Subdivision of Large Introns in Drosophila by Recursive Splicing at Nonexonic Elements (original) (raw)
Abstract
Many genes with important roles in development and disease contain exceptionally long introns, but special mechanisms for their expression have not been investigated. We present bioinformatic, phylogenetic, and experimental evidence in Drosophila for a mechanism that subdivides many large introns by recursive splicing at nonexonic elements and alternative exons. Recursive splice sites predicted with highly stringent criteria are found at much higher frequency than expected in the sense strands of introns >20 kb, but they are found only at the expected frequency on the antisense strands, and they are underrepresented within introns <10 kb. The predicted sites in long introns are highly conserved between Drosophila melanogaster and Drosophila pseudoobscura, despite extensive divergence of other sequences within the same introns. These patterns of enrichment and conservation indicate that recursive splice sites are advantageous in the context of long introns. Experimental analyses of in vivo processing intermediates and lariat products from four large introns in the unrelated genes kuzbanian, outspread, and Ultrabithorax confirmed that these introns are removed by a series of recursive splicing steps using the predicted nonexonic sites. Mutation of nonexonic site RP3 within Ultrabithorax also confirmed that recursive splicing is the predominant processing pathway even with a shortened version of the intron. We discuss currently known and potential roles for recursive splicing.
INTRONS in metazoan genes can be extremely large. About 10% of introns in humans and 5% in Drosophila are >10 kb (Deutsch and Long 1999; Saxonov et al. 2000). The largest in both organisms exceed 100 kb, reaching 3 Mb in the Y-linked dynein β heavy chain genes of Drosophila species (Kurek et al. 2000; Reugels et al. 2000). Exceptionally long introns are found in genes with a wide variety of cellular and developmental functions, including genes with important roles in human disease such as dystrophin, the cystic fibrosis transmembrane conductance regulator CFTR, retinoblastoma, APC and NF1 tumor suppressor genes, and the ABL and PLZF proto-oncogenes.
Large introns can contain transcriptional regulatory elements and nested genes, but intron size itself can influence gene expression and evolution. For example, the time consumed by transcription through a long intron can specify a functionally significant delay in the appearance of processed mRNA and protein products relative to the developmental signals that first activate transcription of the gene (Shermoen and O'Farrell 1991; Rothe et al. 1992; Thummel 1992; Ruden and Jäckle 1995). A striking consequence in Drosophila is the inability to complete transcription of genes like Ultrabithorax (Ubx) or knirps-related during the short cell cycles of the early embryo, when they are first activated in response to maternal and early zygotic factors. Intron size can also affect the balance of alternative splicing through the interplay between transcript elongation rate and splicing kinetics (reviewed by Neugebauer 2002; Proudfoot 2003). By increasing the frequency of recombination, long introns may also reduce interference among sites that are under selection pressure in flanking exons (Comeron and Kreitman 2000, 2002).
Although large introns can provide special opportunities for regulation of gene expression, they may pose problems if they increase the potential for processing errors and premature termination of transcription. The likelihood of errors may be reduced by mechanisms that also operate in smaller genes to ensure the use of correct splice sites; such mechanisms include the activity of splicing enhancers and silencers (Blencowe 2000; Fairbrother and Chasin 2000; Sun and Chasin 2000; Sironi et al. 2004) and interactions among splicing factors across the exons that aid the recognition of 3′ and 5′ splice sites (Berget 1995). Interactions among components of the RNA processing and transcription machineries have also been proposed to enhance the accuracy or efficiency of splicing and 3′-end formation (reviewed in Goldstrohm et al. 2001; Maniatis and Reed 2002; Neugebauer 2002). Specialized mechanisms for large introns could involve the excision of smaller subfragments as they are transcribed; this would avoid the generation of full-length precursors and increase the opportunities for interaction between the splicing and transcription machineries. Removal of nested introns (“intrasplicing”) has been proposed on the basis of bioinformatic analysis (Ott et al. 2003), but it has not been demonstrated experimentally. This mechanism would require that the first 5′ splice site be ignored by the splicing machinery until the complete intron has been transcribed. An alternative is recursive splicing, which has been observed at hybrid elements that function first as 3′ splice sites and then regenerate 5′ splice sites after ligation to an upstream exon (Hatton et al. 1998; Figure 1A). In contrast to intrasplicing, recursive splicing could remove all subfragments cotranscriptionally, beginning at the first nucleotide of the intron, and it could pair all 5′ and 3′ splice sites in the order in which they were transcribed.
Figure 1.—

Recursive splicing. (A) Regeneration of 5′ splice sites. Previously identified recursive splice sites were part of cassette exons (dashed outlines); here we test the hypothesis of nonexonic sites. The overlapping 3′ and 5′ splice-site consensus sequences at the recursive splicing signal are overlined and underlined, respectively. M, A or C; Y, C or U. (B) Nucleotide frequency matrix used to identify potential recursive splice sites in Drosophila. The first row is the ideal sequence. The vertical line indicates the recursive splice site. Numbers indicate the frequencies (as a percentage) of each nucleotide at intron positions adjacent to the standard 3′ and 5′ splice sites (left and right of the vertical line, respectively).
Two instances of recursive splicing have been observed. Both involve tissue-specific cassette exons within a 77-kb intron in the Ubx gene of Drosophila (Hatton et al. 1998). These exons (mI and mII) are spliced constitutively to the upstream exon, but the junctions regenerate 5′ splice sites whose activity is regulated to remove mI and mII in specific tissues and at particular stages during development. This strategy allows mI and mII to subdivide the large intron even when they will not be included in the fully spliced mRNA. Inversion of mII in vivo (Subramaniam et al. 1995) or deletion of mII in transgenes (Hatton et al. 1998) leads to aberrant splicing of the remaining transcript, suggesting that the recursive splicing strategy is important for correct processing of Ubx RNA. The broader significance of recursive splicing has remained unclear, however, because no other natural examples have been described. We hypothesized that this mechanism might be relatively common but that it might have remained undetected if most recursive splice sites are not associated with exons at all, so that their use leaves no trace in any mRNA product. We report bioinformatic, phylogenetic, and experimental analyses that confirm this hypothesis in Drosophila and that indicate that recursive splice sites serve important functions specifically in the context of long introns.
MATERIALS AND METHODS
Identification of potential recursive splice sites:
We used the intron sequences from Release 3 of the annotated genome of Drosophila melanogaster (Celniker et al. 2002; Misra et al. 2002). Potential recursive splice sites were identified by scoring against a postulated nucleotide preference matrix generated by juxtaposing the matrices of observed nucleotide frequencies at each position of the standard 3′ and 5′ splice sites in Drosophila (Figure1B; frequencies were from Mount et al. 1992). Branch sites were predicted using the standard consensus (Mount et al. 1992). Similarity scores were calculated using the following formula (Goodrich et al. 1990):
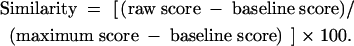
The raw score was calculated by summing the matrix-derived frequencies for the nucleotides observed at each position of the test site. The maximum score is that for an ideal site with the most frequent nucleotide at each position. The baseline score is the matrix length multiplied by 25. A site equivalent to the ideal has a similarity score of 100, whereas a score of 0 indicates that the site resembles the ideal no more than expected for a random sequence. To ensure the presence of AG/GU straddling the splice site (indicated by the slash), the program first identified an exact match to this sequence. A similarity score was then calculated with the flanking nucleotides, and a search for a branch site was conducted in the region from 15 to 115 nt upstream of the AG/GU. Except as noted in the text, we focus here on recursive splice sites with a similarity score ≥80 and branch site scores ≥65. As controls, we also searched the reverse complement of the annotated intron sequences and the sense strands of exons. The program code (MatrixSearch.v1) and sequence files are available upon request.
