Mechanisms of hydralazine induced vasodilation in rabbit aorta and pulmonary artery (original) (raw)
Abstract
- The directly acting vasodilator hydralazine has been proposed to act at an intracellular site in vascular smooth muscle to inhibit Ca2+ release.
- This study investigated the mechanism of action of hydralazine on rabbit aorta and pulmonary artery by comparing its effects on the tension generated by intact and β-escin permeabilized vessels and on the cytoplasmic Ca2+ concentration, membrane potential and K+ currents of isolated vascular smooth muscle cells.
- Hydralazine relaxed pulmonary artery and aorta with similar potency. It was equally effective at inhibiting phasic and tonic contractions evoked by phenylephrine in intact vessels and contractions evoked by inositol 1,4,5 trisphosphate (IP3) in permeabilized vessels.
- Hydralazine inhibited the contraction of permeabilized vessels and the increase in smooth muscle cell Ca2+ concentration evoked by caffeine with similar concentration dependence, but with lower potency than its effect on IP3 contractions.
- Hydralazine had no effect on the relationship between Ca2+ concentration and force generation in permeabilized vessels, but it slowed the rate at which maximal force was developed before, but not after, destroying sarcoplasmic reticulum function with the calcium ionophore, ionomycin.
- Hydralazine had no effect on membrane potential or the amplitudes of K+ currents recorded from isolated smooth muscle cells over the concentration range causing relaxation of intact vessels.
- The results suggest that the main action of hydralazine is to inhibit the IP3-induced release of Ca2+ from the sarcoplasmic reticulum in vascular smooth muscle cells.
Keywords: Hydralazine, IP3, rabbit, aorta, pulmonary artery, smooth muscle, sarcoplasmic reticulum, caffeine, Ca2+, vasodilation
Introduction
Hydralazine has been in clinical use as an anti-hypertensive agent for nearly five decades, but its mechanism of action remains poorly understood. It is a directly acting vasodilator, which at micromolar concentrations relaxes a number of arterial preparations in vitro, including rabbit aorta (Cook et al., 1988; Higashio & Kuroda, 1988; Gurney & Allam, 1995) and renal artery (Khayyal et al., 1981), rat caudal artery (Hermsmeyer et al., 1983), porcine coronary artery (Wei et al., 1997) and human digital artery (Lipe & Moulds, 1981). It continues to relax vessels after removal of the endothelial cell layer (Gurney & Allam, 1995, Wei et al., 1997), implying that it acts directly on vascular smooth muscle.
There is increasing evidence that hydralazine acts at an intracellular site to cause vasorelaxation. It does not act by elevating smooth muscle cyclic nucleotide levels (Diamond & Janis, 1978; Yen et al., 1989). Nor does it act as a calcium antagonist, because it is a poor inhibitor of vasoconstriction caused by elevated extracellular K+ (Khayyal et al., 1981; Lipe & Moulds, 1981; Higashio & Kuroda, 1988; Gurney & Allam, 1995) and its effectiveness at inhibiting phenylephrine-induced contraction is little affected by removing extracellular Ca2+ (McLean et al., 1978; Higashio & Kuroda, 1988; Orallo et al., 1991; Gurney & Allam, 1995). Hydralazine inhibits contractions evoked by caffeine, which directly stimulates the release of Ca2+ from the sarcoplasmic reticulum (SR), and its effects on phenylephrine-constricted vessels are suppressed in conditions that deplete intracellular Ca2+ stores (Gurney & Allam, 1995). The SR is therefore the most likely site of action of hydralazine, through which it causes a fall in the intracellular Ca2+ concentration ([Ca2+]i) available for contraction. Consistent with this, hydralazine reduced [Ca2+]i in ferret aorta without altering the relationship between [Ca2+]i and force generation, although this effect was investigated only at high (mM) drug concentrations (Defeo & Morgan, 1989). Hydralazine was proposed to act by inhibiting the release of Ca2+ from the SR evoked by inositol 1,4,5 trisphosphate (IP3), because it retained its full effect against phenylephrine contractions in Ca2+-free medium when applied in the presence of pre-filled stores (Gurney & Allam, 1995). Moreover, contractions evoked by phenylephrine were more sensitive to hydralazine than those evoked by caffeine (Gurney & Allam, 1995).
The primary aim of this study was to directly determine the influence of hydralazine on the release of Ca2+ from the SR, by investigating its effects on permeabilised vessels and on [Ca2+]i transients evoked by SR Ca2+ release. The possible effects of hydralazine on potassium currents and membrane potential were also investigated, because previous studies suggested that potassium channel activation (Thirstrup & Nielsen-Kudsk, 1992; Bang et al., 1998) and membrane hyperpolarization (Hermsmeyer et al., 1983) might contribute to the vasodilator action of the drug.
Methods
Tissue preparation
Male New Zealand rabbits (2 – 3 kg) were killed with an overdose of sodium pentobarbitone (80 mg kg−1 i.v.) (Ceva, Watford, Hertfordshire, U.K.) and exsanguinated. The descending thoracic aorta or main pulmonary artery was excised into physiological salt solution and cleaned of residual fat and connective tissue. For some experiments the endothelial cell layer was removed by gently rubbing the intimal surface of the vessel with a plastic Pasteur pipette. Successful removal of the endothelium was indicated by the lack of a relaxation response to 1 μM acetylcholine when applied to vessels precontracted with 1 μM phenylephrine. Vessels were cut into 2 – 3 mm wide rings for studies on intact preparations, or transverse strips 200 – 400 μm wide and 10 – 15 mm long for permeabilization. They were suspended in 35 ml (rings) or 300 μl (strips) organ baths for tension recording with an isometric transducer (Harvard Apparatus, model 60-2998). The organ baths contained physiological salt solution of the following composition (mM): NaCl 118, KCl 4.74, NaHCO3 25, KH2PO4 1.19, MgSO4 1.2, glucose 11, CaCl2 1.2. The solution was prepared with 18 MΩ water and was continuously bubbled with 95% O2, 5% CO2 to maintain pH 7.2. Tissues were placed under a predetermined optimal resting tension of 1.25 g (rings) or 350 mg (strips) and allowed to equilibrate at 37°C (rings) or 24°C (strips) for at least 30 min before experiments commenced.
