Origin of the Mutations in the parkin Gene in Europe: Exon Rearrangements Are Independent Recurrent Events, whereas Point Mutations May Result from Founder Effects (original) (raw)
Abstract
A wide variety of mutations in the parkin gene, including exon deletions and duplications, as well as point mutations, result in autosomal recessive early-onset parkinsonism. Interestingly, several of these anomalies were found repeatedly in unrelated patients and may therefore result from recurrent, de novo mutational events or from founder effects. In the present study, haplotype analysis, using 10 microsatellite markers covering a 4.7-cM region known to contain the parkin gene, was performed in 48 families, mostly from European countries, with early-onset autosomal recessive parkinsonism. The patients carried 14 distinct mutations in the parkin gene, and each mutation was detected in more than one family. Our results support the hypothesis that exon rearrangements occurred independently, whereas some point mutations, found in families from different geographic origins, may have been transmitted by a common founder.
Introduction
Parkinson’s disease (PD) is a frequent neurodegenerative disorder with a prevalence of ∼2% in persons >65 years old (Elbaz et al. 1999). The main clinical features are rigidity, bradykinesia, and tremor, associated with a good response to levodopa. The disorder is caused by a massive loss of dopaminergic neurons in the pars compacta of the substantia nigra and is characterized by the presence of Lewy bodies (cytoplasmic eosinophilic hyaline inclusions) (Fearnley and Lees 1991). Genetic risk factors are probably involved in the pathogenesis of Parkinson’s disease (de Silva et al. 2000), and several families with clearly established monogenic inheritance have been reported. The majority of such cases are caused by mutations in the parkin gene, which result in autosomal recessive early-onset parkinsonism (MIM 600116) (Hattori et al. 1998_a_, 1998_b_; Kitada et al. 1998; Leroy et al. 1998_a_; Lücking et al. 1998, 2000; Abbas et al. 1999). The number of patients with mutations in the α-synuclein (Polymeropoulos et al. 1997) or ubiquitin carboxy-terminal hydrolase (UCH)–L1 genes (Leroy et al. 1998_b_) is much smaller. The phenotype associated with parkin gene mutations is variable but is usually characterized by early-onset parkinsonism and slow disease progression (Ishikawa and Tsuji 1996; Lücking et al. 2000). Postmortem examinations reveal massive loss of dopaminergic neurons in the substantia nigra pars compacta and the absence of Lewy bodies, results suggesting that the pathologic process may differ from that of idiopathic Parkinson’s disease (Takahashi et al. 1994; Mori et al. 1998; van de Warrenburg, in press). A wide variety of mutations in the parkin gene have been detected, including exon deletions and duplications, as well as point mutation (Hattori et al. 1998_a_, 1998_b_ ; Kitada et al. 1998; Leroy et al. 1998_a_; Lücking et al. 1998, 2000; Abbas et al. 1999; Klein et al. 2000; Maruyama et al. 2000; Muñoz et al. 2000; Yamamura et al. 2000). The frequency of these mutations in Europe was estimated at 50% in families with early-onset parkinsonism that could have been autosomal recessive inheritance and at 18% in patients who had isolated parkinsonism with onset at age ⩽45 years (Lücking et al. 2000).
Interestingly, several mutations were found repeatedly in index patients (Hattori et al. 1998_a_; Lücking et al. 1998, 2000; Abbas et al. 1999). In our series of patients, the deletion of exon 3 (_n_=11), the Arg275Trp mutation (_n_=8), the c.202-203 delAG mutation (_n_=6), and the c.255delA mutation (_n_=6) were found repeatedly. They may therefore result from recurrent, de novo mutational events or from founder effects. Divergent alleles of markers closely linked to the parkin locus would suggest independent de novo mutations, whereas conservation of alleles would support the hypothesis of a founder effect.
In the present study, haplotype analysis was performed with 10 microsatellite markers covering a 4.7-cM region that contains the parkin gene, which is localized on chromosome 6q25.2-q27. The subjects were members of 48 families with early-onset autosomal recessive parkinsonism who carried 14 different mutations of the parkin gene found in more than one family. Our results support the hypothesis that exon rearrangements occurred independently, whereas there is evidence of founder effects in some families with point mutations.
Patients and Methods
Patients
Forty-eight families with early-onset parkinsonism caused by mutations in the parkin gene, including 69 patients and 49 unaffected relatives, were studied. All but four families (families TRUS and KUZ from Russia, families EGPD 25-95 and PW from Germany) have been described elsewhere (Lücking et al. 1998, 2000; Tassin et al. 1998; Abbas et al. 1999). The families were selected for parkin analysis according to the following criteria: (1) symptoms of parkinsonism (akinesia, rigidity, or tremor), (2) marked improvement resulting from levodopa treatment, (3) age at onset ⩽45 years for at least one affected sib, and (4) family history compatible with autosomal recessive inheritance (except family PW). In 13 of the 48 families, only one member was affected (referred to as isolated cases). These individuals were selected according to the same clinical criteria but had no family history of parkinsonism. They carried parkin gene mutations, on one or both alleles, that were detected at least twice in unrelated index patients (fig. 1B). The numbers and origins of the patients are summarized in table 1.
Figure 1.

Recurrent mutations in the parkin gene and localization of the intragenic microsatellites, showing genetic map of the 10 microsatellites studied (A), exon rearrangements (B, top), and point mutations (B, bottom) that were detected more than once. Exon rearrangements are represented as bars corresponding to their size and position, and they are divided into deletions (del) and duplications (dup). The positions of point mutations are indicated by arrows. The ATG of the initiator methionine codon begins at nucleotide 102 of the published cDNA (Kitada et al. 1998). The number of index patients with the same mutation is indicated in parentheses. The positions of the microsatellites in the parkin gene are indicated by dotted arrows. Note the difference, arising from recent results of chromosome 6 sequencing, between the genetic map and physical map.