Statistical analysis:
We used a Monte Carlo simulation method to estimate the number of patterns matching the recursive splice-site criteria in randomly generated sequences with the same base composition as Drosophila introns. For this we developed a program in C++ called rp_sim, which generates an intronic genome equivalent of random sequence having the same nucleotide frequencies (including the possibility of unidentified bases) as introns in the real genome. The program then searches for recursive splice sites and their corresponding branchpoints using the same MatrixSearch rules that were applied to the real genome. Each simulation consisted of 55 million nucleotides with the following base composition: A, 29.5%; G, 19.7%; C, 20.3%; and T, 30.4%. This base composition was determined using a Perl script to count the number of nucleotides in the introns of genome sequence release 3.2.0 (http://flybase.bio.indiana.edu/annot/). Undefined nucleotides (N) made up 0.79% of the genome sequence, but since MatrixSearch rejects any potential recursive sites containing N, rp_sim was run without specifying the percentage of N (the 0.79% was evenly distributed among the four nucleotides). By repeating the simulation over 3000 intronic genome equivalents, we obtained an estimate of the average frequency of recursive splice-site motifs in random sequences. Using a threshold of 80 for the recursive splice site and 65 for the branch site, this average was 24.3 sites/intronic genome (SD 4.9; range 14–39) or 0.442 sites/Mb of intron sequence.
We also obtained an analytical estimate for the expected number of recursive splice sites meeting the specified criteria in a random intronic genome with the same nucleotide composition as real introns: N ∼ L × P(motif), where L is the length of the genome and P(motif) is the probability of finding a matching motif at a given location. Using a dynamic programming method, rp_sim is able to compute P(rp), the probability for a sequence of length 20 to have a recursive splice-site similarity score above a specified threshold, and P(bp), the probability for a sequence of length 8 to have a branch-site similarity score above a specified threshold. This allows one to estimate the probability of having a recursive splice site preceded by at least one branchpoint within no more than G nucleotides: P(motif) ∼ P(rp) × (1 − (1 − P(bp))G). This is only a very close estimate because it considers the probability of having a branchpoint at position x to be independent of the probability of not having a branchpoint at position x + 1. Using score thresholds of 80 for the recursive splice site and 65 for the branchpoint, P(rp) = 6.01 × 10−7 and P(bp) = 0.012. With L = 55,000,000 and G = 115 (measured from the AG/GU), we obtain N = 24.8, which agrees with the result of the Monte Carlo simulations. The documented rp_sim code is available upon request.
Analysis of RNA lariats and predicted intermediates:
D. melanogaster embryos, larvae, and adults were collected from strain Canton-S raised at 25°. Total RNA was extracted with the RNeasy reagents (QIAGEN, Chatsworth, CA). Reverse transcription by Superscript II (Invitrogen, San Diego) was primed on 1 μg of RNA using 25 ng of random hexamers. The sequences of primers for amplification of lariats, recursive splicing intermediates, and mRNAs are given in Table 1 and their application for specific experiments is indicated in the text and Figures 1–8. After digestion with RNase H, one-tenth of the first-strand cDNA was amplified for 20 cycles (95°, 30 sec; 55°, 30 sec; 72°, 30–120 sec) in a 50-μl reaction containing 10 pmol each of forward and reverse primer, 1.5 mm MgCl2 and 2 units of Platinum Taq (Invitrogen). The amplimers were diluted 1/160 in water and 2 μl were reamplified using nested primers. mRNAs were reamplified for 22 cycles, recursive intermediates for 27 cycles, and lariats for 30–35 cycles. A total of 10 μl from each sample was analyzed by electrophoresis through 2% agarose and staining with GelStar (Cambrex). Photographic images were reversed for printing. For the Ubx mRNAs and intermediates in Figure 6, the nested amplification used primer Ubx.5S4 end-labeled with 32P and was performed for 22 cycles. The amplimers were separated on nondenaturing 8% polyacrylamide gels and detected by autoradiography. The proportion of the amplification reaction loaded was 10 times greater for recursive intermediates than for mRNAs, and the exposure was four times longer. The nucleotide sequences of gel-purified amplification products were obtained as described (Hatton et al. 1998).
TABLE 1.
Primer sequences
| Gene | Primer | Sequence |
|---|---|---|
| Ubx | 5S1 | GCCTGGAATGCCAATTGCACCATC |
| 5S4 | AGCAGTTTACACCAGGCCAGC | |
| mII.3s2 | GCTCACTTCTACCAGACTGG | |
| mII.B5 | GCTGGACATTTTGGAGTGGACG | |
| I3A3 | CTGCTTTAACTGGTTGATGCG | |
| I3A4 | GTTTTTGTTTTGGTTTAAGGGTG | |
| E3.BP5 | ACACTCTGTCACTGCTGCTC | |
| I3.F14 | GGGTGGCAAATAAACCGTAGGAC | |
| Hae3.1 | CATCGTATGGGTAAAAGATGC | |
| 3A1 | GCGGGTCAGATAATGATTCGT | |
| kuz | E2.F3 | GCCCAATAACGCAACCAACTAC |
| RP3.1B18 | ATGGAACCAGTCATCCTCGTCC | |
| RP3.2B1 | TCCAGCTCGATTAAGATGTCTTCC | |
| E4.B3 | CGCTCTATTGTGACTAGCTCGGATG | |
| E4.BP2 | CGAAAAGTAGGGTGTGGAC | |
| RP3.2F3 | AAGGTGTGTGTTCGCTGGGTAAGG | |
| E3L.B1 | GTTTCGCAATCCATTAGCCAAC | |
| osp | E1.F19 | CACGACAATAACAACAACGGTGC |
| E1L.B3 | CAAGAAGCCTGAGATGAGACGAAG | |
| RP1.2F5 | TCTCTACCGCTTGCCGAAC | |
| E2.BP2 | TACGGTCAGGAGATAAGCG | |
| RP1.1B3 | GGTTTCATTCATTATCCACAGG | |
| RP1.1B11 | CATGTAATCGAACGAAGCACGG | |
| RP1.2B1 | AAATGGCTTTATCGCTCGGG | |
| E2L.B1 | GTTAGTCTGCTTTGTTGGGTCCAC | |
| RP2.1B18 | CGCAATCGAGTTTATGCTTACACG | |
| E3L.F6 | TTCCTCACCATTTCTCGTCCCACC | |
| E4.B26 | CACTGGGATTCCTCTGAACTGG |
Figure 8.—

Mutational analysis of recursive splicing mediated by Ubx RP3. (A) Rationale. The diagram shows the competing direct and recursive splicing pathways for the last intron fragment of Ubx. The junction between E5′ and mI is a consensus 5′ splice site whose activity removes mI and mII, thus producing mRNA IVa. In SL2 cells this activity is suppressed to low levels if the downstream 5′ splice site is functional. Production of isoform Ia by transcripts that enter the direct splicing pathway should not be affected by mutations that reduce the activity of the 5′ splice site that is regenerated by RP3 during recursive splicing. In contrast, such mutations should block further processing of transcripts that enter the recursive splicing pathway, thus reducing the yield of mRNA or causing a shift to use of the 5′-ss at the E5′/mI junction, which would produce isoform IVa instead of Ia. (B) Ubx minigene structures. Thin horizontal lines: contiguous intron sequences adjacent to mII and E3′. Thick horizontal lines: contiguous intron sequences flanking RP3. RS, regenerated 5′-ss at the mII/RP3 junction. RP, RP3. The wild-type and mutant sequences at RP3 are shown for Ubx.RP and Ubx.RP*; changes from wild type are underlined. In minigene ΔRP(N), an _Nco_I site replaces the RP3 sequence extending from the first nucleotide of the branch site through position +6 of the recursive splice site (see Figure 4). Arrows show the positions of primers used to detect spliced mRNAs (5S1 + Hae3.1), recursive intermediates (5S1 + I3A3), recursive lariats (I3A4 + I3F14), and direct lariats (mIIB5 + I3F14). (C) Splicing of minigene transcripts in SL2 cells. RT-PCR analysis of processed mRNAs is shown, together with the _lac_Z transfection control and the relevant intermediates and lariats. Amplimers were analyzed by electrophoresis through 2% agarose and staining with GelStar. Identical results were obtained in at least three independent experiments for each construct. The endogenous Ubx gene was not expressed at detectable levels in SL2 cells.