Vessel permeabilization
Vessel strips were permeabilized by bathing them for up to 30 min in solution containing 80 μM β-escin, 9.5 mM creatine phosphate (Na2CP), 5.5 mM Na2ATP, 10 mM ethyleneglycol-_bis_-(β-aminoethyl)-N,N,N′,N′-tetraacetic acid (EGTA), 3 μM calmodulin, 100 mM N,N-bis[2-hydroxyethyl]-2-aminoethane sulphonic acid (BES buffer), 5 mM reduced glutathione, 0.1 mM phenylmethyl-sulphonyl fluoride (to inhibit intracellular proteolysis) and MgCl2 and CaCl2 added to give free Mg2+ and Ca2+ concentrations of 1 mM and 1 μM, respectively. The pH of the solution was adjusted to 7.1 with KOH. When permeabilization was complete, as indicated by contraction of the tissue to a steady level, the tissue was washed for 20 min in relaxing solution, which had the same composition, but without β-escin or Ca2+. In order to load the sarcoplasmic reticulum stores with Ca2+, tissues were incubated for 10 min in a Ca2+ loading solution, which had the same composition as the relaxing solution, but with CaCl2 added to provide a free Ca2+ concentration of 120 nM. The [Ca2+]i-tension relationship of permeabilized tissues was investigated using similar solutions, but with the concentrations of CaCl2 and MgCl2 varied to provide a range of free Ca2+ concentrations while maintaining Mg2+ at 1 mM. The required amounts of CaCl2 and MgCl2 were calculated using the programme Solution VI based on Perrin & Sayce (1967) and checked using Chelator for Windows 1.1 (Steinhardt Software). With the exception of EGTA, all stock solutions used for experiments with permeabilized vessels were passed down a chelex column to remove contaminating Ca2+.
Isolation of vascular smooth muscle cells
Smooth muscle cells were isolated from strips of rabbit aorta or main pulmonary artery as previously described (Clapp & Gurney, 1991; Halliday et al., 1995). Briefly, artery strips were incubated overnight in a refrigerator with 0.25 mg ml−1 papain (Fluka Chemicals, U.K.) and 0.02% bovine serum albumin in dissociation medium (pH 7.0) containing (mM): NaCl 110, KCl 5, NaHCO3 15, CaCl2 0.16, MgCl2 2, NaH2PO4 0.5, KH2PO4 0.5, glucose 10, 5-N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid (HEPES) 15, phenol red 0.04, ethylenediaminetetraacetic acid 0.49 and taurine 10. The next morning, 0.2 mM dithiothreitol (Sigma) was added and the tissue incubated at 37°C for 15 min. Cells were then released by trituration in enzyme-free dissociation medium and stored in a refrigerator for up to 12 h.
Fura-2 loading and measurement of [Ca2+]i
Smooth muscle cells were loaded with fura-2 using the membrane permeable, acetoxymethyl form (fura-2/AM), which was prepared as a 1 mM stock solution in dimethyl sulfoxide (DMSO). Fura-2/AM (10 μM) was added to a 1 ml suspension of cells in an eppendorf tube, which was wrapped in aluminium foil and left at room temperature in a darkened area for 45 – 60 min. The tube was then transferred to a 37°C water bath for 10 min to stimulate hydrolysis of the intracellular indicator, before storing the cells for up to 6 h in a refrigerator. After loading, drops of cell suspension were placed on a glass coverslip forming the base of a 3 ml experimental chamber, mounted on the stage of an epifluorescence microscope. Once cells had adhered to the glass surface, excess fura-2/AM was removed by superfusing the cells with physiological salt solution. Caffeine was applied to cells by pressure ejection (≈#38;2 psi) from a micropipette positioned nearby.
The fluorescence of individual cells was excited alternately at 340 and 380 nm, using a filter wheel spinning at 375 rev min−1. Emitted fluorescence passed through a 510 nm bandpass filter and was detected by a photomultiplier using photon counting. Recording and analysis of fluorescence signals employed PhoCal software (Version 5, Dr J. Dempster, University of Strathclyde). An image mask placed around the cell under investigation minimized background fluorescence. The ratio of fluorescence excited by 340 and 380 nm (R340/380) was determined after subtraction of background fluorescence and converted to [Ca2+]i according to the method of Grynkiewiez et al. (1985), using parameters obtained from in situ calibration with cells exposed at the end of each experiment to ionomycin (10 μM) followed by EGTA (10 mM). The maximum and minimum ratios, R max (2.4±0.1, _n_=12) and R min (0.2±0.02), were measured at saturating and minimum levels of Ca2+, respectively, and used with β (2±0.4), the fluorescence ratio excited at 380 nm in minimum and saturating levels of Ca2+ to estimate the dissociation constant for Ca2+ binding to fura-2, K d (158 nM).
Electrophysiology
Drops of cell suspension were placed on a glass coverslip forming the base of a 2 ml experimental chamber, mounted on the stage of an inverted microscope. Once cells had adhered to the glass surface, they were continually superfused with physiological salt solution. Electrophysiological experiments were carried out on aorta smooth muscle cells as previously described (Halliday et al., 1995). Pipettes for whole-cell recording contained the following (mM): KCl 120, MgCl2 1, EGTA 1, Na2GTP 0.5, HEPES 10; pH 7.2 with KOH. Membrane potentials were measured under current clamp conditions, as the average zero-current potentials during 30 s recording periods. To activate K+ currents, cells were voltage clamped at −80 mV and 80 ms depolarizing steps applied at 5 s intervals. Currents were recorded at test potentials of −70 to 50 mV, incremented in 10 mV steps. Series resistance was less than 10 MΩ before compensation and around 80% compensation was usually achieved. Membrane potential and currents were recorded for at least 5 min before applying hydralazine, to ensure that any current run-up (Clapp & Gurney, 1991) was complete.
Drugs
Hydralazine hydrochloride, phenylephrine hydrochloride, inositol 1,4,5-trisphosphate hydrochloride, caffeine, ionomycin, β-escin, sodium creatine phosphate, ATP, EGTA, BES, glutathione, phenylmethyl-sulphonyl fluoride and chelex 100 were all from Sigma Chemical Company, Poole, Dorset, U.K. Acetylcholine chloride and 1 M stock solutions of CaCl2 and MgCl2 were from BDH Merck Ltd, Poole, Dorset, U.K. Stock solutions of ionomycin were prepared in DMSO (final bath concentration 0.1%). All other stock solutions were prepared with 18 ΩM water and serially diluted with the appropriate solution.
Data analysis
Results are expressed as mean±s.e.mean. Statistical analysis was conducted using Graphpad Prism for Windows 3.1 and Minitab software. Mean data were compared with a paired or unpaired Students two-tailed _t_-test. One- or two-way analysis of variance (ANOVA) was employed for multiple data sets. P<0.05 was considered significant. Inhibitory effects of hydralazine on contractions evoked by phenylephrine, InsP3 or caffeine were measured as the per cent reduction of the maximum contraction recorded before the drug was applied. Since the effects of hydralazine required several hours to recover following washout, it was not possible to routinely check for recovery. The effects of hydralazine were therefore compared with time-controlled preparations, which were treated in the same way except for the omission of hydralazine.