Table 1.
Number and Origin of Index Patients with parkin Mutations Detected More than Once
| No. of Index Patients b | ||||
|---|---|---|---|---|
| Mutationa | Familial | Isolated | No. of Haplotypes | Country of Originc (No.) |
| Del2 | 4 | 0 | 3 | It (3), F (1) |
| Del3 | 9 | 2 | 8 | F (3), N(3), UK (1), It (2), Po (1), R (1) |
| Del3-4 | 3 | 1 | 2 | It (2), F (1), G (1) |
| Del4 | 4 | 0 | 4 | F (1), It (1), UK (1), G (1) |
| Del5 | 3 | 0 | 3 | F (1), It (1), V (1) |
| Del5-6 | 0 | 3 | 2 | L (1), Pa (1), UK (1) |
| Dup3 | 2 | 1 | 2 | It (1), Al (1), S (1) |
| Dup7 | 2 | 0 | 1 | Ar (1), N (1) |
| Lys211Asn | 2 | 0 | 2 | N (2) |
| c.321-322insGT | 2 | 0 | 1 | F (1), G (1) |
| Gly430Asp | 1 | 1 | 1 | UK (1), Ir (1) |
| c.255delA | 4 | 2 | 5 | F (4), S (1), Po (1) |
| Arg275Trp | 4 | 4 | 6 | F (2), It (2), Ir (1), UK (1), G (1), G/Ir (1) |
| c.202-203delAG | 3 | 3 | 4 | UK (2), F (1), N (1), It (1), R (1) |
Genotyping
Blood samples were taken, after written informed consent was obtained, from 69 patients and 49 unaffected relatives, and genomic DNA was extracted using standard procedures. Genotyping was performed by PCR, using the primers specified in the Genome Database (GDB), with the following microsatellite DNA markers: D6S1581 (1 cM), D6S959 (0.2 cM), D6S1579 (0.3 cM), D6S305 (0.1 cM), AFMa155td9 (0 cM), AFMb281wf1 (0.1 cM), D6S411 (0 cM), D6S1550 (1.9 cM), D6S1035 (1.1 cM), and D6S1599 (fig. 1A) (genetic distances according to the Whitehead Institute for Biomedical Research). The physical positions of markers D6S411, AFMa155td9, AFMb281wf1, D6S1550, and D6S1599 differ partly from the genetic map and were determined with the use of clones from the Sanger Center (PAC 292F10 and RP1-45F6). Marker D6S305 was positioned, on the basis of PCR results, in patients with various exon deletions (Leroy et al. 1998_b_; Lücking et al. 2000). Marker D6S1599 was amplified with the following primers: D6S1599 forward, 5′-GGG TGT GCT TGG ATT CCT TCA TG-3′, and D6S1599 reverse, 5′-TAG CAT GTG GAC TGC ATA TCA AC-3′. The primers were labeled by fluorescence, and PCR products were analyzed on an ABI 377 automated sequencer with the GENESCAN 3.1 and GENOTYPER 1.1.1 software programs (all from Applied Biosystems). To ensure accurate sizing of the alleles, a control DNA sample of individual 1347.02, from the Centre d’Etude du Polymorphisme Humain, was tested for each marker, and allele numbers were assigned in increasing order from the smallest to the largest PCR product.
Haplotypes and Linkage Disequilibrium
Haplotypes were constructed manually to include a minimum number of recombinations. For each mutation, the disease-associated haplotypes (DHs) were compared among families, to detect (1) common parkin haplotypes that would indicate that the families were related and (2) a common ancestral DH. The haplotypes constructed in families with known consanguinity were considered as a single haplotype for statistical analysis.
The difference in allele distribution between normal and carrier chromosomes was evaluated by χ2 and two-tailed Fisher's exact tests, with Yates correction when appropriate. The most frequent allele on disease-bearing chromosomes was defined as a single allele, and the others were pooled to form a second allele. The presence of linkage disequilibrium was tested by 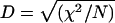 , where N is the total number of DHs and control chromosomes used. A P value <.01 was considered to be statistically significant. The proportion of carrier chromosomes bearing the original associated allele was calculated with the equation δ=(P _D_-P N)/(1-P N), where PD and PN are the frequencies of carrier and normal chromosomes, respectively. The control population comprised 140 chromosomes from normal, white subjects.
, where N is the total number of DHs and control chromosomes used. A P value <.01 was considered to be statistically significant. The proportion of carrier chromosomes bearing the original associated allele was calculated with the equation δ=(P _D_-P N)/(1-P N), where PD and PN are the frequencies of carrier and normal chromosomes, respectively. The control population comprised 140 chromosomes from normal, white subjects.
Results
The origins of the 69 patients and 49 relatives from 48 families with early-onset parkinsonism are shown in table 1. DHs were constructed in the 38 families in which unaffected relatives were available (tables 2–4).
Table 2.
Genotypes and Haplotypes of Index Patients with an Exon 2 Deletion in the parkin Gene[Note]
| IT31 (F) | IT22 (F) | F155 (F) | IT67 (F) | |
|---|---|---|---|---|
| Origin | Italy | Italy | France | Italy |
| Consanguinity | No | No | No | No |
| Mutations | del2/del2-4 | del2/del3 | del2/del3 | del2/ND |
| Marker: | ||||
| D6S1581 | 71 | 83 | 77 | [37] |
| D6S959 | 11 | 22 | 12 | 22 |
| D6S1579 | 25 | 72 | 55 | 22 |
| D6S1599 | del/del | del/1 | del/12 | [313] |
| D6S305 | 71 | 31 | 97 | [13] |
| D6S411 | 34 | 33 | 43 | 33 |
| AFMa155td9 | 13 | 11 | 22 | [12] |
| AFMb281wf1 | 33 | 32 | 33 | 33 |
| D6S1550 | 23 | 41 | 44 | [23] |
| D6S1035 | 26 | 54 | 43 | [45] |
Table 3.
