann K S, Kuhlmann J, Lessmann V, Herrmann L, Eulenburg V, Muller O, Heumann R. The Adenomatous Polyposis Coli-protein (APC) interacts with the protein tyrosine phosphatase PTP-BL via an alternatively spliced PDZ domain. Oncogene. 2000;19:3894–3901. doi: 10.1038/sj.onc.1203725. [DOI] [PubMed] [Google Scholar]
- 7.Fontes M, Teh T, Kobe B. Structural basis of recognition of monopartite nuclear localization sequences by mammalian importin-α. J Mol Biol. 2000;297:1183–1194. doi: 10.1006/jmbi.2000.3642. [DOI] [PubMed] [Google Scholar]
- 8.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, Sargeant L, Krapcho K, Wolff E, Burt R, Hughes J P, Warrington J, McPherson J, Wasmuth J, Le Paslier D, Abderrahim H, Cohen D, Leppert M, White R. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 9.Hart M J, de los Santos R, Albert I N, Rubinfeld B, Polakis P. Downregulation of β-catenin by human Axin and its association with the APC tumor suppressor, β-catenin and GSK3β. Curr Biol. 1998;8:573–581. doi: 10.1016/s0960-9822(98)70226-x. [DOI] [PubMed] [Google Scholar]
- 10.He T C, Sparks A B, Rago C, Hermeking H, Zawel L, da Costa L T, Morin P J, Vogelstein B, Kinzler K W. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 11.Henderson B. Nuclear-cytoplasmic shuttling of APC regulates β-catenin subcellular localization and turnover. Nat Cell Biol. 2000;2:653–660. doi: 10.1038/35023605. [DOI] [PubMed] [Google Scholar]
- 12.Hughes T A, Cook P R. Mimosine arrests the cell cycle after cells enter S-phase. Exp Cell Res. 1996;222:275–280. doi: 10.1006/excr.1996.0035. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3β and β-catenin and promotes GSK-3β-dependent phosphorylation of β-catenin. EMBO J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Issinger O G. Casein kinases: pleiotropic mediators of cellular regulation. Pharmacol Ther. 1993;59:1–30. doi: 10.1016/0163-7258(93)90039-g. [DOI] [PubMed] [Google Scholar]
- 15.Itoh K, Krupnik V E, Sokol S Y. Axis determination in Xenopus involves biochemical interactions of axin, glycogen synthase kinase 3 and β-catenin. Curr Biol. 1998;8:591–594. doi: 10.1016/s0960-9822(98)70229-5. [DOI] [PubMed] [Google Scholar]
- 16.Ivics Z, Izsvak Z. Family of plasmid vectors for the expression of β-galactosidase fusion proteins in eukaryotic cells. BioTechniques. 1997;22:254–256. doi: 10.2144/97222bm12. , 258. [DOI] [PubMed] [Google Scholar]
- 17.Jans D A, Ackermann M J, Bischoff J R, Beach D H, Peters R. p34cdc2-mediated phosphorylation at T124 inhibits nuclear import of SV-40 T antigen proteins. J Cell Biol. 1991;115:1203–1212. doi: 10.1083/jcb.115.5.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jans D A, Hubner S. Regulation of protein transport to the nucleus: central role of phosphorylation. Physiol Rev. 1996;76:651–685. doi: 10.1152/physrev.1996.76.3.651. [DOI] [PubMed] [Google Scholar]
- 19.Jans D A, Jans P. Negative charge at the casein kinase II site flanking the nuclear localization signal of the SV40 large T-antigen is mechanistically important for enhanced nuclear import. Oncogene. 1994;9:2961–2968. [PubMed] [Google Scholar]
- 20.Joslyn G, Carlson M, Thliveris A, Albertsen H, Gelbert L, Samowitz W, Groden J, Stevens J, Spirio L, Robertson M, Sargeant L, Krapcho K, Wolff E, Burt R, Hughes J P, Warrington J, McPherson J, Wasmuth J, Le Paslier D, Abderrahim H, Cohen D, Leppert M, White R. Identification of deletion mutations and three new genes at the familial polyposis locus. Cell. 1991;66:601–613. doi: 10.1016/0092-8674(81)90022-2. [DOI] [PubMed] [Google Scholar]
- 21.Karam S M. Lineage commitment and maturation of epithelial cells in the gut. Front Biosci. 1999;4:D286–D298. doi: 10.2741/karam. [DOI] [PubMed] [Google Scholar]
- 22.Kennelly P J, Krebs E G. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J Biol Chem. 1991;266:15555–15558. [PubMed] [Google Scholar]
- 23.Kinzler K, Nilbert M, Vogelstein B, Bryan T, Levy D, Smith K, Presinger A C, Hamilton S R, Hedge. Markham P A, et al. Identification of a gene located at chromosome 5q21 that is mutated in colorectal cancers. Science. 1991;251:1366–1370. doi: 10.1126/science.1848370. [DOI] [PubMed] [Google Scholar]
- 24.Lee S, Chen D Y, Humphrey J S, Gnarra J R, Linehan W M, Klausner R D. Nuclear/cytoplasmic localization of the von Hippel-Lindau tumor suppressor gene product is determined by cell density. Proc Natl Acad Sci USA. 1996;93:1770–1775. doi: 10.1073/pnas.93.5.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCartney B M, Peifer M. Teaching tumour suppressors new tricks. Nat Cell Biol. 2000;2:E58–E60. doi: 10.1038/35008685. [DOI] [PubMed] [Google Scholar]
