SOCS3: an essential regulator of LIF receptor signaling in trophoblast giant cell differentiation (original) (raw)
Abstract
Suppressor of cytokine signaling 3 (SOCS3) binds cytokine receptors and thereby suppresses cytokine signaling. Deletion of SOCS3 causes an embryonic lethality that is rescued by a tetraploid rescue approach, demonstrating an essential role in placental development and a non-essential role in embryo development. Rescued SOCS3-deficient mice show a perinatal lethality with cardiac hypertrophy. SOCS3-deficient placentas have reduced spongiotrophoblasts and increased trophoblast secondary giant cells. Enforced expression of SOCS3 in a trophoblast stem cell line (Rcho-1) suppresses giant cell differentiation. Conversely, SOCS3-deficient trophoblast stem cells differentiate more readily to giant cells in culture, demonstrating that SOCS3 negatively regulates trophoblast giant cell differentiation. Leukemia inhibitory factor (LIF) promotes giant cell differentiation in vitro, and LIF receptor (LIFR) deficiency results in loss of giant cell differentiation in vivo. Finally, LIFR deficiency rescues the SOCS3-deficient placental defect and embryonic lethality. The results establish SOCS3 as an essential regulator of LIFR signaling in trophoblast differentiation.
Keywords: cardiac hypertrophy/LIF receptor/SOCS3/trophoblast differentiation
Introduction
Cytokines regulate a variety of aspects of cell growth and differentiation through their interaction with receptors of the cytokine receptor superfamily, the associated Janus kinases (JAKs) and, in part, by the activation of members of the signal transduction and activators of transcription (STATs) (Ihle, 1996; Darnell, 1997). The suppressor of cytokine signaling (SOCS) proteins are characterized by the presence of an SH2 domain and a conserved motif termed the SOCS box (Starr and Hilton, 1998; Yoshimura, 1998). They suppress cytokine signaling through interaction of the SH2 domain with sites of tyrosine phosphorylation on receptors or on the JAKs. These interactions either compete for the recruitment of other signaling proteins, directly block the catalytic activity of the kinases and/or target the proteins for degradation through recruitment of ubiquitylation complexes through the SOCS box.
The physiological functions of the SOCS genes have been addressed through the derivation of strains of mice lacking individual genes. SOCS1 deficiency results in a perinatal lethality that is associated with altered T-cell development and interferon signaling (Alexander et al., 1999; Marine et al., 1999b). SOCS2 deficiency results in a phenotype consistent with a primary role in growth hormone signaling (Metcalf et al., 2000). SOCS6-deficient mice are normal, with the exception that they weigh 10% less than wild-type littermates (Krebs et al., 2002). SOCS3, unlike many of the SOCS genes, is induced by a broad spectrum of cytokines including interleukin (IL)-10 (Ito et al., 1999), leptin (Bjorbaek et al., 1998), ciliary neurotrophic factor (CNTF) (Bjorbaek et al., 1999) and leukemia inhibitory factor (LIF) (Starr et al., 1997), as well as by factors such as lipopolysaccharide (Stoiber et al., 1999). As with the other SOCS proteins, overexpression of SOCS3 can suppress responses to a number of cytokines including growth hormone (Hansen et al., 1999), erythropoietin (Sasaki et al., 1999), LIF (Nicholson et al., 1999), CNTF (Bjorbaek et al., 1999) and leptin (Bjorbaek et al., 1998). In the case of cytokines that utilize gp130, inhibition is associated with the binding to receptor sites of tyrosine phosphorylation. The potential physiological roles of SOCS3 have also been examined through the derivation of mice lacking the gene (Marine et al., 1999a; Roberts et al., 2001). In both studies, disruption of the SOCS3 gene resulted in an embryonic lethality, although the timing of the lethality varied. In one case, embryos were obtained beyond mid-gestation and frequently were found to exhibit an erythrocytosis phenotype (Marine et al., 1999a). Reconstitution experiments did not reveal defects in definitive erythropoiesis, leading to the conclusion that SOCS3 may play a role in specifically regulating a stage of fetal erythropoiesis. In the second study (Roberts et al., 2001), no embryos were found beyond mid-gestation, and analysis of erythropoiesis at this stage failed to identify any phenotypic alterations. However, histologically, there were detectable phenotypic alterations in placental morphology, suggesting a role for SOCS3 in placental development.
Placental development is essential for providing the exchange of maternal factors to sustain embryo growth. Among the trophoblast lineage-derived cells, the trophoblast giant cells are unique in undergoing extensive endoduplication to give rise to large, polyploid cells with functions such as the secretion of placental lactogen-1 (PL-1). A number of genes have been identified that control trophoblast stem cell differentiation and serve as markers of the process (Cross, 2000). For example, the basic helix–loop–helix (bHLH) transcription factor Mash2 is required to maintain diploid trophoblasts of the spongiotrophoblast layer at the expense of giant cells (Guillemot et al., 1994). In contrast, Hand1, another bHLH transcription factor, promotes giant cell differentiation (Riley et al., 1998). Importantly, these genes, and a number of other genes associated with placental development, create an embryonic lethal phenotype when disrupted (Ihle, 2000; Hemberger and Cross, 2001).
Potential roles for cytokine signaling in placental development have also been identified. Maternally derived LIF is essential for early implantation, although its precise target of action, and whether the target is maternal or embryonic, has not been identified (Stewart et al., 1992). In addition, the disruption of the LIF receptor (LIFR) results in a decrease in embryo viability and, histologically, the placentas lack well-organized spongiotrophoblast and labyrinthine layers, suggesting a role for LIF at a later stage of placental differentiation (Ware et al., 1995). The studies described here were designed to explore further the role of SOCS3 in placental development and to assess whether these functions contribute to the embryonic lethality. The results demonstrate that the embryonic lethality is due to altered trophoblast differentiation and implicate LIF signaling as an important regulator of trophoblast giant cell differentiation.
