Key stages in mammary gland development: The mammary end bud as a motile organ (original) (raw)
Abstract
In the rodent, epithelial end buds define the tips of elongating mammary ducts. These highly motile structures undergo repeated dichotomous branching as they aggressively advance through fatty stroma and, turning to avoid other ducts, they finally cease growth leaving behind the open, tree-like framework on which secretory alveoli develop during pregnancy. This review identifies the motility of end buds as a unique developmental marker that represents the successful integration of systemic and local mammotrophic influences, and covers relevant advances in ductal growth regulation, extracellular matrix (ECM) remodeling, and cell adhesion in the inner end bud. An unexpected growth-promoting synergy between insulin-like growth factor-1 and progesterone, in which ducts elongate without forming new end buds, is described as well as evidence strongly supporting self-inhibition of ductal elongation by end-bud-secreted transforming growth factor-β acting on stromal targets. The influence of the matrix metalloproteinase ECM-remodeling enzymes, notably matrix metalloproteinase-2, on end bud growth is discussed in the broader context of enzymes that regulate the polysaccharide-rich glycosaminoglycan elements of the ECM. Finally, a critical, motility-enabling role for the cellular architecture of the end bud is identified and the contribution of cadherins, the netrin/neogenin system, and ErbB2 to the structure and motility of end buds is discussed.
Introduction
The mammary gland develops in two structurally and functionally distinct phases: ductal and secretory. In the ductal phase a branched 'tree' arises as the epithelial mammary anlagen invades fatty stroma and, between parturition and adolescence, undergoes repeated dichotomous branching, creating the tubes that deliver milk to the nipple (Fig. 1a). The resulting 'open' ductal architecture permits infilling of the interductal stroma by secretory alveoli during the second phase of development initiated by pregnancy. The open mammary duct system is unique in comparison with other branched organs such as the lung and kidney, whose epithelial elements are tightly packed.
Figure 1.
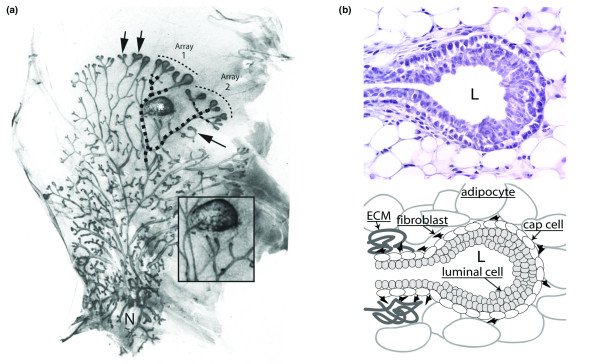
Photomicrographs illustrating motility and histoarchitecture of end buds. (a) Natural and experimentally induced motility 'behavior' of end buds in the mammary ductal system of a 5-week-old nulliparous mouse. The 'open' ductal architecture of the mammary tree leaves 80% or more of the gland epithelium-free. Large terminal end buds identify the most actively growing region of the gland (top arrows), and progressively smaller lateral end buds extend to each side of the center, indicating slowed forward growth as the end bud encounters a thinning fat pad. End buds may also reverse direction to grow back into accommodating stroma (side arrow). Bifurcating end buds (top arrows) are arrayed along the growth front. Original magnification approx. ×12. (b) Cross-section through end bud with accompanying diagram. End buds are bilayered structures; an outer layer of myoepithelial progenitor cells (cap cells) overlays a multilayered mass of luminal cells fated to form the walls of the ductal lumen (L). Stained with hematoxylin and eosin. Original magnification approx. ×300.
As we will discuss in this review, the development of the mammary ductal tree presents fascinating and challenging problems to the developmental biologist. Beyond this, there is a true urgency to improve understanding of ductal growth because 90% or more of human mammary cancers are ductal in origin. The mammary end bud is the icon of the ductal phase of mammary development in the rodent, at one and the same time the most familiar and the most mysterious of structures. Terminal end buds are familiar in rodents as the bulbous, epithelial structures at the tips of ducts strategically located facing an open expanse of fat pad (Fig. 1a, top arrows). This location and their sensitivity to mammotrophic hormones such as estrogen and growth hormone (GH) marked them early on as the engines of ductal elongation, and their implied motility was understood to be the basis for the open architecture of the mammary ductal system [1,2]. In contrast, lateral buds develop along mature ducts and are constrained in growth by the lack of open territory (Fig. 1a, side arrow). Apart from location, the general architecture and function of terminal and lateral end buds are the same.
