Posttranslational Modifications in Cu,Zn-Superoxide Dismutase and Mutations Associated with Amyotrophic Lateral Sclerosis (original) (raw)
. Author manuscript; available in PMC: 2006 Nov 2.
Published in final edited form as: Antioxid Redox Signal. 2006;8(5-6):847–867. doi: 10.1089/ars.2006.8.847
Abstract
Activation of the enzyme Cu,Zn-superoxide dismutase (SOD1) involves several posttranslational modifications including copper and zinc binding, as well as formation of the intramolecular disulfide bond. The copper chaperone for SOD1, CCS, is responsible for intracellular copper loading in SOD1 under most physiological conditions. Recent in vitro and in vivo assays reveal that CCS not only delivers copper to SOD1 under stringent copper limitation, but it also facilitates the stepwise conversion of the disulfide-reduced immature SOD1 to the active disulfide-containing enzyme. The two new functions attributed to CCS, (i.e., O2-dependent sulfhydryl oxidase- and disulfide isomerase-like activities) indicate that this protein has attributes of the larger class of molecular chaperones. The CCS-dependent activation of SOD1 is dependent upon oxygen availability, suggesting that the cell only loads copper and activates this enzyme when O2-based oxidative stress is present. Thiol/disulfide status as well as metallation state of SOD1 significantly affects its structure and protein aggregation, which are relevant in pathologies of a neurodegenerative disease, amyotrophic lateral sclerosis (ALS). The authors review here a mechanism for posttranslational activation of SOD1 and discuss models for ALS in which the most immature forms of the SOD1 polypeptide exhibits propensity to form toxic aggregates.
PHYSIOLOGICAL ROLES OF CU,ZN-SUPEROXIDE DISMUTASE
Reactive oxygen species are frequently proposed to exert a toxic effect on cells via oxidation of a wide range of biomolecules including DNA, proteins, and lipids. Oxidative modifications of these biomolecules often lower enzymatic activities and/or induce mutations in DNA, and can lead to cell death through necrosis or apoptosis (39). Hydrogen peroxide (H2O2) and hydroxyl radical exhibit high redox potential and have been shown to damage biomolecules in vitro and in vivo. While superoxide anion (O2−) is not a strong oxidant and readily acts as a reductant, it can attack and disassemble the iron–sulfur cluster in the enzymes such as dehydratases and release Fe2+ ion, which produces hydroxyl radical via the Fenton reaction (102). Furthermore, reaction of O2− with nitric oxide rapidly forms the potent and versatile oxidant peroxynitrite (78). The toxic effects of superoxide anion are employed in host–pathogen interactions: O2− is generated by NADPH oxidase in phagocytes to kill infectious bacteria and fungi (37). Although the mechanisms underlying reactive oxygen species toxicity need further investigation, it is clear that uncontrolled oxidative stress damages many essential components in the cell.
One of the cellular defense systems for oxidative insults is an antioxidant enzyme, Cu,Zn-superoxide dismutase (SOD1) (111). SOD1 is a cuproenzyme (Fig. 1) and catalyzes the disproportionation reaction of superoxide anion to oxygen and hydrogen peroxide at a bound copper ion (111);
Cu2+,Zn2+−SOD1+O2−↔Cu1+,Zn2+−SOD1+O2
Cu1+,Zn2+−SOD1+O2−+2H+↔Cu2+,Zn2+−SOD1+H2O2
SOD1 is a very robust protein with respect to physical and chemical denaturation; catalytic activity can be observed in the presence of 10 M urea or 4% SDS and at 80°C (51). While nonenzymatic dismutation of O2− to O2 and H2O2 occurs with a rate constant, 2 × 105 _M_−1s−1, at pH 7.4, SOD1-catalyzed dismutation is significantly accelerated to an extent that it is diffusion-controlled (2 × 109 M_−1s−1) (53). Interestingly, this enzymatic rate of disproportionation is very similar to that of copper salts; however, given that intracellular free copper concentrations are quite low under normal aerobic growth (133), there has been a strong selection for organisms to elaborate a more efficient defense. Given negative charge of the substrate, O2−, positive characters of the charge distribution near the SOD1 active site (Arg 143 and Lys 136) enhance the dismutation reaction through the electrostatic steering mechanism (19, 170) (Fig. 1). The intracellular concentration of SOD1 is high (ranging from 10 to 100 μ_M (92, 101)), and apparently sufficient to consume physiological levels of superoxide radical.