Results
Histological changes in the placentas of SOCS3-deficient embryos
We initially reported that SOCS3 deficiency resulted in a mid-gestation embryonic lethality associated with erythrocytosis (Marine et al., 1999a). More recently, another SOCS3-deficient strain was reported (Roberts et al., 2001) with a more severe embryonic lethal phenotype and placental abnormalities. To determine whether placental abnormalities were a common feature of SOCS3 deficiency, we examined placentas from E9.5 to E12.5 embryos (Supplementary figure 1, available at The EMBO Journal Online). Consistent with other studies (Roberts et al., 2001), there were histological differences in the placentas of SOCS3-deficient embryos. At E9.5, there were increased numbers of trophoblast giant cells and decreased numbers of trophoblast stem cells in the ectoplacental cone. Mutant primary trophoblast giant cells were larger in cell size and the nuclei were larger (Supplementary figure 1C and D). Fusion of the chorion and alantois occurred normally. At E11.5 (Supplementary figure 1I–L), the spongiotrophoblast layer was decreased, with an increase in secondary trophoblast giant cells. These changes continued through E12.5 (Supplementary figure 1M–P). These results suggest that the absence of SOCS3 is associated with changes in the differentiation of trophoblast giant cells and spongiotrophoblasts in the developing placenta.
Tetraploid rescue of SOCS3-deficient embryos
To determine whether the changes were due to intrinsic properties of SOCS3-deficient trophoblasts and responsible for the embryonic lethality, we utilized a tetraploid rescue approach (Nagy and Rossant, 1993). Morula stage embryos from a SOCS3 heterozygous intercross were used for aggregation. Among 73 embryos that were analyzed at term, or that were Cesarean derived and foster nursed, 10 were SOCS3 deficient and viable. These results demonstrate that the embyronic lethality is attributable to the intrinsic defects in extraembryonic, SOCS3-deficient cells.
As previously described (Marine et al., 1999a), SOCS3-deficient embryos frequently had a visible expansion of red blood cells. However, tetraploid rescued embryos did not exhibit this phenotype, demonstrating that it was secondary to the placental defects (Figure 1A). Consistent with this, histological analysis of the embryos, including fetal livers, failed to detect any abnormalities (data not shown). Moreover, the numbers of CFU-E and BFU-E were comparable with wild-type (Figure 1B), as were the sizes of the colonies from tetraploid rescued embryos (data not shown). Lastly, histological analysis of the placentas of E12.5 tetraploid rescued embryos showed that they were indistinguishable from wild-type placentas (data not shown). At term, tetraploid rescued embryos generally were not distinguishable from wild-type embryos. However, the SOCS3 deficiency in tetraploid rescued pups was associated with a perinatal lethality (Figure 2A), and all SOCS3-deficient embryos died within 3 weeks after birth. In contrast, comparably manipulated wild-type embryos, or embryos heterozygous for SOCS3, could be Cesarean derived and foster nursed, and survived. All the SOCS3-deficient pups became growth retarded (Figure 2B) and lethargic. Histological analysis of SOCS3-deficient pups revealed that the hearts of the pups were grossly enlarged (Figure 2C), there was a consistent increase in the diameter of myocardial fibers (Figure 2D and E) and they were aligned irregularly (Figure 2F). The high level of expression of SOCS3 in normal heart tissue (Figure 2G) is consistent with the hypothesis that the changes are due to SOCS3 deficiency. Histological analysis of brain, lung, liver, spleen, kidney and intestine showed no other changes in SOCS3-deficient mice (data not shown). Also, no hematological abnormalities including the numbers of red blood cells were detected in the rescued SOCS3-deficient mice (data not shown).

Fig. 1. Tetraploid rescue experiments demonstrate that SOCS3 is dispensable in embryonic development. Aggregation with wild-type tetraploid embryos rescues SOCS3–/– embryos. (A) Rescued SOCS3–/– embryos (right) on E12.5 were visually comparable with wild-type (left) and did not display the erythrocytosis seen in SOCS3-deficient embryos. (B) Colony assay using fetal liver from E12.5 rescued wild-type and mutant embryos. There was no difference in the numbers of CFU-E and BFU-E between wild-type and mutant cells from the fetal livers.

Fig. 2. Rescued SOCS3–/– mice demonstrated perinatal lethality associated with cardiac defects. (A) Rescued SOCS3–/– mice displayed a perinatal lethality and died within 25 days after birth. (B) Compared with the wild type (upper), mutant mice (lower) were lethargic and growth retarded. (C) Hearts from SOCS3–/– offspring were markedly enlarged in size and displayed wall thickening. (D) Hypertrophy of cardiomyocyte in the mutant heart. (E) The diameter of the myocardial fiber was measured using NIH image software and was increased in SOCS3-deficient mice. (F) The wild-type and mutant heart at day 7, demonstrating the myocardial fiber disarray in SOCS3-deficient hearts. (G) Northern blotting of SOCS3 in adult tissues. SOCS3 was expressed in the heart, spleen, lung and liver. Scale bars: (C) 5 mm; (D) 50 µm; (F) 100 µm.
Increased expression of markers for trophoblast giant cells in mutant placentas
To define better the changes in trophoblast differentiation in mutant embryos, the expression of trophoblast markers was examined (Figure 3A). Mash2 is expressed in trophoblast stem cells and in intermediate trophoblasts of the ectoplacental cone, but not in trophoblast giant cells, and functions to maintain spongiotrophoblasts at the expense of giant cell differentiation (Guillemot et al., 1994). In contrast, Hand1 is not expressed in stem cells but is expressed in intermediate trophoblasts and in trophoblast giant cells, and is required for trophoblast giant cell differentiation. Hand1 is more highly expressed in the mutant placentas, while Mash2 is diminished, consistent with increased numbers of committed trophoblast giant cells and decreased numbers of stem cells and spongiotrophoblasts. There is a dramatically increased number of _PL1_-expressing cells, which is an endocrine marker for giant cells consistent with an expansion of differentiated giant cells. Similarly, the serine/threonine protein kinase Limk is expressed most abundantly in trophoblast giant cells, (Cheng and Robertson, 1995). As illustrated, the expression of Limk is also increased in mutant placentas. In addition, whereas the expression of Limk is restricted in wild-type embryos, its expression extends into the labyrinthine layer in the mutants.