End buds, like the ducts they give rise to, have a 'tube within a tube' structure with an outer layer of undifferentiated cap cells and inner layers of luminal epithelial cells (Fig. 1b); both layers of the end bud have high rates of mitosis, consistent with a motile organ dedicated to ductal morphogenesis (Fig. 2) [3]. From these and other studies, forward extension rates in end buds of up to 0.5 mm per day have been estimated during maximal growth in puberty (GB Silberstein and CW Daniel, unpublished data). The purpose of this review is threefold: first, to draw attention to complex 'motility behaviors' in end buds, such as bifurcation, turning, and growth cessation; second, to revisit ductal morphogenesis and epithelial stromal interactions from the point of view of the end bud; and third, to review the mechanisms mediating adhesion between cells within the end bud and their crucial role in organizing forward movement. We will first address recent advances in our understanding of ductal growth regulation, then consider extracellular matrix (ECM) remodeling, and conclude with a consideration of the molecular mechanisms that ensure the integrity of the internal layers of the end bud as it moves through the fat pad.
Figure 2.
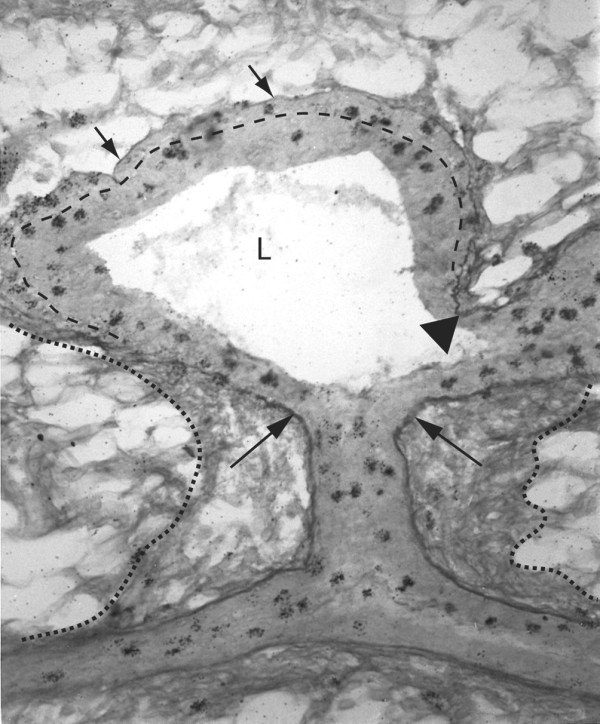
Photomicrograph of a longitudinal section through an end bud and its subtending duct. A lateral end bud stained to reveal sulfated glycosaminoglycans and mitotic cells with the cap cell layer is indicated by the dashed line. Constriction of the end bud to ductal dimensions coincides with induction of a collagenous extracellular matrix along the end bud flank. Note that this sheath is continuous with the subtending duct as well as the duct of origin (dotted lines). The basal lamina along the end bud flank (large arrows) as well as in the cleft of a bifurcation (triangle) stained deeply for sulfated glycosaminoglycans (Alcian blue stain). This contrasts with weakly stained basal lamina around the end bud tip indicative of non-sulfated hyaluronate (short arrows). Silver grains (dark spots) are from tritiated thymidine autoradiography and mark mitotic cells in the end bud and subjacent ducts. Original magnification approx. ×300.
End bud motility
The mystery of the end bud has always been in the details of its motility. Over the past decade these have begun to come into focus and are now known to encompass endocrine and local growth-regulatory signals, stromal–epithelial interactions, ECM remodeling, and dynamic adhesions within the end bud that maintain the bilayered structure. Relatively recent reviews have addressed the growth and morphogenesis of the end bud as part of the broader picture of postnatal mammary development without focusing on motility itself [4,5]. In the absence of real-time photographic evidence, the motility of end buds has always been inferred from static photomicrographs. To demonstrate the complexity of end bud motility experimentally, we surgically placed a plastic obstacle into the fat pad of a mouse in the path of advancing end buds a week before killing (Fig. 1a, asterisk). The resulting branching pattern of two affected ducts (dashed lines) illustrates how a combination of end bud forward movement, avoidance of nearby obstacles, and bifurcation was adapted to achieve open architecture. End bud array no. 1 arose from an end bud that grew around the obstacle and then bifurcated twice; array no. 2 arose from an end bud that passed beneath the plastic, sending two lateral branches upward before leaving the proximity of the plastic and twice bifurcating. Despite the obstacle, therefore, end bud motility achieved ductal spacing similar to the unaffected part of the gland. While these types of behavior can be inferred by experiments such as these, the development of methods in vitro to culture mammary explants containing end buds such that extending end buds could be imaged in real time would constitute a major advance.