Genotypes and Haplotypes of Index Patients with an Exon 3 Deletion in the parkin Gene[Note]
| F141 (F) | F155 (F) | F431 (F) | F192 (F) | F195 (F) | UK57 (F) | F711 (F) | IT22 (F) | KUS (F) | JMP10 (I) | UK10551 (I) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Origin | France | France | France | N | N | N | Portugal | Italy | Russia | Italy | UK |
| Consanguinity | Yes | No | No | No | No | No | Yes | No | No | No | No |
| Mutations | Del3/Del3 | Del3/Del2 | Del3/ND | Del3/Dup7 | Del3/Lys211Asn | Del3/Del4 | Del3/Del3 | Del3/Del2 | Del3/Del7 | Del3/Del2-3 | Del3/Gly430Asp |
| Marker: | |||||||||||
| D6S1581 | 34 | 77 | 37 | 18 | 83 | 77 | 77 | 38 | [78] | [17] | 33 |
| D6S959 | 12 | 21 | 22 | 22 | 35 | 22 | 11 | 22 | [12] | [25] | [23] |
| D6S1579 | 22 | 55 | 22 | 52 | 22 | 22 | 22 | 27 | [25] | [26] | [25] |
| D6S1599 | 22 | 12/del | 32 | 23 | 32 | 32 | 22 | 1/del | [412] | 13/del | 22 |
| D6S305 | 18 | 79 | 14 | 76 | 73 | 37 | 33 | 13 | 33 | 11 | [37] |
| D6S411 | 33 | 34 | 35 | 33 | 34 | 33 | 33 | 33 | 33 | [25] | 33 |
| AFMa155td9 | 11 | 22 | 22 | 22 | 22 | 12 | 22 | 11 | 11 | [12] | [12] |
| AFMb281wf1 | 22 | 33 | 33 | 33 | 23 | 23 | 33 | 23 | 33 | 33 | [23] |
| D6S1550 | 22 | 44 | 24 | 22 | 22 | 12 | 22 | 14 | [24] | [34] | 22 |
| D6S1035 | 33 | 34 | 35 | 64 | 44 | 52 | 44 | 45 | [35] | [35] | [24] |
Table 4.
Genotypes and Haplotypes of Index Patients with an Exon 4 Deletion in the parkin Gene[Note]
| F29 (F) | DE25 (F) | UK57 (F) | IT05 (F) | |
|---|---|---|---|---|
| Origin | France | Germany | United Kingdom | Italy |
| Consanguinity | No | No | No | No |
| Mutations | del4/ND | del4/ND | del4/del3 | del4/del4 |
| Marker: | ||||
| D6S1581 | 33 | 84 | 77 | 93 |
| D6S959 | 25 | 61 | 22 | 51 |
| D6S1579 | 23 | 23 | 22 | 25 |
| D6S1599 | 103 | 41 | 23 | 33 |
| D6S305 | 35 | 33 | 73 | 11 |
| D6S411 | 33 | 33 | 33 | 44 |
| AFMa155td9 | 21 | 22 | 21 | 32 |
| AFMb281wf1 | 33 | 33 | 32 | 13 |
| D6S1550 | 24 | 23 | 21 | 22 |
| D6S1035 | 25 | 33 | 25 | 62 |
Exon Rearrangements
Genotypes or haplotypes of patients with deletion of exon 2, 3, or 4 are shown in tables 2, 3, and 4, respectively. Patients with exon 2 deletions did not share common haplotypes, and alleles at markers D6S1579 and D6S305, which flank the deletion, also differed (table 2). Although allele 3 at marker AFMb281wf1 was present on 100% of the disease-causing chromosomes, the association was not statistically significant, because this allele was present in 80% of the control population. Interestingly, marker D6S1599, located in intron 2, was deleted with exon 2 in 3 of 4 families (table 2). This observation indicated the existence of at least two distinct breakpoints and supported the hypothesis of independent mutational events. Haplotypes of the two patients with deletions of exons 3 and 4, only one of which was associated with a deletion of marker D6S1599, also differed (data not shown).
Four of the five haplotypes that segregated with exon 4 deletions were identical at markers AFMa155td9, AFMb281wf1, and D6S1550 (table 4). However, the association was not statistically significant, because these were the most frequent alleles in the control population (50%, 82%, and 46%, respectively). Furthermore, the haplotypes for the markers closest to the deletion (D6S1599 and D6S305) differed, suggesting independent recurrent mutations (table 4). However patient IT05, who was homozygous for an exon 4 deletion and who had no known consanguinity, was homozygous for markers D6S1599, D6S305, and D6S411, which flank the deletion. Although the patient’s parents were not known to be related, a common founder for the exon 4 deletion on both chromosomes may be suspected.
Eleven index patients had exon 3 deletions, the most frequent mutation in the parkin gene, but no significant association was observed between allele markers and the DHs (table 3). Allele 2 at marker D6S1599, the marker closest to the mutation, was present on 4 of 10 disease-bearing chromosomes but was also the most frequent allele in the control population (43%). Furthermore, patients F155 and JMP10 carried alleles 12 and 13, respectively, (whose sizes were very different from those of alleles 2 and 3), which probably arose from independent events. Patients F141 and F711 shared alleles 1-2-2 at markers D6S959, D6S1579, and D6S1599, but this association was not statistically significant, because these alleles were very frequent in the control population (32%, 50%, and 43%, respectively). This is also the case for patients F431 and UK57, who shared alleles 2-2-3 for the same markers, and for patient F195, who had alleles 2-3 for markers D6S1579 and D6S1599. Alleles 2-2-3 were present in 57%, 50%, and 24% of the control population, respectively. Patients F155 and KUS shared alleles 2-5-12 at markers D6S959, D6S1579, and D6S1599; however, because the phase transmission for patient KUS was not known, we do not know whether these alleles were carried by the chromosome associated with the mutation. Because alleles 5 and 12 are rare in the control population (8% and 6%, respectively), these two patients may share a common haplotype, but no definite conclusion can be made.