Results
Hydralazine was previously shown to evoke a slowly developing, long-lived relaxation in rabbit aorta rings. As shown in Figure 1A, it had the same effect on rabbit main pulmonary artery. When applied to pulmonary artery rings precontracted with 1 μM phenylephrine, hydralazine reduced the developed tone by 78±2 % (_n_=5) at 100 μM, with the response taking around 20 min to reach maximum and several hours to recover after removing the drug. The results illustrated in Figure 1 were obtained in vessels from which the endothelium had been removed, thus demonstrating that, as previously found in rabbit aorta, the ability of hydralazine to relax pulmonary artery was independent of the endothelium. Therefore, in both vessel types, hydralazine causes vasodilation as a result of a direct action on vascular smooth muscle cells. Hydralazine was equipotent in relaxing pulmonary artery and aorta as shown by the superimposed concentration-response curves in Figure 1B. The concentration producing 50% of the maximum effect (EC50) was 16±2 μM (_n_=5) in pulmonary artery and 20±1 μM (_n_=7) in aorta. A similar maximum relaxation, equivalent to around 90% of the phenylephrine-induced tone, was also observed in each vessel and in both cases the maximum was achieved at hydralazine concentrations above 100 μM.
Figure 1.
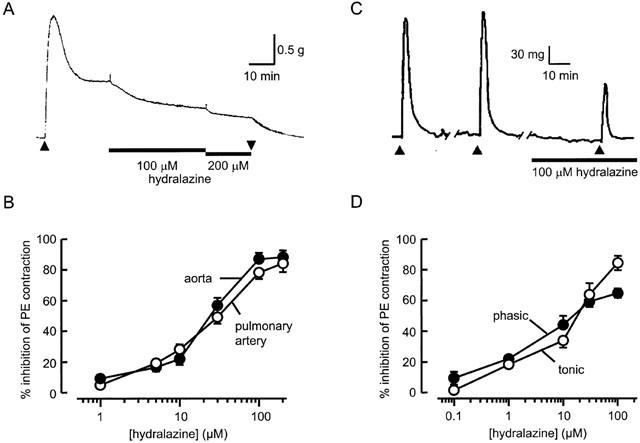
Influence of hydralazine on tension generation by intact arteries. (A) Hydralazine was added as indicated by bars to a rabbit pulmonary artery ring precontracted with 1 μM phenylephrine, present during the period indicated by the arrows. (B) Relationship between hydralazine concentration and relaxation in endothelium-denuded rabbit aorta (_n_=7) and pulmonary artery (_n_=7) precontracted with 1 μM phenylephrine. (C) Influence of hydralazine applied for 30 min (bar) on phasic responses of aorta to 10 μM phenylephrine, applied for 5 min at 30 min intervals. (D) Comparison of concentration-effect curves for hydralazine constructed for inhibition of tonic (_n_=7) and phasic (_n_=5) contractions evoked in endothelium-denuded rabbit aorta by 10 μM phenylephrine.
Tonic tension developed in the continued presence of phenylephrine is thought to be sustained by Ca2+ influx carried through Ca2+-permeable channels in the smooth muscle cell membrane (Lodge & van Breemen, 1985), although it may pass through the SR before activating contraction (van breemen et al., 1995; Gurney & Allam, 1995). The release of SR Ca2+, mediated by intracellular IP3 in response to agonist-induced activation of phospholipase C, is thought to underlie the initial, usually transient phase of tension, developed in response to phenylephrine exposure (van breemen & Saida, 1989). If hydralazine promotes vasodilation by interfering with the release of Ca2+ from SR stores, it would therefore be expected to inhibit the initial, phasic response to phenylephrine as effectively as it inhibits tonic tension. Brief (5 min) applications of phenylephrine (10 μM) in normal, Ca2+-containing solution, repeated at 30 min intervals, evoked phasic contractions that were reproducible over several hours, but were suppressed after 30 min exposure to hydralazine (Figure 1C). In the presence of 100 μM hydralazine the amplitude of the phasic contraction was reduced by 65±3% (_n_=5), which is not significantly different from its effect on tonic tension (82±7%, _n_=6). Overall, the concentration-effect curves for inhibition of tonic and phasic contractions produced by 10 μM phenylephrine were almost superimposed (Figure 1D), indicating a similar potency for inhibition of both phases of the response.
Influence of hydralazine on permeabilized vessels
To determine whether or not the vasodilator effect of hydralazine requires an intact cell membrane, we investigated its ability to interfere with the contraction of thin strips of aorta after disrupting cell membranes with β-escin. Strips of aorta exposed to 80 μM β-escin for up to 30 min became permeabilized, as indicated by the reversible, spontaneous development of tension when the permeabilizing solution contained 1 μM Ca2+. Permeabilized vessels were relaxed when bathed in Ca2+-free relaxing solution, but they contracted transiently in response to 5 min applications of 100 μM IP3 (Figure 2A) or 20 mM caffeine (Figure 2B). Since these agents act directly on the SR to stimulate Ca2+ release through IP3 and Ca2+-activated ion channels, respectively, the results indicate that β-escin permeabilized the plasma membrane without disrupting the SR membrane, as previously reported (Kobayashi et al., 1989; Savineau & Marthan, 1994). IP3 produced monophasic contractile responses that were reproducible over several hours during repeated applications at 30 – 40 min intervals, provided Ca2+ stores were refilled between applications by exposure to Ca2+-containing, loading solution. Contractile responses to caffeine were also reproducible provided the stores were refilled between applications. Otherwise, a second application failed to induce a response, indicating that a single application of 20 mM caffeine was sufficient to empty the stores. The transient contraction evoked by caffeine was often followed by brief relaxation to below the basal level (Figure 2b).
Figure 2.
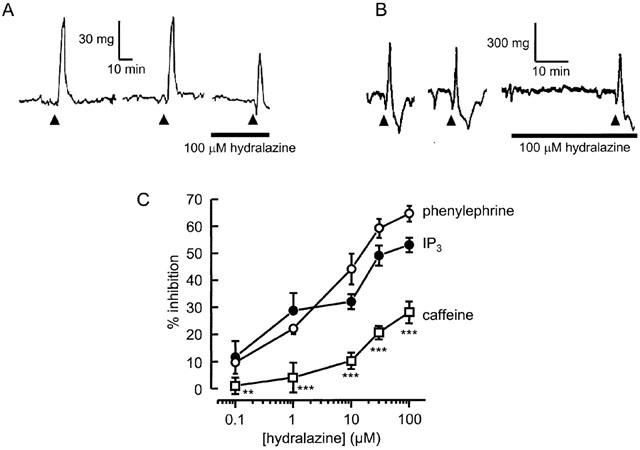
Influence of hydralazine on tension generation by permeabilized arteries. Application of 100 μM IP3 (A) or 20 mM caffeine (B), indicated by arrows, evoked transient contractile responses in strips of aorta permeabilized with β-escin. Ca2+ stores were refilled between applications by incubating the tissues in Ca2+ loading solution for 10 min. Exposure to hydralazine for 30 min (bar) reduced the amplitude of contraction. (C) Comparison of the concentration-effect relationships for hydralazine inhibition of contractions activated by IP3 (_n_=5) or caffeine (_n_=5) in permeabilized aorta and phasic contractions to phenylephrine in intact vessels (_n_=5).