Growth stimulation and motility of end buds
Although motility clearly includes subtle 'behaviors' such as turning and bifurcation, the crucial consideration is forward movement. In broad terms this could be due to two mechanisms: end buds pulling themselves forward by cellular extensions such as filopodia, or pushing forward due to mitosis and an increase in cell mass. Electron microscopy studies show the imprints of adipocytes compressed onto an unbroken basal lamina covering the tip of the end bud [3]. This, and the absence of filopodia that might pull the duct forward or any evidence of enzymatic 'clearing' of a stromal path, indicates that forward movement of the end bud must depend on channeling the internal force of dividing cells in a forward direction. Motility and ductal growth of the end bud are therefore synonymous and must be wholly explainable by the balance of positive and negative growth regulation.
The endocrine hormones estrogen and GH were the only known ductal mammogens until genetically engineered mice lacking ovarian hormone receptors or specific growth factors enabled crucial experiments demonstrating that these hormones did not act directly on the duct, but rather through stroma-derived growth factors (reviewed in [5]). In seminal experiments, Cuhna and colleagues co-transplanted estrogen receptor knockout (ERKO) mammary epithelium with wild-type mammary stroma and discovered that stromal, not epithelial, estrogen receptors were necessary for ductal development; estrogen therefore stimulated ductal growth through secondary, paracrine effectors [6]. EGF can substitute for estrogen in stimulating end bud growth and was a candidate estrogen intermediary; however, when EGF receptor knockout mammary tissue was used in epithelial–stromal co-transplant experiments, EGF receptor-null stroma could not support ductal growth by wild-type epithelium. Estrogen-stimulated stromal EGF must therefore generate other stromal mammogens whose identity is unknown [7].
During the same period as the estrogen-centered investigations, evidence was developed by the Kleinberg group that GH also stimulated end bud growth through a stromal intermediary, in this case insulin-like growth factor-1 (IGF-1). In hypophysectomized mice, exogenous GH stimulated stromal, not epithelial, IGF-1 mRNA expression, and a targeted deletion of IGF-1 ablated end buds that could then be restored by exogenous IGF-1 but not by GH [8,9]. The stimulatory effects of both estrogen and GH on end buds with each hormone acting through a different stromal intermediary suggested that the two pathways might normally synergize. Synergy was in fact demonstrated by Ruan and colleagues with ovariectomized, IGF-1-knockout mice supplemented with IGF-1 alone or in combination with estrogen; the combined treatments stimulated significant end bud development, whereas estrogen alone was not stimulatory [10]. Interestingly, the pathway governing end bud bifurcation may also be linked to IGF-1 because branching, but not elongation, was reduced in IGF-1-deficient (IGF-1m/m) mammary glands [11]. The effect of progesterone on ductal growth was also investigated in the IGF-1-null model with surprising and important results [10]. As with estrogen, progesterone alone had no effect on end bud development in ovariectomized mice. In combination with IGF-1, however, progesterone stimulated ductal elongation and branching equivalent to that seen with IGF-1 plus estrogen, but without an increase in the number of end buds. The finding that progesterone can have a role in ductal elongation in the mouse mammary gland could help to explain very rapid ductal development in puberty when estrous-cycle progesterone and estrogen could act together to optimize growth.
Inhibiting ductal elongation: the case for transforming growth factor-β
The mammary stroma strongly promotes ductal growth; the smallest fragment of duct transplanted anywhere within an epithelium-free fat pad vigorously grows into a full ductal tree [12]. Logic dictates that if the stromal background is growth-promoting, then achieving patterned growth requires ducts to adjust their own extension locally by focal inhibition. Strong evidence now implicates transforming growth factor-β (TGF-β) as the primary local inhibitor of both ductal elongation and lateral branching. The inhibition of lateral branching by TGF-β secreted by ductal epithelium and acting on periductal stromal targets has been well documented and can account, in part, for the open architecture of the gland (reviewed in [4]). More recently, epithelial TGF-β was also shown to inhibit the forward movement of terminal end buds. In wild-type mammary glands, activated TGF-β was localized in the end bud by IHC; when this expression was reduced through engineered heterozygosity (TGF-β+/- epithelium), ductal elongation into wild-type stroma was accelerated [13]. Expression of a dominant-negative, TGF-β type-II receptor in the mammary stroma also caused accelerated ductal growth during puberty, confirming the stromal target for TGF-β [14].