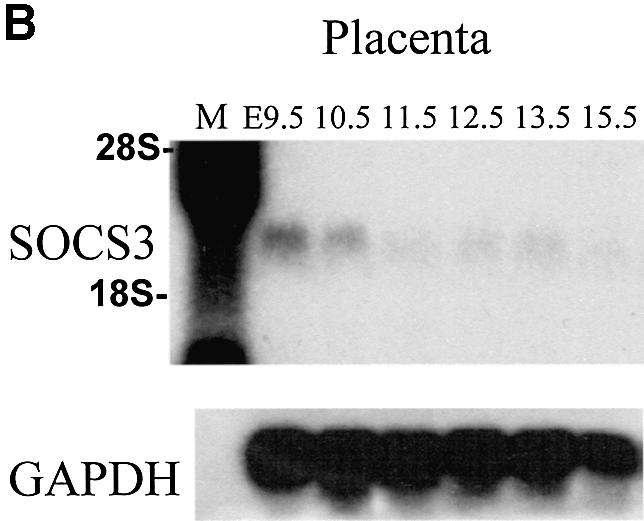

Fig. 3. Expression of trophoblast-specific markers and SOCS3 in the placenta. (A) In situ hybridization of trophoblast-specific markers in E12.5 placenta. In situ hybridization was performed using E12.5 wild-type and SOCS3-deficient placental serial sections. The expression of trophoblast giant cells markers (Hand1, PL1 and Limk1) was enhanced in SOCS3–/– placenta. 4311, a specific marker for expression of spongiotrophoblast cells, was markedly reduced, which was comparable with the histological analysis. The expression of the markers for stem cells (Mash2 and Cdx2) was reduced. Scale bars: 500 µm. (B) SOCS3 expression in the placenta. SOCS3 expression was observed in E9.5 and E10.5 placenta. (C) In situ hybridization of SOCS3 in E9.5 placenta. SOCS3 was expressed in all lineages of trophoblasts, in the ectoplacental cone, chorion and giant cells.
In contrast to the above markers, 4311 is expressed specifically in spongiotrophoblasts (Lescisin et al., 1988). As illustrated, there is a dramatic reduction in the number of _4311_-expressing cells in the mutant placentas. Similarly Cdx2 is expressed in trophoblast stem cells found in the spongiotrophoblast and labyrinthine layers (Beck et al., 1995). As illustrated in Figure 3A, there was a marked reduction in cells expresssing Cdx2 in the mutant placenta. The results are consistent with the hypothesis that there is a shift in the cellular differentiation in the mutant placenta that results in decreased numbers of spongiotrophoblasts and their precursors, and a dramatically increased differentiation of trophoblast secondary giant cells.
SOCS3 is expressed in trophoblast in the placenta
To elucidate the expression pattern of SOCS3 in the placenta, northern blot analysis was performed using each stage of placentas (Figure 3B). Abundant expression of SOCS3 was observed in E9.5 and E10.5 placenta. We next analyzed SOCS3 expression in E9.5 placenta using in situ hybridization (Figure 3C). In E9.5 placenta, SOCS3 was expressed in all trophoblast lineage including the trophoblast stem cell population in ectoplacental cone, chorion and primary giant cells.
SOCS3 suppresses trophoblast giant cell differentiation in vitro
The Rcho-1 cell line was derived from a rat choriocarcinoma and consists of trophoblast stem cells that can be induced to differentiate to trophoblast giant cells in vitro (Faria and Soares, 1991). We therefore utilized these cells to assess further the direct role of SOCS3 in regulating trophoblast giant cell differentiation. The pattern of expression of SOCS3 in differentiating Rcho-1 cells is illustrated in Figure 4A. As illustrated, prior to inducing differentiation, both Hand1 and PL1 are expressed at low or non-detectable levels. Following induction of differentiation, both markers increase and, by day 6, expression of both markers is increased dramatically. Conversely, SOCS3 expression is high in undifferentiated cells and decreases with the induction of differentiation as assessed by RNA (Figure 4A) or protein levels (Figure 4B). The differentiation of Rcho-1 cells results in a morphological change in which the cells spread and become more firmly attached to the substratum as assessed by resistance to release with trypsin. Differentiation is also associated with the reorganization of actin filaments and DNA endoduplication, resulting in cells with ploidies of 4N–128N. To determine whether the decreased expression of SOCS3 was necessary for differentiation, a series of clones of cells were established that were transfected with a SOCS3 expression construct. As illustrated in Figure 4C, these clones expressed levels of SOCS3 that were ∼5- to 10-fold higher than control cells. Expression of SOCS3 resulted in the suppression of differentiation as evidenced by the lack of morphological change (Figure 4D, top), including the increased nuclear size and the lack of actin filament reorganization (Figure 4D, bottom). In addition, SOCS3-expressing clones retained sensitivity to trypsin release from the substratum (Figure 4E) and demonstrated impaired DNA endoduplication (Figure 4F). These results support the hypothesis that downregulation of SOCS3 expression is essential for the full differentiation of trophoblast giant cells.




Fig. 4. SOCS3 is a negative regulator of giant cell differentiation in vitro. (A) Northern blotting. SOCS3 was expressed in undifferentiated Rcho-1 cells and decreased with differentiation. Hand1 and PL1 expression on day 4 and 6 is a marker of giant cell differentiation. (B) Western blotting. SOCS3 protein levels decrease with differentiation. Protein lysates from stable SOCS3-expressing cells were used as a control. (C) Western blotting indicating the levels of SOCS3 in each stable clone transfected with expression constructs for SOCS3. (D) SOCS3 stable transfectant Rcho-1 cells (right panel) were impaired in giant cell differentiation compared with control cells (left panel). Photographed at day 2. The lower panels show nuclear (blue) and actin stress fiber reorganization (green), demonstrating that in SOCS3 transfectants, nuclei were smaller and actin stress fiber reorganization was undeveloped. Representative pictures are shown. The other clones demonstrated similar results. (E) Analysis of trypsin-resistant cells in the stable transfectant. The trypsin-resistant cells were counted in each clone 2 days after incubation with 10% HS. SOCS3 stable transfectants demonstrated significantly decreased numbers of differentiated giant cells. Representative data from four independent experiments are shown. (F) DNA endoreduplication was impaired in SOCS stable tranfectants. Representative data from three independent experiments are shown. The other clones demonstrated similar results. (G) Trophoblast stem cells in conditioned medium in the presence of FGF4. Wild-type trophoblast stem cells showed few differentiated giant cells (left panel) under these conditions. In contrast, SOCS3-deficient trophoblast stem cells differentiated more readily into giant cells (right panel). The lower panels show nuclear staining by DAPI, demonstrating enhanced differentiation to giant cells. (H) SOCS3-deficient trophoblast stem cells showed significantly increased expression of PL1 and decreased expression of 4311 after removal of the condition medium and FGF4. Representative data from three independent experiments are shown. The other clones demonstrated similar results.