When permeabilized vessel strips were exposed to hydralazine for 30 min, the amplitudes of responses to both IP3 and caffeine were reduced in a concentration-dependent manner (Figure 2). This contrasts with the responses of paired strips, which were unaltered over the same period when treated in the same way but without exposure to hydralazine. At 100 μM hydralazine, the mean inhibition of the IP3-induced contraction was 52±3% (_n_=5), which is significantly less (P<0.05) than that observed for phasic contractions to phenylephrine. Nevertheless, comparison of the hydralazine concentration-effect curves for inhibition of IP3 contractions in permeabilized strips and phenylephrine-activated contractions in intact aorta rings indicates only a small difference in effectiveness, limited to the highest hydralazine concentrations (Figure 2c). In contrast, at all concentrations tested, hydralazine was significantly less effective at inhibiting contractile responses to caffeine (Figure 2c). At 100 μM it reduced the response by only 32±5% (_n_=5), which is less than half of the inhibition observed with IP3 or phenylephrine mediated contractions.
The ability of hydralazine to reduce contractile responses to agents that release SR Ca2+ could reflect an inhibitory action on the SR, to reduce the Ca2+ available for contraction, or reduced sensitivity of the contractile proteins to the released Ca2+. To differentiate between these possible mechanisms, the effects of hydralazine were investigated on the contractile responses evoked directly by Ca2+ applied to vessel strips permeabilized with β-escin (Figure 3). When the solution bathing permeabilized strips was changed from the Ca2+-free relaxing solution to a solution containing 500 nM Ca2+, the muscle contracted to a plateau level that was reached in 18±2 min (_n_=6) and was sustained as long as the Ca2+ concentration remained elevated (Figure 3A). Cumulative increases in Ca2+ concentration above 100 nM produced concentration-dependent contraction (Figure 3B), with the maximum tension reached at about 1 μM Ca2+ and 50% of maximum at 400±50 nM (_n_=3). Repeating the cumulative increases in [Ca2+] after washing the tissue with Ca2+-free relaxing solution, which promoted complete relaxation, evoked nearly identical responses.
Figure 3.
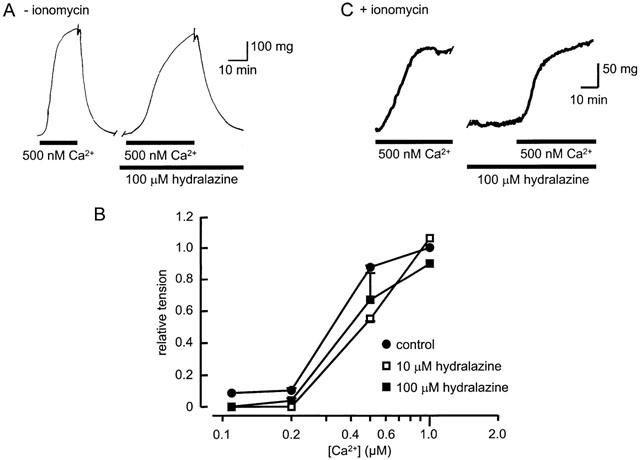
Hydralazine does not inhibit tension activation by Ca2+. Application of 500 nM Ca2+ to strips of aorta permeabilized with β-escin induced a sustained contraction in the absence (A) or presence (C) of 10 μM ionomycin. The vessels were washed with Ca2+-free solution and exposed to hydralazine (100 μM) for 30 min before the second application of Ca2+. (B) The Ca2+-tension relationship measured in permeabilized rings from five aortae in the absence and presence of 10 and 100 μM hydralazine. At each [Ca2+], tension was measured relative to the response evoked at 1 μM Ca2+ under control conditions.
In the presence of 100 μM hydralazine, the application of 500 nM Ca2+ evoked contraction that reached a similar plateau level to that observed in the absence of the drug (Figure 3A). Over a range of concentrations, hydralazine had no significant effect on the [Ca2+]-tension relationship of permeabilized aorta (Figure 3B). Hydralazine did however slow the rate of rise of the Ca2+-induced contraction (Figure 3A), the time taken to reach the plateau level increasing to 30±5 min (_n_=3; P<0.05). This could be due to an action on the SR, which could have influenced the activation of tension as a consequence of altered Ca2+ buffering and/or Ca2+-induced Ca2+ release. To test this hypothesis, vessel strips were exposed to the Ca2+ ionophore, ionomycin (10 μM), and the experiments repeated. In the presence of ionomycin, permeabilised aorta contracted in response to Ca2+, but failed to respond to IP3 (100 μM) or caffeine (20 mM), indicating the loss of functional Ca2+ stores due to collapse of the [Ca2+] gradient across the SR membrane. Under these conditions, the rate of rise of the contractile response to 500 nM Ca2+, as well as its amplitude, was unaffected by hydralazine (Figure 3C). The mean time to peak of the contraction in the presence of ionomycin was 20±3 min (_n_=3) in control conditions and 18±3 min after exposing the tissue to 100 μM hydralazine.
Influence of hydralazine on [Ca2+]i
Isolated smooth muscle cells loaded with the Ca2+ indicator fura-2 were used to investigate the effect of hydralazine on Ca2+ released from the SR by caffeine. As illustrated in Figure 4, the application of 20 mM caffeine to smooth muscle cells from aorta or pulmonary artery caused a transient increase in [Ca2+]i, as indicated by an increase in R340/380 recorded from intracellular fura-2. The response reached a peak in around 10 s and returned to the basal level after around 45 s. Caffeine applied at 10 min intervals evoked responses that were reproducible over several hours. On average, caffeine increased R340/380 from a baseline of 0.45±0.02 to a peak of 1.4±0.1 (_n_=13), equivalent to a rise in [Ca2+]i from a resting level of 40 to 380 nM at the peak of the response. The increase was sometimes followed by a reduction of [Ca2+]i to below the resting level (Figure 4A), consistent with the relaxation phase that was often seen to follow the contractile response of intact vessels to caffeine. In the presence of 100 μM hydralazine, the amount of Ca2+ released by caffeine was reduced, the peak of the [Ca2+]i transient, measured as R340/380, declining by around 50%. There was no significant difference between the effects of hydralazine on the [Ca2+]i transient recorded from aorta or pulmonary artery smooth muscle cells and in both cases the effect took 20 – 30 min to fully develop (Figure 4A). Inhibition was concentration-dependent, with the maximum effect observed at around 100 μM hydralazine (Figure 4B). In order to compare the effect of hydralazine on caffeine-induced [Ca2+]i transients with its effect on caffeine-activated tension, for each response the per cent reduction was normalized against the reduction produced by 100 μM hydralazine and plotted in Figure 4C. This shows that hydralazine reduced the Ca2+ and tension responses to caffeine over a similar concentration range. Although hydralazine may be slightly more potent at inhibiting the [Ca2+]i transient, the difference was small. It was consistently found that, after exposure to hydralazine, the basal [Ca2+]i was reduced and the fall in [Ca2+]i that normally followed the caffeine-induced increase in [Ca2+]i was prevented (Figure 4A).