The above results demonstrate that TGF-β must limit ductal elongation through secondary effectors, and a case can be made that these unknown factors act by inhibiting hepatocyte growth factor. Mammary hepatocyte growth factor is negatively regulated by TGF-β and its overexpression in retrovirally transduced mouse mammary epithelial cells resulted not only in increased lateral branching but also in a marked increase in the number and size of end buds [15]. A role for parathyroid hormone-related protein must also be considered in this context because it is positively regulated by TGF-β and inhibits ductal elongation when overexpressed in pubertal mice [16]. Collectively, these findings place the inhibition of each type of ductal extension, lateral branches and end buds, under what may be a common, TGF-β-dependent, circuit. This has two interesting implications. First, ductal growth stimulation, whether lateral branching or end bud extension, probably shares mechanisms designed to inhibit TGF-β action locally, possibly by modulating the activation of latent growth factor. Second, the inhibition observed when an end bud approaches the edge of the fat pad or another duct is likely to come from the advancing end bud itself and not, as speculated in earlier reviews, from nearby tissues [5].
These recent studies build on a history of research in which genetically modified mammary epithelium and stroma are recombined with wild-type counterparts in all permutations to evaluate potential ductal mammogens and also to define whether their primary site of action is the epithelium or stroma. In this way, stromal sites of action for TGF-β, parathyroid hormone releasing protein as well as estrogen and EGF receptors have been defined (reviewed in [17]).
Although these recombination experiments are powerful, they cannot define the action of particular stromal cells because they use an intact fat pad. The histologically complex fat pad comprises many cell types, not just adipocytes and fibroblasts but also endothelial and migratory white blood cells, all of which influence the dynamic ECM. A new and very creative generation of experiments has begun to address this subtlety by focusing on individual stromal components. For example, a role for eosinophils and macrophages in end bud growth has been identified by the ablation of white blood cells with radiation [18]. In addition, a novel transplant system that combines purified mouse fibroblasts with mammary epithelium will enable the testing of specific fibroblast mutations on ductal growth [17]. The ultimate challenge, then, will be to relate findings from these experiments to mechanisms controlling the cell proliferation driving end bud extension and to identify putative motility factors that affect turning and bifurcation.
End bud 'behaviors' and ECM remodeling
The forward movement of an end bud is inseparable from the drastic remodeling of the ECM that occurs along its flank (Fig. 2). There, the induction of sulfated glycosaminoglycans (SGAGs) in the basal lamina is accompanied by thickening of the ECM by type I collagen [19]. Because these changes coincide with the constriction of the end bud to ductal dimensions it seems likely that the relatively inelastic ECM girdle ultimately channels the pressure developed by cell division within the end bud forward, where no type I collagen constrains expansion. This model could also account, in part, for end bud bifurcation, in which focal induction of SGAGs in the end bud basal lamina, followed by the deposition of type I collagen, retards advancement at the induction point. To either side of focal type-1 collagen induction, newly formed lobes advance in different directions (Fig. 2). Although speculative, the turning of an end bud could depend on asymmetric deposition and remodeling of SGAG and type I collagen; collagen deposition on one side of an end bud might channel extension in the opposite direction. A possible role for asymmetric mitosis as a turning mechanism has also been considered; however, when mitotic patterns were examined with the use of thymidine autoradiography no obvious correlations between mitosis and turning were found (GB Silberstein and CW Daniel, unpublished data).
The molecular mechanisms that affect ECM remodeling around the end bud are coming into focus and fall broadly into two categories: ECM-active growth factors and matrix-modifying enzymes. TGF-β1 remains the primary candidate for inducing the matrix remodeling described above. It is well known for its 'matrix sparing' activity because it inhibits matrix-degrading proteases and induces SGAGs and other matrix elements. When TGF-β was delivered by slow-release implants in the vicinity of end buds, it induced SGAG and type I collagen around end bud tips that was indistinguishable from that seen on the flank [4,20].
Research to understand the role of ECM-remodeling enzymes in the motility of end buds is in its infancy. In addition to proteinases, such as the matrix metalloproteinases (MMPs), matrix glycosaminoglycan (GAG)-degrading glycolytic enzymes (for example β-glucuronidase) and polysaccharide synthetases (glycosyltransferases) must also have crucial functions. MMPs are by far the best studied of the matrix-remodeling enzymes and comprise a diverse family of stromal enzymes with overlapping substrates that collectively can degrade all protein and glycoprotein elements of the ECM [21]. Inhibition of MMPs by a small-molecule inhibitor (GM6001) or by transgenic overexpression of a natural MMP inhibitor, tissue inhibitor of metalloproteinase (TIMP), retarded the extension of end buds and reduced their total number [22]. MMP-2 can degrade basal lamina proteins of end buds, such as type IV collagen and laminin, and can also modulate the activation of latent TGF-β. MMP-2 is expressed in the stroma in front of end buds, where it can affect ductal growth and, indeed, genetic ablation of MMP-2 retarded end bud extension. Interestingly, this effect was not attributable to a build-up of undegraded ECM proteins, nor did MMP-2 ablation affect the number of end buds or their cell proliferation, but rather rates of apoptosis were approximately doubled, indicating that MMP-2 normally supports cell survival in the end bud.