To explore further the possibility that the absence of SOCS3 promotes secondary giant cell differentiation, we established trophoblast stem cell cultures by placing individual blastocysts from a heterozygous cross in the presence of fibroblast growth factor 4 (FGF4) and mouse embryo fibroblasts (MEFs) (Tanaka et al., 1998). When wild-type cells from such cultures were placed in medium containing FGF4 and conditioned medium from MEFs, only minimal differentation was seen (Figure 4G, left). In contrast, cultures containing SOCS3-deficient cells morphologically differentiated to giant cells, as indicated by the cell body and nuclear size (Figure 4G, right). The analysis of DNA ploidy clearly demonstrated more differentiation to giant cells (Figure 4G, lower panel). Increased expression of PL1 and decreased expression of 4311 similarly demonstrated enhanced giant cell differentiation from stem cells (Figure 4H).
LIF promotes giant cell differentiation in vitro and in vivo
A number of growth factors [FGF, epidermal growth factor (EGF) and hepatocyte growth factor (HGF)] or cytokines (LIF) have been implicated in placental development through the analysis of gene-deficient strains of mice (Threadgill et al., 1995; Uehara et al., 1995; Ware et al., 1995; Arman et al., 1998; Tanaka et al., 1998). The involvement of LIF is of particular importance since SOCS3 has been shown to suppress strongly signaling through LIFR (Duval et al., 2000; Yasukawa et al., 2001). Therefore, it might be proposed that LIF induces the differentiation of trophoblast giant cells and that the extent and timing of differentiation are controlled by the levels of SOCS3 and the levels of maternally or embryonically produced LIF. To test this possibility, we initially examined the effect of LIF on giant cell differentiation using Rcho-1 cells in vitro. LIF significantly promoted giant cell differentiation, evident from the numbers of trypsin-resitant cells (Figure 5A), actin stress fiber reorganization (Figure 5B) and DNA endoduplication (Figure 5B and C). Moreover, forced expression of SOCS3 in Rcho-1 cells completely blocked LIF-induced giant cell differentiation (Figure 5).

Fig. 5. LIF promotes trophoblast giant cell differentiation in vitro. (A) LIF promoted giant cell differentiation in Rcho-1 cells. Rcho-1 cells were incubated in the indicated concentration of LIF for 2 days in the presence of 2% FCS, and trypsin-resistant cells were counted. LIF treatment significantly increased trypsin-resistant cells, while SOCS3-overexpressing cells showed no effect of LIF treatment. Representative data from three independent experiments are shown. Three independent clones demonstrated similar results. (B) LIF promoted actin stress fiber reorganization and DNA endoduplication in Rcho-1 cells. In control Rcho-1 cells, LIF treatment stimulated actin stress fiber reorganization (green) and increased the numbers of enlarged nuclei stained by DAPI (blue); however, SOCS3-overexpressing cells showed no effect. Representative data from three independent experiments are shown. The other clones demonstrated similar results. (C) DNA content was analyzed using NIH image software.
The above results supported the concept that LIF signaling could dramatically affect the status of giant cell differentiation. If correct, it can be hypothesized that the levels of LIFR would also be a critical determinant in placental development. Importantly, the initial description of the LIFR deficiency noted that there was a significant embryonic lethality. The only grossly detectable embryonic defects were in the placentas, which lacked organization into distinct spongiotrophoblasts and labyrinthine layers (Ware et al., 1995). Therefore, we next examined the placental defect in LIFR-deficient embryos in detail. As illustrated in Figure 6A, we observed placental defects that were comparable with those previously reported. Importantly, whereas wild-type placentas contain a distinct zone of secondary giant cells (arrows) that are dramatically expanded in SOCS3-deficienct placentas, secondary giant cells are greatly reduced in LIFR-deficient placentas (Figure 6A). These characteristics were verified by in situ hybridization of the trophoblast giant cell marker, PL1 (Figure 6B). The signal of PL1 in LIFR-deficient placenta was reduced and the numbers of PL1-positive cells were significantly decreased (Figure 6B). The impaired production of giant cells was observed from E9.5 in LIFR-deficient placenta (data not shown).

Fig. 6. LIFR is important for trophoblast giant cell differentiation in vivo. (A) Trophoblast giant cell defect in E12.5 LIFR-deficient placenta. In the wild-type placenta, there was a distinct layer of trophoblast giant cells between the decidua and spongiotrophoblast layer (arrows). In contrast, there is a marked reduction in the numbers of giant cells in LIFR-deficient placenta. Wild-type and LIFR–/– placenta were taken from the same litter. In the SOCS3-deficient placenta, enlarged giant cells occupied the spongiotrophoblast layer. (B) PL1 expression in E12.5 LIFR-deficient placenta. The numbers of PL-1-positive cells in LIFR-deficient placenta were significantly decreased.
LIFR-null or hetrozygous background rescues SOCS3-null placental defect and embryonic lethality
The requirement for LIFR for the generation of trophoblast secondary giant cells suggested the possibility that LIFR deficiency might rescue the embryonic lethality associated with SOCS3 deficiency. To explore this possibility, the SOCS3 deficiency was crossed onto a LIFR deficiency. The LIFR deficiency results in a perinatal lethality within hours after birth (Ware et al., 1995), and consequently we crossed SOCS3 and LIFR double heterozygous mice to obtain embryos that would be deficient for both genes. Because of the perinatal lethality, embryos from the cross were recovered at term by Cesarean section and analyzed. As illustrated in Table I, among the embryos that were homozygously wild-type for the LIFR, no viable SOCS3-deficient embryos were obtained when eight would have been anticipated. However, among the embryos that were heterozygous for the LIFR deficiency, 15 term embryos were SOCS3 deficient, although four of these were not viable. Based on the numbers of +/+ or +/– embryos, we would have anticipated 26–37 embryos with a SOCS3–/– genotype. This is striking since we have not obtained SOCS3-deficient embryos in analysis of 144 embryos from a SOCS3 heterozygous cross. Lastly, the embryonic lethality associated with LIFR deficiency was evidenced by the few embryos obtained that were LIFR deficient and wild-type for SOCS3 (five) or heterozygous for the SOCS3 deficiency (six). However, five embryos were deficient for both the LIFR and SOCS3, demonstrating the dramatic ability of LIFR deficiency to rescue SOCS3-deficient embryos.
