PTEN tumor suppressor associates with NHERF proteins to attenuate PDGF receptor signaling (original) (raw)
Abstract
PTEN, a tumor suppressor frequently inactivated in many human cancers, directly antagonizes the activity of phosphatidylinositol-3-OH kinase (PI3K) by dephosphorylating phosphoinositides. We show here that PTEN interacts directly with the NHERF1 and NHERF2 (Na+/H+ exchanger regulatory factor) homologous adaptor proteins through the PDZ motif of PTEN and the PDZ1 domain of NHERF1 or both PDZ domains of NHERF2. NHERFs were shown to interact directly with platelet-derived growth factor receptor (PDGFR), and we demonstrate the assembly of a ternary complex between PTEN, NHERFs and PDGFR. The activation of the PI3K pathway after PDGFR stimulation was prolonged in NHERF1(−/−) mouse embryonic fibroblasts as compared to wild-type cells, consistent with defective PTEN recruitment to PDGFR in the absence of NHERF1. Depletion of NHERF2 by small interfering RNA similarly increased PI3K signaling. Phenotypically, the loss of NHERF1 enhanced the PDGF-induced cytoskeletal rearrangements and chemotactic migration of the cells. These data indicate that, in normal cells, NHERF proteins recruit PTEN to PDGFR to restrict the activation of the PI3K.
Keywords: cancer, EBP50, NHERF, PDGFR, PTEN
Introduction
PTEN is a tumor suppressor gene frequently inactivated in human cancers (Li et al, 1997; Steck et al, 1997). In addition, germline mutations in the PTEN gene are the cause of Cowden and Bannayan–Riley–Ruvalcaba syndromes (Waite and Eng, 2002). PTEN is formed of an amino-terminal (NT) phosphatase domain, a phospholipid-binding C2 domain and a carboxy-terminal (CT) tail region that contains Ser-Thr phosphorylation sites and a PDZ (PSD-95/Disc-large/ZO-1)-binding motif (Lee et al, 1999; Vazquez et al, 2000). PTEN antagonizes the activity of phosphatidylinositol-3-OH kinase (PI3K) by dephosphorylating phosphatidylinositol 3,4,5-trisphosphate (PIP3) to phosphatidylinositol 4,5-bisphosphate (Maehama and Dixon, 1998). PIP3 recruits and activates pleckstrin-homology-containing molecules such as the Ser-Thr kinase Akt/PKB, triggering the activation of multiple downstream effectors. These control cell survival, growth, proliferation and other cellular processes. The PI3K pathway is activated by a plethora of extracellular stimuli. Platelet-derived growth factor (PDGF) is a potent activator of the PI3K in many cell types. PDGF family comprises four members, A, B, C and D, that are secreted as dimers, with roles in both normal growth and pathologies associated with tissue overgrowth such as atherosclerosis, rheumatoid arthritis and cancer (Heldin and Westermark, 1999). PDGFs bind and signal through two cell-surface receptor tyrosine kinases, PDGFRα and PDGFRβ. PDGF-dependent receptor dimerization is prerequisite for receptor autophosphorylation and subsequent signal propagation. Through tyrosine residues located in the intracellular domain, the receptor dimers recruit and activate several Src homology 2-domain-containing proteins, such as the p85 subunit of PI3K, phospholipase C-γ, SHP2 phosphatase and Src family kinases. PDGFRs also contain a CT PDZ-binding motif that was described to bind the adaptor molecule NHERF1/EBP50 (Na+/H+ exchanger isoform 3 (NHE3) regulatory factor 1/ezrin–radixin–moesin (ERM)-binding phosphoprotein 50) (Maudsley et al, 2000). NHERF1 was independently identified as a partner for ERM proteins (Reczek et al, 1997) and for NHE3 (Weinman et al, 1998). NHERF2, also identified as an NHE3-interacting protein (Yun et al, 1997), shares 52% identity and conserved domain architecture with NHERF1. Both NHERFs contain two NT PDZ domains and a CT ERM-binding region and were shown to interact with PDGFR via their PDZ1 domain (Maudsley et al, 2000; Jung Kang et al, 2004). However, the significance of this association in the activation of the extracellular signal-regulated kinase (Erk) and PI3K by PDGF is controversial (Maudsley et al, 2000; Demoulin et al, 2003; Jung Kang et al, 2004).
If the function of PTEN as phosphoinositide phosphatase is well documented, the regulation of its activity is still poorly understood. Our previous work indicated that the recruitment of PTEN to the plasma membrane is crucial for its tumor suppressor function (Georgescu et al, 2000; Sumitomo et al, 2004). Several PTEN-interacting proteins were proposed to recruit PTEN at the membrane (Wu et al, 2000a, 2000b; Sumitomo et al, 2004; Kotelevets et al, 2005). Of these, the scaffolding membrane-associated guanylate kinase with inverted orientation (MAGI) proteins 1b, 2 and 3 (Wu et al, 2000a, 2000b; Kotelevets et al, 2005) were reported to interact with PTEN via PTEN's PDZ motif. However, their role in regulating PTEN tumor suppressor function is not yet clearly defined. In this study, we found that PTEN interacts with the NHERF adaptor proteins. We show the formation of a ternary complex between PTEN, NHERF proteins and PDGFR. By using NHERF1(+/+) and (−/−) mouse embryonic fibroblasts (MEFs) in combination with small interfering RNA (siRNA) depletion of NHERF2, we showed specific enhancement of the PI3K pathway activation after PDGFR stimulation in cells lacking NHERF proteins. These data are consistent with our model in which PTEN is recruited by NHERF proteins to restrict the PI3K signaling downstream of PDGFR.
Results
NHERF proteins directly associate with PTEN
To identify proteins that associate and potentially regulate PTEN, we performed yeast two-hybrid screen with PTEN as bait. After nutritional selection and β-galactosidase reporter screening, a positive cDNA was further characterized. This cDNA encoded a splice variant of NHERF1, containing the PDZ1 domain followed by 109 residues encoded by a sequence from the 3′UTR. Because we subsequently identified other splice variants of NHERF1 (Morales et al, 2004), we named this variant I5 (isoform 5) (Figure 1A). The presence of a PDZ domain in I5 suggested that the interaction with PTEN takes place between the CT PDZ motif of PTEN and the PDZ domain of I5. PTEN wild type and PTEN-401 mutant lacking the PDZ motif (Figure 1A) were purified as glutathione _S_-transferase (GST)-fusion proteins and incubated with extracts from 293T cells overexpressing I5 or the ZO-1 PDZ-containing protein, as negative control (Figure 1B). PTEN associated with I5 and the association was severely reduced for PTEN-401. We next purified each PDZ domain of NHERF1 (Figure 1A) and tested their binding affinity for PTEN. Only PDZ1 was able to precipitate PTEN (Figure 1C, upper panel). Taken together, these data indicated that the interaction between NHERF1 and PTEN takes place between NHERF1-PDZ1 domain and the PDZ motif of PTEN.
Figure 1.