No common haplotype, or even common marker alleles, were observed for the other exon deletions (exons 3 and 4, exons 5 and 6, exon 5) or duplications (exon 3 and exon 7) (data not shown).
Point Mutations
Genotypes or haplotypes of patients with the c.255delA, c.202-203delAG, and Arg275Trp mutations are shown in tables 5, 6, and 7, respectively. It is possible that F744 and S70 (alleles 3-2-2-7 at markers D6S1581, D6S959, D6S1579, and D6S1599) or S70 and UK17275 (alleles 2-7-3-3-1 at markers D6S1579, D6S1599, D6S305, D6S411, and AFMa155td9) share a common haplotype for the c.255delA mutation. No definite conclusion can be drawn, however, because the phase of transmission could not be determined for all markers or patients (table 5). However, genotype analysis revealed that, in most cases, the mutation segregated with allele 7 of marker D6S1599 (table 5), which is located 24 kb downstream of exon 2. This allele was not found in the 62 patients who did not carry the c.255delA mutation (not determined in one patient) (6/6 vs. 0/62; P<.0001).
Table 5.
Genotypes and Haplotypes of Index Patients with c.255delA Mutations in the parkin Gene[Note]
| F171 (F) | F744 (F) | F96 (F) | S70 (I) | UK17275 (F) | S74 (I) | |
|---|---|---|---|---|---|---|
| Origin | France | France | France | Spain | Portugal | France |
| Consanguinity | No | No | No | No | Yes | No |
| Mutations | c.255delA/Dup6 | c.255delA/Del5 | c.255delA/c.255delA | c.255delA/c.255delA | c.255delA/c.255delA | c.255delA/Del3-4 |
| Marker: | ||||||
| D6S1581 | 38 | 33 | 33 | [38] | [38] | [17] |
| D6S959 | 15 | 21 | 12 | 22 | [16] | 22 |
| D6S1579 | 36 | 25 | 77 | [23] | [23] | [25] |
| D6S1599 | 72 | 74 | 79 | [79] | 77 | [27] |
| D6S305 | 89 | 63 | 71 | [38] | [36] | 33 |
| D6S411 | 43 | 33 | 43 | 33 | 33 | [34] |
| AFMa155td9 | 21 | 22 | [12] | 11 | 11 | 22 |
| AFMb281wf1 | 33 | 23 | 33 | [23] | 33 | 22 |
| D6S1550 | 42 | 24 | 32 | [24] | [12] | [23] |
| D6S1035 | 54 | 52 | 32 | [34] | [34] | [34] |
Table 6.
Genotypes and Haplotypes of Index Patients with the c.202-203delAG Mutation in the parkin Gene[Note]
| IT20 (F) | UK86 (F) | NL24 (F) | TRUZ (F) | S749 (I) | UK4720 (I) | |
|---|---|---|---|---|---|---|
| Origin | Italy | United Kingdom | Netherlands | Russia | France | United Kingdom |
| Consanguinity | No | No | No | No | Yes | No |
| Mutations | c.202-203delAG/Lys161Asn | c.202-203delAG/ND | c.202-203delAG/Lys211Asn | c.202-203delAG/ND | c.202-203delAG/c.202-203delAG | c.202-203delAG/c.202-203delAG |
| Marker: | ||||||
| D6S1581 | 33 | 44 | 31 | 33 | 33 | 33 |
| D6S959 | [12] | 61 | 25 | [12] | [12] | [12] |
| D6S1579 | [25] | 34 | 22 | [68] | 44 | 33 |
| D6S1599 | [23] | 32 | 23 | [24] | 22 | 22 |
| D6S305 | [39] | 88 | 98 | [56] | 33 | [14] |
| D6S411 | 33 | 31 | 33 | 33 | [34] | 33 |
| AFMa155td9 | 22 | [12] | 21 | 22 | [12] | [12] |
| AFMb281wf1 | 33 | 33 | 32 | 33 | [23] | [23] |
| D6S1550 | 22 | 14 | 22 | 22 | [24] | 22 |
| D6S1035 | [36] | [35] | 34 | [23] | [45] | [25] |
Table 7.
Genotypes and Haplotypes of Index Patients with the Arg275Trp Mutation[Note]
| IT15 (F) | IT63 (F) | UK 2329 (I) | S92 (I) | S96 (I) | UK 4823 (I) | PW (F) | EGPD 25-95 (F) | |
|---|---|---|---|---|---|---|---|---|
| Origin | Italy | Italy | Ireland | France | France | United Kingdom | Germany | Germany/Ireland |
| Consanguinity | No | No | No | No | No | No | No | No |
| Mutation | Arg275Trp/ND | Arg275Trp/ND | Arg275Trp/Gly430 | Arg275Trp/del3-6 | Arg275Trp/ND | Arg275Trp/del5-6 | Arg275Trp/40pbdel3 | Arg275Trp/ND |
| Marker: | ||||||||
| D6S1581 | 48 | 73 | 38 | 33 | 33 | 33 | 33 | 73 |
| D6S959 | [12] | 11 | 25 | 21 | [12] | [12] | ND | 12 |
| D6S1579 | 25 | 27 | 25 | 22 | [25] | 33 | 22 | ND |
| D6S1599 | 22 | 24 | 1013 | 44 | [34] | 22 | ND | 32 |
| D6S305 | 48 | 43 | [43] | 48 | [45] | [41] | 43 | 49 |
| D6S411 | 33 | 33 | 33 | 33 | 33 | 33 | 21 | [46] |
| AFMa155td9 | [12] | [12] | 11 | [12] | 11 | [12] | 32 | 11 |
| AFMb281wf1 | [23] | [23] | 22 | [23] | 22 | [23] | 22 | [23] |
| D6S1550 | 22 | 24 | ND | 22 | 22 | ND | [34] | 33 |
| D6S1035 | 73 | 35 | 32 | 45 | [24] | [25] | 54 | 64 |
Regarding allele frequencies (independent of the transmission phase), all index patients, whether homozygous or heterozygous for the c.202-203delAG mutation in exon 2, carried allele 2 at marker D6S1599 (table 6), but about half the patients with other mutations also carried this allele (6/6 vs. 29/62; _P_>.01). Furthermore, in family UK86, allele 2 did not segregate with the c.202-203delAG mutation (table 6). Patients IT20, NL24, and TRUZ shared alleles 3-2-3-2 at markers D6S411, AFMa155td9, AFMb281wf1, and D6S1550, but this association was not significant because of the high frequency of these alleles in the control population (59%, 50%, 82%, and 46%, respectively). Although it cannot be proved, IT20 and NL24 may share a common haplotype over the entire region.