- 26.Morin P J, Vogelstein B, Kinzler K W. Apoptosis and APC in colorectal tumorigenesis. Proc Natl Acad Sci USA. 1996;93:7950–7954. doi: 10.1073/pnas.93.15.7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neufeld K L, Nix D A, Bogerd H, Kang Y, Beckerle M C, Cullen B R, White R L. Adenomatous polyposis coli protein contains two nuclear export signals and shuttles between the nucleus and cytoplasm. Proc Natl Acad Sci USA. 2000;97:12085–12090. doi: 10.1073/pnas.220401797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neufeld K L, White R L. Nuclear and cytoplasmic localizations of the adenomatous polyposis coli protein. Proc Natl Acad Sci USA. 1997;94:3034–3039. doi: 10.1073/pnas.94.7.3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neufeld K L, Zhang F, Cullen B R, White R L. APC-mediated down-regulation of β-Catenin activity involves nuclear sequestration and nuclear export. EMBO Rep. 2000;6:519–523. doi: 10.1093/embo-reports/kvd117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P, Markham A, Krush A J, Petersen G, Hamilton S R, Nilbert M C, Levy D, Bryan T M, Preisinger A C, Smith K J, Su L-K, Kinzler K, Vogelstein B. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665–668. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 31.Ostermeyer A G, Runko E, Winkfield B, Ahn B, Moll U M. Cytoplasmically sequestered wild-type p53 protein in neuroblastoma is relocated to the nucleus by a C-terminal peptide. Proc Natl Acad Sci USA. 1996;93:15190–15194. doi: 10.1073/pnas.93.26.15190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouhibi N, Fulka J, Jr, Kanka J, Moor R M. A reversible block at the G1/S border during cell cycle progression of mouse embryos. Int J Dev Biol. 1994;38:731–736. [PubMed] [Google Scholar]
- 33.Plafker K, Macara I G. Facilitated nucleocytoplasmic shuttling of the Ran binding protein RanBP1. Mol Cell Biol. 2000;20:3510–3521. doi: 10.1128/mcb.20.10.3510-3521.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Potten C S, Loeffler M. Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development. 1990;110:1001–1020. doi: 10.1242/dev.110.4.1001. [DOI] [PubMed] [Google Scholar]
- 35.Powell S M, Zilz N, Beazer-Barclay Y, Bryan T M, Hamilton S R, Thibodeau S N, Vogelstein B, Kinzler K W. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 36.Rihs H P, Jans D A, Fan H, Peters R. The rate of nuclear cytoplasmic protein transport is determined by the casein kinase II site flanking the nuclear localization sequence of the SV40 T-antigen. EMBO J. 1991;10:633–639. doi: 10.1002/j.1460-2075.1991.tb07991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez J A, Henderson B R. Identification of a functional nuclear export sequence in BRCA1. J Biol Chem. 2000;275:38589–38596. doi: 10.1074/jbc.M003851200. [DOI] [PubMed] [Google Scholar]
- 38.Rosin-Arbfeld R, Townsley F, Bienz M. The APC tumour suppressor has a nuclear export function. Nature. 2000;406:1009–1012. doi: 10.1038/35023016. [DOI] [PubMed] [Google Scholar]
- 39.Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain S H, Masiarz F R, Munemitsu S, Polakis P. Association of the APC gene product with β-catenin. Science. 1993;262:1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- 40.Sakanaka C, Weiss J B, Williams L T. Bridging of beta-catenin and glycogen synthase kinase-3β by axin and inhibition of β-catenin-mediated transcription. Proc Natl Acad Sci USA. 1998;95:3020–3023. doi: 10.1073/pnas.95.6.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sherr C J. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 42.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spink K E, Polakis P, Weis W I. Structural basis of the Axin-adenomatous polyposis coli interaction. EMBO J. 2000;19:2270–2279. doi: 10.1093/emboj/19.10.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su L K, Vogelstein B, Kinzler K W. Association of the APC tumor suppressor protein with catenins. Science. 1993;262:1734–1737. doi: 10.1126/science.8259519. [DOI] [PubMed] [Google Scholar]
- 45.Tetsu O, McCormick F. β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 46.Urbani L, Sherwood S W, Schimke R T. Dissociation of nuclear and cytoplasmic cell cycle progression by drugs employed in cell synchronization. Exp Cell Res. 1995;219:159–168. doi: 10.1006/excr.1995.1216. [DOI] [PubMed] [Google Scholar]
- 47.Zhang F, White R, Neufeld K. Phosphorylation near nuclear localization signal regulates nuclear import of APC protein. Proc Natl Acad Sci USA. 2000;97:12577–12582. doi: 10.1073/pnas.230435597. [DOI] [PMC free article] [PubMed] [Google Scholar]