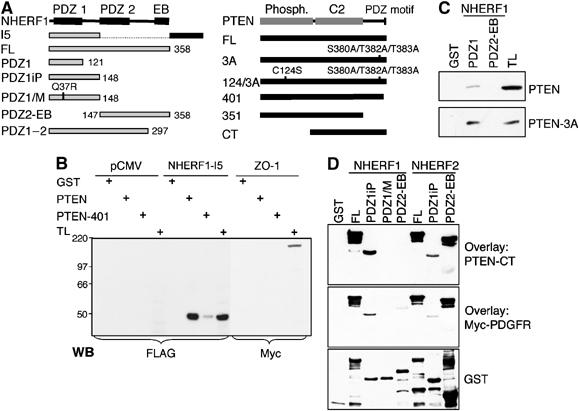
In vitro direct binding between PTEN and NHERF proteins. (A) Diagram of NHERF1 (358 residues) and PTEN (403 residues) constructs. I5, isoform 5; FL, full length; CT, carboxy terminus. (B) PTEN with intact PDZ motif binds to NHERF1-I5. GST pull-down assay with GST, GST-PTEN and GST-PTEN-401 of proteins (0.3 mg) from lysates of 293T cells transfected with vector (pCMV), FLAG-tagged NHERF1-I5 or Myc-tagged ZO-1 protein. TL, control total lysate (30 μg). The filter was re-probed with anti-GST antibody to confirm that equal amounts of GST-fusion proteins were used (not shown). (C) NHERF1-PDZ1 domain binds preferentially to PTEN-3A phosphorylation-deficient mutant. Proteins (0.3 mg) from lysates of 293T cells transfected with FLAG-tagged PTEN wild type or PTEN-3A mutant were incubated with GST-NHERF1 fusion proteins (5 μg) as indicated. The filter was probed with the anti-FLAG M2 antibody. TL, control total lysate (30 μg). (D) Direct binding of PTEN and PDGFR to NHERFs. A 0.5 μg portion of GST-NHERF1 or NHERF2 fusion proteins was separated on gel, transferred to filter and overlaid with 10 μg/ml of either GST-PTEN-CT or GST-Myc-PDGFR-CT. The filters were subsequently probed with PTEN and Myc antibodies, respectively, and then stripped and re-probed with GST antibody.
An increased affinity of unphosphorylated PTEN for the PDZ-containing protein MAGI-2 has been reported previously (Tolkacheva et al, 2001; Vazquez et al, 2001). We analyzed the affinity of unphosphorylated PTEN for NHERF1-PDZ1 by GST-PDZ1 precipitation of the phosphorylation-defective mutant PTEN-3A relatively to wild-type PTEN (Figure 1C). PTEN-3A had a significantly higher affinity than wild-type PTEN for NHERF1-PDZ1, indicating that the PDZ-based interaction between PTEN and NHERF1 is decreased by the phosphorylation of the S380/T382/T383 site situated in the proximity of the PDZ-binding motif.
To investigate if there is direct association between PTEN and NHERF1, we performed overlay assays using filter-immobilized GST-NHERF1 fusion proteins (Figure 1D). Similarly, we analyzed the interaction between PTEN and NHERF2. Overlaid GST-PTEN-CT (Figure 1A) bound directly to NHERF1-PDZ1 domain and to both PDZ domains of NHERF2 (Figure 1D). To ensure that the domain structure is preserved in filter-immobilized proteins, we analyzed the binding between PTEN and the Q37R NHERF1-PDZ1/M point mutant (Figure 1A). The residue Q37 is conserved in NHERF proteins from various species and is situated in the PDZ1-βC strand, which does not make a direct contact with the peptide ligand (Doyle et al, 1996). The mutant protein did not bind PTEN (Figure 1D), indicating that the binding is specific and dependent on the correct folding of the PDZ1 domain. A known binding partner of NHERFs is PDGFRβ. We confirmed that PDGFRβ binds to NHERF1-PDZ1 domain (Maudsley et al, 2000) but we found that it associates with NHERF2 through the NHERF2-PDZ2 domain (Figure 1D).
PTEN associates with NHERF2 in vivo
To show in vivo association between NHERF proteins and PTEN, we expressed GST-tagged NHERF2 and FLAG-tagged PTEN-124/3A in 293T cells (Figure 2A). The phosphatase-inactivating C124S mutation was previously noticed to increase the stability of overexpressed PTEN proteins (Das et al, 2003). By introducing it in PTEN-3A, we increased the expression level of PTEN-3A that otherwise presents very low expression levels (see Figure 1C). Western blot analysis with anti-FLAG antibody of proteins precipitated with glutathione beads showed that PTEN co-immunoprecipitated with both human and mouse NHERF2. The reciprocal co-immunoprecipitation experiment confirmed the complex formation between NHERF2 and PTEN in vivo (Figure 2A). The associations between NHERF1 and its partners have been reportedly difficult to prove in vivo (Maudsley et al, 2000; Lau and Hall, 2001; Li et al, 2004). We have also failed to reproducibly show in vivo the association between NHERF1 and PTEN (not shown).
Figure 2.
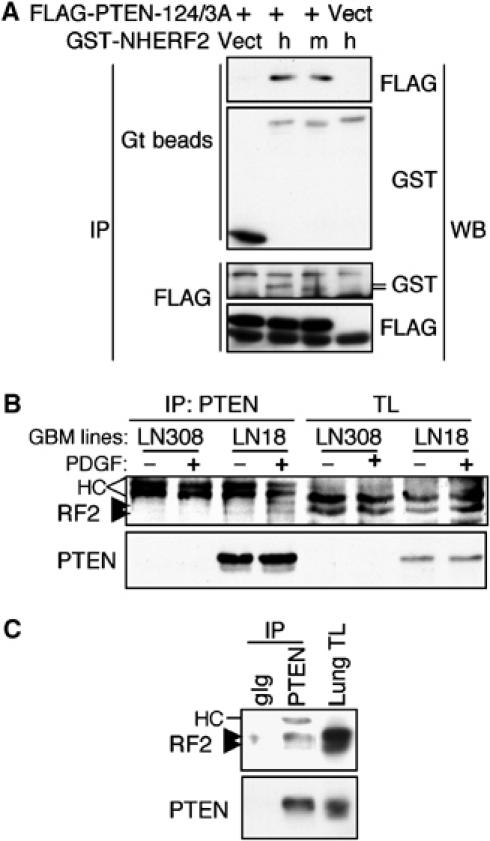
In vivo binding between PTEN and NHERF2. (A) FLAG-tagged PTEN-124/3A or pCMV vector was coexpressed in 293T cells with GST-tagged human (h) and mouse (m) NHERF2 or pEBG vector control (that expresses 29 kDa GST protein), as shown. Upper panels show co-immunoprecipitation of FLAG-PTEN with GST-NHERF2 pulled with glutathione (Gt) beads directly from the cell lysates. Lower panels show the reciprocal co-immunoprecipitation where the human and mouse NHERF2 proteins detected by anti-GST antibodies are indicated by lines. (B) Co-immunoprecipitation of endogenous NHERF2 (arrowheads) with PTEN only in the LN18 glioblastoma (GBM) cells that express endogenous PTEN and not in the LN308 PTEN-deficient cells. Both cell lines express PDGFR and were treated (+) or not (−) for 20 min with 50 ng/ml PDGF-BB. (C) Co-immunoprecipitation of endogenous NHERF2 with PTEN in normal mouse lung tissue. TL, total lysate; HC, heavy chain; gIg, matched goat Ig control.
To show the association between endogenous PTEN and NHERF2, PTEN was immunoprecipitated from LN308 PTEN-deficient and LN18 PTEN-expressing glioblastoma cell lines (Ishii et al, 1999). Endogenous NHERF2 co-immunoprecipitated with PTEN only in LN18 PTEN-expressing cells (Figure 2B). The association between endogenous NHERF2 and PTEN was also detected in normal lung tissue where NHERF2 is highly expressed (Figure 2C). We observed that NHERF2 appears as a doublet by Western blotting. On searching the expressed sequence tag (EST) database, we found that NHERF2 may have two splice variants in both humans and mice in addition to the full-length protein of 338 residues. The first isoform has 317 residues and lacks exon 6 that encodes residues 264–286 (e.g. GenBank accession numbers CK020840 and BC070458). This deletion is in the linker separating the PDZ2 domain from the EB region and is not predicted to affect the interactions of NHERF2 with its ligands. The second isoform of 227 residues lacks exon 1 that encodes the PDZ1 domain and has an ectopic transcription initiation in intron 1 that adds an initial stretch of 28 residues (e.g. AK002557, BC065778 and CR613953).