Table I. The genotypic distribution of SOCS3+/–, LIFR+/– double heterozygous intercrosses at term.
| | SOCS3 | | | | | ----------------------------- | --- | --- | ----------- | | LIFR | +/+ | +/– | –/– | | +/+ | 19 | 22 | 0 | | +/– | 34 | 122 | 15 (4 dead) | | –/– | 5 | 6 | 5 | | SOCS3 heterozygous intercross | 46 | 98 | 0 |
The appearances of the embryos from the above cross are illustrated in Figure 7A. As shown, the embryos that were deficient for both the LIFR and SOCS3 were indistinguishable from the wild-type embryos. Full histological and hematological analysis of the neonatal SOCS3–/–LIFR–/– mice failed to detect any abnormalities (data not shown). In contrast, the viable term embryos that were heterozygous for the LIFR deficiency and were SOCS3 deficient were generally smaller than wild-type controls. Lastly, the placentas were examined histologically. As is evident in Figure 7B, the double deficiency of LIFR and SOCS3 significantly restored placental histology to that of LIFR-deficient placentas. In these placentas, there still were characteristics of LIFR deficiency; however, the characteristics of SOCS3 deficiency were completely restored, suggesting that SOCS3 localized downstream of LIFR signaling. SOCS3 deficiency and heterozygousity for the LIFR deficiency was partially restored to that of LIFR-deficient placentas. The degree of restoration was correlated with the maturity (size) seen in embryos of the SOCS3–/–LIFR+/– genotype.

Fig. 7. LIFR-null or heterozygous background rescues the SOCS3-null placental defect and embryonic lethality. (A) Rescued SOCS3–/–LIFR–/– embryos were comparable in size with wild-type or LIFR–/– embryos. However, SOCS3–/–LIFR+/– embryos were significantly smaller, and some embryos were no longer viable. (B) Histological analysis of the placentas. SOCS3–/–LIFR–/– placentas had a restored labyrinthine layer similar to LIFR-deficient placenta. SOCS3–/–LIFR+/– placentas had enlarged giant cells with some intact labyrinth, suggesting a partial rescue by the LIFR+/– background. Scale bars: 100 µm.
Discussion
The results establish a critical role for SOCS3 in placental development and demonstrate that the previously described embryonic lethality is due to the placental defects. This is established most definitively through the ability to rescue embryonic development by providing wild-type extraembryonic cells with a tetraploid rescue approach. Moreover, the lack of an erythrocytosis phenotype in the rescued embryos strongly suggests that the previously described erythrocytosis (Marine et al., 1999a) was a consequence of placental insufficiency and not an inherent consequence of SOCS3 deficiency in erythroid cells. One possibility is that the hypoxia caused by the placental failure resulted in an increased production of erythropoeitin that, in turn, caused the associated erythrocytosis. The absence of any further defects in embryonic development in rescued embryos further demonstrates that SOCS3 does not have any additional, essential functions during embryonic development, although it is widely expressed. Although we observed erythrocytosis of embryos, this phenotype was not seen in the other strain of SOCS3-deleted mice (Roberts et al., 2001). However, in this strain, there were no SOCS3–/– embryos beyond E13.5, and therefore they did not reach an age at which the phenotype was manifested.
Although the tetraploid approach rescued SOCS3-deficient embryos, there was a striking perinatal lethality. Histological analysis of the SOCS3-deficient offspring identified, as the only obvious phenotypic change, evidence for heart failure consisting of myocyte hypertrophy and disarray similar to the manifestations of hypertrophic cardiomyopathy (HCM) (Marian and Roberts, 2001). In this regard, the common receptor component of the IL-6 family of cytokines, gp130, has been demonstrated to play an important role in cardiac hypertrophy and heart failure (Hirota et al., 1999; Negoro et al., 2001; Uozumi et al., 2001). Consistent with this, both cardiotrophin-1 (CT-1) and LIF are potent inducers of cardiomyocyte hypertrophy, and the hypertrophy induced by LIF was shown to be suppressed by forced expression of SOCS3. In addition, SOCS3 is induced in the heart following injection of CT-1 as well as by pressure overload. Taken together, the previous results have led to the hypothesis that CT-1 and LIF signaling in cardiomyocytes is negatively regulated by induced SOCS3 expression. The consequences of SOCS3 deficiency on heart development described here would support such a role.
Collectively, our results support a role for SOCS3 in regulating trophoblast differentiation by specifically suppressing the differentiation of secondary giant cells during development of the placental organ. This is supported by the overproduction of secondary giant cells in SOCS3-deficient embryos as assessed by histological analysis or by the changes in the expression of markers of giant cell differentiation. It is supported further by the inverse correlation of SOCS3 expression with giant cell differentiation of Rcho-1 cells and the ability of SOCS3 over expression to suppress the differentiation of Rcho-1 trophoblast stem cells to giant cells. Lastly, it is supported by the increased in vitro differentiation of giant cells of trophoblast stem cell cultures from SOCS3-deficient embryos relative to wild-type cells.
The mechanisms by which SOCS3 suppresses the differentiation of trophoblast giant cells is not known precisely, but our results and previous studies would suggest a role in regulating LIFR signaling. LIF has been proposed to control trophoblast differentiation by increasing cell adhesive properties in vitro and ex vivo through inhibiting secretion of metalloproteinases and by increasing deposition of fibronectin and inhibiting the differentiation of cytotrophoblast cells into syncytium (Bischof et al., 1995). The potential role for LIF signaling in inducing trophoblast giant cell differentiation or function is indicated by the evidence that LIF promotes giant cell differentiation in vitro and by the greatly reduced numbers of trophoblast secondary giant cells in LIFR-deficient embryos. Lastly, the ability of LIFR deficiency to rescue the SOCS3 deficiency strongly supports the proposed role in negatively regulating LIF signaling.
The source of LIF during placental development at mid-gestation is not known. However, embryonic LIF deficiency is not associated with placental abnormalities or embryonic lethality (Stewart et al., 1992). This would suggest that LIF is maternally derived. Unfortunately, maternal deficiency of LIF results in a very early embryonic lethality, precluding assessment of its role in the later stages of placental development. The role of maternally derived LIF is unclear. Since LIFR deficiency does not cause a similar, early embryonic lethality, it has been concluded that maternally derived LIF is controlling the function of maternal cells at the site of implantation. However, it is also possible that the other cytokines may contribute to the giant cell differentiation, since LIFR has as ligand not only LIF, but also CT-1 and CNTF.