The turnover of polysaccharides in the basal lamina and ECM around the tip of the end bud has been well documented, and enzymatic degradation of hyaluronate in situ disrupts cellular organization in the end bud [19,23]. Localization of β-glucuronidase and _N_-acetylglucosaminidase by activity staining in situ with chromogenic substrates revealed strong concentrations of these GAG-degrading enzymes in the cap and myoepithelial cells (GB Silberstein and CW Daniel, unpublished data). This suggests a role for these and possibly other glycosidases in basal lamina dynamics and suggests that further investigation of these enzymes during ductal extension will be fruitful. Finally, the incorporation of polysaccharides into elements of the basal lamina–ECM complex depends on specific glycosyltransferases. Although nothing is currently known about the biosynthetic roles of these enzymes in ECM dynamics in the mammary gland, the fact that they that can also serve as cell-surface receptors for ECM carbohydrate substrates is intriguing. Mice lacking cell-surface β1,4-galactosyltransferase, for example, exhibited increased branching accompanied by perturbation of laminin and the expression of certain MMPs [24].
Dynamic integrity: adhesion within the end bud
In addition to growth regulation and ECM remodeling, end bud motility and ductal outgrowth rely on the integrity of the end bud because perturbations that disrupt cell adhesion within an end bud inhibit ductal extension. There are at least two examples in the literature of different ways in which disorganized end bud structure can compromise forward growth. In one example, the disruption of cell–cell contacts within an end bud inhibited cell proliferation and ductal extension, suggesting that normal cell contacts are required to mediate the growth signals from mammogenic hormones such as estrogen and GH. In two other examples, disruption of cell–cell contacts inhibited forward growth independently of cell proliferation, suggesting that additional mechanisms, involving tissue integrity, also control end bud outgrowth. Together these examples highlight the idea that, in addition to the essential relay of information that occurs between the epithelial and stromal compartments, proper communication between cells within the epithelium is crucial for normal ductal development.
One of the first examples demonstrating the essential nature of proper cell–cell contacts involved the perturbation of cadherin function. Cadherins are calcium-dependent cell adhesion proteins that mediate interactions between cells of the end bud. At least two types of cadherin are expressed in the end bud: E-cadherin (Cdh1) is expressed by luminal cells that constitute the body of the end bud, and P-cadherin (Cdh3) is expressed by cap cells that form the outer layer (Fig. 3) [25]. Because homozygous mutations in the Cdh1 gene resulted in early embryonic lethality, functional studies were performed by surgically inserting slow-release implants containing function-blocking antibodies ahead of advancing end buds. Within 12 hours of implantation, luminal cells in the body of the end bud exhibited disrupted cell–cell adhesion and epithelial DNA synthesis abruptly declined. After 72 hours the function-blocking antibodies were depleted, leading to the restoration of normal tissue morphology and recovered rates of DNA synthesis. These results showed that normal cell contacts are required for the maintenance of tissue architecture. Moreover, these experiments demonstrated that the high rates of DNA synthesis responsible for driving cell proliferation rely on cadherin-mediated cell–cell contacts. The authors also used function-blocking antibodies directed against CDH3 [25]. Interestingly, this treatment resulted in only a modest disruption in the cap cell layer of the end bud that was accompanied by only a slight decline in DNA synthesis. Studies on genetically engineered mice lacking Cdh3 yielded a similar result by showing that loss of CDH3 had no discernible affect on ductal outgrowth or branching morphogenesis [26]. Together, these results suggest that tissue integrity is required for epithelial proliferation, but modest perturbations in end bud structure can be accommodated.
Figure 3.