Figure 4.
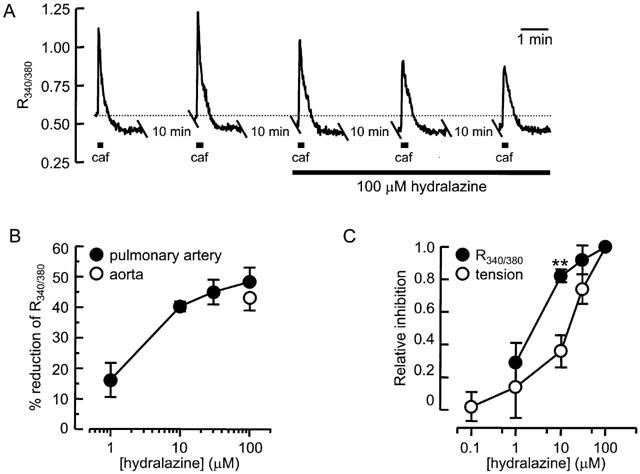
Hydralazine inhibits Ca2+ release evoked by caffeine. (A) Application of 20 mM caffeine (caf) to isolated aorta smooth muscle cells caused a transient increase in [Ca2+]i, as indicated by R340/380 reported by fura-2. Caffeine was applied at 10 min intervals and fluorescence recorded only during the response to caffeine. Gaps in the record represent 10 min periods between applications. Hydralazine (100 μM) was applied for 30 min after obtaining reproducible responses to caffeine and caused a gradual decline in the peak amplitude of the response. (B) Relationship between the hydralazine concentration and the reduction of peak R340/380 in smooth muscle cells from pulmonary artery (_n_=6) and aorta (_n_=4). (C) Comparison of the concentration-effect relationships for inhibition by hydralazine of the caffeine-induced increase in R340/380 in isolated cells (_n_=6) and the increase in tension in permeabilized vessels (_n_=5). For each response, the effect of hydralazine at each concentration is normalized against the inhibition measured at 100 μM hydralazine.
Influence of hydralazine on membrane potential and K+ current
The resting membrane potential of isolated rabbit aorta smooth muscle cells, recorded using the whole-cell patch-clamp technique with the membrane current clamped at zero, was −40±2 mV (_n_=20). As shown in Figure 5A, the membrane potential remained at that level during up to 1 h of recording and it was unaffected by hydralazine applied to the cell for up to 30 min. Figure 5B shows that hydralazine had no effect on membrane potential over a range of concentrations (1 – 100 μM) that produce vasodilation. Consistent with this finding, hydralazine, at concentrations up to 100 μM, also failed to alter voltage-activated K+ currents recorded from the same cells under voltage-clamp conditions (Figure 5C).
Figure 5.
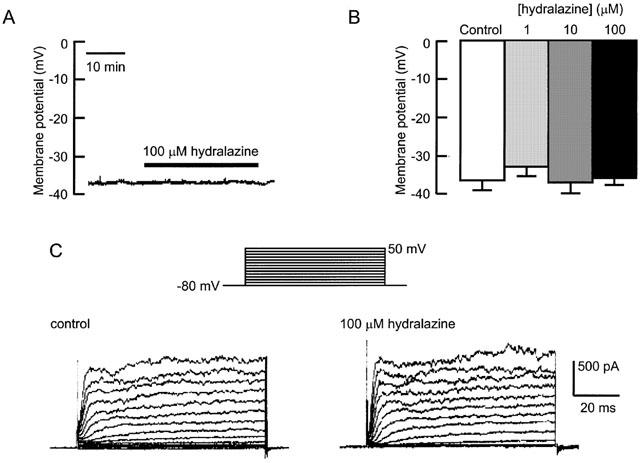
Hydralazine does not affect membrane potential or voltage-activated potassium currents. (A) A continuous record of resting membrane potential before and during the application of 100 μM hydralazine. (B) Histogram showing mean measurements of resting potential in the absence and presence of 1, 10 and 100 μM hydralazine. (C) Records of whole-cell potassium currents before and after 30 min exposure to 100 μM hydralazine. The cell was voltage clamped at −80 mV and currents activated by 80 ms steps to potentials between −70 and 50 mV, applied at 5 s intervals.
Discussion
Hydralazine was equally effective at relaxing rabbit main pulmonary artery and aorta. As found in some (Khayyal et al., 1981; Kreye, 1984; Gurney & Allam, 1995) but not all previous studies on isolated arteries, it was consistently effective at low micromolar concentrations. At concentrations of 1 – 2 μM, which are reached in human serum following a 25 mg dose (Talseth, 1976), hydralazine inhibited phenylephrine- and IP3-activated responses by between 10 and 30%, suggesting that these effects contribute to its anti-hypertensive action. In both pulmonary artery and aorta the EC50 for hydralazine was around 20 μM and a maximum relaxation of 80 – 90% of the phenylephrine-induced contraction was observed at 200 μM. Hydralazine also displayed the same slow onset of action and recovery in pulmonary artery as previously reported in aorta (Gurney & Allam, 1995) and in agreement with earlier studies (e.g. Moína et al., 1994; Gurney & Allam, 1995), its relaxant effect did not require the presence of endothelium. The results further show that the relaxant effect of hydralazine did not require an intact cell membrane, but was mediated by an action on the SR of vascular smooth muscle, to inhibit Ca2+ release. This directly confirms previous suggestions that hydralazine has an intracellular site of action (Gurney & Allam, 1995; Orallo et al., 1991; Higashio & Kuroda, 1988) and is consistent with a report that cultured vascular smooth muscle cells accumulate tritiated hydralazine at intracellular sites (Baker et al., 1992).
The SR as the site of hydralazine action
The present finding that hydralazine was equally effective at inhibiting phasic and tonic contractile responses to phenylephrine, its effectiveness in the absence of extracellular Ca2+ (McLean et al., 1978; Ebeigbe & Aloamaka, 1985; Higashio & Kuroda, 1988; Orallo et al., 1991; Gurney & Allam, 1995), its lack of effect on contractions stimulated by Ca2+ influx (Higashio & Kuroda, 1988; Gurney & Allam, 1995) and the loss of its effectiveness when SR Ca2+ stores are pharmacologically depleted (Gurney & Allam, 1995) all implicate the SR as the main site of hydralazine action. Relaxation must result from reduced [Ca2+]i, because hydralazine did not interfere with the ability of Ca2+ to activate tension in permeabilized vessels. It also failed to affect the response of saponin-skinned renal artery to Ca2+-calmodulin and ATP (Kreye et al., 1983) or the [Ca2+]i-force relationship in ferret aorta (Defeo & Morgan, 1989). Moreover, hydralazine reduced the rise in [Ca2+]i evoked by caffeine in isolated smooth muscle cells over the same concentration range that it inhibited caffeine and phenylephrine-induced contractions of intact and permeabilized vessels.