The downstream targets of LIF signaling that are responsible for induction of giant cell differentiation are not known. A role for a STAT transcription factor could be proposed based on the general role that STATs play in cytokine signaling. However, with the exception of STAT3, deficiency of any of the other STATs is not associated with an embryonic lethality. Deficiency of STAT3 causes a very early embryonic lethality (Takeda et al., 1997; Akira, 2000), in the critical stage for func tional trophoblast differentiation. STAT3 is highly expressed in the extra embryonic tissues, suggesting a role in these tissues (Takeda et al. 1997). Unfortunately, it is not known whether a tetraploid rescue approach would rescue embryonic development. However, because of the early lethality, it is not known whether STAT3 function is required for secondary giant cell differentiation at later stages of placental development. One potential downstream gene target is the bHLH transcription factor Hand1. Deficiency in Hand1 results in placental defects associated with the absence of giant cell differentiation (Riley et al., 1998). Similarly l-mfa deficiency results in placental defects including reduced giant cells (Kraut et al., 1998), and therefore is also a potential target.
In summary, the data support an integral role for SOCS3-regulated LIF signaling in trophoblast differentiation and/or function. Specifically, it can be proposed that LIFR signaling is essential for promoting embryonic trophoblast stem cells to differentiate to terminal secondary giant cells during the formation of the placental organ. Induction of SOCS3 in stem cells, by an unknown pathway, suppresses LIF signaling and thereby suppresses secondary giant cell differentiation.
Materials and methods
Mice
Generation of SOCS3-disrupted mice was described previously (Marine et al., 1999a). Genotyping was performed by PCR using tail biopsies. Mutant phenotypes were analyzed in mixed 129/Sve × C57Bl/6 background. LIFR-disrupted mice were obtained from the Jackson Laboratory. Genotyping was performed as described previously (Ware et al., 1995).
Tetraploid rescue experiments
E1.5 embryos at the two-cell stage were flushed and collected in M2 medium from the oviduct of CD-1 females and subjected to electrofusion using the CF150B electrofusion apparatus (Biochemical Laboratory Service, Ltd) as described (Nagy and Rossant, 1993). Briefly, the two-cell stage embryos were aligned in a 1.6 V AC field and fused with two 75 V DC pulses of 40 µs each using a 500 µm electrode in 0.3 M mannitol drops. Successfully fused embryos were cultured overnight in KSOM medium (Specialty Media Inc.). Eight-cell embryos were recovered from E2.5 pregnant SOCS3 heterozygous females mated to SOCS3 heterozygous males. After zona removal, two four-cell tetraploid wild-type embryos and a diploid eight-cell stage embryo from a heterozygous cross were aggregated overnight, and successfully developed embryos were transferred to the uterus of E2.5 pseudopregnant B6CBAF1 recipients. Recipients were sacrificed at term to recover the embryos. Embryos were genotyped and submitted to histological analysis. To recover viable fetuses, Cesarean section was performed on day E19.5 of gestation and the embyros were foster nursed. Colony assays were performed as described previously (Marine et al., 1999a) using tetraploid rescued embryos at E12.5.
Histological analysis of heart
To evaluate cardiac hypertrophy, transverse diameters of the ventricular myocytes were measured in histological sections as described (Tullio et al., 1997). The transverse diameters of the myocytes were measured (n = 50 for each ventricle) at the level of the nuclei. The mean ± SD was calculated for each, and the data were analyzed by Student’s _t_-test.
Cell culture, transfection and analysis of trophoblast giant cell differentiation
The Rcho-1 cell line, a rat choriocarcinoma (Faria and Soares, 1991), was obtained from M.Soares. Cells were maintained in NCTC135 supplemented with 0.1 mg/ml sodium pyruvate, 2 mM glutamine, penicillin, streptomycin and 20% fetal calf serum (FCS) (Hyclone). At confluence, cells were cultured in NCTC135 + 10% horse serum (HS) (Hyclone) to induce the differentiation. pcDNA3-SOCS3 was constructed by ligating an _Eco_RI–_Xho_I fragment of full-length murine SOCS3 cDNA into the pcDNA3 expression vector. Stable Rcho-1 transfectants were derived by transfecting linearized pcDNA3-SOCS3 vector using Fugene (Roche), and selected for 10 days in 250 µg/ml G418 as described (Kraut et al., 1998). G418-resistant clones were established by limiting dilution. Four clones of control (pcDNA3-empty vector) and five clones of SOCS3 stable transfectant were obtained. SOCS3 expression was verified by western blotting. For giant cell differentiation assays, 2 days after the induction in 10% HS, stem cells were removed by trypsinization [0.25% trypsin and 1 mM EDTA, incubation for 5 min and washing with phosphate-buffered saline (PBS) twice], and the adherent cells in 20 random high magnification fields for each clone were counted. Values were reported as the mean ± SE, and significant differences between values were determined by Student’s _t_-test. Nuclear DNA content was estimated by the area of 4′,6-diamidino-2-phenylindole (DAPI) staining using NIH image software as described previously (MacAuley et al., 1998) with >200 cells from random fields. To analyze the effect of LIF, 2% FCS was used instead of 10% HS. Recombinant hLIF was from Carbiochem. The trophoblast stem cells were established from blastocysts as described (Tanaka et al., 1998). The cells were cultured on MEFs in the presence of FGF4 and heparin. From 32 blastocysts, 12 trophoblast stem cell lines were established (four wild type, five heterozygous and three SOCS3 deficient). These were cultured in feeder-conditioned medium and photographed.
Histological analysis, in situ hybridization and immunofluorescence
Freshly isolated placenta and heart were fixed in 4% paraformaldehyde (PFA), dehydrated, embedded in paraffin and sectioned (4 µm). Sections were analyzed by hematoxylin/eosin staining. RNA in situ hybridization was carried out as described (Marine et al., 1999a) using [γ-33P]UTP-labeled antisense cRNA probes. Serial cryosections of E12.5 placentas of each genotype were used for in situ hybridization for trophoblast marker analysis. For the evaluation of giant cell numbers in LIFR-deficient placentas, five wild-type and five LIFR-deficient placentas were used. PL-1-positive cells per section were counted using dark field microscopy, and analyzed by Student’s _t_-test. For immunofluoresence, cells on coverslips were fixed in 4% PFA for 10 min and permeabilized in PBS/0.1% Triton X-100 for 5 min. Actin stress fiber staining was performed using Oregon green-labeled phalloidin (1:40, Molecular Probes). Sections were then washed, counterstained with DAPI (1:1000, Sigma), mounted and visualized by Leica DM-IRBE confocal microscopy.