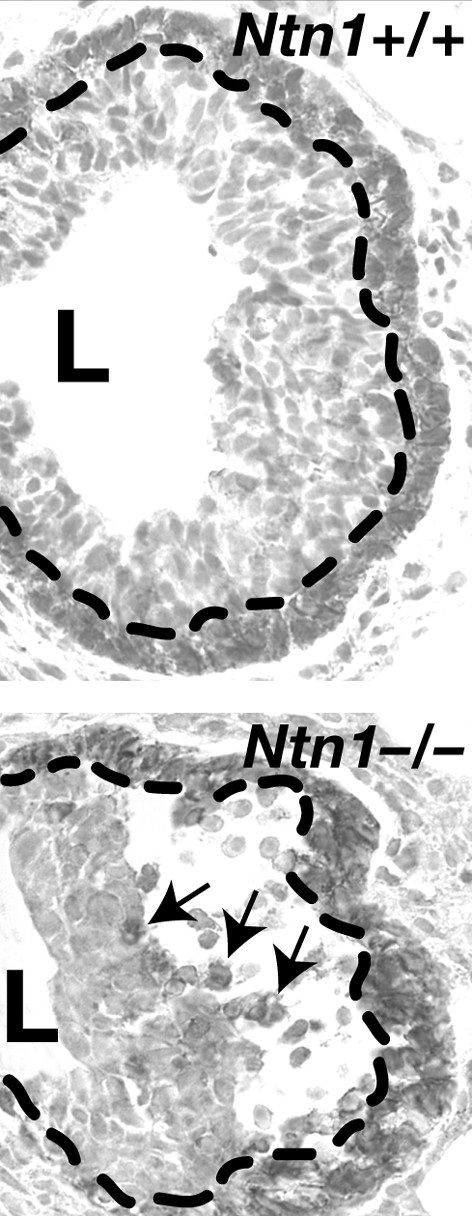
Loss of Ntn1 disorganizes end buds. The Ntn1+/+ end bud (top) displays normal CDH3 staining of the cap cell layer at the tip of the end bud (delineated by the dashed line). In contrast the _Ntn1_-/- end bud (bottom) displays a loss of adhesion between the cap and luminal cell layers, with a large space forming under the cap cell layer (delineated by the dashed line). This space fills with dissociated cap cells (arrows show three examples) that either die by apoptosis or migrate inappropriately into the body of the end bud. Original magnification approx. ×300.
Although CDH1 and CDH3 mediate interactions between cells of the luminal and cap cell compartments, respectively, recent data suggest that Netrin-1 (Ntn1) mediates interactions between these two compartments. NTN1 was originally identified as a guidance cue for developing neurons [27], but in the mammary gland this cue seems to function adhesively at short range, rather than instructively at long range [28]. NTN1 is expressed by luminal cells [28]; although a secreted protein, it is not freely diffusible but is instead immobilized in association with cell membranes or components of the ECM [29]. The NTN1 receptor, Neogenin (Neo1), is expressed in a complementary pattern by overlying cap cells [28]. Glands harboring homozygous loss-of-function mutations in either gene displayed slower growth that corresponded to significantly disorganized end buds [28]. These growth structures exhibited loss of adhesion between the cap and luminal cell layers, with large spaces forming under the cap cell layer (Fig. 3). These spaces filled with dissociated cap cells that either died by apoptosis or migrated inappropriately either into the body of the end bud or into the stromal compartment. Rates of DNA synthesis in mutant and wild-type glands were similar, suggesting that disrupting contacts between the cap and luminal cell layers inhibited mammary outgrowth by elevating cell death, not by inhibiting cell proliferation. These data suggest that NTN1, signaling though NEO1, maintains the integrity of the end bud by mediating contacts between cap and luminal epithelial cells, and these contacts are required for robust forward growth of the mammary tree.
A second example demonstrating that disruption of cell–cell contacts inhibits forward growth, independently of cell proliferation, is in glands harboring homozygous loss-of-function mutations in Erbb2. ERBB2 is an orphan receptor that forms heteromers with ligand-activated EGFR, ERBB3, and ERBB4, allowing it to respond to EGF and neuregulin-like growth factors. In _Erbb2_-/- mammary glands, there was a marked reduction in ductal outgrowth into the mammary fat pad that was accompanied by increased lateral branching [30]. There were no apparent changes in cell proliferation or apoptosis to account for this lack of mammary outgrowth; it therefore seemed that lateral branching occurred at the expense of forward penetration of the end bud into the fat pad [30]. Because end buds are the structure responsible for generating forward growth, the authors examined end bud structure and discovered that _Erbb2_-/- end buds were small and disorganized. Although they had a normal cap cell layer, exaggerated spaces developed between the cap and luminal cell layers. The small number of luminal cells present in the bodies of end buds were loosely packed and seemed disorganized, probably as a result of the inappropriate influx of cap cells into this compartment. The apparent phenotypic similarities between _Erbb2_-/-, _Ntn1_-/-, and _Neo1_-/- glands suggested that loss of ERBB2 leads to downregulation of NTN1 or NEO1, but expression of these proteins seemed normal in _Erbb2_-/- glands [30]. Taken together, these results suggest that multiple pathways regulate adhesion between cap and luminal epithelial cells, and that disrupting the integrity of these layers severely compromises forward growth of the mammary tree.
Thus, numerous signaling pathways must interact to drive and channel the forward growth and motility of end buds and this, in turn, is responsible for establishing the primary structure of the mammary tree. Although mammogenic hormones stimulate the cell proliferation required to generate enough epithelial mass to force end buds forward, the examples above highlight a second requirement. The structural integrity of the end bud is essential for both receiving mammogenic growth signals and organizing forward movement of the end bud into the fat pad. Together these examples demonstrate how function follows form, and generating a mammary ductal tree requires proper cell–cell contacts within end buds coordinated with integrated epithelial–stromal and epithelial–ECM communication.