Figure 6.—

Splicing of upstream exons to recursive splice sites. (A–C) RT-PCR analyses of recursive splicing intermediates and mRNAs below a diagram of the relevant exon/intron structures in the corresponding gene. In the diagrams, vertical lines represent recursive splice sites. Arrows indicate the positions of forward and reverse primers. In the gel figures, the reverse primers used are indicated above each lane. (A) kuzbanian and (B) outspread: amplimers were analyzed on agarose gels stained with GelStar. The expected size of each amplimer is indicated at the right, size markers at the left. The results shown are for RNA from embryos at 3–6 hr after egg laying (25°), but the same was observed at all embryonic and adult stages. The rightmost lane in each case is the final, completely spliced product: No cassette exons that might be associated with the recursive splice sites were detected at any stage. The mRNAs were amplified for 30 cycles, whereas the ratcheting intermediates were amplified for 35 cycles. (C) Ultrabithorax. The 32P end-labeled amplification products were analyzed on 8% acrylamide gels. The primers used and the developmental stage (hours after egg laying at 25°) are indicated above each lane. The sizes and structures of the alternative mRNA isoforms are labeled at the right. The corresponding recursive splicing intermediates are labeled at the left; R5 marks the 5′-ss regenerated at the junction with RP3. Cassette exons mI and mII are each 51 nt long. Exon E5′ has two alternative 5′ splice sites separated by 27 nt, giving rise to a and b variants of the mRNAs. The mRNAs and ratcheting intermediates were amplified for the same number of cycles, but the reaction sample loaded was 10 times greater for ratcheting intermediates than for mRNAs and the exposure was four times longer. The relative band intensities are thus consistent with accumulation of intermediates to ∼3% of mRNA levels. No additional cassette exon that might be associated with RP3 was detected at any other stage.
Construction of Ubx minigenes:
Minigenes were constructed by ligating appropriate DNA fragments from genomic Ubx clones, from a cDNA clone of Ubx mRNA isoform Ia, and from RT-PCR amplimers corresponding to the type Ia RP3 intermediate (Figure 5). The constructs included most of the 5′- and 3′-untranslated sequences from Ubx, beginning 353 nt downstream of the transcription initiation site and ending 288 bp downstream of the first cleavage/polyadenylation signal (O'Connor et al. 1988; Kornfeld et al. 1989). Nucleotides 24–151 in exon E3′ were substituted with a 70-bp synthetic fragment that serves as the targeting site for PCR primer Hae3.1 (Hatton et al. 1998). In Ubx.RI, the unprocessed intron segment (26 kb) was shortened to 1092 bp by deleting the _Cla_I fragment from 902 bp downstream of RP3 to 200 bp upstream of exon E3′. In Ubx.RI-S, the same segment was shortened to 1077 bp by deleting from 24 bp downstream of RP3 to the _Pst_I site 1053 bp upstream of exon E3′. In Ubx.RP the unprocessed intron (52 kb) was shortened to 2692 bp by deleting the _Mun_I fragment from 616 bp downstream of mII to 971 bp upstream of RP3 plus the _Cla_I fragment from 902 bp downstream of RP3 to 200 bp upstream of E3′. The RP3* mutation was generated in Ubx.RP using the Altered Sites II system (Promega, Madison, WI). The ΔRP(N) deletion was generated in Ubx.RP by using PCR to substitute an _Nco_I site for the RP3 recursive splice-site sequences extending from the first nucleotide of the branch site through position +6 (see Figure 4 for the deleted sequence). The minigenes were fused to the Drosophila polyubiquitin promoter in plasmid pPUb (Lee et al. 1988) by replacing all polyubiquitin sequences downstream of the _Mun_I site at +6 relative to the transcription start site.
Figure 5.—

Spacing between predicted recursive splice sites. For each predicted recursive splice site in D. melanogaster, the distance to the subsequent 3′ splice site (exon or RP) is plotted against the distance from the preceding 5′ splice site (exon or RP). Both distances are measured in nucleotides. Descriptive statistics are given in the text.
Figure 4.—
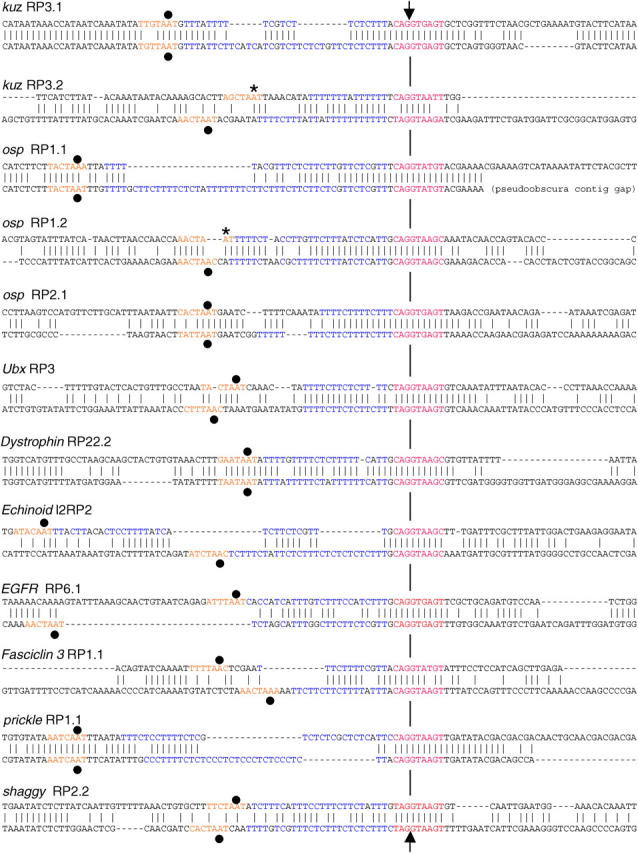
Examples of recursive splice-site conservation. Twelve representative examples are shown. The top 6 have been confirmed experimentally in this study (see results). The bottom 6 have not been tested, but are as good or better predictions as the confirmed sites. In each case the top sequence is from D. melanogaster, and the lower sequence is from D. pseudoobscura. A maximum of 112 nucleotides flanking each splice site are shown.Vertical ticks mark identical nucleotides, and horizontal dashes are gaps introduced to optimize the alignment. The arrows mark the point at which recursive splicing has been shown to occur (top 6 examples) or is predicted to occur (lower 6 examples). The hybrid 3′-ss/5′-ss sequence is shown in red; the polypyrimidine tract in blue; and the closest potential branch site in orange with the branchpoint nucleotide marked by a dot (if predicted) or an asterisk (if verified experimentally; see Figure 6). The score and location of each recursive splice site within the intron are shown in supplementary Table S1 at http://www.genetics.org/supplemental/. Note that the splice-site signals have been conserved despite extensive nucleotide substitutions, insertions, and deletions. The polypyrimidine tracts are generally longer than the search criteria require (see Figure 1), and conserved and verified sites include examples with the infrequent T at position +4 (osp RP1.1 and Fas3 RP1.1).
Analysis of minigene products in SL2 cells:
To facilitate lariat analysis, we used Drosophila SL2 cells in which lariat debranching enzyme had been knocked down by stable transfection with an RNA interference construct under control of the metallothionein promoter (J. Conklin and A. J. Lopez, unpublished results). The results, as judged by expression of minigene mRNA isoforms and recursive intermediates, were identical to those using standard SL2 cells. The cells were transfected with supercoiled DNA as described (Hatton et al. 1998). Total RNA was extracted 40 hr after transfection and 1 μg was used for reverse transcription primed with oligo(dT) (to analyze mRNAs) or random hexamers (to analyze recursive intermediates). After treatment with RNase H, one-tenth of the product was amplified by PCR as described above, except that mRNAs were amplified for 26 cycles and recursive intermediates or lariats were amplified for 35 cycles, without nesting. Amplifications were determined to be within the exponential range by testing aliquots at different cycles. Amplimer identities were confirmed by size, sequencing and digestion with _Not_I, _Bgl_II, and _Dde_I, which cleave in E5′, mI and mII, respectively.