Northern blot analysis
Total RNA was isolated using Trizol (Invitrogen). Rcho-1 cells were lysed by Trizol 3 h after changing the medium. A 10 µg aliquot of total RNA was loaded in each lane. Trophoblast stem cells were induced to differentiate by removal of FGF4 and conditioned medium. RNA was isolated on the indicated days. Full-length SOCS3 cDNA was used as a cDNA probe. After hybridization of SOCS3, the membrane was stripped and reprobed with a mouse PL-1, Hand-1, 4311 and GAPDH cDNA probe, respectively. The multiple tissue blot was obtained from Clontech.
Western blot analysis
Western blotting analysis was performed as described (MacAuley et al., 1998). Anti-SOCS3 antibody was raised in the rabbit using the peptide CYEKVTQLPGPIREFLDQYDAPL corresponding to the C-terminal portion of SOCS3. The specificity of the antibody was verified using SOCS3–/– placenta and SOCS3-overexpressing cells. Equal amounts of protein from each lysates were immnunoprecipitated with anti-SOCS3 antibody and separated by SDS–PAGE using 12% separating gels. Proteins were transferred to PVDF membranes and the membranes were incubated with anti-SOCS3 antibody after blocking. The membranes were washed using Tris-buffered saline–Tween-20 (TBS-T), then incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody. The signal was visualized by enhanced chemiluminescence (ECL; Amersham). The same amount of protein was loaded and blotted using anti-β-actin antibody (Sigma) for the loading control.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We would like to thank the laboratory members for helpful discussions and advice, ARC members Lisa Emmons, John Raucci, Christie Nagy, John Swift and Gerard Grosveld for assistance, Marina Gertsenstein and Andras Nagy for technical advice in tetraploid rescue experiments, Kelli Boyd for pathological analysis of the heart, and Jyh-Rong Chao, Toshihiko Iizuka, Catriona McKay, Kristen Rothammer, Neena Carpino, Linda Snyder, Juliana Nune, Fran Allen, Dorothy Bush, Yasuhumi Shigeyoshi and Satoshi Tanaka for technical support and advice. Y.T. especially thanks S.T.Jou, H.Iwakabe and M.Takahashi for support and encouragement. This work is supported by the Uehara Memorial Foundation to Y.T, the Cancer Center CORE grant to J.N.I and by the American Lebanese Syrian Associated Charities (ALSAC).
References
- Akira S. (2000) Roles of STAT3 defined by tissue-specific gene targeting. Oncogene, 19, 2607–2611. [DOI] [PubMed] [Google Scholar]
- Alexander W.S. et al. (1999) SOCS1 is a critical inhibitor of interferon γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell, 98, 597–608. [DOI] [PubMed] [Google Scholar]
- Arman E., Haffner-Krausz,R., Chen,Y., Heath,J.K. and Lonai,P. (1998) Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc. Natl Acad. Sci. USA, 95, 5082–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck F., Erler,T., Russell,A. and James,R. (1995) Expression of Cdx-2 in the mouse embryo and placenta: possible role in patterning of the extra-embryonic membranes. Dev. Dyn., 204, 219–227. [DOI] [PubMed] [Google Scholar]
- Bischof P., Haenggeli,L. and Campana,A. (1995) Effect of leukemia inhibitory factor on human cytotrophoblast differentiation along the invasive pathway. Am. J. Reprod. Immunol., 34, 225–230. [DOI] [PubMed] [Google Scholar]
- Bjorbaek C., Elmquist,J.K., Frantz,J.D., Shoelson,S.E. and Flier,J.S. (1998) Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol. Cell, 1, 619–625. [DOI] [PubMed] [Google Scholar]
- Bjorbaek C., Elmquist,J.K., El-Haschimi,K., Kelly,J., Ahima,R.S., Hileman,S. and Flier,J.S. (1999) Activation of SOCS-3 messenger ribonucleic acid in the hypothalamus by ciliary neurotrophic factor. Endocrinology, 140, 2035–2043. [DOI] [PubMed] [Google Scholar]
- Cheng A.K. and Robertson,E.J. (1995) The murine LIM-kinase gene (limk) encodes a novel serine threonine kinase expressed predominantly in trophoblast giant cells and the developing nervous system. Mech. Dev., 52, 187–197. [DOI] [PubMed] [Google Scholar]
- Cross J.C. (2000) Genetic insights into trophoblast differentiation and placental morphogenesis. Semin. Cell Dev. Biol., 11, 105–113. [DOI] [PubMed] [Google Scholar]
- Darnell J.E. Jr (1997) STATs and gene regulation. Science, 277, 1630–1635. [DOI] [PubMed] [Google Scholar]
- Duval D., Reinhardt,B., Kedinger,C. and Boeuf,H. (2000) Role of suppressors of cytokine signaling (Socs) in leukemia inhibitory factor (LIF)-dependent embryonic stem cell survival. FASEB J., 14, 1577–1584. [DOI] [PubMed] [Google Scholar]
- Faria T.N. and Soares,M.J. (1991) Trophoblast cell differentiation: establishment, characterization and modulation of a rat trophoblast cell line expressing members of the placental prolactin family. Endocrinology, 129, 2895–2906. [DOI] [PubMed] [Google Scholar]
- Guillemot F., Nagy,A., Auerbach,A., Rossant,J. and Joyner,A.L. (1994) Essential role of Mash-2 in extraembryonic development. Nature, 371, 333–336. [DOI] [PubMed] [Google Scholar]
- Hansen J.A., Lindberg,K., Hilton,D.J., Nielsen,J.H. and Billestrup,N. (1999) Mechanism of inhibition of growth hormone receptor signaling by suppressor of cytokine signaling proteins. Mol. Endocrinol., 13, 1832–1843. [DOI] [PubMed] [Google Scholar]
- Hemberger M. and Cross,J.C. (2001) Genes governing placental development. Trends Endocrinol. Metab., 12, 162–168. [DOI] [PubMed] [Google Scholar]
- Hirota H., Chen,J., Betz,U.A., Rajewsky,K., Gu,Y., Ross,J.,Jr, Muller,W. and Chien,K.R. (1999) Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell, 97, 189–198. [DOI] [PubMed] [Google Scholar]
- Ihle J.N. (1996) Janus kinases in cytokine signalling. Philos. Trans. R. Soc. Lond. B. Biol. Sci., 351, 159–166. [DOI] [PubMed] [Google Scholar]