At marker D6S305, in intron 7, all patients with the Arg275Trp mutation in exon 7 carried allele 4 (table 7). This allele was observed in three other patients who did not carry the Arg275Trp mutation (8/8, vs. 3/61; P<.0001). Furthermore, a common haplotype was observed between patients IT15 and IT63, who shared alleles 2-2-4-3 at markers D6S1579, D6S1599, D6S305, and D6S411; however, these alleles were the most frequent alleles in the control population (50%, 43%, and 59%, for D6S1579, D6S1599, and D6S411, respectively), except for allele 4 at marker D6S305, which was observed in only 9% in the control population. A common haplotype was also observed in UK2329 and S96 who shared alleles 3-1-2 at markers D6S411, AFMa155td9, and AFMb281wf1, with allele frequencies of 59%, 38%, and 9%, respectively. Patients IT15, IT63, S92, and UK4823 may share this haplotype, but the phase transmission was unknown for these markers.
For the other point mutations (Lys211Asn, c.321-322 insGT, and Gly430Asp), no common haplotypes or allelic associations were observed (data not shown).
Linkage Disequilibrium
We observed a strong linkage disequilibrium (LD) of 0.75 between the c.255delA mutation and allele 7 of marker D6S1599 (table 8). Allele 7 was present on five of seven independent chromosomes associated with the mutation, in patients from France, Portugal, and Spain (table 5, excluding patient S74, in whom the allele cannot be attributed to the mutation) but was absent from the control population (P<.0001) (table 8). However, patient F96 (from France) and patient S70 (from Spain), both of whom were homozygous for the c.255delA mutation, carry alleles 7 and 9 at marker D6S1599 (table 5). This observation could be explained by the existence of two distinct founder mutations or by a recombination between the mutation and marker D6S1599. Alternatively, a mutation in marker D6S1599 could have changed allele 7 to 9.
Table 8.
Linkage Disequilibrium for Three-Point Mutations Found Two or More Times in the parkin Gene
| Mutation | Locus | Allele | PDa | PNb | χ2 | P | _P_correctedc | Dd | δd |
|---|---|---|---|---|---|---|---|---|---|
| c.255delA | D6S1599 | 7 | 5/7 | 0/124 | 73.65 | <.0001 | <.001 | .75 | .75 |
| c.202-203delAG | D6S1599 | 2 | 4/5 | 53/124 | 1.40 | >.01 | >.1 | ND | ND |
| Arg275Trp | D6S305 | 4 | 5/5 | 39/140 | 8.71 | <.003 | <.03 | .24 | 1 |
At locus D6S1599, allele 2 was present on 4 of 5 of the chromosomes associated with the c.202-203delAG mutation for whom the phase transmission was known. Because allele 2 of marker D6S1599 was also very frequent in the control population (43%), this association was not statistically significant (_P_>.01) (table 8).
At locus D6S305, (_LD_=0.24) was observed between allele 4 and the Arg275Trp mutation (table 8). Allele 4 of marker D6S305 was present in five of five patients from France, Germany, and Italy on chromosomes carrying the mutation for which the phase transmission was known (table 7). This association was statistically significant since allele 4 was present in only 28% of the control chromosomes (P<.003). LD for marker AFMb281wf1 could not be tested, because the phase transmission was known in only three subjects. However, the frequency of allele 2 was much higher in the eight patients with the Arg275Trp mutation compared with the control population (68% and 9%, respectively) (P<.0001). It is possible that allele 2 is carried by all the chromosomes carrying the Arg275Trp mutation.
Discussion
To discriminate between single founder effects and independent recurrent events, DHs in the parkin region were established for intragenic and tightly flanking markers in 48 families with repeatedly observed mutations in the parkin gene.
The disease-associated alleles and haplotypes in families with the same exonic rearrangement were discordant in most of the families, even for markers located close to the rearrangements. Furthermore, genotypes of marker D6S1599 revealed at least two distinct breakpoints for exon 2 deletions in four unrelated families. These results suggest that these recurrent mutations originated independently, although we cannot exclude a very ancient founder effect associated with recombinations or mutations involving tightly linked markers. The hypothesis of independent recurrent events was recently confirmed by molecular analysis of the breakpoints of the parkin gene in patients with some of the deletions analyzed here (Asakawa et al. 2000). These deletions were classified into 18 types. However, one type of exon 4 deletion was commonly found among six families. This may reflect the case of our patient IT05, who is homozygous, without known consanguinity, for an exon 4 deletion, as well as for three markers flanking the deletion. Thus, even if de novo mutations occur regularly, a given exon deletion that is transmitted to subsequent generations becomes a new founder for this deletion.