RESULTS
The frequency of predicted recursive splice sites in large introns exceeds the expectation for a random distribution:
We searched computationally through all annotated introns in the genome of D. melanogaster to identify potential recursive splice sites, which were defined by scoring against a matrix representing the juxtaposed sequence preferences at standard 3′ and 5′ splice sites (Figure 1B). We symbolize recursive splice sites as RPs (_r_atchetting _p_oints) to distinguish them from the corresponding _r_egenerated 5′ _s_plice sites (RSs). We demanded that predicted RPs contain the core sequence AG/GU (where the slash represents the phosphodiester bond involved in splicing) because AG and GU are almost invariant at the 3′- and 5′-ends of introns, respectively (Weir and Rice 2004). We also demanded an RP score ≥80 (which exceeds that of the known recursive splice site associated with the Ubx cassette exon mI) and the presence of a potential branch site within 115 nt upstream of the AG/GU core. Using these stringent criteria, we identified 165 potential recursive splice sites distributed among 124 introns (the complete documentation of sequences, locations, and scores is presented in supplementary Table S1 at http://www.genetics.org/supplemental/). Eleven of these recursive splice sites correspond to the 5′-ends of known alternatively spliced exons, and one corresponds to the 3′-end of such an exon (Table 2). As shown below and in supplementary Table S1 at http://www.genetics.org/supplemental/, these exons and their associated recursive splice sites are conserved between D. melanogaster and D. pseudoobscura. Hence, the inclusion or exclusion of these exons in mRNA is probably regulated by recursive splicing, like cassette exons mI and mII in Ubx (Hatton et al. 1998).
TABLE 2.
Recursive splice sites associated with known alternative exons
| Gene | RPa | Comments |
|---|---|---|
| Recursive splice site at 5′-end | ||
| multiple edematous wings | 3.1 | Cassette (noncoding) |
| muscleblind | 2.1 | Alternative terminal exonb |
| smooth | 12.1 | Cassette |
| bric-a-brac 1 | 1.1 | Alternative terminal exonb |
| bruno-3 | 3.1 | Cassette |
| frizzled | 4.1 | Alternative terminal exonb |
| pumilio | 8.6 | Cassette |
| pasilla | 1.1 | Cassette |
| Ultrabithorax | mI | Cassette |
| Ultrabithorax | mII | Cassette |
| hephaestus | 3.1 | Cassette |
| Recursive splice site at 3′-end | ||
| CG6014 | 1.1 | Cassette |
As we had hypothesized, the great majority of predicted RPs (155 of the 165 high-scoring sites) are not associated with any known or predicted exon, so their use would not be detected by analysis of mRNAs. In addition, there is a pronounced bias in the distribution of predicted RPs, which are found primarily within large introns (Figure 2, A and B). Only four are in introns <10 kb (which compose a total of 29.4 Mb), whereas 161 are in introns >10 kb (which compose a total of 26.1 Mb). This bias could reflect selection for RPs in large introns, selection against RPs in shorter introns, or both. We distinguished among these possibilities by performing Monte Carlo simulations to estimate the expected random frequency of recursive splice sites that would meet the same threshold criteria. We performed 3000 simulations, each generating an intronic genome equivalent of random sequence with the same base composition as the real introns. This yielded an average of 24.3 recursive splice sites/intronic genome (SD 4.9; range 14–39), which agreed with the analytical estimate of 24.8 sites (see materials and methods). Thus, the observed overall frequency of predicted recursive splice sites is seven times greater than expected. Furthermore, comparison of the distributions with respect to intron size shows that the frequency of observed sites is enhanced over random expectation in very large introns, but is reduced below expectation in small and moderate introns (Figure 2B). The observed frequency is 10-fold higher than expected in introns >20 kb, but is only one-fifth of that expected in introns <10 kb. Additional controls are provided by analysis of the antisense strands of real introns and the sense strands of exons. We observed only 32 recursive splice sites with scores ≥80 on the noncoding strands of real introns (discounting RPs within nested genes in the opposite orientation); this was comparable to the random expectation and should represent the functionally neutral situation. The only RPs with scores in this range within annotated exons corresponded to the 5′- or 3′-ends of the known alternatively spliced exons listed in Table 2; this agrees with the expectation that RPs should not be found within exons (except for regulatory purposes), because they could interfere with correct splicing at the nearby exon boundaries. These results indicate that the predicted recursive splice sites are under positive selection in large introns. In contrast, they appear to be selected against in introns <10 kb, possibly because they interfere with normal processing in that context.
Figure 2.—

Frequency and distribution of predicted recursive splice sites. (A) The total nucleotide content in each intron size class is plotted, along with the frequency of recursive splice sites in the same intron size classes. Although introns <20 kb account for most of the nucleotides, there is a strong and opposite bias in the distribution of recursive splice sites, which are found mostly in introns >20 kb. (B) Comparison of expected and observed occurrence of recursive splice sites. The expected numbers and distribution were calculated from the frequency expected if recursive splice-site signals occur at random. This frequency was estimated analytically and by Monte Carlo simulations as described in the text. The observed recursive splice sites occur at much higher frequency than expected overall and their distribution is skewed in two ways: they occur at lower-than-expected frequency in introns <10 kb and at greater-than-expected frequency in introns >20 kb.
Conservation of predicted recursive splice sites in large introns:
If the predicted recursive splice sites are functional and provide a selective advantage in large introns, we might expect them to be conserved between D. melanogaster and D. pseudoobscura, whose nearly complete genome sequences are available. It is estimated that D. melanogaster and D. pseudoobscura diverged ∼25 MY ago (Powell 1997), providing sufficient time for many changes in nonessential DNA while retaining enough similarity for reliable alignment. We investigated the possible conservation of recursive splice sites using the VISTA browser for whole-genome alignment (Couronne et al. 2003). We found that 150 of the predicted recursive splice sites were located in regions that had been sequenced in both species and could thus be informative. Of these sites, at least 138 (92%) have been conserved (or replaced by equivalent sites) and are located at the same relative positions within the introns in both species (Figure 3 and supplementary Table S1 at http://www.genetics.org/supplemental/). This is remarkable in light of otherwise extensive sequence divergence within the same introns, including large insertions and deletions. Figure 4 shows detailed alignments for 12 representative examples, 6 of which have been confirmed to mediate recursive splicing in the experiments described below. Although substantial variation in nucleotide sequence is evident even in these local alignments, the changes clearly have maintained or regenerated the splicing signals. We have not performed an extensive analysis of RPs across larger evolutionary distances, but we have traced several sites (e.g., nonexonic site RP3 in Ubx) as far as the honeybee, Apis melifera. In contrast to the strong conservation of recursive splice sites in large introns, only one of the four RPs predicted in introns <10 kb was found to be conserved in D. pseudoobscura. Together with the analysis of frequency and distribution with respect to intron size, the observed conservation of recursive splice sites indicates that they confer a selective advantage in the context of large introns.
Figure 3.—

Evolutionary conservation of predicted recursive splice sites in D. melanogaster and D. pseudoobscura. (A) Length comparison for introns that contain recursive splice sites predicted in D. melanogaster with a score ≥80. The exact length of each intron is given in supplementary Table S1 at http://www.genetics.org/supplemental/. (B) Conservation of position for recursive splice sites predicted in D. melanogaster with a score ≥80. Positions are indicated as a percentage of intron length, measured from the upstream exon-intron boundary. The exact location of each recursive splice site is given in supplementary Table S1 at http://www.genetics.org/supplemental/.
Spacing between recursive splice sites:
Figure 3B shows that strongly predicted RPs are not constrained to specific relative positions within their host introns, although they are less likely to be found near the ends. Figure 5 illustrates the absolute spacing (in nucleotides) between the predicted RPs and their flanking splice sites, which can be standard 5′ and 3′ splice sites or other RPs. The distances cluster around 18,000–20,000 nt, but there is wide variation in these values. The average distance to the preceding 5′ splice site (exon or RP) is 18,009 nt (range 352–59,560 nt; SD 11,091), whereas the average distance to the next 3′ splice site (exon or RP) is 20,666 nt (range 2389–103,624 nt; SD 13,695). The actual distribution may be substantially narrower if many of the large gaps are filled by functional RPs that do not meet our stringent cutoff score. Previous studies on the recursively spliced Ubx cassette exons (Hatton et al. 1998) and the experimental analyses described below support this proposal, as they demonstrate the function of at least two recursive splice sites with lower scores (Ubx mI, with a score of 74, and outspread (osp) RP1.1, with a score of 77).