Complex formation between PTEN, NHERF proteins and PDGFR
GST-NHERF proteins were tested for their ability to precipitate endogenous PTEN and PDGFRβ from mouse brain total lysate (Figure 3A). The affinity of NHERF2 for PTEN was much higher than that of NHERF1, confirming the in vivo binding data (Figure 2). The affinity for endogenous mouse PDGFRβ was equivalent among human NHERF proteins, but significantly increased for mouse NHERF2, suggesting intra-species preference. In agreement with the in vitro data from Figure 1D, NHERF2-PDZ1 was required for PTEN but not for PDGFRβ association, whereas NHERF1-PDZ1 domain was required for PDGFRβ association (Figure 3A). Although this experiment suggested that NHERF proteins complex with both PTEN and PDGFRβ, it did not exclude the possibility of separate complexes with these partners. To ascertain that NHERF proteins bridge a complex between PTEN and PDGFRβ, we performed a sandwich-overlay assay in which immobilized PTEN was overlaid with NHERF proteins, in a first step, and with PDGFRβ, in a second step (Figure 3B). Recombinant PTEN-CT, PTEN-351 (see Figure 1A), control GST and full-length NHERF1 were immobilized on filters and overlaid either with recombinant human NHERF1, NHERF2 or with buffer (Figure 3B, first, second and third panels, respectively). The filters were washed and incubated with Myc-tagged PDGFRβ-CT, which was further revealed by subsequent incubation with Myc antibody. PDGFRβ-CT did not bind PTEN-CT directly (Figure 3B, buffer panel) but bound PTEN when either NHERF1 or NHERF2 was initially incubated with PTEN (Figure 3B, upper two panels). Similar results were obtained when the whole intracellular domain of PDGFRβ (550 residues) was used instead of the PDGFRβ-CT (not shown). No association was observed with PTEN-351 that lacks the PDZ motif (Figure 3B). Because NHERF1 interacts only through its PDZ1 domain with the PDZ motifs of PTEN and PDGFR (Figure 1D), we analyzed the molecular requirements for the ternary complex formation. Filter-immobilized PTEN-CT was overlaid with PDZ1, PDZ2 or both PDZ domains of NHERF proteins, followed by Myc-PDGFRβ-CT incubation (Figure 3C). Both NHERF PDZ domains were able to bridge PDGFRβ to PTEN. Except for a very weak association in the case of NHERF1-PDZ1 domain, the isolated PDZ domains were not sufficient for assembling the complex. This observation, coupled with published results showing oligomerization of NHERF proteins through PDZ–PDZ interactions (Fouassier et al, 2000; Lau and Hall, 2001; Shenolikar et al, 2001), suggested that the complex between PTEN and PDGFR is mediated by dimers or oligomers of NHERF proteins that require both PDZ domains in order to form.
Figure 3.
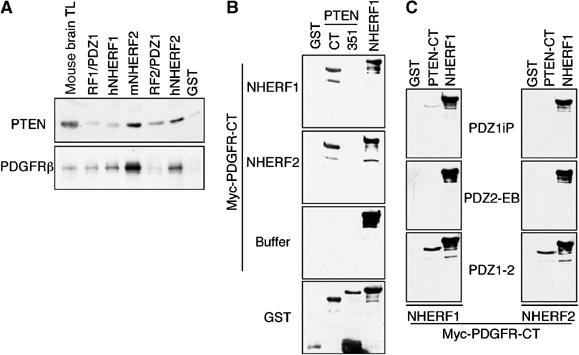
Complex formation between PTEN, NHERF proteins and PDGFR. (A) Pull-down of endogenous PTEN and PDGFRβ from mouse brain total lysate (TL) with GST-NHERF1 and NHERF2 mouse (m) and human (h) fusion proteins. (B) Bridging experiments in which the GST-PTEN-CT and GST-PTEN-351 filter-immobilized proteins shown on top of the gels were overlaid in a first step with GST-NHERF1, NHERF2 or buffer, and in a second step with GST-Myc-PDGFR-CT. The binding of PDGFR-CT was revealed by further incubation with Myc antibody, followed by the corresponding secondary antibody. (C) Same experiment as in (B), except that the full-length NHERF1 and NHERF2 used for the first-step overlay were replaced by their corresponding domains schematically shown in Figure 1A. Note bridging of a complex between PTEN and PDGFR by full-length or both PDZ domains of NHERF proteins.
Because the bridging overlay assay revealed that both NHERF proteins could link PDGFRβ to PTEN (Figure 3B), whereas the GST pull-down assay showed a higher affinity of NHERF2 for endogenous PTEN (Figure 3A), we analyzed the in vivo formation of the complexes containing NHERF proteins, PTEN and PDGFRβ by gel filtration (Figure 4A). In addition to the two elution peaks previously detected (Vazquez et al, 2001; Sumitomo et al, 2004), corresponding to the monomeric form and to the form engaged in high molecular weight (MW) complexes, PTEN from mouse brain lysates also eluted in intermediate MW peaks (Figure 4A and C). PDGFRβ and NHERF2, but not NHERF1 or MAGI-1, co-eluted with PTEN in the high MW peak (around 669 kDa), suggesting that NHERF2 is more likely to bridge PTEN and PDGFRβ in mouse brain. Recently, antibodies to PTEN's CT were shown to reduce PTEN's ability to interact with PDZ domains (Andres-Pons et al, 2005). Immunodepletion of PTEN from total brain lysates with an antibody against PTEN's CT (Figure 4B, depletion 1) resulted in the depletion of the monomeric peaks and one intermediate MW (232 kDa) peak but not of the high MW peak (Figure 4C). This suggested that PTEN's CT is involved in intermolecular interactions in the high MW complexes, most likely through the PDZ motif. Subsequent depletion of PTEN with an NT antibody from the lysate clarified first with the CT antibody (Figure 4B, depletion 2) showed partial depletion of PTEN from the high MW complex (Figure 4C), suggesting participation of PTEN's NT in protein interactions. This procedure also enabled us to detect the complex between PTEN, NHERF2 and PDGFRβ in mouse brain (Figure 4D). As expected, this complex was immunoprecipitated only by PTEN NT antibody after enrichment of complex-engaged PTEN by prior depletion of the monomeric PTEN by the CT antibody.
Figure 4.
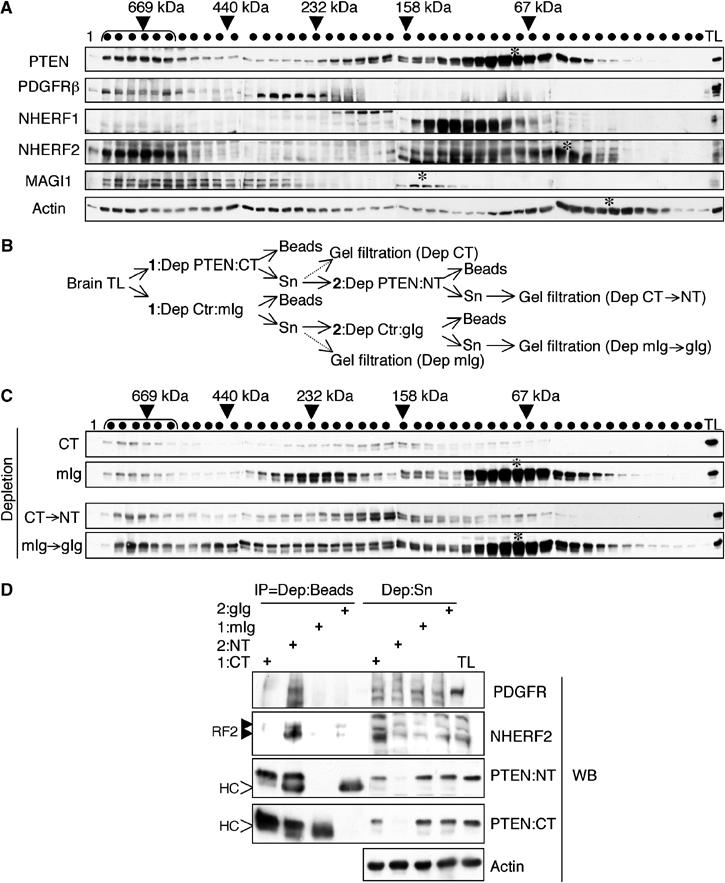
Association of PTEN, PDGFRβ and NHERF2 in a high MW complex in normal brain. (A) Gel filtration of mouse brain total lysate (TL) from NHERF1(+/+) mice. Shown on top are the MWs of eluted fractions (shown with dots) that were calculated using standard proteins (Amersham). Western blotting was performed using antibodies indicated on the left. Bracket and stars indicate high MW peak and monomeric protein peaks, respectively. (B) Flow chart showing the immunodepletion (Dep) of PTEN from brain TL. In a first step (1), half of the TL was depleted with an antibody against PTEN's CT and half with a matched mouse Ig control (Ctr:mIg). The resulting supernatants (Sn) were depleted in a second step (2) with an antibody against PTEN's NT or with matched goat Ig control (Ctr:gIg). (C) Gel filtration PTEN profile after CT antibody depletion or sequential CT followed by NT (CT → NT) antibody depletion. Same amount of brain total lysate (TL) was loaded on each membrane and matched PTEN antibody and control Ig membranes were processed concomitantly. Note partial depletion of PTEN from the high MW peak (bracket) only by NT antibody. (D) Western analysis with antibodies indicated on the right of proteins immunoprecipitated on beads (IP) or remaining in the supernatants (Sn) after brain TL immunodepletion with the antibodies indicated on top. Note co-immunoprecipitation of NHERF2 and PDGFR with PTEN NT antibody.