Unequal inter- or intrachromosomal crossovers that result from the misalignment of two homologous flanking sequences may account for the existence of deletions and for duplications of the same regions of the parkin gene, as has been reported elsewhere in several genes, such as the steroid sulfatase gene (Yen et al. 1990) or the α-globin locus (Nicholls et al. 1987). Unequal crossovers, which result in duplications and deletions of a 1.5-Mb region of chromosome 17p11.2, give rise, respectively, to Charcot-Marie-Tooth Type 1A (CMT1A) (Lupski et al. 1991) and hereditary neuropathy with liability to pressure palsies (HNPP) (Chance et al. 1993). Excisions of an intrachromatid loop, resulting exclusively in deletions, also occur in this region of 17p11.2 (Kiyosawa et al. 1995; Lopes et al. 1998; Lopes et al. 1999). If both these mechanisms also cause exon rearrangements in the parkin gene, this would explain why deletions appear to be more frequent than duplications (29 vs. 5 in our series) in the parkin gene.
Transposable elements (TEs), such as Alu elements, are also involved in recombination. Almost 0.5% of known genetic disorders result from TE insertions or TE-mediated recombination (Deragon and Capy 2000; Deininger and Batzer 1999). TEs are relatively abundant in the genome (∼1 Alu/6 kb). The density of Alu elements in the first 130 kb of intron 2 of the parkin gene, which is involved in the deletion of exons 2 and 3, reaches 1 Alu/2.5 kb. This may explain the frequency of deletions of exons 2, 3, and 3–4, duplications of exon 3, and triplication of exon 2.
Unlike the exon rearrangements, point mutations in the patients studied may be accounted for by a limited number of founders. Strong linkage disequilibrium was observed between the c.255delA mutation in exon 2 and allele 7 of marker D6S1599 in intron 2 (table 5) and between the Arg275Trp mutation in exon 7 and allele 4 of marker D6S305 in intron 7 (table 7). These associations may reflect the transmission of a single ancestral mutation or may be caused by recurrent mutations on a predisposing haplotype, as shown in spinocerebellar ataxia 7 (Stevanin et al. 1999). Interestingly, alleles at markers D6S1579 and D6S305, which cover a 0.3-cM interval that includes D6S1599, were not conserved in patients with the c.255delA mutation in exon 2. These observations indicate that multiple recombinations and/or mutations have modified the alleles that segregate with the closely flanking markers, suggesting that this mutation can be attributed to very ancient founder effects. The geographic diversity of the patients carrying the same allele also supports this hypothesis: patients with the c.255delA mutation come from France, Spain, and Portugal (tables 5–7).
In contrast, the Arg275Trp mutation may be more recent, because a common ancestral haplotype between markers D6S1579 and D6S1550 is suspected. Patients IT15 and IT63 shared a haplotype between markers D6S1579 and D6S1550 (not proved for markers AFMa155td9 and AFMb281wf1, because the phase was not determined). Furthermore, patients UK2329 and S96 (who were from Ireland and France, respectively) and probably patients S92 and UK4823 (who were from France and the United Kingdom) also shared part of this haplotype between marker D6S411 and marker AFMb281wf1. Although these associations were not statistically significant for the majority of the markers, associations between the Arg275Trp mutation and the markers D6S305 and AFMb281wf1 were significant (P<.001). The ancestral founder haplotype could thus have been 2-2-4-3-1-2-2 for markers D6S1579 to D6S1550 before a recombination separated haplotype 2-2 (marker D6S1579 and D6S1599) from haplotype 4-3-1-2-2 (markers D6S305 to D6S1550). Because the suspected ancestral haplotype was observed in two Italian patients, the mutation may have arisen in Italy.
Allele 2 of marker D6S1599 was present in the six index patients with the c.202-203delAG mutation in exon 2 and on four of the five chromosomes associated with the mutation. The association was not statistically significant, because allele 2 is also very frequent in the control population. Because of the absence of other known markers in the introns flanking this mutation, we cannot distinguish between a single founder effect on a chromosome carrying allele 2 or recurrent mutations on chromosomes that carry this allele by chance.
It is also difficult to draw conclusions concerning the three other point mutations analyzed in this study (Lys211Asn, c.321-322insGT and Gly430Asp). No allelic associations were observed. It is difficult, however, either to demonstrate or to exclude a founder effect because of (1) the small number of index patients, (2) the lack of polymorphic markers in flanking introns (except for mutation c.321-322insGT in exon 3), and (3) the absence of relatives who, if available, would enable us to determine the phase of transmission.
The release of sequence data for the parkin region of chromosome 6, which is expected in the near future, should allow us to clarify some of the patients in this study. The sequencing of a contig of BACs that contain the parkin gene is in progress, and some of the data are already available. It will therefore be possible to define new microsatellite markers in each intron of the parkin gene and to detect ancient founder effects. In addition, single-nucleotide polymorphism (SNP) databanks, which help in the reconstruction of haplotypes, are also being created. Only one SNP in the parkin gene, localized in intron 8, has been identified to date. The known SNPs in the coding region of the parkin gene (Abbas et al. 1999) were not informative for the present study.
In conclusion, a wide variety of mutations in the parkin gene have been found repeatedly in patients from many European populations. Interestingly, the majority of exon rearrangements seem to result from distinct mutational events, whereas at least two point mutations may have arisen from a small number of founders. Therefore, the mechanisms underlying these two groups of mutations appear to be different. According to this hypothesis, the frequency of exon rearrangements would be expected, in the absence of selection, to increase with the passage of time because of new mutational events, whereas the frequency of point mutations would be expected to remain stable, because new mutations would be rare.