Selection of recursive splice sites for experimental tests:
The sections that follow describe experiments performed on a sample of six predicted nonexonic RPs to test whether they actually mediate recursive splicing of their host introns. The selected sites were derived from three genes that are unrelated in sequence, expression, or function (Table 3). The genes are kuzbanian (kuz), which encodes a metalloprotease implicated in Notch signaling; osp, which encodes a cytoskeletal component of muscle; and Ubx, which encodes a homeodomain transcription factor that specifies segmental identity. Exon-intron structures have been defined experimentally for all three genes (FlyBase Consortium 2003). The recursive splice sites that we analyzed are the highest-scoring sites identified in their introns (Table 3). One of these sites (osp RP1.1) scored below the cutoff for our original bioinformatic analysis, but it was included in the experimental analysis to test all likely RPs found within these introns. The recursive splice sites selected for these experimental tests are not part of any cassette exons represented in EST and cDNA collections for the host genes (FlyBase Consortium 2003), and we have not detected associated exons at any stage of development using extensive RT-PCR analyses (Figure 6 and data not shown).
TABLE 3.
Recursive splice sites selected for experimental analysis
| Gene | Intron | Length (kb) | RP positiona | Score | Name |
|---|---|---|---|---|---|
| kuz | 3 | 51.0 | 19.5 | 85.4 | RP3.1 |
| 35.4 | 83.4 | RP3.2 | |||
| osp | 1 | 52.3 | 10.9 | 77.0 | RP1.1 |
| 32.0 | 80.4 | RP1.2 | |||
| 2 | 24.2 | 8.9 | 89.1 | RP2.1 | |
| Ubx | 1a | 77.5 | 48.0b | 84.6 | RP3b |
Analysis of recursive splicing intermediates:
The initial use of an RP as a 3′ splice site would produce a recursive splicing intermediate in which the preceding exon is juxtaposed with the region immediately downstream of the RP. We used RT-PCR assays to probe for the production of such intermediates, using primers in upstream exons paired with primers downstream of the RPs (Figure 6). For example, use of RP1.1 in osp (Figure 6B) should generate a recursive splicing intermediate in which exon 1 is spliced to the RP, and this should yield an amplimer of 446 bp with primers F19 (in exon 1) and B11 (downstream of RP1.1); this was the observed result (Figure 6B). We readily detected all of the intermediates predicted for each of the six RPs, and their identities were verified by sequencing. The amplified intermediates from kuzbanian and outspread are shown in Figure 6, A and B; the same intermediates were detected throughout development. Analysis of the RP3 intermediates from Ubx was more complex because the three upstream exons are involved in combinatorial alternative splicing (O'Connor et al. 1988; Kornfeld et al. 1989). The Ubx results are presented in Figure 6C for early and late embryos, which express different isoform ratios. We observed the predicted RP3 intermediates that correspond to each of the five alternative splicing patterns. The proportions of the intermediates were consistent with the proportions of the mRNAs and they changed during development in the same manner, as expected.
Although the polarity and kinetics of transcription make it most likely that RPs function initially only as 3′ splice sites (as detected in the above experiments), in principle they could also serve first as 5′ splice sites. We tested this second possibility using analogous RT-PCR assays with forward primers upstream of each RP paired with reverse primers within the corresponding downstream exon or 3′ of the next RP. In contrast to the results described above, we did not detect any of the intermediates predicted for initial use of RPs as 5′ splice sites (not shown). This agrees with the proposal that RPs mediate removal of intron subfragments in 5′–3′ order, as they are transcribed.
Although the conservation of RPs makes it unlikely that their use represents rare cryptic or aberrant splicing, we also used quantitative RT-PCR with preamplified standards to determine whether the levels of the putative recursive splicing intermediates are consistent with a significant role in processing of the host introns. On the basis of signal intensity and number of cycles required for detection, each of the predicted intermediates for kuz, osp, and Ubx accumulated to a steady-state level equivalent to ∼3% of the corresponding mRNAs. A similar estimate (2%) was obtained independently for Ubx by cloning and sequence analysis of 3′ RACE products generated by randomly primed reverse transcription followed by amplification with a forward primer targeting the first exon and a reverse primer targeting an adapter ligated to the 3′-end of the cDNA. Thus, the putative recursive intermediates are produced at significant levels that are consistent with a productive role in pre-mRNA processing. These high levels of predicted intermediates are also consistent with the relatively long half-lives predicted by the elongation rate of RNA polymerase (∼1.4 kb/min) and the distances (16–26 kb) between each RP and the next downstream 3′ splice site (Table 3).
Analysis of recursive splicing lariats:
The activities of the regenerated 5′ splice sites cannot be detected by direct analysis of spliced intermediates or mRNAs except in special circumstances (illustrated by the mutational analysis of Ubx RP3, described below). However, this activity should be manifested by the generation of a lariat intermediate and product in which the regenerated 5′ splice site is ligated to the branchpoint nucleotide of the next recursive splice site or exon (Figure 7, A and B). We took advantage of this to confirm whether recursive splicing occurs at the six sites identified in kuz, osp, and Ubx.
Figure 7.—

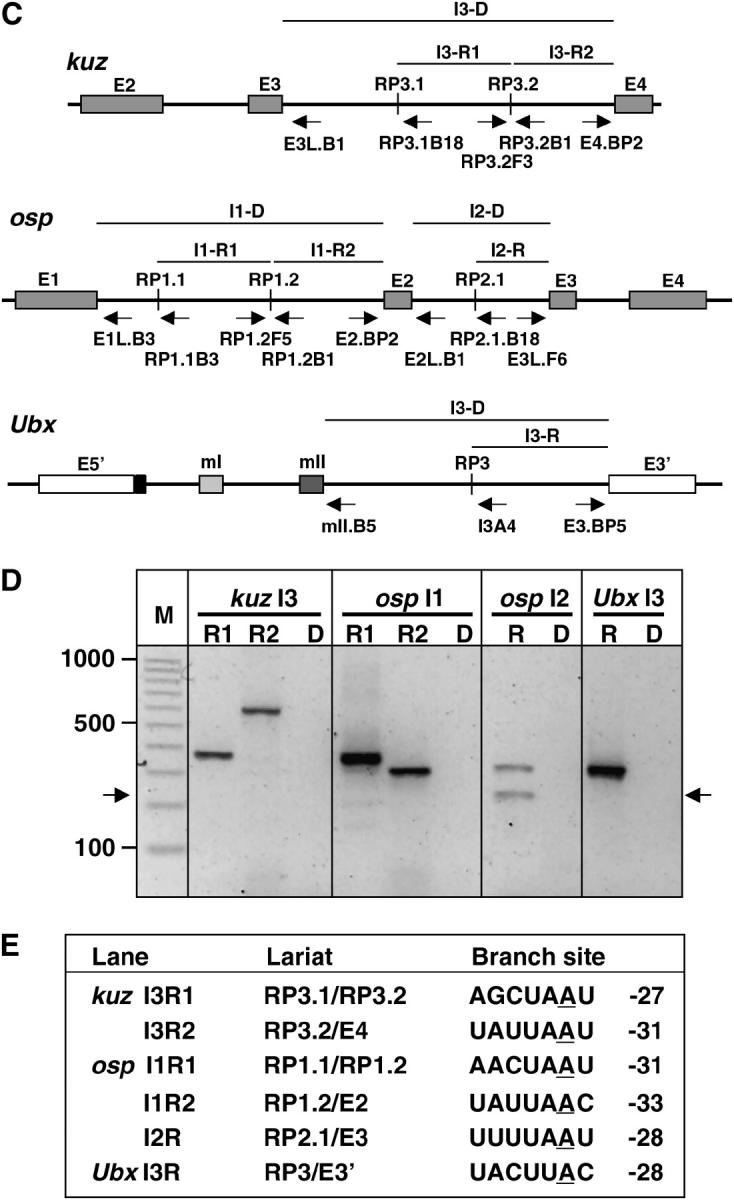
Lariat analysis of recursive splicing. (A) Rationale of lariat analysis. Direct splicing between the 5′-ss at the exon/intron boundary and the 3′-ss at the intron/exon boundary should generate a single lariat spanning the entire intron, with the 5′-ss ligated to the 2′-OH of the branchpoint nucleotide. In contrast, recursive splicing should generate a series of lariats, each spanning an intron subfragment. Although the activity of the recursively generated 5′ splice sites cannot be detected by direct analysis of spliced intermediates or mRNAs, each should be detected as a lariat in which the recursive 5′-ss is ligated to the branchpoint nucleotide of the next recursive splice site or exon. (B) RT-PCR strategy for lariat analysis. Reverse transcription was performed on total RNA using random hexamers or gene-specific primers. Subsequent PCR used gene-specific primers Y and Z, which prime convergently across the branch junction on lariat-derived cDNA. (C) Relevant gene structures. Flanking exons and recursive splice sites (vertical lines) are identified. Arrows indicate the position and orientation of primers used to detect the predicted recursive or direct splicing lariats. The horizontal lines above each gene diagram identify the intron segments whose removal corresponds to the recursive splicing lariats (designated R) or direct splicing lariats (designated D) being tested. The primers used in each test are the divergent primers at the extreme ends of the indicated segment (primer pairs are also identified in E). (D) Electrophoretic analysis of amplimers. The relevant splicing event is identified above each lane, as in C. R lanes are tests for recursive splicing lariats in 5′–3′ order for each intron. Lanes designated “D” are tests for direct splicing lariats of the same intron; all D amplimers were expected to be <800 bp. The M lane contains markers, with the size in base pairs shown at left. Arrows at left and right mark the true lariat band in lane osp I2 R1; sequencing revealed that the upper band in this lane is a novel circular splicing product in which the 5′-ss of osp exon 3 is joined to the 3′-ss of RP2.1. (E) Branch sites determined from sequence analysis of each amplimer. The branchpoint nucleotide is underlined; this was always an A in the genome but was replaced by T in the amplimers. The position of each branchpoint nucleotide is indicated relative to the 3′-ss. All lariat sequences revealed the predicted ligation of the recursive 5′-ss to the next branchpoint nucleotide. The branch sites conformed to the consensus for standard splicing.