Increased PI3K pathway activation by PDGF in NHERF-depleted cells
PDGFRβ activation results in the potent stimulation of the PI3K pathway by direct recruitment of the PI3K regulatory subunit to the receptor. Because PTEN antagonizes PI3K, we investigated whether PTEN is functionally recruited to PDGFRβ by the NHERF proteins to confine the activation of the PI3K. NHERF1(+/+) and (−/−) MEFs were stimulated with PDGF-BB and analyzed comparatively for the activation of Akt and p70 S6 kinase downstream effectors of PI3K. PDGF stimulation resulted in higher levels of activation of Akt and p70 S6 kinase in NHERF1(−/−) compared to (+/+) MEFs (Figure 5A). There was a decrease in total Akt upon PDGF stimulation, which might represent a compensatory mechanism for the positive feedback loop of Akt activation by Rictor–mTOR complex (Sarbassov et al, 2005). The Akt basal activity was higher in non-stimulated NHERF1(−/−) MEFs than in (+/+) MEFs and this ratio significantly increased at later time points (Figure 5A, lower graph), indicating persistent activation of PI3K in NHERF1(−/−) MEFs. As expected, expression of exogenous NHERF1 in NHERF1(−/−) MEFs by retroviral infection decreased the activation of Akt by approximately two-fold (Figure 5B). Conversely, the expression of dominant-negative NHERF1-PDZ1 in NHERF1(+/+) MEFs increased the activation of Akt by approximately two-fold (Figure 5C). Equal amounts of p85, p55 and p50 PI3K regulatory subunits were recruited to stimulated PDGF (Supplementary Figure 1A), whereas PTEN expression levels were slightly elevated in NHERF1(−/−) MEFs (Figure 5A). By cellular fractionation, PTEN's cytoplasmic levels were higher in NHERF1(−/−) cells compared to (+/+) cells (Figure 5D). However, the phosphatase activity of immunoprecipitated cytoplasmic PTEN (Georgescu et al, 1999) was undetectable compared to negative control (not shown), suggesting that the cytoplasmic pool of PTEN was inactive. PTEN membrane levels were barely detectable in both cell types, suggestive of rapid turnover of PTEN at the membrane (Das et al, 2003). It is likely that PTEN's enhanced cytoplasmic level in the absence of NHERF1 might be due to a reduced recruitment to the membrane compartment where PTEN is rapidly degraded.
Figure 5.
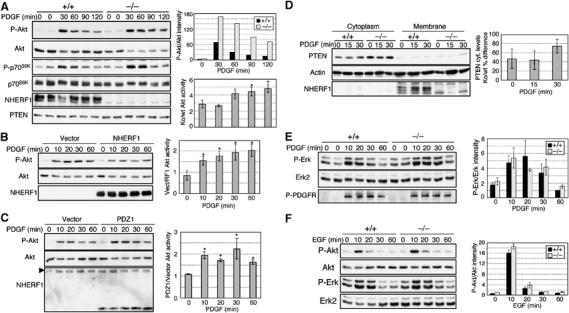
Activation of the PI3K pathway in NHERF1(−/−) MEFs. (A) Time-course experiment showing activation of Akt and p70 S6 kinase by stimulation with PDGF-BB (0.5 ng/ml) of NHERF1(+/+) and (−/−) MEFs. The activation shown with phospho-specific antibodies P-Akt and P-p70S6K was normalized to the corresponding Akt and p70S6K total protein levels, respectively. The upper graph shows the normalized activation of Akt in NHERF1(+/+) and (−/−) MEFs from one representative experiment. The lower graph shows the fold difference of normalized Akt activation in NHERF1(−/−) (Ko) versus NHERF1(+/+) (wt) cells from three similar experiments. Asterisks mark statistically significant differences (P<0.01) between the fold-difference activation at various time points and the basal fold-difference activation at time 0. (B) Reintroduction of NHERF1 in NHERF1(−/−) MEFs reduces Akt phosphorylation compared to vector control. The graph quantifies the fold difference of normalized Akt activation in control versus NHERF1-reconstituted cells. Mean values±standard deviations are computed from four experiments in three different NHERF1(−/−) cell lines (P<0.0001). (C) Expression of NHERF1-PDZ1 in NHERF1(+/+) MEFs increases Akt activation. The arrowhead indicates endogenous NHERF1. The fold difference of normalized Akt activation in NHERF1-PDZ1-expressing versus control cells is shown as means±standard deviations from three experiments in two different NHERF1(+/+) cell lines (P<0.01). (D) Membrane–cytoplasm fractionation showing increased cytoplasmic PTEN in NHERF1(−/−) MEFs. The graph shows the percent increase of PTEN cytoplasmic (cyt.) levels normalized to actin in three different matched pairs of NHERF1(−/−) (ko) and (+/+) (wt) cells. (E) PDGF stimulation of the same cells as in (A) does not reveal significant differences in the activation of Erk, as detected by phospho-specific P-Erk antibody. (F) Stimulation of NHERF1(+/+) and (−/−) MEFs with 20 ng/ml EGF shows similar activation of the PI3K and Erk pathways in both cell types. The normalized activation of Erk in (E) and Akt in (F) is shown as mean±standard deviation of values from three similar experiments.
PDGF stimulation resulted in a milder activation of Erk than of Akt in MEFs (Figure 5E). No clear difference between the levels of Erk activation in NHERF1(−/−) and (+/+) cells was apparent (Figure 5E), suggesting that NHERF1 is important for the modulation of the PI3K rather than of the Erk pathway following PDGF stimulation. Control phosphorylated PDGFRβ on Tyr751 that represents the docking site for the p85 PI3K on PDGFRβ showed equal levels in NHERF1(+/+) and (−/−) MEFs, indicating that receptor autophosphorylation is similar in both cell types (Figure 5E). Although the epidermal growth factor receptor (EGFR) was reported not to bind to NHERF1 (Maudsley et al, 2000), a recent report showed association of the two proteins through internal PDZ motifs in the EGFR-CT (Lazar et al, 2004). We also tested their association by overlay assay and found a significantly lower affinity of NHERF1 for EGFR than for PDGFR (Supplementary Figure 1B). By stimulating NHERF1(+/+) and (−/−) MEFs with EGF, no difference was detected for the activation of the PI3K and Erk pathways between the two cell types (Figure 5F), indicating that NHERF1 specifically restricts the activation of the PI3K pathway downstream of PDGFR in these cells.