FIG. 1.
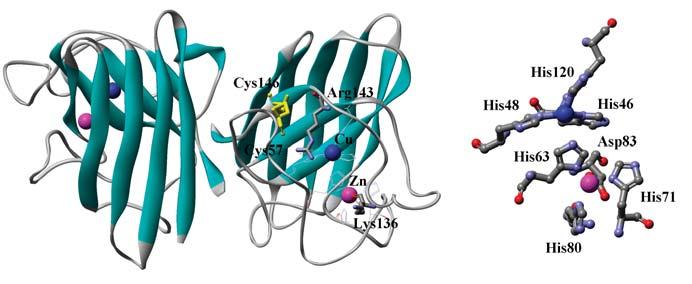
Crystal structure of human SOD1 shown in a ribbon model (PDB ID: 1HL5). Cu, Zn ion and intramolecular disulfide bond are shown on ball and stick, respectively. Coordination structure around Cu and Zn ion is also shown in the right panel.
Primary sequence as well as three-dimensional structure of SOD1 is highly conserved from prokaryotes to eukaryotes (21). SOD1 forms a tight dimer with a dissociation constant, ∼1.0 × 10−10 _M_−1 (90), and each subunit has an immunoglobulin-like β-sandwich fold with an intrasubunit disulfide bond (Fig. 1). A copper and zinc ion are embedded in each subunit (Fig. 1); Cu2+ ion is ligated with four histidines (His 46, 48, 63, 120), forming a distorted square plane, and binds water as a fifth ligand. One of the histidines, His 63, is also a ligand for binding a Zn2+ ion with the other three amino acid residues, His 71, 80, and Asp 83, in a distorted tetrahedral arrangement. Binding of Zn2+ ion is not essential for the dismutation reaction but confers high protein stability to SOD1. Several structural variations have been found in bacterial SOD1 proteins: a monomeric form in Escherichia coli (12), a heme binding site in Haemophilus ducreyi (126), no Zn binding site in Mycobacterium tuberculosis (160).
Eukaryotic SOD1 is mainly localized in cytosol with a smaller fraction at the intermembrane space of mitochondria (49, 101, 122, 166) and has been also found in nuclei, lysosomes, and peroxisomes, using immunocytochemical methods (30). While superoxide is produced in cytosol by several enzymes such as xanthine oxidase (114), respiration process in mitochondria is a major source of O2− generation (27). In fact, >95% of our daily oxygen consumption is reduced to water in the respiratory chain. It has been estimated that ca 0.1% of the O2 consumed is converted to O2− in the electron transport chain, corresponding to production of ca. 30 mmol O2− in a 60–80 kg person on a daily basis (27). A good deal of mitochondrial O2− is disproportionated in the mitochondrial matrix by Mn-superoxide dismutase (SOD2) which is present there in high levels (ca. 1 × 10−5 M) (181). While the possible presence of copper, zinc enzyme (SOD1) in the mitochondrial matrix is still debated (184), its localization in the intermembrane space of mitochondria (IMS) is now well established (49, 166). Thus, superoxide produced via adventitious reactions of O2 with components of the respiratory chain can be disproportionated by SOD1 in the intermembrane space (71). Interestingly, in gram-negative bacteria, SOD1 is localized in a compartment that is distantly related to the IMS, namely the periplasmic space (91). SOD1 is important for survival of prokaryotes in relatively late/stationary phase and contributes to the ability to grow aerobically (161). The periplasmic sources of O2− have not yet been characterized (16), but it is likely that periplasmic SOD1 protects against both endogenous and exogenous sources of O2−, for instance those arising from host-parasite responses.