The rationale for this analysis is illustrated in Figure 7A. Direct removal of an intron in a single step (or in multiple steps by intrasplicing) generates a lariat in which the first nucleotide of the intron is linked to a branchpoint near the end of the intron. Recursive splicing would not generate such a lariat; instead, it would generate a series of lariats in which the first nucleotide downstream of each preexisting or regenerated 5′ splice site is linked to the branchpoint upstream of the next 3′ splice site, which may belong to an exon or another recursive splice site. Although lariats are short lived in vivo, they can be detected and characterized using an RT-PCR strategy with sense primers upstream of the branch site and antisense primers downstream of the 5′ splice site (Figure 7B) (Vogel et al. 1997). We were able to detect the lariats for each of the predicted recursive splicing steps in the large introns from kuz, osp, and Ubx (Figure 7, C–E; lariat identities and branchpoint locations were verified by sequencing of the amplimers). In contrast, even using more extensive cycling and multiple primer combinations, we were unable to detect the lariats that would correspond to removal of these introns in a single step or by intrasplicing (Figure 7, C–E). This difference was not due to amplimer size, since the expected amplimers were in the same size range for direct and recursive lariats. Neither was the difference due to unusual stability of the recursive lariats, because we detected lariats for direct splicing when we analyzed eight different introns that lack RPs, including introns up to 15 kb long and lariats for alternative splicing events from Sxl, Rbp1, and Rbp1-like (not shown). Most conclusive, however, was the finding that the lariat for direct splicing could be detected readily when recursive splice site RP3 was deleted from Ubx in minigene constructs, but not when RP3 was present in those constructs (experiments described in Mutational tests of recursive splicing at Ubx RP3 below).
Together, the results from analysis of recursive intermediates and lariats support two conclusions. First, the lariats detected from the large introns of kuz, osp, and Ubx result from the activity of 5′ splice sites that are regenerated after the RPs have been spliced to the upstream exon. Second, the abundance of the recursive intermediates and the detection of recursive but not direct lariats suggest that recursive splicing is the predominant processing pathway for these introns. Additional support for this conclusion is provided by the analysis of Ubx minigenes described below.
Mutational tests of recursive splicing at Ubx RP3:
Ubx is ideal for experimental tests of recursive splicing at a nonexonic site because the ratios of natural alternative mRNAs can be used to assess the activity of RP3. This is a consequence of the functional consensus 5′ splice site (CAG/GUAAGU) that separates exon E5′ from exons mI and mII in the partially spliced intermediates that are precursors to isoform Ia (Hatton et al. 1998; Figure 8A). If mII splices to RP3 and this does not regenerate a functional 5′ splice site (ss), the competing 5′-ss at the E5′/mI junction can be used to process the last intron fragment, but this will remove mI and mII from the mRNA (Figure 8A). We designed two tests based on this property: one in which the 5′ splice site activity of a preformed mII/RP3 junction was assessed and a more stringent test of the recursive splicing cycle by mutation of unprocessed RP3.
The first test used a Ubx minigene corresponding to the RP3 recursive splicing intermediate for isoform Ia (Ubx.RI in Figure 8B): exons E5′, mI, and mII were already spliced together, and mII was already spliced to RP3 (this corresponds to the type Ia intermediate detected in Figure 6C and depicted in Figure 8A). The proportion of mRNAs from this minigene that retain mI and mII is a measure of splicing efficiency at the mII/RP3 junction vs. the E5′/mI junction (Figure 8A). We expressed Ubx.RI in SL2 cells and analyzed the processed mRNAs by semiquantitative RT-PCR (Figure 8C). The mRNAs produced were almost exclusively isoform Ia, which retains exons mI and mII. This demonstrates that the mII/RP3 junction competes effectively with the 5′-ss at the E5′/mI junction. The result should be compared with that for Ubx.RI-S, which deletes intron sequences extending from 24 to 902 nt downstream of the mII/RP3 junction (Figure 8B). In striking contrast to Ubx.RI, Ubx.RI-S produced exclusively type IVa mRNAs, which lack mI and mII (Figure 8C). This confirms that the competing 5′-ss at the E5′/mI junction is functional in SL2 cells and suggests that preferential use of the mII/RP3 junction requires downstream sequences.
We used minigenes that represent an earlier processing intermediate to test whether RP3 actually mediates an efficient recursive splicing cycle. In these minigenes, exons E5′, mI, and mII are already spliced together as in Ubx.RI, but mII has not been spliced yet to RP3. Version Ubx.RP contains the wild-type RP3 sequence, but Ubx.RP* contains a mutated RP3 that should still function as a 3′-ss but cannot regenerate a consensus 5′-ss (Figure 8B). Splicing of mII to wild-type RP3 should regenerate a functional 5′-ss and thus allow retention of mI and mII in mRNAs (Figure 8A), as observed with Ubx.RI. Splicing of mII to RP3* should instead lead to use of the E5′/mI junction to complete processing of the intron (Figure 8A), removing mI and mII as observed with Ubx.RI-S. Both predictions were confirmed by analysis of the mRNAs, recursive intermediates, and lariats. With Ubx.RP, almost all the mRNAs retained mI and mII (Figure 8C). With Ubx.RP* there was a dramatic shift to produce almost exclusively isoform IVa, as predicted by the recursive splicing hypothesis (Figure 8C). Nevertheless, cells expressing Ubx.RP or Ubx.RP* exhibited similar levels of recursive intermediates corresponding to the use of RP3 as a 3′ splice site (Figure 8C); this was also as predicted. Cells transfected with Ubx.RP did not express direct splicing lariats at detectable levels; instead they expressed the predicted recursive splicing lariat corresponding to retention of mI + mII, with the first nucleotide downstream of RP3 ligated to the branchpoint upstream of exon E3′ (Figure 8C). Cells transfected with Ubx.RP* expressed the predicted lariat corresponding to removal of mI + mII after splicing to RP3*, with the first nucleotide of mI ligated to the branchpoint upstream of E3′ (Figure 8C). Together, the results confirm that most or all of the Ubx mRNAs are made via a pathway that uses RP3 as a recursive splice site.