To determine the contribution of NHERF2 to the activation of the PI3K by PDGF, we depleted NHERF2 in MEFs by using siRNA. A mixture of two siRNA sequences successfully decreased by more than 80% the expression of NHERF2 in comparison with control, scrambled siRNA (Figure 6A). Similar to our findings in NHERF1(+/+) and (−/−) MEFs (Figure 5A), the depletion of NHERF2 in NHERF1(−/−) MEFs resulted in a prolonged PI3K activation after PDGF stimulation (Figure 6B). These data suggested that both NHERF1 and NHERF2 restrict the PI3K activation downstream of PDGFR.
Figure 6.
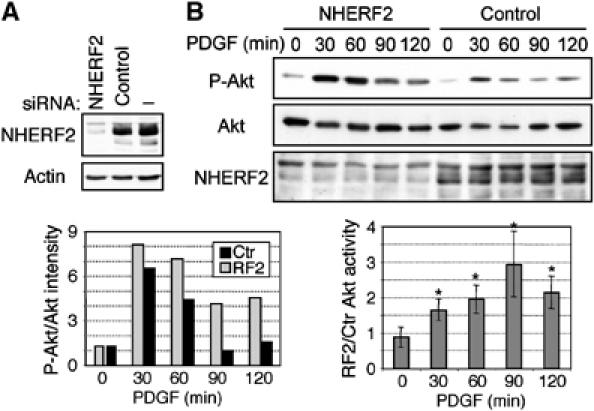
siRNA depletion of NHERF2 increases the activation of the PI3K pathway. (A) Efficient depletion of NHERF2 in NHERF1(−/−) MEFs by specific siRNA treatment as compared to control scrambled siRNA. (B) Time-course experiment showing the activation of Akt in NHERF1(−/−) MEFs pretreated for 96 h with NHERF2 siRNA or control siRNA. The graphs show the normalized activation of Akt in one representative experiment (left), and the fold difference of normalized Akt activation in NHERF2-depleted and control (Ctr) siRNA-treated cells from similar experiments in three different NHERF1(−/−) MEF lines (right). Asterisks mark statistically significant differences (P<0.005).
NHERF1 loss enhances PDGF-induced actin reorganization and chemotaxis
PDGF is known to induce rapidly actin reorganization in stimulated cells with loss of stress fibers and peripheral and circular dorsal ruffle formation (Heldin and Westermark, 1999). After PDGF-BB stimulation, we observed the formation of both peripheral and circular dorsal ruffles in both NHERF1(+/+) and (−/−) MEFs. However, the circular dorsal ruffles were more numerous and with more prominent actin ring reorganization in NHERF1(−/−) cells than in the (+/+) controls (Figure 7A). PDGF also induces chemotactic migration of cells (Heldin and Westermark, 1999). A Transwell assay where NHERF1(+/+) and (−/−) MEFs migrated across fibronectin-coated filters was used to assess PDGF- and EGF-driven chemotaxis (Figure 7B). PDGF but not EGF determined enhanced chemotaxis in NHERF1(−/−) cells as compared to (+/+) cells. These results indicate that NHERF1 suppresses PDGF-induced cytoskeletal rearrangements and chemotactic migration of cells.
Figure 7.
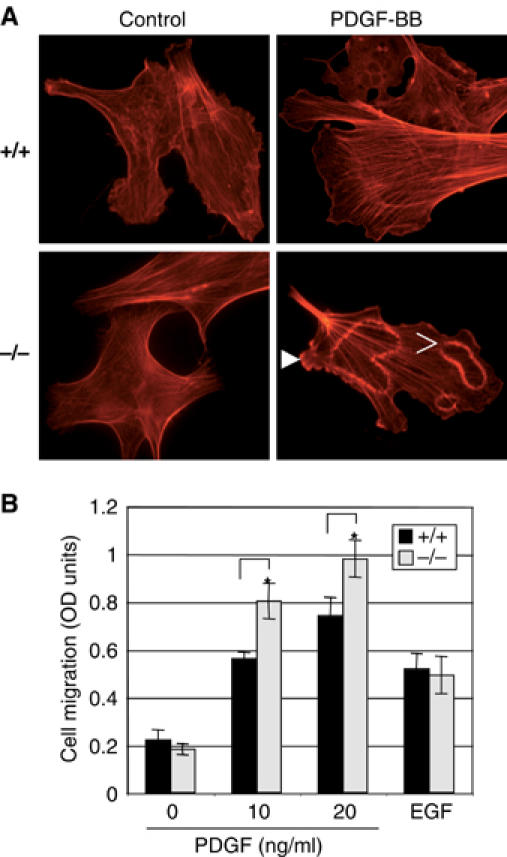
Enhanced ruffle formation and migration of NHERF1(−/−) MEFs stimulated with PDGF. (A) Immunofluorescence analysis of NHERF1(+/+) and (−/−) MEFs showing actin reorganization after treatment with control serum-free medium or 5 ng/ml PDGF-BB for 10 min. Images (× 400) were taken with an Axioskop 40 Zeiss microscope equipped with AxioVision Rel.4.2 software. Solid and open arrowheads indicate peripheral ruffles and circular dorsal ruffles, respectively. (B) Chemotactic migration of NHERF1(+/+) and (−/−) MEFs toward PDGF-BB (concentrations indicated) or EGF (20 ng/ml) gradients. On ordinate, the number of migratory cells, processed as described in Materials and methods, is represented as the photometric intensity of the hematoxylin–eosin staining. The means±standard deviations were calculated from triplicates within the same experiment and asterisks mark statistically significant differences (P<0.05). This experiment was repeated three times with similar results.
Discussion
In this study, we addressed the question of how PTEN, a cytoplasmic protein, contacts its enzymatic substrate in the membrane. Candidate proteins that can recruit PTEN to the membrane have been identified previously, and these include the MAGI proteins and NEP (Wu et al, 2000a, 2000b; Sumitomo et al, 2004; Kotelevets et al, 2005). We described here the interaction of PTEN with the two homologous NHERF proteins, which are localized at the plasma membrane in physiological conditions (Ingraffea et al, 2002). Similarly to MAGI proteins, NHERF proteins interacted via their PDZ domains with the PDZ motif of PTEN. This interaction was increased by dephosphorylation of S380/T382/T383 in the tail of PTEN that most likely exposes PTEN's PDZ motif. Whereas the MAGI proteins that contain six PDZ domains have not been shown to interact with PDGFR, both NHERF proteins were shown to associate with PDGFR through a PDZ-domain-mediated interaction with the CT PDZ motif of PGDFR (Maudsley et al, 2000; Jung Kang et al, 2004). We showed by in vitro overlay assays that both NHERF proteins assembled a complex between PTEN and the intracellular domain of PDGFR. Unlike a recent report that indicated direct association between PTEN and PDGFR (Mahimainathan and Ghosh Choudhury, 2004), we found that PTEN and PDGFR did not interact directly and required the presence of NHERF proteins for complex formation (Figure 8). We also showed the assembly of a ternary complex between PTEN, NHERF2 and PDGFR in mouse brain lysate that appeared to fractionate in a high MW (>669 kDa) elution peak by gel filtration.
Figure 8.
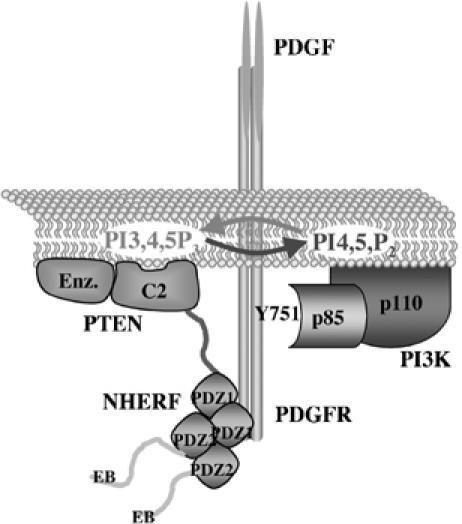
Model of the ternary complex between PDGFR, NHERF1 and PTEN. NHERF1 oligomerizes through PDZ domain interactions and is presented dimerized in this scheme. NHERF1 interacts through its PDZ1 domain with the PDZ motifs of both PDGFR and PTEN. PTEN dephosphorylates the second messenger PIP3 that is produced by PI3K. PI3K p85 regulatory subunit in complex with the p110 catalytic subunit is directly recruited to the phospho-tyrosine 751 of PDGFR upon PDGF stimulation. Other proteins recruited to activated PDGFR are not shown.