At a tissue level, SOD1 is widely expressed with high in liver and kidney (7). It is also abundant in motor neurons (127). This correlates with high expression of xanthine oxidase in liver and with high susceptibility of neuronal cells to oxidative stress. Knockout studies indicate that elimination of the SOD1 gene results in the widespread oxidative damage and has been shown to induce various symptoms/phenotypes in rodent models including alcohol-induced liver injury (89, 129), hepatocarcinogenesis (43), infertility in female mice (110), age-related hearing loss through cochlear hair cell degeneration (112), vulnerability to motor neuron loss after axonal injury (140), and the life span decrease (43). Also in humans, the level of glycated SOD1 is increased in the erythrocytes of patients with diabetes mellitus (5). Given that glycation of SOD1 causes fragmentation of the enzyme (124), reduction of SOD1 activity may lead to the altered redox status in diabetic patients (72). Elevated levels of SOD1 activity have been discussed in relation to Down's syndrome, which results from triplication of human chromosome 21 (45). SOD1 is encoded by a gene localized to 21q22.1, and the expressed levels of this protein are ca. 1.5-fold increased in Down's patients. Although there is no clear evidence for the pathogenic role of SOD1 in Down's syndrome, elevated SOD1 activity has been reported in some adult patients (45).
As we will discuss later in detail, over 100 types of point mutations in SOD1 have been reported to cause familial form of amyotrophic lateral sclerosis (ALS) (26). Many of the mutant proteins exhibit a normal level of the SOD1 activity, which leads to the idea that some aberrant ‘gain-of-function’ is associated with the neurodegeneration observed in ALS patients.
CELLULAR CONTROL OF SOD1 ACTIVITY
SOD1 needs a copper ion for its enzymatic activity; in fact, copper deficiency in diet results in reduction of both SOD1 protein and activity levels in a rat model (131), and this loss in activity is proportional to the degree of copper deficiency (130). Interestingly, the converse is true for the copper chaperone for SOD1: CCS levels are significantly elevated under conditions of copper deficiency in rodents (17, 188). In response to elevated copper ion, SOD1 transcription is induced to a small extent by a Cu-binding protein transcriptional factor, ACE1, in yeast (63). To maintain the copper ion homeostasis in the face of excess copper (133), many eukaryotic organisms express the metal-binding protein, metallothionein (87). Interestingly, overexpression of SOD1 can suppress copper toxicity in yeast cells lacking metallothionein (35). While physiological connections between these various pools of cellular copper are not well understood, these studies reveal the delicate interplay between antioxidant defense and metal homeostasis machinery.
Consistent with the fact that SOD1 is involved in the oxygen metabolism, SOD1 activity is also augmented upon increase of O2 concentration in yeast (66) and E. coli (65). Anaerobiosis of yeast cells is reported to result in ca. 4- and 10-fold decrease in SOD1 protein and activity level, respectively (56, 57). Oxygen-dependent control of SOD1 activity has been also suggested in animal models; uptake of O2 in active muscles is markedly facilitated during exercise, leading to an increased generation of reactive oxygen species. SOD1 activity is upregulated in rat diaphragm muscle during endurance training at the post-transcriptional level (121). Chronic exercise training in pig also increases SOD1 mRNA, protein, and activity levels (148, 149). Physiological regulation of SOD1 activity thus appears to involve transcriptional, post-transcriptional, and posttranslational mechanisms. In this review article, we will overview posttranslational regulation of SOD1 activation and introduce several recent findings in our lab.