- Ihle J.N. (2000) The challenges of translating knockout phenotypes into gene function. Cell, 102, 131–134. [DOI] [PubMed] [Google Scholar]
- Ito S., Ansari,P., Sakatsume,M., Dickensheets,H., Vazquez,N., Donnelly,R.P., Larner,A.C. and Finbloom,D.S. (1999) Interleukin-10 inhibits expression of both interferon α- and interferon γ-induced genes by suppressing tyrosine phosphorylation of STAT1. Blood, 93, 1456–1463. [PubMed] [Google Scholar]
- Kraut N., Snider,L., Chen,C.M., Tapscott,S.J. and Groudine,M. (1998) Requirement of the mouse I-mfa gene for placental development and skeletal patterning. EMBO J., 17, 6276–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs D.L. et al. (2002) SOCS-6 binds to insulin receptor substrate 4 and mice lacking the SOCS-6 gene exhibit mild growth retardation. Mol. Cell Biol., 22, 4567–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescisin K.R., Varmuza,S. and Rossant,J. (1988) Isolation and characterization of a novel trophoblast-specific cDNA in the mouse. Genes Dev., 2, 1639–1646. [DOI] [PubMed] [Google Scholar]
- MacAuley A., Cross,J.C. and Werb,Z. (1998) Reprogramming the cell cycle for endoreduplication in rodent trophoblast cells. Mol. Biol. Cell, 9, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marian A.J. and Roberts,R. (2001) The molecular genetic basis for hypertrophic cardiomyopathy. J. Mol. Cell Cardiol., 33, 655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine J.C. et al. (1999a) SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell, 98, 617–627. [DOI] [PubMed] [Google Scholar]
- Marine J.C., Topham,D.J., McKay,C., Wang,D., Parganas,E., Stravopodis,D., Yoshimura,A. and Ihle,J.N. (1999b) SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell, 98, 609–616. [DOI] [PubMed] [Google Scholar]
- Metcalf D., Greenhalgh,C.J., Viney,E., Willson,T.A., Starr,R., Nicola,N.A., Hilton,D.J. and Alexander,W.S. (2000) Gigantism in mice lacking suppressor of cytokine signalling-2. Nature, 405, 1069–1073. [DOI] [PubMed] [Google Scholar]
- Nagy A. and Rossant,J. (1993) Production of completely ES cell-derived fetuses. In Joyner,A. (ed.), Gene Targeting: A Practical Approach. IRL Press at Oxford University Press, Oxford, UK, pp. 147–149.
- Negoro S. et al. (2001) Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation, 104, 979–981. [DOI] [PubMed] [Google Scholar]
- Nicholson S.E. et al. (1999) Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J., 18, 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley P., Anson-Cartwright,L. and Cross,J.C. (1998) The Hand1 bHLH transcription factor is essential for placentation and cardiac morphogenesis. Nat. Genet., 18, 271–275. [DOI] [PubMed] [Google Scholar]
- Roberts A.W., Robb,L., Rakar,S., Hartley,L., Cluse,L., Nicola,N.A., Metcalf,D., Hilton,D.J. and Alexander,W.S. (2001) Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc. Natl Acad. Sci. USA, 98, 9324–9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A., Yasukawa,H., Suzuki,A., Kamizono,S., Syoda,T., Kinjyo,I., Sasaki,M., Johnston,J.A. and Yoshimura,A. (1999) Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells, 4, 339–351. [DOI] [PubMed] [Google Scholar]
- Starr R. and Hilton,D.J. (1998) SOCS: suppressors of cytokine signalling. Int. J. Biochem. Cell Biol., 30, 1081–1085. [DOI] [PubMed] [Google Scholar]
- Starr R. et al. (1997) A family of cytokine-inducible inhibitors of signalling. Nature 387, 917–921. [DOI] [PubMed] [Google Scholar]
- Stewart C.L., Kaspar,P., Brunet,L.J., Bhatt,H., Gadi,I., Kontgen,F. and Abbondanzo,S.J. (1992) Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature, 359, 76–79. [DOI] [PubMed] [Google Scholar]
- Stoiber D., Kovarik,P., Cohney,S., Johnston,J.A., Steinlein,P. and Decker,T. (1999) Lipopolysaccharide induces in macrophages the synthesis of the suppressor of cytokine signaling 3 and suppresses signal transduction in response to the activating factor IFN-γ. J. Immunol., 163, 2640–2647. [PubMed] [Google Scholar]
- Takeda K., Noguchi,K., Shi,W., Tanaka,T., Matsumoto,M., Yoshida,N., Kishimoto,T. and Akira,S. (1997) Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl Acad. Sci. USA, 94, 3801–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S., Kunath,T., Hadjantonakis,A.K., Nagy,A. and Rossant,J. (1998) Promotion of trophoblast stem cell proliferation by FGF4. Science, 282, 2072–2075. [DOI] [PubMed] [Google Scholar]
- Threadgill D.W. et al. (1995) Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science, 269, 230–234. [DOI] [PubMed] [Google Scholar]
- Tullio A.N., Accili,D., Ferrans,V.J., Yu,Z.X., Takeda,K., Grinberg,A., Westphal,H., Preston,Y.A. and Adelstein,R.S. (1997) Nonmuscle myosin II-B is required for normal development of the mouse heart. Proc. Natl Acad. Sci. USA, 94, 12407–12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara Y., Minowa,O., Mori,C., Shiota,K., Kuno,J., Noda,T. and Kitamura,N. (1995) Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature, 373, 702–705. [DOI] [PubMed] [Google Scholar]
- Uozumi H. et al. (2001) gp130 plays a critical role in pressure overload-induced cardiac hypertrophy. J. Biol. Chem., 276, 23115–23119. [DOI] [PubMed] [Google Scholar]
- Ware C.B. et al. (1995) Targeted disruption of the low-affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development, 121, 1283–1299. [DOI] [PubMed] [Google Scholar]
- Yasukawa H. et al. (2001) Suppressor of cytokine signaling-3 is a biomechanical stress-inducible gene that suppresses gp130-mediated cardiac myocyte hypertrophy and survival pathways. J. Clin. Invest., 108, 1459–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A. (1998) The CIS family: negative regulators of JAK–STAT signaling. Cytokine Growth Factor. Rev., 9, 197–204. [DOI] [PubMed] [Google Scholar]