The data describing the effects of the association between NHERF proteins and PDGFR on PDGFR signaling are controversial in the literature (Maudsley et al, 2000; Demoulin et al, 2003; Jung Kang et al, 2004). These differences could be due to the fact that most of these studies were performed with overexpressed proteins that can induce experimental artifacts. To avoid these drawbacks, we used normal MEFs in which the endogenous NHERF1 was depleted by gene inactivation (Morales et al, 2004) and NHERF2 by direct siRNA treatment. In contrast to overexpressed NHERF1 that is detected in both membrane and cytoplasmic compartments, we detected NHERF1 only in the membrane fractions from MEFs, suggesting a physiological subcellular localization for NHERF1 in these cells. We found that in NHERF1(−/−) as compared to (+/+) MEFs, the activation of the PI3K pathway and not of Erk was increased after stimulation with PDGF. These results indicated that NHERF1 is necessary for the attenuation of the PI3K pathway, most likely in complex with PDGFR and PTEN (Figure 8). In support of this model, it was reported that stimulation of a PDZ motif mutant PDGFR that was unable to bind NHERF1 induced higher Akt but not Erk activation in comparison to the wild-type PDGFR (Demoulin et al, 2003).
The depletion of NHERF2 by siRNA treatment in NHERF1(−/−) MEFs raised also the level of PI3K activation after PDGF stimulation, suggesting functional redundancy between NHERFs. Although NHERF1 and NHERF2 are coexpressed in MEFs, their expression is generally not overlapping in the same cells from adult tissues (Ingraffea et al, 2002), arguing against redundancy in the living organism. NHERF1 and NHERF2 exhibited also molecular differences in complex formation with PTEN and PDGFR. The PDZ1 domain of NHERF1 was the exclusive interactor of both PTEN and PDGFR. For NHERF2, the PDZ2 domain interacted with both PDGFR and PTEN, and PDZ1 interacted weakly with PTEN. Nevertheless, both PDZ1 and PDZ2 domains of NHERF proteins were necessary for bridging PTEN to PDGFR. NHERF proteins were shown to self-associate through PDZ–PDZ interactions (Fouassier et al, 2000; Lau and Hall, 2001; Shenolikar et al, 2001) and the requirement for both PDZ domains most likely reflects the necessity for oligomerized NHERF proteins in the ternary complex formation (Figure 8). The ability to engage in protein complexes in vivo appeared different for NHERF2 and NHERF1. The analysis of complexes from brain extracts separated by gel filtration showed participation of NHERF2 in a high MW complex where PTEN and PDGFR also co-eluted. NHERF1 was not detected in this complex. We are currently investigating whether NHERF1 may be artificially dissociated from complexes by detergent solubilization of membranes or whether it participates to a very limited extent in protein complexes, as reported in the case of its association with cystic fibrosis transmembrane conductance regulator (Li et al, 2004).
We found that only a small fraction of PTEN from brain extracts eluted in the high MW complex. Vazquez et al (2000) reported that this fraction is not phosphorylated on Ser-380 and presumably can engage in interactions with PDZ-domain-containing proteins. By depleting PTEN from brain lysate with antibodies directed to PTEN's CT or NT, we found that only the NT antibody partially depleted PTEN from this high MW complex. This clearly indicates that PTEN associates through its CT to proteins from the high MW complexes. Although PTEN's PDZ motif is not required for PTEN-mediated growth suppression of PTEN-deficient cancer cell lines (Georgescu et al, 1999), it was elegantly shown to be necessary for the inhibition by PTEN of PDGF-induced membrane ruffling (Leslie et al, 2000). We have found that NHERF1 is also required for inhibiting PDGF-induced membrane ruffling and chemotactic migration. As PTEN interacts through its PDZ motif with NHERF proteins, the absence of either the PDZ motif of PTEN or of NHERF proteins would prevent the formation of the PTEN–NHERF–PDGFR complex with inhibitory effect on the activation of the PI3K. Several studies indicated that pharmacological inhibition of PI3K blocked the cytoskeletal rearrangements and chemotaxis induced by PDGF (Wymann and Arcaro, 1994; Hooshmand-Rad et al, 1997). From our study, these processes appear to be physiologically regulated in normal fibroblasts by PTEN in complex with NHERF proteins and PDGFR.
This study clarifies that NHERF proteins suppress PI3K signaling and PDGF-induced cell motility and points to a helper role for NHERF proteins in PTEN tumor suppressor activity. Unlike PTEN-deficient mice, NHERF1-deficient animals do not present a significantly higher rate of tumor development compared to controls (data not shown), but crosses between PTEN and NHERF1-deficient mice will indicate whether NHERF1 has a helper role for oncogenesis in vivo. In humans, point mutations and loss of heterozygosity of NHERF1 were recently described in advanced breast cancers (Dai et al, 2004). A careful examination of the NHERFs' status in tumors harboring wild-type PTEN but increased PI3K activation will further elucidate the importance of the PTEN–NHERF connection for cancer progression.
Materials and methods
Yeast two-hybrid library screening
Human PTEN cDNA (403 amino acids) was inserted in-frame with LexA in the pLex9 vector (gift of Stanley Hollenberg). The L40 yeast strain carrying the HIS3 and LacZ genes under the control of four and eight tandem LexA operators, respectively (Hollenberg et al, 1995), was transformed first with pLex-PTEN (pLEX9 carries the TRP1 gene as a selectable marker) and subsequently with a human Jurkatt T-cell cDNA library (Clontech) cloned in pACT2 vector in fusion with GAL4 activation domain (pACT2 carries the LEU2 gene as a selectable marker). Double transformed yeast cells were grown on plates in selection medium lacking tryptophan, leucine and histidine. Resistant colonies were isolated and processed for the identification of positive clones by β-galactosidase filter assay. Target cDNAs were subjected to sequence analysis.
Plasmid construction
Wild-type PTEN and CT deletion mutants PTEN-401 and PTEN-351 inserted in pGEX-6P-1, FLAG-tagged PTEN and the phosphorylation-defective PTEN-3A mutant, in which S380, T382 and T383 were replaced with alanines, were described elsewhere (Georgescu et al, 1999; Sumitomo et al, 2004). The CT half of PTEN (residues 186–403), PTEN-CT, was inserted in-frame with the GST moiety in the pGEX-6P-1 vector. The double phosphorylation-defective and phosphatase-inactive mutant PTEN-124/3A in pCMV-2-FLAG was generated by introducing the C124S mutation in PTEN-3A. Human NHERF1 (encoding 358 residues) was obtained from the clone CS0DE007YB08 (Invitrogen). NHERF1 deletion mutants in pGEX-6P-1 vector were obtained by PCR or enzyme restriction: PDZ1 (121 residues), PDZ1iP (inter-PDZ, 148 residues), PDZ2-EB (from residue 147-end) and PDZ1-2 (297 residues) (see Figure 1A). The mutation Q37R was introduced by PCR in PDZ1iP, yielding the mutant PDZ1/M. NHERF1 and PDZ1 were also cloned in pCXb and pCXp retroviral vectors (Georgescu et al, 1999), respectively. Human (h) and mouse (m) NHERF2 cDNAs (encoding 337 residues) were obtained from ESTs 2958186 (ATCC) and GI001B12 (Open Biosystems), respectively, and were inserted in-frame with an NT GST moiety in the vectors pGEX-6P-1 and pEBG (gift of Bruce Meyer) for bacterial and mammalian expression, respectively. NHERF2 deletion mutants in pGEX-6P-1 vector were obtained by PCR or enzyme restriction: hPDZ1iP (145 residues), mPDZ2-EB (from residue 47-end) and hPDZ1-2 (315 residues). pGEX-Myc vector was generated by inserting a Myc tag in-frame after the GST moiety of pGEX-6P-1 vector. A fragment encoding the CT 50 residues of hPDGFRβ was obtained by reverse transcription and PCR from placenta mRNA (Stratagene) and inserted in pGEX-Myc vector. Similarly, the whole intracellular domain of PDGFRβ (residues 557–1106), obtained by PCR from hPDGFRβ (gift of Andrius Kazlauskas), and the CT 196 residues of EGFR obtained by PCR from hEGFR (gift of Oliver Bögler) were inserted in pGEX-Myc vector. The Myc-tagged ZO-1 construct containing the two NT PDZ domains of ZO-1 was a gift of Alan Fanning. More information on the cloning of these constructs is available on request.