Phasic contractions activated by phenylephrine are thought to result from the IP3-dependent release of SR Ca2+. Since hydralazine inhibited responses to both IP3 and caffeine, its ability to inhibit phenylephrine-induced tone is most likely due to reduced effectiveness of IP3 at activating Ca2+ release, rather than altered IP3 production. The greater effectiveness of hydralazine at blocking responses of permeabilized vessels to IP3 compared with caffeine suggests that it acts on the release process, rather than by stimulating Ca2+ accumulation and increasing store filling. This conclusion is further supported by the maintained effectiveness of hydralazine at inhibiting phenylephrine contractions in Ca2+-free medium when applied in the presence of pre-filled Ca2+ stores (Gurney & Allam, 1995). The results therefore suggest that hydralazine interacts primarily with the IP3 receptor or channel to inhibit SR Ca2+ release, but that this action is reinforced by lower efficacy interactions with the caffeine-sensitive, Ca2+-activated Ca2+-release (CICR) channel. It is not unusual for agents acting at one of these channels to interact with the other (Ehrlich et al., 1994).
Inhibition of Ca2+ release channels can explain all of the effects of hydralazine. For example, based on the evidence that spontaneous Ca2+ release via CICR channels contributes to resting [Ca2+]i and basal vascular tone (Knot et al., 1998), inhibition of CICR by hydralazine could account for the lowering of resting [Ca2+]i observed in isolated smooth muscle cells. The slowing of Ca2+-activated tension in permeabilized vessels by hydralazine can also be explained by an inhibitory action on CICR. That this effect involves an action on the SR is suggested by the finding that it was prevented by ionomycin, which caused functional disruption of the SR. Any explanation for the slowing of Ca2+-induced contraction must also account for the lack of effect of hydralazine on the time courses of phasic responses to phenylephrine, IP3 or caffeine, which ultimately depend on Ca2+-activated contraction. A clear distinction is that, while responses to the latter agents depend on Ca2+ being released from an existing store in the smooth muscle cell, the time course of the response of permeabilized vessels to Ca2+ depends largely on the rate at which the buffered Ca2+ solution reaches and equilibrates with the smooth muscle cells in the tissue and activates the contractile proteins. In control conditions, the SR can accumulate Ca2+ from the solution as it reaches the cells, but simultaneous CICR would tend to maintain the SR in a depleted state and prevent it from acting as a Ca2+ buffer. Inhibition of CICR by hydralazine would promote SR Ca2+ buffering, which in turn would increase the extraction of Ca2+ and reduce its effective concentration as the solution reaches the smooth muscle cells. It would therefore take longer for the solution to equilibrate with the tissue and generate tension.
The inhibitory effect of hydralazine on tonic tension can also be accounted for by reduced activity of IP3-receptor channels and SR Ca2+ release. Although tonic contraction is sustained by Ca2+ influx (Lodge & van Breemen, 1985), hydralazine was found to be a poor inhibitor of contractions mediated by Ca2+ influx (Higashio & Kuroda, 1988; Gurney & Allam, 1995). Moreover, the depletion of SR Ca2+ stores was found to prevent the effect of hydralazine on tonic contractions (Gurney & Allam, 1995). Along with the equal potency of hydralazine against phasic and tonic contractions, this suggests that inhibition of SR Ca2+ release underlies the effects of hydralazine on tonic responses and implies further that SR Ca2+ stores play a major role in tonic as well as phasic contractions. There is increasing evidence to support this conclusion. Thus although Ca2+ influx is required to maintain tension in the long term, a recent study found that tonic contraction was due to Ca2+ oscillations mediated by the cyclical accumulation and release of SR Ca2+, which could be seen even in the absence of extracellular Ca2+ (Ruehlmann et al., 2000). The authors proposed that the SR acts to guide Ca2+ entering the cell from the extracellular space towards the myofilaments. This extends the superficial buffer barrier hypothesis (van breemen et al., 1995), which proposes a major role for the superficial SR in buffering Ca2+ as it enters the cell, effectively separating the subsarcolemmal cytosol from the deeper, bulk cytosol containing the myofilaments. The study by Ruehlmann et al. (2000) was unable to distinguish between the involvement of IP3 and CICR in the Ca2+ oscillations underlying tonic contraction. The present results suggest that IP3 receptors play the major role, because hydralazine inhibited tonic contractions to phenylephrine with a potency equal to its effect on IP3-activated contraction of permeabilized vessels, but greater than its effect on responses to caffeine. Synthesis of IP3 has been shown to continue for several minutes in rabbit aorta exposed to agonist (Coburn et al., 1988; Kajikuri & Kuriyama, 1990), further supporting a role for IP3 in tonic contractions. Therefore, in agreement with earlier studies (van breemen et al., 1995; Gurney & Allam, 1995; Ruehlmann et al., 2000), we propose that tonic tension in the presence of phenylephrine is mediated by the continual cycling of Ca2+ from the subsarcolemmal space through the IP3-sensitive SR to the myofilaments. As illustrated in Figure 6, by inhibiting Ca2+ release through IP3 receptors, hydralazine is proposed to abrogate this cycling and prevent Ca2+ from reaching the contractile proteins. Since in the model of Figure 6, blockade of SR Ca2+ release by hydralazine could lead to saturation of SR Ca2+ buffering, additional mechanisms must contribute to the removal of Ca2+ from the subsarcolemmal to the extracellular space. These are likely to include sarcolemmal Na+ – Ca2+ exchange and Ca2+ ATPase proteins located in close proximity to the superficial SR, because this arrangement is thought to be important for the unloading of SR Ca2+ to the extracellular space (van breemen et al., 1995). Unloading of Ca2+ from the SR requires its release into the subsarcolemmal space, probably through IP3 or CICR channels (van breemen et al., 1995). This unloading is essential for the buffer function of the SR to continually prevent Ca2+ entering the cell from reaching the myofilaments directly. Unloading must persist in the presence of hydralazine, or it would not be able to maintain the bulk cytosolic Ca2+ concentration at a sufficiently low level to inhibit tonic contraction. Perhaps this occurs through CICR, since inhibition of caffeine responses amounted to only 30% even at the highest concentrations of hydralazine.
Figure 6.