Acknowledgments
Supported by Assistance Publique-Hôpitaux de Paris, the Association France-Parkinson, a grant from the European Community Biomed 2 (BMHCT960664), the Prinses Beatrix Fund, and Aventis-Pharma. V.B. and G.M. were supported by the Italian Ministry for University, Scientific and Technological Research. G.D.M. was supported by grants from the Italian Ministry of Health and the Italian Ministry for University, Scientific, and Technological Research (Italy). J.R.V. and N.W.W. were supported by the Brain Research Trust, the United Kingdom Parkinson’s Disease Society, and the Doris Hillier Award (British Medical Association). C.B.L. and T.G. were supported by the Deutsche Forschungsgemeinschaft. We are indebted to P. Bejjani, F. Dubas, J. P. Ferroir, I. A. Ivanova-Smolenskaya, J. W. Langston, D. Maraganore, R. Marconi, S. Medjbeur, D. Nicholl, N. Quinn, A. Schrag, and N. Vanacore for directing patients to the study; to the families for their participation; to C. Penet and Y. Pothin for expert technical assistance; and to N. Abbas and M. Ruberg for helpful discussions.
Appendix
Members of the French Parkinson’s Disease Genetics Study Group are as follows: Y. Agid, A.-M. Bonnet, M. Borg, A. Brice, E. Broussolle, P. Damier, A. Destée, A. Dürr, F. Durif, J. Feingold, G. Fénelon, F. Gasparini, M. Martinez, C. Penet, P. Pollak, O. Rascol, F. Tison, C. Tranchant, M. Vérin, F. Viallet, M. Vidailhet, and J.-M. Warter.
Members of the European Consortium on Genetic Susceptibility in Parkinson’s Disease are as follows: N. W. Wood and J. R. Vaughan (United Kingdom); A. Brice, A. Dürr, M. Martinez, and Y. Agid (France); T. Gasser and B. Müller-Myhsok (Germany); M. Breteler, S. Harhangi, and B. Oostra (The Netherlands); V. Bonifati, M. deMari, G. De Michele, E. Fabrizio, A. Filla, and G. Meco (Italy).
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Centre d’Etude du Polymorphisme Humain, http://www.cephb.fr (for the control DNA sample)
- DNA Databank of Japan, http://www.ddbj.nig.ac.jp (for the cDNA sequence of the parkin gene [accession number AB009973])
- Genome Database, http://www.gdb.org (for primer sequences)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for early-onset parkinsonism [MIM 600116])
- Sanger Center, http://www.sanger.ac.uk (for the clone 292F10 and RP1-45F6 sequences [accession numbers 2760544 and 5924005, respectively])
- Whitehead Institute for Biomedical Research, http://www.genome.wi.mit.edu (for integrated map of chromosome 6)
References
- Abbas N, Lücking CB, Ricard S, Durr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R, Broussolle E, Brefel-Courbon C, Harhangi BS, Oostra BA, Fabrizio E, Bohme GA, Pradier L, Wood NW, Filla A, Meco G, Denefle P, Agid Y, Brice A (1999) A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Hum Mol Genet 8:567–574 [DOI] [PubMed] [Google Scholar]
- Asakawa S, Hattori N, Shintani A, Sasaki T, Kawasaki K, Shimizu A, Kitada T, Matsumine H, Minoshima S, Shimizu Y, Mizuno Y, Shimizu N (2000) Molecular analysis of the deletion breakpoints of parkin gene. Am J Hum Genet Suppl 67:398 [Google Scholar]
- Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsunami N, Smith B, Swanson PD, Odelberg SJ, Disteche CM, Bird TD (1993) DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 72:143–151 [DOI] [PubMed] [Google Scholar]
- de Silva HR, Khan NL, Wood NW (2000) The genetics of Parkinson's disease. Curr Opin Genet Dev 10:292–298 [DOI] [PubMed] [Google Scholar]
- Deininger PL, Batzer MA (1999) Alu repeats and human disease. Mol Genet Metab 67:183–193 [DOI] [PubMed] [Google Scholar]
- Deragon JM, Capy P (2000) Impact of transposable elements on the human genome. Ann Med 32:264–273 [DOI] [PubMed] [Google Scholar]
- Elbaz A, Grigoletto F, Baldereschi M, Breteler MM, Manubens-Bertran JM, Lopez-Pousa S, Dartigues JF, Alperovitch A, Tzourio C, Rocca WA (1999) Familial aggregation of Parkinson's disease: a population-based case-control study in Europe. Europarkinson Study Group. Neurology 52:1876–1882 [DOI] [PubMed] [Google Scholar]
- Fearnley JM, Lees AJ (1991) Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain 114:2283–2301 [DOI] [PubMed] [Google Scholar]
- Hattori N, Kitada T, Matsumine H, Asakawa S, Yamamura Y, Yoshino H, Kobayashi T, Yokochi M, Wang M, Yoritaka A, Kondo T, Kuzuhara S, Nakamura S, Shimizu N, Mizuno Y (1998_a_) Molecular genetic analysis of a novel parkin gene in Japanese families with autosomal recessive juvenile parkinsonism: evidence for variable homozygous deletions in the parkin gene in affected individuals. Ann Neurol 44:935–941 [DOI] [PubMed] [Google Scholar]
- Hattori N, Matsumine H, Asakawa S, Kitada T, Yoshino H, Elibol B, Brookes AJ, Yamamura Y, Kobayashi T, Wang M, Yoritaka A, Minoshima S, Shimizu N, Mizuno Y (1998_b_) Point mutations (Thr240Arg and Gln311Stop [correction of Thr240Arg and Ala311Stop]) in the parkin gene. Biochem Biophys Res Commun 249:754–758 [DOI] [PubMed] [Google Scholar]
- Ishikawa A, Tsuji S (1996) Clinical analysis of 17 patients in 12 Japanese families with autosomal-recessive type juvenile parkinsonism. Neurology 47:160–166 [DOI] [PubMed]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608 [DOI] [PubMed] [Google Scholar]