COPPER CHAPERONE: POSTTRANSLATIONAL ACTIVATION OF SOD1
While copper and zinc insertion into the apo-SOD1 polypeptide can be achieved by addition of the corresponding metal salts in vitro (18), the situation inside the cell is more complex. Many but not all organisms employ one or more accessory proteins for copper insertion. The copper chaperone protein for SOD1, CCS, was first identified in the yeast strain Saccharomyces cerevisiae using genetic approaches (36) and has emerged as an important posttranslational regulator of SOD1 structure, function, and physiology. Yeast cells lacking the CCS gene, formerly known as LYS7, exhibit the same phenotypes as _sod1_Δ null mutant, which is auxotrophic for methionine and lysine in aerobic conditions. In yeast _ccs_Δ null mutant, any SOD1 activity is not observed, whereas SOD1 expression remains unchanged relative to wild type. SOD1 activation in yeast cells is thus strictly dependent upon the existence of CCS. CCS is highly specific for SOD1 and cannot deliver a copper ion to other proteins, such as cytochrome c oxidase or copper transport ATPase, that have different metallochaperones (120). CCS proteins have been also identified in various species including humans (29), rodents (193), insects (159), and plants (191). While CCS is 15- to 30-fold and fivefold less abundant than SOD1 in mammalian (147) and yeast cells (133), respectively, cellular distribution and expression of CCS appear to parallel those of SOD1. Recently, however, CCS-knockout mouse and CCS−/− fibroblast mouse cells have been shown to retain a certain degree of SOD1 activity, suggesting the CCS-independent pathway of SOD1 activation in mammalian cells (28, 167). Reduced glutathione appears involved in CCS-independent activation of SOD1 in mammalian cells (28). It is interesting to note that the nematode Caenorhabditis elegans does not have a CCS homologue and that this organism uses some type of CCS-independent pathway for activation of intracellular Cu,Zn-SOD proteins (83). The apo-form of mammalian SOD1 has also been proposed to acquire a copper ion from metallothionein (MT) and ceruloplasmin in vitro (38, 105, 168, 169); however, the latter protein is extracellular and is unlikely to donate copper to SOD1 in vivo. Further experiments will be required to delineate in vivo roles of MT and other proteins in SOD1 activation.
We have recently found that posttranslational activation of apo-SOD1 by CCS requires oxygen (23). Air-exposure of anaerobically grown yeast triggers the protein expression and activation of SOD1 within an hour. Interestingly, even after inhibiting the protein synthesis using cycloheximide, the preexisting pool of apo-SOD1 in anaerobically grown yeast is spontaneously (within 5 min) activated upon exposure of the yeast cells to air. Furthermore, in vitro experiments have shown that CCS can activate apo-SOD1 under aerobic conditions but not in the absence of oxygen (23). The cellular protection against oxidative stress is thus multitiered; existing pools of apo-SOD1 are activated by CCS in the early response to a sudden elevation of oxygen availability, followed by increasing expression of SOD1 protein with persistent posttranslational regulation of SOD1 activity.
MECHANISTIC FUNCTIONS OF CCS IN SOD1 ACTIVATION
To better understand the posttranslational mechanism of SOD1 activation, several groups, including us, have characterized the copper chaperone protein, CCS. CCS is a 28 kDa protein and comprised of three domains, while a fourth domain at C-terminus showing homology with metallothionein has been reported in Schizosaccharomyces pombe CCS (93). Crystal structures are available for human (97) and S. cerevisiae (96) CCS, although some parts of the protein are disordered or truncated during crystallization. The N-terminus region (∼8 kDa) has a similar structure to a secretory pathway copper chaperone, ATX1, and contains the “MXCXXC” Cu-binding motif (Fig. 2A). Domain I alone of human CCS retains a copper ion when overexpressed in E. coli with supplement of CuSO4 (134). While EXAFS studies on human CCS have also suggested that two Cys residues in the MXCXXC motif are involved in copper binding (41), Domain I is not essential to SOD1 activation in the cell under normal conditions; CCS mutant protein, in which Domain I is truncated, can still activate SOD1, albeit at much reduction in the activation (151). Besides, the Cys to Ser mutations in the MXCXXC motif still exhibit 70% metallochaperone activity compared to the wild-type protein in vitro (162). Domain I-truncated CCS, however, cannot complement the aerobic lysine auxotrophy of _ccs_Δ null yeast strain in the presence of a copper(I) chelator, bathocuproine sulfonate, implying that Domain I plays some roles in acquiring a copper ion when intracellular availability of a copper ion is limited (151). Interestingly, however, two cysteine residues of Domain I Cu-binding motif is not conserved in D. melanogaster CCS, which further complicates exact roles of Domain I in SOD1 activation.
FIG. 2.