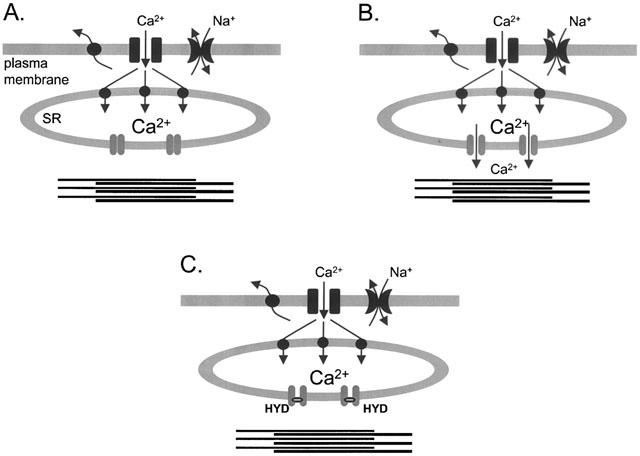
Simplified model illustrating the buffer barrier role of the SR and its modulation by agonists and hydralazine. In resting conditions (A), the Ca2+ ATPase (SERCA) in the membrane of the superficial SR accumulates Ca2+ entering the cell through leak channels. Sarcolemmal Ca2+ transport proteins, such as the Na+-Ca2+ exchanger and Ca2+-ATPase, may also extrude Ca2+ from the subsarcolemmal space, e.g. after unloading from the SR. In the presence of agonist (B), IP3 opens Ca2+ channels in the deeper SR membrane, releasing Ca2+ in the vicinity of the contractile proteins. This Ca2+ may be re-sequestered by the SR to maintain Ca2+ oscillations and tonic contraction, or extruded to the extracellular space. Maintenance of Ca2+ oscillations may additionally involve Ca2+ influx through receptor-operated channels (not shown). When hydralazine is added (C), Ca2+ release through IP3-activated channels is blocked, so Ca2+ is prevented from reaching the myofilaments to maintain contraction.
Hydralazine is not a K-channel opener
Hydralazine-induced dilation of porcine coronary arteries was proposed to involve the opening of large conductance Ca2+-activated potassium (BKCa) channels (Bang et al., 1998), which would lead to membrane hyperpolarization and reduced Ca2+ influx. Despite a previous report that hydralazine hyperpolarized the smooth muscle cells of rat caudal artery by around 4 mV (Hermsmeyer et al., 1983), we could find no evidence for an effect of the drug on membrane potential in smooth muscle cells from aorta. Hydralazine also failed to alter the amplitude of potassium currents recorded from isolated smooth muscle cells, even after 30 min exposure to concentrations causing near maximal relaxation of intact vessels. With the voltage protocols and conditions used, the recorded currents arose from BKCa and delayed rectifier channels (Halliday et al., 1995). Thus neither of these channels is likely to act as a mediator of hydralazine-induced vasodilation. It is possible that hydralazine acts differently in other vascular preparations, because in porcine coronary artery and rabbit femoral artery its vasodilator effects were antagonized by the BKCa channel blockers, tetraethylammonium and iberiotoxin (Thirstrup & Nielsen-Kudsk, 1992; Bang et al., 1998). A more likely explanation is, however, that potassium channel blockade indirectly influenced the response to hydralazine, possibly by promoting membrane depolarisation and enhancing Ca2+ influx through voltage-operated calcium channels. The lack of effect of hydralazine on Ca2+ influx (Orallo et al., 1991) and contractile responses mediated by Ca2+ influx (Higashio & Kuroda, 1988; Gurney & Allam, 1995) argues strongly against potassium channels as mediators of the vasodilator response.
Summary
In summary, the results indicate that the main mechanism of hydralazine-induced vasodilation is to inhibit the IP3-dependent release of Ca2+ from the SR, although inhibition of CICR through caffeine-sensitive channels may also contribute. In this way, hydralazine inhibits the rise in cytoplasmic Ca2+ concentration required for phasic contraction and prevents cycling of Ca2+ through IP3 receptors in the SR, which appears to maintain tonic contraction induced by vasoconstrictor agents.
Acknowledgments
The authors wish to thank Dr J.C. Kentish for help with vessel permeabilization and programmes for calculating ion concentrations and Prof J.M. Ritter for helpful discussions. D.C. Ellershaw was funded by a British Heart Foundation PhD studentship.
Abbreviations
Caf
caffeine
[Ca2+]i
intracellular Ca2+ concentration
CICR
Ca2+-activated Ca2+ release
DMSO
dimethyl-sulphoxide
EC50
concentration producing 50% of the maximal effect
EGTA
ethyleneglycol-bis-β-aminoethyl-N,N,N′,N′-tetraacetic acid
fura-2/AM
fura-2 acetoxymethyl ester
HEPES
5-N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid
IP3
inositol 1,4,5 trisphosphate
R340/380
ratio of fluorescence excited by 340 and 380 nm
SR
sarcoplasmic reticulum
References
- BAKER J.R., HEDWALL P.R., HERMSMEYER K. Subcellular distribution of hydralazine in rat single vascular smooth muscle cells. Cell Biol. Inter. Reports. 1992;16:1023–1039. doi: 10.1016/s0309-1651(06)80055-x. [DOI] [PubMed] [Google Scholar]
- BANG L., NIELSEN-KUDSK J.E., GRUHN N., TRAUTNER S., THEILGAARD S.A., OLESEN S.P., BOESGAARD S, ALDERSHVILE J. Hydralazine-induced vasodilation involves opening of high conductance Ca2+-activated K+ channels. Eur. J. Pharmacol. 1998;361:43–49. doi: 10.1016/s0014-2999(98)00701-8. [DOI] [PubMed] [Google Scholar]
- CLAPP L.H., GURNEY A.M. Outward currents in rabbit pulmonary artery cells dissociated with a new technique. Pflügers Archiv. 1991;418:682–470. doi: 10.1113/expphysiol.1991.sp003535. [DOI] [PubMed] [Google Scholar]
- COBURN R.F., BARON C, PAPADOPOULOS M.T. Phosphoinositide metabolism and metabolism-contraction coupling in rabbit aorta. Am. J. Physiol. 1988;255:H1476–H1483. doi: 10.1152/ajpheart.1988.255.6.H1476. [DOI] [PubMed] [Google Scholar]
- COOK N.S., WEIR S.W, DANZEISEN M.C. Anti-vasoconstrictor effects of the K+ channel opener cromakalim on the rabbit aorta - comparison with the calcium antagonist isradipine. Br. J. Pharmacol. 1988;95:741–752. doi: 10.1111/j.1476-5381.1988.tb11700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEFEO T.T., MORGAN K.G. Calcium-force coupling mechanism during vasodilator-induced relaxation of ferret aorta. J. Physiol. 1989;412:123–133. doi: 10.1113/jphysiol.1989.sp017607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIAMOND J., JANIS R.A. Increases in cyclic GMP levels may not mediate relaxant effects of sodium nitroprusside, verapamil and hydralazine in rat vas deferens. Nature. 1978;271:472–473. doi: 10.1038/271472a0. [DOI] [PubMed] [Google Scholar]
- EBEIGBE A.B., ALOAMAKA C.P. Mechanism of hydralazine-induced relaxation of arterial smooth muscle. Cardiovasc. Res. 1985;19:400–405. doi: 10.1093/cvr/19.7.400. [DOI] [PubMed] [Google Scholar]