Overlay assays
GST-fusion proteins were purified from bacterial cultures and used for GST pull-down assays as described (Georgescu et al, 1999). For overlay assay, 0.5 μg of recombinant GST-fusion proteins was separated on gel, transferred to nitrocellulose filter and overlaid overnight at 4°C with either GST-PTEN-CT or GST-Myc-PDGFR-CT (10 μg protein/ml blocking buffer). The filters were washed and processed for Western blot analysis by incubation with PTEN (A2B1) or Myc antibodies, followed by incubation with a secondary antibody. Bridging overlay assays were performed similarly to the overlay assays, except that the filter-immobilized proteins were overlaid with various recombinant proteins in two consecutive steps, separated by filter washing, before incubating the filters with the primary Myc antibody directed against the last overlaid recombinant protein.
Cells
293T human embryonic kidney cells, Bosc cells, LN308 and LN18 glioblastoma cells (gift of Erwin Van Meir) were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum (FCS) (complete growth medium). MEFs were isolated from 14-day-old embryos resulting from NHERF1(+/−) crosses (Morales et al, 2004). The heads and limbs of the embryo were removed under microscope and prepared for genotyping, and the bodies of the embryos emptied of the bowels, heart and liver were homogenized and incubated with trypsin. Each homogenate was plated on gelatinized plates in DMEM supplemented with 10% FCS, resulting in individual MEFs at passage 0. In this study, MEFs derived from at least five cell populations from each NHERF1(−/−) and (+/+) MEF were expanded and used for subsequent experiments.
Protein analysis
The protocols for transfection, retroviral infection, cell lysis, protein immunoprecipitation and Western blotting were previously described (Georgescu et al, 1999). All buffers were supplemented with 1 mM phenylmethylsulfonyl fluoride, 21 μg/ml aprotinin, 1 mM sodium orthovanadate and 0.1 mM sodium molybdate. For fractionation, MEFs were homogenized in buffer 1 (320 mM sucrose, 1 mM MgCl2, 0.5 mM CaCl2 and 4 mM Hepes (pH 7.1)). The lysates were clarified at 1500 g for 10 min at 4°C and further centrifuged at 14 000 g for 15 min at 4°C. The supernatant was saved as the cytoplasmic fraction and the pellet was washed with buffer 1 and resuspended in membrane solubilization buffer (500 mM NaCl, 10 mM Tris (pH 7.4), 1 mM EDTA and 1% Triton X-100).
Antibodies used were PTEN (mouse A2B1 and goat N-19 raised against epitopes in the CT and NT, respectively), Myc tag (9E10), GST tag (B-14), PDGFRβ (958), Erk2 (C-14), p85 PI3K (Z8) (Santa Cruz Biotechnology), phospho-Akt-S473, Akt/PKB, phospho-p44-p42-T202/Y204-MAP kinase (P-Erk), phospho-PDGFR-Y751, phospho-p70-S6 kinase-T389, p70-S6 kinase (Cell Signaling), FLAG tag (M2, Sigma-Aldrich), NHERF1 (Calbiochem), NHERF2 (Morales et al, 2004) and actin (Chemicon).
The gel filtration analysis of proteins from brain total extracts was performed as described (Sumitomo et al, 2004). Each brain from 9-month-old NHERF1(+/+) mice was homogenized in 2 ml of buffer (50 mM Hepes (pH 7.5), 150 mM NaCl, 5 mM EDTA and 0.5% NP-40). After clarification, the brain lysate was crosslinked with 5 mM dithiobis(succinimidyl)propionate (Pierce) for 2 h on ice. Tris–HCl (20 mM, pH 7.5) was added to quench the reaction. The sample was filtered and added to a Sephacryl S-300 column (Amersham). The crosslinking was verified by resuspending the precipitated proteins from fractions in non-reducing or reducing Laemmli sample buffer. For the depletion studies, the crosslinked brain lysate containing approximately 25 mg proteins was divided equally and incubated with 100 μl Dynabeads protein G (Dynal Biotech) and either 20 μg PTEN CT antibody or control mouse Ig for 4 h at 4°C. The supernatants were used for gel filtration (CT depletion) or further incubated with 50 μl Dynabeads and either 10 μg PTEN NT antibody or control goat Ig overnight at 4°C. The remaining supernatants were applied to gel filtration column (CT → NT depletion).
Small interfering RNA depletion
A total of 8 × 104 MEFs/well in six-well plates were transfected with 4 μl DharmaFECT4 and 100 nM of a 1:1 combination of #2 and #3 individual siRNAs from the siGENOME SMARTpool siRNA for mouse NHERF2 or with scrambled siCONTROL non-targeting siRNA pool (Dharmacon, Lafayette, CO). For the time-course experiments, the cells treated with siRNA for 72 h were placed in DMEM supplemented with 0.5% serum for 24 h. Cells were stimulated with 0.5 ng/ml of PDGF-BB (Sigma-Aldrich) for various periods of time and promptly lysed in cell lysis buffer.
Actin reorganization and chemotaxis
The immunofluorescence analysis of formaldehyde-fixed cells was previously described (Georgescu et al, 2000). Briefly, NHERF1(+/+) and (−/−) MEFs plated overnight on poly-D-lysine-coated coverslips in complete growth medium were serum-deprived for 8 h, stimulated with 5 ng/ml PDGF-BB for 10 min and fixed. The actin cytoskeleton was stained by incubation with rhodamine-labeled phalloidin (Molecular Probes) for 30 min. For chemotaxis, a modified Boyden chamber assay was used. The lower surface of Transwell inserts that fit 24-well plates and support an 8-μm-pore-size polycarbonate filter (Corning Incorporated Costar) was coated with 10 μg/ml fibronectin (Gibco BRL) in 200 μl DMEM overnight at 4°C. The inserts were transferred to new wells that contained 400 μl of either serum-free DMEM or various concentrations of growth factors. A suspension of 5 × 104 serum-deprived cells was added to 100 μl of serum-free DMEM in the inserts. The cells were allowed to migrate across the pores for 6 h at 37°C. The inserts were removed and the upper surface of the filter was wiped with a cotton tip to remove non-migratory cells. Migratory cells on the lower surface were fixed and stained using HEMA 3 stain set (Biochemical Sciences Inc.). To remove excess stain, filters were washed extensively with water and air-dried. The dye was extracted from the fixed cells with 500 μl of 2% (wt/vol) sodium dodecyl sulfate solution. The intensity of the dye was quantified by measuring the absorbance at 630 nm with a Beckman spectrophotometer DU640.
Supplementary Material
Supplementary Figure 1
Acknowledgments
We are indebted to TJ Liu, Kathrin Kirsch and Tomoo Iwakuma for helpful discussions. This work was supported by awards from the Texas Business and Professional Women, Goldhirsh Foundation and NCI-CA107201 to MMG, the American Brain Tumor Association to FCM and the NCI-CA09299-26 to ELK. The animal breeding was partially supported by NCI-CA16672.