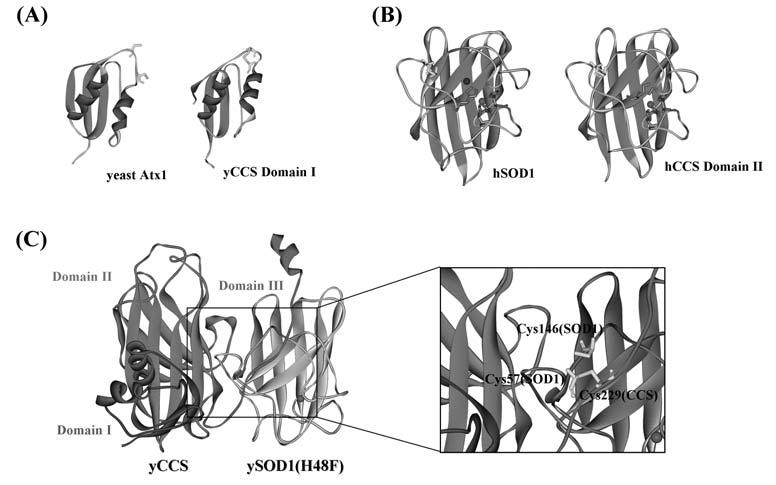
Crystal structures of CCS. (A) Domain I of CCS (PDB ID: 1QUP) has a homologous structure with another copper chaperone, Atx1 (PDB ID: 1FES). (B) Domain II of human CCS (PDB ID: 1DO5) is similar to human SOD1 (PDB ID: 1HL5). (C) Heterodimer structure between yeast SOD1 with the mutation, H48F, and yeast CCS (PDB ID: 1JK9). All three domains in CCS are identified in this crystal structure. Magnified image of the interfacial region containing intermolecular disulfide bond between SOD1 and CCS is also shown.
Domain II of CCS (∼16 kDa) is highly homologous to SOD1 (Fig. 2B); 47% identity between human SOD1 and human CCS Domain II. While no metal binding sites are found in yeast CCS Domain II, all of the SOD1 zinc binding ligands are conserved in the human counterpart. In fact, an equimolar amount of zinc ion is bound in human CCS Domain II when overexpressed in E. coli (134), and Zn binding is essential to human CCS function possibly through protein stabilization (44). Three of four histidine residues in the copper binding site are also conserved in Domain II of human CCS, and the fourth histidine residue is replaced by an aspar-tate (Asp 200). Copper ion is not held at this site, and SOD1 activation by CCS is still observed after mutations of these His residues to Ala (151). Potential copper binding site in Domain II would thus be not required for copper activation of SOD1.
Given the similarity of the SOD1 and CCS dimer interfaces, CCS has been proposed to interact with SOD1 through SOD1-like Domain II by mimicking SOD1 dimerization. In fact, when dimerization of yeast SOD1 is retarded by mutations at the subunit interface, F50E/G51E, this mutant SOD1 is not activated by yeast CCS (150). Similarly, when the corresponding amino acid residues in yeast CCS Domain II are mutated (F136E/G137E), in vivo activation of yeast SOD1 is not observed (150). These findings have supported the idea that CCS Domain II interacts with the SOD1 subunit interface. On the other hand, SOD1 forms a tightly associating dimer, and monomeric forms of the WT holo protein have not been observed. Based upon high dimer stability of either SOD1 (_K_d, 1 × 10−10 _M_−1) (90) or CCS (_K_d, 3.0 × 10−6 _M_−1 in yeast) (69), the hetero-tetramer model, in which both proteins do not need to dissociate into monomers, has been proposed (69). We have recently shown that the SOD1 quaternary structure is dependent on the degree of posttranslational modifications and that the most immature form (i.e., the disulfide reduced apo-SOD1 polypeptide) favors the monomeric state (6, 55). In fact, the Cu-bound yeast CCS can only activate the disulfide-reduced apo-yeast SOD1 monomeric form: a dimeric apo-form of SOD1 that already possesses an intramolecular disulfide bond cannot accept copper from the chaperone and cannot support the heterodimeric interaction between SOD1 and CCS (55).