Abbreviations
ECM = extracellular matrix; GAG = glycosaminoglycan; GH = growth hormone; IGF = insulin-like growth factor; MMP = matrix metalloproteinase; SGAG = sulfated glycosaminoglycan; TGF-β = transforming growth factor-β.
Competing interests
The author(s) declare that they have no competing interests.
Note
This article is part of a review series on Key stages in mammary gland development, edited by Charles Streuli.
Other articles in the series can be found online at http://breast-cancer-research.com/articles/review-series.asp?series=bcr_keystages.
Acknowledgments
Acknowledgements
This work was supported by a research scholar grant no. RSG0218001MGO from the American Cancer Society (LH), research grant 10PB-0188 from the California Breast Cancer Research Program (LH) and research grant NIH CA091996 (GBS).
References
- Nandi S. Endocrine control of mammary gland development and function in the C3H/He Crgl mouse. J Natl Cancer Inst. 1958;21:1039–1063. [PubMed] [Google Scholar]
- Lyons WR. Hormonal synergism in mammary growth. Proc R Soc Lond B. 1958;149:303–325. doi: 10.1098/rspb.1958.0071. [DOI] [PubMed] [Google Scholar]
- Williams JM, Daniel CW. Mammary ductal elongation: differentiation of myoepithelium during branching morphogenesis. Dev Biol. 1983;97:274–290. doi: 10.1016/0012-1606(83)90086-6. [DOI] [PubMed] [Google Scholar]
- Silberstein GB. Postnatal mammary gland morphogenesis. Microsc Res Tech. 2001;52(2):155–162. doi: 10.1002/1097-0029(20010115)52:2<155::AID-JEMT1001>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Silberstein G. Role of the stroma in mammary development. Breast Cancer Res. 2001;3:218–224. doi: 10.1186/bcr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha GR, Young P, Hom YK, Cooke PS, Taylor JA, Lubahn DB. Elucidation of a role for stromal steroid hormone receptors in mammary gland growth and development using tissue recombinations. J Mamm Gland Biol Neoplasia. 1997;2:393–402. doi: 10.1023/A:1026303630843. [DOI] [PubMed] [Google Scholar]
- Wiesen JF, Young P, Werb Z, Cunha GR. Signaling through the stromal epidermal growth factor receptor is necessary for mammary ductal development. Development. 1999;126:335–344. doi: 10.1242/dev.126.2.335. [DOI] [PubMed] [Google Scholar]
- Walden PD, Ruan W, Feldman M, Kleinberg DL. Evidence that the mammary fat pad mediates the action of growth hormone in mammary gland development. Endocrinology. 1998;139:659–662. doi: 10.1210/en.139.2.659. [DOI] [PubMed] [Google Scholar]
- Ruan W, Powell-Braxton L, Kopchick JJ, Kleinberg DL. Evidence that insulin-like growth factor I and growth hormone are required for prostate gland development. Endocrinology. 1999;140:1984–1989. doi: 10.1210/en.140.5.1984. [DOI] [PubMed] [Google Scholar]
- Ruan W, Monaco ME, Kleinberg DL. Progesterone stimulates mammary gland ductal morphogenesis by synergizing with and enhancing insulin-like growth factor-I action. Endocrinology. 2005;146:1170–1178. doi: 10.1210/en.2004-1360. [DOI] [PubMed] [Google Scholar]
- Richards RG, Klotz DM, Walker MP, Diaugustine RP. Mammary gland branching morphogenesis is diminished in mice with a deficiency of insulin-like growth factor-I (IGF-I), but not in mice with a liver-specific deletion of IGF-I. Endocrinology. 2004;145:3106–3110. doi: 10.1210/en.2003-1112. [DOI] [PubMed] [Google Scholar]
- Daniel CW, DeOme KB, Young JT, Blair PB, Faulkin LJ. The in vivo lifespan of normal and preneoplastic mouse mammary glands: a serial transplantation study. Proc Natl Acad Sci USA. 1968;61:52–60. doi: 10.1073/pnas.61.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewan KB, Shyamala G, Ravani SA, Tang Y, Akhurst R, Wakefield L, Barcellos-Hoff MH. Latent transforming growth factor-beta activation in mammary gland: regulation by ovarian hormones affects ductal and alveolar proliferation. Am J Pathol. 2002;160:2081–2093. doi: 10.1016/s0002-9440(10)61158-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley MR, Bowtell D, Serra R. TGF-β, c-Cbl, and PDGFR-α in the mammary stroma. Dev Biol. 2005;279:58–72. doi: 10.1016/j.ydbio.2004.11.034. [DOI] [PubMed] [Google Scholar]