An unanticipated result with Ubx.RP* was the production of direct splicing lariats in addition to recursive splicing lariats (Figure 8C). A clue to the cause for these direct lariats comes from the fact that they remove mI + mII, indicating that the RP3* mutation in the middle of the intron affects splice-site choice during pairing between the 5′- and 3′-ends of the intron. A possible explanation is that RP3*, acting in its normal capacity as a 3′ splice site, forms the usual commitment complex with the 5′ splice site at the mII/intron boundary, but the mutation slows progression to the catalytically active spliceosome (in addition to blocking subsequent recursive splicing); this delay may allow productive pairing between the normally disfavored 5′ splice site at the E5′/mI junction and the 3′ splice site at the end of the intron, leading some of the time to removal of mI + mII by the direct pathway before mII can splice to RP3*. Thus, the appearance of direct lariats in RP3* can also be explained as a consequence of an aborted recursive splicing cycle.
Analysis of RP3 deletion in the minigene context:
RP3 was deleted in construct Ubx.ΔRP(N) (Figure 8B). The deletion replaced an _Nco_I site for the sequence extending from the first nucleotide of the branch site through the last nucleotide of the recursive splice site signal. As expected, Ubx.ΔRP(N) expressed the direct splicing lariat (Figure 8C), confirming that the failure to detect this lariat in the presence of wild-type RP3 was due to the predominance of the recursive splicing pathway. Ubx.ΔRP(N) expressed Ubx mRNAs at apparently normal levels, like its parent Ubx.RP, although it consistently produced a slightly higher ratio of isoform Ia to isoform IVa. This may be because recursive splicing allows two opportunities for use of the 5′ splice site at the E5′/mI boundary, whereas direct splicing allows only one (Figure 8A). Thus, although RP3 is conserved between D. melanogaster and D. pseudoobscura and recursive splicing is the predominant processing pathway for endogenous and minigene transcripts, RP3 appears not to be essential for expression or processing of the minigene. This result is consistent with the conclusion, based on the recursive splice-site distribution and conservation described above, that RPs play an important role specifically in the context of the long native introns.
DISCUSSION
Our results confirm the subdivision of four long introns by recursive splicing at nonexonic sites and suggest very strongly that many long introns in Drosophila are processed in a similar way. The bioinformatic and phylogenetic analyses generated strong predictions for recursive splicing in 124 introns, with up to seven potential steps identified in a given intron (the 133-kb intron of pumilio). These are probably underestimates because two of the conserved recursive splice sites that we verified experimentally have scores below the minimum demanded in the computational analysis; these were the nonexonic site RP1.1 in osp (this study) and the cassette exon mI in Ubx (Hatton et al. 1998). Only 12 of the 165 top-scoring recursive splice sites identified in this study are associated with known alternatively spliced exons; this agrees with our initial hypothesis and it implies that most cases of recursive splicing would not be detected by standard analyses of cDNAs and expressed sequence tags (ESTs). This result also indicates that the documented role of recursive splice sites in excluding some alternatively spliced exons constitutes only a relatively minor specialization (as in Ubx and potentially in other genes listed in Table 2). Other functions must account for the enrichment and conservation of nonexonic RPs in long introns.
Some evidence suggests additional roles for RPs in regulation or stimulation of splicing, possibly by juxtaposing upstream splice sites with regulatory elements or enhancers that are distant in the primary transcript. Thus, elimination of recursively spliced cassette exon mII appears to be responsible for aberrant splicing of upstream exons in Ubx transcripts derived from the endogenous gene or from minigenes transfected into SL2 cells (Subramaniam et al. 1995; Hatton et al. 1998). The alternative circular splicing product generated between RP2.1 and downstream exon E3 in osp (Figure 7D) may reflect a mechanism for regulation of gene expression by spliting the transcript: the other product of this circular splicing event would be a T-shaped branched RNA, which upon debranching should release the 5′ and 3′ halves of the transcript in nontranslatable forms due to lack of a poly(A) tail or 5′-cap, respectively. In principle, roles like these need not be limited to very large introns. However, RPs might interfere with efficient or accurate processing when the time required for transcription is shorter than the recursive splicing cycle, and this could contribute to the underrepresentation of recursive splice sites in introns <10 kb.
Recursive splicing could provide specific advantages in very large introns through a number of mechanisms. Recursive splicing reduces precursor length and the time between successive splicing events across large introns; both effects could help to avoid the formation of secondary structures or hnRNP complexes that might affect the accuracy or efficiency of processing. The predicted recursive splice sites exhibit an irregular distribution within and between introns (Figure 3 and supplementary Table S1 at http://www.genetics.org/supplemental/), but their relative positions are conserved between species; although some of the gaps may be filled by lower-scoring RPs, the positional conservation of high-scoring RPs suggests that their function is not simply to maintain the splicing interval below some defined maximum. The advantage derived from recursive splicing may vary with position within and between genes depending on sequence or structural features that may interfere with processing or transcript elongation. Local suppression of cryptic splicing and polyadenylation signals may be one of these advantages. For example, the use of a cryptic exon that truncates the open reading frame in the human ATM gene is normally suppressed by an intronic element that resembles a recursive splice site and interacts with U1 snRNP (Pagani et al. 2002). Deletion of this intronic element has been associated with a case of ataxia telangiectasia. Similarly, binding of U1 snRNP at downstream 5′ splice sites is known to inhibit the cleavage step of pre-mRNA 3′-end processing and may suppress premature polyadenylation before synthesis of terminal exons (Ashe et al. 1997; Vagner et al. 2000). At some long introns it may be necessary to engage the 5′ splice site (and therefore to regenerate a functional 5′ splice site at the same position) multiple times during the period required for transcription to prevent cleavage at cryptic polyadenylation sites within the upstream exons. An apparently opposite effect has been observed in the mammalian RNA for calcitonin/CGRP, where interaction of U1 snRNP and SRp20 with a hybrid element that resembles an RP activates 3′-end formation at an alternative upstream polyadenylation site (Lou et al. 1998); however, in this case additional sequence features and interactions with pyrimidine-tract-binding protein subvert the function of the element in splicing and convert it into an enhancer of polyadenylation (Lou et al. 1999).
These effects alone do not explain why selection would maintain recursive splice sites rather than eliminate interfering signals, except for regulatory purposes and in cases where cryptic sites might be maintained by selection for other functions, such as coding within exons. An intriguing possibility is that recursive splicing also helps to ensure transcription through long introns by recruiting or stimulating the activity of elongation factors for RNA polymerase II. A stimulatory effect of introns on gene expression has been observed in yeast and mice (Ares et al. 1999), and increasing evidence indicates that transcript initiation, elongation, and processing are coupled functionally (reviewed in Goldstrohm et al. 2001; Maniatis and Reed 2002; Neugebauer 2002; Kornblihtt et al. 2004). A specific precedent is the finding that addition of a functional intron stimulates elongation on an HIV-1 in vitro transcription template (Fong and Zhou 2001). This effect is mediated by the binding of snRNPs to TAT-SF1 and is likely to involve the interaction between TAT-SF1 and positive elongation factor P-TEFb, which phosphorylates the carboxy-terminal domain of RNA polymerase II (Fong and Zhou 2001).
Detailed hypotheses for the functions of recursive splicing can be investigated now that conserved RPs have been identified in many Drosophila genes. Furthermore, the results presented here make it clear that studies of gene prediction, expression, mutation, and evolution should take into account the possible subdivision of introns and the existence of productive splicing events that are not reflected in mRNA structure.
Acknowledgments
We thank Neil Christopher for programming assistance and Eric Xing for discussions. Supported by National Institutes of Health grants RO1-HD28664 and K02-HD01155 and by a grant from the Pennsylvania Department of Health (project 017394). J.C. was a Beckman Foundation Undergraduate Research Scholar. E.M.-S. was a visiting student supported by the National Science Foundation Science and Technology Center for Light Microscope Imaging and Biotechnology at Carnegie Mellon University.