Domain III of CCS is a short polypeptide (30–40 amino acids) without any preference to form the secondary structures but is essential to the CCS function; deletion of Domain III cannot complement the lysine auxotrophy in _ccs_Δ null yeast cells (151). Domain III shows a weak homology with a part of prolyl cis-trans isomerase, and the CXC motif found in this domain is highly conserved among all species. Domain III polypeptide alone is sufficient to bind Cu1+ ion, and the Cys to Ser mutations in CXC motif dramatically reduces the metallochaperone activity in vivo and in vitro (151). Although it is possible for Cu1+ ion to be bound at the CXC site with an exogenous ligand (137), recent EXAFS studies have suggested that the trigonal copper-thiolate cluster is formed between two Domain III CXC sites within a human CCS dimer (162). Depending upon the experimental conditions, more than one copper ion can be bound in both yeast and human CCS proteins (94, 134). Electronic absorption spectra of cobalt-substituted CCS have also implied formation of copper-thiolate cluster between CXXC of Domain I and CXC of Domain III (196). These data have shown that domain III plays essential roles in Cu binding and SOD1 activation, but the copper coordination chemistry in CCS has still been controversial.
The crystal structure of the yeast SOD1–yeast CCS heterodimeric complex (Fig. 2C) has strengthened several conclusions from biochemical studies on copper transfer from CCS to SOD1 (95). SOD1 dimer-like interaction between SOD1 monomer and CCS Domain II is observed, and functionally essential Domain III is well positioned to interact with SOD1. Given that crystallization is performed using a mutant SOD1 (H48F), which cannot bind a copper ion, it is still speculative how a copper ion changes its coordination structure when transferred from CCS to SOD1. In another metallochaperone system, Atx1 and Ccc2, a Cu1+ ion rapidly partitions between the surface-exposed metal-binding sites of these two proteins via formation and decay of two- and three-coordinate Cu1+-thiolate intermediates (132). The thermodynamic gradient for metal exchange between Atx1 and Ccc2 is quite shallow with a Cu1+ exchange equilibrium constant of 1.4, where the driving force for the copper transfer is considered to be an ATP hydrolysis to remove Cu1+ from the cytosol to the Golgi compartment. In the SOD1–CCS system, in contrast, the copper binding site in SOD1 is relatively buried in the protein interior, and there is no cellular compartmentalization of the transferred copper ion. Compared to Atx1/Ccc2 partnership, therefore, a more complex mechanism of copper transfer by CCS is expected to be exerted. In fact, one of the important aspects in the SOD1–CCS copper transfer has been implicated in the heterodimer structure (Fig. 2C): introduction of a conserved intramolecular disulfide bond in SOD1. In the SOD1–CCS heterodimer, a disulfide bond forms between Cys 57 (SOD1) and Cys 229 (CCS) (Fig. 2C). Instead, the active SOD1 has the intramolecular disulfide bond between Cys 57 and 146 (Fig. 1). This finding has implied the involvement of CCS in the disulfide formation in SOD1 and recently highlighted essential roles of this disulfide in the SOD1 activation mechanism.
CCS-DEPENDENT FORMATION OF ESSENTIAL DISULFIDE IN SOD1
The intramolecular disulfide bond of SOD1 was characterized in 1974 (1) and is highly conserved in SOD1 proteins from all species found so far. Disulfide formation generally stabilizes protein structure by lowering entropy of the unfolded polypeptide chain (174), and we have recently found essential characters of the intramolecular disulfide bond in enzymatic activity of yeast SOD1 (55). A structural role of the disulfide between Cys 57 and Cys 146 has been implied as formation of a hydrogen bond between the backbone amide of Cys 57 and Arg 143 (50), by which the side-chain of Arg 143 is optimally oriented for uptake of the superoxide anion (Fig. 1). It is, however, enigmatic how this essential disulfide bond in SOD1 forms and survives under reducing environment of the cytosol.
An important feature of the SOD1 disulfide bond is its high stability in the cytosolic environment; however, we have found that redox potential of thiol/disulfide pair in SOD1 is about −0.23 eV (55). This value is comparable to the ones of other disulfide-containing proteins and the cytosol (59). Maintenance of the SOD1 disulfide bond in cytosol is thus not due to its thermodynamic stability but is possibly kinetically controlled. Kinetic stabilization of a disulfide bond has been proposed in immunoglobulin light chain, a disulfide bond of which is completely buried in the protein interior and is unreactive toward reductants (60-62). In the holo-form of human SOD1, solvent accessible area of the disulfide-bonding cysteines, Cys 57 and 146, is small (2), implying that reducing agents are not accessible to the disulfide bond. In addition, location of the disulfide bond near the dimerization interface (Fig. 1) would also play an important role in stabilizing the disulfide bond. We have suggested that dimerization sterically limits access of cellular reductants to the disulfide bond and thereby contributes to kinetic stabilization of the disulfide bond. To examine effects of dimerization on the disulfide stability, the kinetic parameters of the disulfide reduction need to be compared between SOD1 monomeric and dimeric states in future studies.