- EHRLICH B.E., KAFTAN E., BEZPROZVANNAYA S, BEZPROZVANNY I. The pharmacology of intracellular Ca2+-release channels. Trends Pharmacol. Sci. 1994;15:145–149. doi: 10.1016/0165-6147(94)90074-4. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M, TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- GURNEY A.M., ALLAM M. Inhibition of calcium release from the sarcoplasmic reticulum of rabbit aorta by hydralazine. Br. J. Pharmacol. 1995;114:238–244. doi: 10.1111/j.1476-5381.1995.tb14931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALLIDAY F.C., AARONSON P.I., EVANS A.M, GURNEY A.M. The pharmacological properties of K+ currents from rabbit isolated aortic smooth muscle cells. Br. J. Pharmacol. 1995;116:3139–3148. doi: 10.1111/j.1476-5381.1995.tb15116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERMSMEYER K., TRAPINI A., ABEL P.W., WORCEL M. Effects of hydralazine on tension and membrane and membrane potential in rat caudal artery. J. Pharmacol. Exp. Ther. 1983;227:322–326. [PubMed] [Google Scholar]
- HIGASHIO T., KURODA K. Effects of cadralazine on contractions induced by Ca2+ and norepinephrine in isolated rabbit aortic strips. Drugs Res. 1988;38:346–349. [PubMed] [Google Scholar]
- KAJIKURI J., KURIYAMA H. Inhibitory action of α-human atrial natriuretic peptide on noradrenaline-induced synthesis of myo-inositol 1,4,5-trisphosphate in the smooth muscle cells of rabbit aorta. Br. J. Pharmacol. 1990;99:536–540. doi: 10.1111/j.1476-5381.1990.tb12964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHAYYAL M., GROSS F, KREYE V.A.W. Studies on the direct vasodilator effect of hydralazine in the isolated rabbit renal artery. J. Pharmacol. Exp. Ther. 1981;216:390–394. [PubMed] [Google Scholar]
- KNOT H.J., STANDEN N.B, NELSON M.T. Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+ channels. J. Physiol. 1998;508:211–221. doi: 10.1111/j.1469-7793.1998.211br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOBAYASHI S., KITAZAWA T., SOMLYO A.V, SOMLYO A.P. Cytosolic heparin inhibits muscarinic and -adrenergic Ca2+ release in smooth muscle. Physiological role of inositol 1,4,5-trisphosphate in pharmacomechanical coupling. J. Biol. Chem. 1989;264:17997–18004. [PubMed] [Google Scholar]
- KREYE V.A. Direct vasodilators with unknown modes of action: the nitro-compounds and hydralazine. J. Cardiovasc. Pharmacol. 1984;6:S646–S655. doi: 10.1097/00005344-198406004-00011. [DOI] [PubMed] [Google Scholar]
- KREYE V.A., RUEGG J.C., HOFMANN F. Effect of calcium-antagonist and calmodulin-antagonist drugs on calmodulin-dependent contractions of chemically skinned vascular smooth muscle from rabbit renal arteries. Naunyn Schmied. Arch. Pharmacol. 1983;323:85–89. doi: 10.1007/BF00634253. [DOI] [PubMed] [Google Scholar]
- LIPE S., MOULDS R.F. In vitro differences between human arteries and veins in their responses to hydralazine. J. Pharmacol. Exp. Ther. 1981;217:204–208. [PubMed] [Google Scholar]
- LODGE N.J., VAN BREEMEN C. Mobilisation of extracellularly bound Ca2+ during high K+ and norepinephrine stimmulation of the rabbit aorta. Blood Vessels. 1985;22:234–243. doi: 10.1159/000158607. [DOI] [PubMed] [Google Scholar]
- MCLEAN A.J., BARRON K., DU SOUICH P., HAEGELE K.D., MCNAY J.L., CARRIER O., BRIGGS A.H. Interaction of hydralazine and hydrazone derivatives with contractile mechanisms in rabbit aortic smooth muscle. J. Pharmacol. Exp. Ther. 1978;205:418–425. [PubMed] [Google Scholar]
- MOÍNA M.J., BARDAN B., CAMPOS M., GIL-LONGO J., VERDE I, ORALLO F. Effects of hydralazine on contractile responses to alpha1 and alpha2 adrenoceptor agonists in isolated rat aorta. Gen. Pharmacol. 1994;25:165–172. doi: 10.1016/0306-3623(94)90028-0. [DOI] [PubMed] [Google Scholar]
- ORALLO F., GILONGO J., BARDON B, CALLEJA J.M. Comparison of the effects of hydralazine and nifedipine on contractions and 45Ca influx in rat aorta. J. Pharm. Pharmacol. 1991;43:356–359. doi: 10.1111/j.2042-7158.1991.tb06704.x. [DOI] [PubMed] [Google Scholar]
- PERRIN D.D., SAYCE I.G. Computer calculation of equilibrium concentrations in mixtures of metal ions and complexing species. Talanta. 1967;14:833–842. doi: 10.1016/0039-9140(67)80105-x. [DOI] [PubMed] [Google Scholar]
- RUEHLMANN D.O., LEE C.-H., POBURKO D, VAN BREMMEN C. Asynchronous Ca2+ waves in intact venous smooth muscle. Circ. Res. 2000;86:e72–e79. doi: 10.1161/01.res.86.4.e72. [DOI] [PubMed] [Google Scholar]
- SAVINEAU J.P., MARTHAN R. Activation properties of chemically skinned fibres from human isolated bronchial smooth muscle. J. Physiol. 1994;474:433–438. doi: 10.1113/jphysiol.1994.sp020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TALSETH T. Studies on hydralazine. I. Serum concentrations of hydralazine in man after a single dose and ar steady state. Eur. J. Clin. Pharmacol. 1976;10:183–187. [Google Scholar]
- THIRSTRUP S., NIELSEN-KUDSK J.E. Effects of K+ channel blockers on the relaxant action of dihydralazine, cromakalim and nitroprusside in isolated rabbit femoral arteries. Eur. J. Pharmacol. 1992;215:177–183. doi: 10.1016/0014-2999(92)90026-z. [DOI] [PubMed] [Google Scholar]
- VAN BREEMEN C., CHEN Q, LAHER I. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. Trends Pharmacol. Sci. 1995;16:98–105. doi: 10.1016/s0165-6147(00)88990-7. [DOI] [PubMed] [Google Scholar]
- VAN BREEMEN C., SAIDA K. Cellular mechanisms regulating intracellular calcium concentration in smooth muscle. Annu. Rev. Physiol. 1989;51:315–329. doi: 10.1146/annurev.ph.51.030189.001531. [DOI] [PubMed] [Google Scholar]
- WEI S., KASUYA Y., YANAGISAWA M., KIMURA S., MASAKI T, GOTO K. Studies on endothelium-dependent vasorelaxation by hydralazine in porcine coronary artery. Eur. J. Pharmacol. 1997;321:307–314. doi: 10.1016/s0014-2999(96)00972-7. [DOI] [PubMed] [Google Scholar]
- YEN M.H., WU C.C., CHIOU W.F., LIAO C.H. Effects of hydralazine on guanosine cyclic 3′, 5′-monophosphate levels in rat aorta. Proc. Natl. Sci. Counc. B. Repub. China. 1989;13:83–88. [PubMed] [Google Scholar]