- Kiyosawa H, Lensch MW, Chance PF (1995) Analysis of the CMT1A-REP repeat: mapping crossover breakpoints in CMT1A and HNPP. Hum Mol Genet 4:2327–2334 [DOI] [PubMed] [Google Scholar]
- Klein C, Pramstaller PP, Kis B, Page CC, Kann M, Leung J, Woodward H, Castellan CC, Scherer M, Vieregge P, Breakefield XO, Kramer PL, Ozelius LJ (2000) Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: expanding the phenotype. Ann Neurol 48:65–71 [PubMed] [Google Scholar]
- Leroy E, Anastasopoulos D, Konitsiotis S, Lavedan C, Polymeropoulos MH (1998_a_) Deletions in the parkin gene and genetic heterogeneity in a Greek family with early onset Parkinson's disease. Hum Genet 103:424–427 [DOI] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH (1998_b_) The ubiquitin pathway in Parkinson's disease. Nature 395:451–452 [DOI] [PubMed] [Google Scholar]
- Lopes J, Ravise N, Vandenberghe A, Palau F, Ionasescu V, Mayer M, Levy N, Wood N, Tachi N, Bouche P, Latour P, Ruberg M, Brice A, LeGuern E (1998) Fine mapping of de novo CMT1A and HNPP rearrangements within CMT1A-REPs evidences two distinct sex-dependent mechanisms and candidate sequences involved in recombination. Hum Mol Genet 7:141–148 [DOI] [PubMed] [Google Scholar]
- Lopes J, Tardieu S, Silander K, Blair I, Vandenberghe A, Palau F, Ruberg M, Brice A, LeGuern E (1999) Homologous DNA exchanges in humans can be explained by the yeast double-strand break repair model: a study of 17p11.2 rearrangements associated with CMT1A and HNPP. Hum Mol Genet 8:2285–2292 [DOI] [PubMed] [Google Scholar]
- Lücking CB, Abbas N, Durr A, Bonifati V, Bonnet AM, de Broucker T, De Michele G, Wood NW, Agid Y, Brice A (1998) Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. The European Consortium on Genetic Susceptibility in Parkinson's Disease and the French Parkinson's Disease Genetics Study Group. Lancet 352:1355–1356 [DOI] [PubMed] [Google Scholar]
- Lücking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A (2000) Association between early-onset Parkinson's disease and mutations in the parkin gene. French Parkinson's Disease Genetics Study Group. N Engl J Med 342:1560–1567 [DOI] [PubMed] [Google Scholar]
- Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA (1991) DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66:219–232 [DOI] [PubMed] [Google Scholar]
- Maruyama M, Ikeuchi T, Saito M, Ishikawa A, Yuasa T, Tanaka H, Hayashi S, Wakabayashi K, Takahashi H, Tsuji S (2000) Novel mutations, pseudo-dominant inheritance, and possible familial effects in patients with autosomal recessive juvenile parkinsonism. Ann Neurol 48:245–250 [PubMed] [Google Scholar]
- Mori H, Kondo T, Yokochi M, Matsumine H, Nakagawa-Hattori Y, Miyake T, Suda K, Mizuno Y (1998) Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 51:890–892 [DOI] [PubMed] [Google Scholar]
- Muñoz E, Pastor P, Marti MJ, Oliva R, Tolosa E (2000) A new mutation in the parkin gene in a patient with atypical autosomal recessive juvenile parkinsonism. Neurosci Lett 289:66–68 [DOI] [PubMed] [Google Scholar]
- Nicholls RD, Fischel-Ghodsian N, Higgs DR (1987) Recombination at the human alpha-globin gene cluster: sequence features and topological constraints. Cell 49:369–378 [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047 [DOI] [PubMed] [Google Scholar]
- Stevanin G, David G, Durr A, Giunti P, Benomar A, Abada-Bendib M, Lee MS, Agid Y, Brice A (1999) Multiple origins of the spinocerebellar ataxia 7 (SCA7) mutation revealed by linkage disequilibrium studies with closely flanking markers, including an intragenic polymorphism (G3145TG/A3145TG). Eur J Hum Genet 7:889–896 [DOI] [PubMed] [Google Scholar]
- Takahashi H, Ohama E, Suzuki S, Horikawa Y, Ishikawa A, Morita T, Tsuji S, Ikuta F (1994) Familial juvenile parkinsonism: clinical and pathologic study in a family. Neurology 44:437–441 [DOI] [PubMed] [Google Scholar]
- Tassin J, Durr A, de Broucker T, Abbas N, Bonifati V, De Michele G, Bonnet AM, Broussolle E, Pollak P, Vidailhet M, De Mari M, Marconi R, Medjbeur S, Filla A, Meco G, Agid Y, Brice A (1998) Chromosome 6–linked autosomal recessive early-onset Parkinsonism: linkage in European and Algerian families, extension of the clinical spectrum, and evidence of a small homozygous deletion in one family. The French Parkinson's Disease Genetics Study Group, and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Am J Hum Genet 63:88–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Warrenburg BPC, Lammens M, Lücking CB, Denefle P, Wesseling P, Booij J, Praamstra P, Quinn N, Brice A, Horstink MWIM. Parkinsonism associated with parkin gene mutations: clinical and pathological abnormalities in a Dutch family. Neurology (in press) [DOI] [PubMed] [Google Scholar]
- Yamamura Y, Hattori N, Matsumine H, Kuzuhara S, Mizuno Y (2000) Autosomal recessive early-onset parkinsonism with diurnal fluctuation: clinicopathologic characteristics and molecular genetic identification. Brain Dev (Suppl) 22:87–91 [DOI] [PubMed]
- Yen PH, Li XM, Tsai SP, Johnson C, Mohandas T, Shapiro LJ (1990) Frequent deletions of the human X chromosome distal short arm result from recombination between low copy repetitive elements. Cell 61:603–610 [DOI] [PubMed] [Google Scholar]