References
- Andres-Pons A, Valiente M, Torres J, Gil A, Rogla I, Ripoll F, Cervera J, Pulido R (2005) Functional definition of relevant epitopes on the tumor suppressor PTEN protein. Cancer Lett 223: 303–312 [DOI] [PubMed] [Google Scholar]
- Dai JL, Wang L, Sahin AA, Broemeling LD, Schutte M, Pan Y (2004) NHERF (Na+/H+ exchanger regulatory factor) gene mutations in human breast cancer. Oncogene 23: 8681–8687 [DOI] [PubMed] [Google Scholar]
- Das S, Dixon JE, Cho W (2003) Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci USA 100: 7491–7496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demoulin JB, Seo JK, Ekman S, Grapengiesser E, Hellman U, Ronnstrand L, Heldin CH (2003) Ligand-induced recruitment of Na+/H+-exchanger regulatory factor to the PDGF (platelet-derived growth factor) receptor regulates actin cytoskeleton reorganization by PDGF. Biochem J 376: 505–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R (1996) Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell 85: 1067–1076 [DOI] [PubMed] [Google Scholar]
- Fouassier L, Yun CC, Fitz JG, Doctor RB (2000) Evidence for ezrin–radixin–moesin-binding phosphoprotein 50 (EBP50) self-association through PDZ–PDZ interactions. J Biol Chem 275: 25039–25045 [DOI] [PubMed] [Google Scholar]
- Georgescu MM, Kirsch KH, Akagi T, Shishido T, Hanafusa H (1999) The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc Natl Acad Sci USA 96: 10182–10187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu MM, Kirsch KH, Kaloudis P, Yang H, Pavletich NP, Hanafusa H (2000) Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res 60: 7033–7038 [PubMed] [Google Scholar]
- Heldin CH, Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79: 1283–1316 [DOI] [PubMed] [Google Scholar]
- Hollenberg SM, Sternglanz R, Cheng PF, Weintraub H (1995) Identification of a new family of tissue-specific basic helix–loop–helix proteins with a two-hybrid system. Mol Cell Biol 15: 3813–3822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooshmand-Rad R, Claesson-Welsh L, Wennstrom S, Yokote K, Siegbahn A, Heldin CH (1997) Involvement of phosphatidylinositide 3′-kinase and Rac in platelet-derived growth factor-induced actin reorganization and chemotaxis. Exp Cell Res 234: 434–441 [DOI] [PubMed] [Google Scholar]
- Ingraffea J, Reczek D, Bretscher A (2002) Distinct cell type-specific expression of scaffolding proteins EBP50 and E3KARP: EBP50 is generally expressed with ezrin in specific epithelia, whereas E3KARP is not. Eur J Cell Biol 81: 61–68 [DOI] [PubMed] [Google Scholar]
- Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Van Meir EG (1999) Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol 9: 469–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Kang Y, Su Jeon E, Jin Lee H, Oh YS, Suh PG, Sup Jung J, Donowitz M, Ho Kim J (2004) NHERF2 increases platelet-derived growth factor-induced proliferation through PI-3-kinase/Akt-, ERK-, and Src family kinase-dependent pathway. Cell Signal 16: 791–800 [DOI] [PubMed] [Google Scholar]
- Kotelevets L, van Hengel J, Bruyneel E, Mareel M, van Roy F, Chastre E (2005) Implication of the MAGI-1b/PTEN signalosome in stabilization of adherens junctions and suppression of invasiveness. FASEB J 19: 115–117 [DOI] [PubMed] [Google Scholar]
- Lau AG, Hall RA (2001) Oligomerization of NHERF-1 and NHERF-2 PDZ domains: differential regulation by association with receptor carboxyl-termini and by phosphorylation. Biochemistry 40: 8572–8580 [DOI] [PubMed] [Google Scholar]
- Lazar CS, Cresson CM, Lauffenburger DA, Gill GN (2004) The Na+/H+ exchanger regulatory factor stabilizes epidermal growth factor receptors at the cell surface. Mol Biol Cell 15: 5470–5480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP (1999) Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99: 323–334 [DOI] [PubMed] [Google Scholar]
- Leslie NR, Gray A, Pass I, Orchiston EA, Downes CP (2000) Analysis of the cellular functions of PTEN using catalytic domain and C-terminal mutations: differential effects of C-terminal deletion on signalling pathways downstream of phosphoinositide 3-kinase. Biochem J 346: 827–833 [PMC free article] [PubMed] [Google Scholar]
- Li C, Roy K, Dandridge K, Naren AP (2004) Molecular assembly of cystic fibrosis transmembrane conductance regulator in plasma membrane. J Biol Chem 279: 24673–24684 [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275: 1943–1947 [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273: 13375–13378 [DOI] [PubMed] [Google Scholar]
- Mahimainathan L, Ghosh Choudhury G (2004) Inactivation of platelet-derived growth factor receptor by the tumor suppressor PTEN provides a novel mechanism of action of the phosphatase. J Biol Chem 15: 15258–15268 [DOI] [PubMed] [Google Scholar]
- Maudsley S, Zamah AM, Rahman N, Blitzer JT, Luttrell LM, Lefkowitz RJ, Hall RA (2000) Platelet-derived growth factor receptor association with Na(+)/H(+) exchanger regulatory factor potentiates receptor activity. Mol Cell Biol 20: 8352–8363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales FC, Takahashi Y, Kreimann EL, Georgescu M-M (2004) Ezrin–radixin–moesin (ERM)-binding phosphoprotein 50 organizes ERM proteins at the apical membrane of polarized epithelia. Proc Natl Acad Sci USA 101: 17705–17710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczek D, Berryman M, Bretscher A (1997) Identification of EBP50: a PDZ-containing phosphoprotein that associates with members of the ezrin–radixin–moesin family. J Cell Biol 139: 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor–mTOR complex. Science 307: 1098–1101 [DOI] [PubMed] [Google Scholar]
- Shenolikar S, Minkoff CM, Steplock DA, Evangelista C, Liu M, Weinman EJ (2001) N-terminal PDZ domain is required for NHERF dimerization. FEBS Lett 489: 233–236 [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV (1997) Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 15: 356–362 [DOI] [PubMed] [Google Scholar]
- Sumitomo M, Iwase A, Navarro D, Zheng R, Kaminetzky D, Shen R, Georgescu M-M, Nanus D (2004) Synergy in tumor suppression by direct interaction of neutral endopeptidase with PTEN. Cancer Cell 5: 67–78 [DOI] [PubMed] [Google Scholar]
- Tolkacheva T, Boddapati M, Sanfiz A, Tsuchida K, Kimmelman AC, Chan AM (2001) Regulation of PTEN binding to MAGI-2 by two putative phosphorylation sites at threonine 382 and 383. Cancer Res 61: 4985–4989 [PubMed] [Google Scholar]
- Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR (2001) Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem 276: 48627–48630 [DOI] [PubMed] [Google Scholar]
- Vazquez F, Ramaswamy S, Nakamura N, Sellers WR (2000) Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol 20: 5010–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite KA, Eng C (2002) Protean PTEN: form and function. Am J Hum Genet 70: 829–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinman EJ, Steplock D, Tate K, Hall RA, Spurney RF, Shenolikar S (1998) Structure–function of recombinant Na/H exchanger regulatory factor (NHE-RF). J Clin Invest 101: 2199–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Hepner K, Castelino-Prabhu S, Do D, Kaye MB, Yuan XJ, Wood J, Ross C, Sawyers CL, Whang YE (2000a) Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc Natl Acad Sci USA 97: 4233–4238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dowbenko D, Spencer S, Laura R, Lee J, Gu Q, Lasky LA (2000b) Interaction of the tumor suppressor PTEN/MMAC with a PDZ domain of MAGI3, a novel membrane-associated guanylate kinase. J Biol Chem 275: 21477–21485 [DOI] [PubMed] [Google Scholar]
- Wymann M, Arcaro A (1994) Platelet-derived growth factor-induced phosphatidylinositol 3-kinase activation mediates actin rearrangements in fibroblasts. Biochem J 298: 517–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun CH, Oh S, Zizak M, Steplock D, Tsao S, Tse CM, Weinman EJ, Donowitz M (1997) cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci USA 94: 3010–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1