- Kamalati T, Niranjan B, Yant J, Buluwela L. HGF/SF in mammary epithelial growth and morphogenesis: in vitro and in vivo models. J Mamm Gland Biol Neoplasia. 1999;4:69–77. doi: 10.1023/A:1018756620265. [DOI] [PubMed] [Google Scholar]
- Dunbar ME, Dann P, Brown CW, Van Houton J, Dreyer B, Philbrick WP, Wysolmerski JJ. Temporally regulated overexpression of parathyroid hormone-related protein in the mammary gland reveals distinct fetal and pubertal phenotypes. J Endocrinol. 2001;171:403–416. doi: 10.1677/joe.0.1710403. [DOI] [PubMed] [Google Scholar]
- Parmar H, Cunha GR. Epithelial–stromal interactions in the mouse and human mammary gland in vivo. Endocr Relat Cancer. 2004;11:437–458. doi: 10.1677/erc.1.00659. [DOI] [PubMed] [Google Scholar]
- Gouon-Evans V, Rothenberg ME, Pollard JW. Postnatal mammary gland development requires macrophages and eosinophils. Development. 2000;127:2269–2282. doi: 10.1242/dev.127.11.2269. [DOI] [PubMed] [Google Scholar]
- Silberstein GB, Daniel CW. Glycosaminoglycans in the basal lamina and extracellular matrix of the developing mouse mammary duct. Dev Biol. 1982;90:215–222. doi: 10.1016/0012-1606(82)90228-7. [DOI] [PubMed] [Google Scholar]
- Silberstein GB, Strickland P, Coleman S, Daniel CW. Epithelium-dependent extracellular matrix synthesis in transforming growth factor-β1-growth-inhibited mouse mammary gland. J Cell Biol. 1990;110:2209–2219. doi: 10.1083/jcb.110.6.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman BS, Sternlicht MD, Lund LR, Alexander CM, Mott J, Bissell MJ, Soloway P, Itohara S, Werb Z. Site-specific inductive and inhibitory activities of MMP-2 and MMP-3 orchestrate mammary gland branching morphogenesis. J Cell Biol. 2003;162:1123–1133. doi: 10.1083/jcb.200302090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein GB, Daniel CW. Elvax 40P implants: sustained, local release of bioactive molecules influencing mammary ductal development. Dev Biol. 1982;93:272–278. doi: 10.1016/0012-1606(82)90259-7. [DOI] [PubMed] [Google Scholar]
- Steffgen K, Dufraux K, Hathaway H. Enhanced branching morphogenesis in mammary glands of mice lacking cell surface β1,4-galactosyltransferase. Dev Biol. 2002;244:114–133. doi: 10.1006/dbio.2002.0599. [DOI] [PubMed] [Google Scholar]
- Daniel CW, Strickland P, Friedmann Y. Expression and functional role of E- and P-cadherins in mouse mammary ductal morphogenesis and growth. Dev Biol. 1995;169:511–519. doi: 10.1006/dbio.1995.1165. [DOI] [PubMed] [Google Scholar]
- Radice GL, Ferreira-Cornwell MC, Robinson SD, Rayburn H, Chodosh LA, Takeichi M, Hynes RO. Precocious mammary gland development in P-cadherin-deficient mice. J Cell Biol. 1997;139:1025–1032. doi: 10.1083/jcb.139.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy TE. Cellular mechanisms of netrin function: long-range and short-range actions. Biochem Cell Biol. 2000;78:569–575. doi: 10.1139/bcb-78-5-569. [DOI] [PubMed] [Google Scholar]
- Srinivasan K, Strickland P, Valdes A, Shin GC, Hinck L. Netrin-1/neogenin interaction stabilizes multipotent progenitor cap cells during mammary gland morphogenesis. Dev Cell. 2003;4:371–382. doi: 10.1016/S1534-5807(03)00054-6. [DOI] [PubMed] [Google Scholar]
- Kappler J, Franken S, Junghans U, Hoffmann R, Linke T, Muller HW, Koch KW. Glycosaminoglycan-binding properties and secondary structure of the C-terminus of netrin-1. Biochem Biophys Res Commun. 2000;271:287–291. doi: 10.1006/bbrc.2000.2583. [DOI] [PubMed] [Google Scholar]
- Jackson-Fisher AJ, Bellinger G, Ramabhadran R, Morris JK, Lee KF, Stern DF. ErbB2 is required for ductal morphogenesis of the mammary gland. Proc Natl Acad Sci USA. 2004;101:17138–17143. doi: 10.1073/pnas.0407057101. [DOI] [PMC free article] [PubMed] [Google Scholar]