References
- Ares, M., L. Grate and M. H. Pauling, 1999. A handful of intron-containing genes produces the lion's share of yeast mRNA. RNA 9**:** 1138–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe, M. P., L. H. Pearson and N. J. Proudfoot, 1997. The HIV-1 5′ LTR poly(A) site is inactivated by U1 snRNP interaction with the downstream major splice donor site. EMBO J. 16**:** 5752–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berget, S. M., 1995. Exon recognition in vertebrate splicing. J. Biol. Chem. 270**:** 2411–2414. [DOI] [PubMed] [Google Scholar]
- Blencowe, B. J., 2000. Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem. Sci. 25**:** 106–110. [DOI] [PubMed] [Google Scholar]
- Celniker, S. E., D. A. Wheeler, B. Kronmiller, J. W. Carlson, A. Halpern et al., 2002 Finishing a whole-genome shotgun: release 3 of the Drosophila melanogaster euchromatic genome sequence. Genome Biol. 3: research0079.1–0079.14. [DOI] [PMC free article] [PubMed]
- Comeron, J. M., and M. Kreitman, 2000. The correlation between intron length and recombination in Drosophila: dynamic equilibrium between mutational and selective forces. Genetics 156**:** 1175–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron, J. M., and M. Kreitman, 2002. Population, evolutionary and genomic consequences of interference selection. Genetics 161**:** 389–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couronne, O., A. Poliakov, N. Bray, T. Ishkhanov, D. Ryaboy et al., 2003. Strategies and tools for whole-genome alignments. Genome Res. 13**:** 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch, M., and M. Long, 1999. Intron-exon structures of eukaryotic model organisms. Nucleic Acids Res. 27**:** 3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother, W. G., and L. A. Chasin, 2000. Human genomic sequences that inhibit splicing. Mol. Cell. Biol. 20**:** 6816–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FlyBase Consortium 2003. The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 31**:** 172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong, Y. W., and Q. Zhou, 2001. Stimulatory effect of splicing factors on transcriptional elongation. Nature 414**:** 929–933. [DOI] [PubMed] [Google Scholar]
- Goldstrohm, A. C., T. R. Albrecht, C. Sune, M. T. Bedford and M. A. Garcia-Blanco, 2001. The transcription elongation factor CA150 interacts with RNA polymerase II and the pre-mRNA splicing factor SF1. Mol. Cell. Biol. 21**:** 7617–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich, J. A., M. L. Schwartz and W. R. McClure, 1990. Searching for and predicting the activity of sites for DNA binding proteins: compilation and analysis of the binding sites for Escherichia coli integration host factor (IHF). Nucleic Acids Res. 18**:** 4993–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton, A. R., V. Subramaniam and A. J. Lopez, 1998. Generation of alternative Ultrabithorax isoforms and stepwise removal of a large intron by resplicing at exon exon junctions. Mol. Cell 2**:** 787–796. [DOI] [PubMed] [Google Scholar]
- Kornblihtt, A. R., M. De La Mata, J. P. Fededa, M. J. Muñoz and G. Nogues, 2004. Multiple links between transcription and splicing. RNA 10**:** 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld, K., R. B. Saint, P. A. Beachy, P. J. Harte, D. A. Peattie et al., 1989. Structure and expression of a family of Ultrabithorax mRNAs generated by alternative splicing and polyadenylation in Drosophila. Genes Dev. 3**:** 243–258. [DOI] [PubMed] [Google Scholar]
- Kurek, R., A. M. Reugels, U. Lammermann and H. Bunemann, 2000. Molecular aspects of intron evolution in dynein encoding mega-genes on the heterochromatic Y chromosome of Drosophila sp. Genetica 109**:** 113–123. [DOI] [PubMed] [Google Scholar]
- Lee, H., J. A. Simon and J. T. Lis, 1988. Structure and expression of ubiquitin genes of Drosophila melanogaster. Mol. Cell. Biol. 8**:** 4727–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou, H., K. M. Neugebauer, R. F. Gagel and S. M. Berget, 1998. Regulation of alternative polyadenylation by U1 snRNPs and SRp20. Mol. Cell. Biol. 18**:** 4977–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou, H., D. M. Helfman, R. F. Gagel and S. M. Berget, 1999. Polypyrimidine tract-binding protein positively regulates inclusion of an alternative 3′-terminal exon. Mol. Cell. Biol. 19**:** 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis, T., and R. Reed, 2002. An extensive network of coupling between gene expression machines. Nature 416**:** 499–506. [DOI] [PubMed] [Google Scholar]
- Misra, S., M. A. Crosby, C. J. Mungall, B. B. Matthews, K. S. Campbell et al., 2002 Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biol. 3: research0083.1–0083.22 [DOI] [PMC free article] [PubMed]
- Mount, S.M., C. Burks, G. Hertz, G. D. Stormo, O. White et al., 1992. Splicing signals in Drosophila: intron size, information content, and consensus sequences. Nucleic Acids Res. 20**:** 4255–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer, K. M., 2002. On the importance of being co-transcriptional. J. Cell Sci. 115**:** 3865–3871. [DOI] [PubMed] [Google Scholar]
- O'Connor, M. B., R. L. Binari, A. Perkins and W. Bender, 1988. Alternative RNA products from the Ultrabithorax domain of the bithorax complex. EMBO J. 7**:** 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott, S., Y. Tamada, H. Bannai, K. Nakai and S. Miyano, 2003 Intrasplicing—analysis of long intron sequences. Pac. Symp. Biocomput. 2003: 339–350. [DOI] [PubMed]
- Pagani, F., E. Buratti, C. Stuani, R. Bendix, T. Dörk et al., 2002. A new type of mutation causes a splicing defect in ATM. Nat. Genet. 30**:** 426–429. [DOI] [PubMed] [Google Scholar]
- Powell, J. R., 1997 Progress and Prospects in Evolutionary Biology: The Drosophila Model. Oxford University Press, New York.
- Proudfoot, N. H., 2003. Dawdling polymerases allow introns time to splice. Nat. Struct. Biol. 10**:** 876–877. [DOI] [PubMed] [Google Scholar]
- Reugels, A. M., R. Kurek, U. Lammermann and H. Bunemann, 2000. Mega-introns in the dynein gene DhDhc7(Y) on the heterochromatic Y chromosome give rise to the giant threads loops in primary spermatocytes of Drosophila hydei. Genetics 154**:** 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothe, M., M. Pehl, H. Taubert and H. Jäckle, 1992. Loss of gene function through rapid mitotic cycles in the Drosophila embryo. Nature 359**:** 156–159. [DOI] [PubMed] [Google Scholar]
- Ruden, D. M., and H. Jäckle, 1995. Mitotic delay dependent survival identifies components of cell cycle control in the Drosophila blastoderm. Development 121**:** 63–73. [DOI] [PubMed] [Google Scholar]
- Saxonov, S., I. Daizadeh, A. Fedorov and W. Gilbert, 2000. EID: the exon-intron database: an exhaustive database of protein-coding intron-containing genes. Nucleic Acids Res. 28**:** 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shermoen, A. W., and P. H. O'Farrell, 1991. Progression of the cell cycle through mitosis leads to abortion of nascent transcripts. Cell 67**:** 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi, M., G. Menozzi, L. Riva, R. Cagliani, G. P. Comi et al., 2004. Silencer elements as possible inhibitors of pseudoexon splicing. Nucleic Acids Res. 32**:** 1738–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam, V., H. M. Bomze and A. J. Lopez, 1995. Functional differences between Ultrabithorax protein isoforms in Drosophila melanogaster: evidence from elimination, substitution and ectopic expression of specific isoforms. Genetics 136**:** 979–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, H., and L. A. Chasin, 2000. Multiple splicing defects in an intronic false exon. Mol. Cell. Biol. 20**:** 6414–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel, C. S., 1992. Mechanisms of transcriptional timing in Drosophila. Science 255**:** 39–40. [DOI] [PubMed] [Google Scholar]
- Vagner, S., U. Ruegsegger, S. I. Gunderson, W. Keller and I. W. Mattaj, 2000. Position-dependent inhibition of the cleavage step of pre-mRNA 3′-end processing by U1 snRNP. RNA 6**:** 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, J., W. R. Hess and T. Borner, 1997. Precise branch point mapping and quantification of splicing intermediates. Nucleic Acids Res. 25**:** 2030–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, M., and M. Rice, 2004. Ordered partitioning reveals extended splice-site consensus information. Genome Res. 14**:** 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]