In addition to its high stability, the disulfide formation in SOD1 needs to be explained under the reducing environment of cytosol. Transient disulfide formation in cytoplasm is possible as seen in redox cycles of Cys pairs of thioredoxins and glutaredoxins (75); however, stable and persistent disulfide bonds are usually found in extracellular proteins, and the presence of cytoplasmic proteins with stable disulfide bonds is rare except several viral proteins (106, 152). It has been reported that disulfide bond in prokaryotic SOD1 is introduced by the oxidative folding machinery called Dsb proteins (11), which is consistent with the fact that prokaryotic SOD1 localizes at an oxidizing cellular compartment, periplasm (91). In eukaryotes, disulfide bonds are generally formed at endoplasmic reticulum (ER), which involves a set of enzymes for disulfide formation, Ero1 and PDI (179). Compared to the reducing environment of the cytosol, ER is maintained in a significantly more oxidized state by preferential transport of the oxidized glutathione over its reduced counterpart from cytosol (79). Interestingly, there is little evidence that the ER-based thiol-oxidases play a role in SOD1 disulfide formation. For instance, immunocytochemistry studies show that ER does not contain significant amount of SOD1, which is otherwise abundant in the cytosol (30). Other compartments may have specialized mechanisms for SOD1 maturation. It has recently been shown that the disulfide-reduced apo-form of SOD1 is imported from cytosol into the intermembrane space (IMS) of mitochondria (49). The discovery that this form is unfolded (54, 55) and has a monomeric structure (6) compared to the dimeric holo-SOD1 places it well within paradigms for the mechanism of import of other immature polypeptides into the IMS. Very recently, it has been shown that Mia40 and Erv1 catalyze disulfide formation in proteins in the IMS of mitochondria (113). The disulfide bond may thus be introduced in the reduced SOD1 protein imported into the IMS via Mia40 and Erv1. Given that the SOD1-CCS heterodimer contains the intermolecular disulfide bond (Fig. 2C) and that SOD1 is colocalized with CCS at the cytosol (29), Cu-CCS is a good candidate to introduce the disulfide bond in cytosolic SOD1. In fact, our in vitro and in vivo experiments have recently shown that Cu-CCS regulates the SOD1 disulfide formation in the cytosol (55).
The disulfide formation in SOD1 also begins to explain the requirement of O2 for SOD1 activation by CCS (Fig. 3). Under anaerobic conditions, the cysteine residues in SOD1 remain reduced even after incubation with Cu-CCS, and the protein remains inactive. Oxygen itself can function as an oxidant to form a disulfide bond, but it is kinetically sluggish for SOD1 disulfide formation (> 6 h). Rather, oxygen will be the ultimate source of oxidizing equivalents for disulfide formation in SOD1 and would first oxidize the thiol groups in CCS that ligate Cu1+ ion. The bound copper ion may then be released from CCS to apo-SOD1 upon oxidation of the thiol groups into a disulfide (Fig. 3). Metal release upon disulfide formation has been proposed in the Hsp33 activation mechanism; the thiol groups binding a Zn ion are oxidized to form the disulfide bond, leading to release of Zn ion from those thiol groups (64, 82). The disulfide formed in CCS is then transferred to SOD1 through formation of inter-molecular disulfide between SOD1 and CCS (Fig. 3). Alternatively, Cu1+ in CCS would be first attacked by oxygen to form Cu2+. In the copper transfer from CCS to SOD1, the coordination environment around copper ion is changed from Cys-rich (CCS) to His-rich (SOD1). According to the empirical hard-soft chemistry, oxidation of Cu1+ ion may be required to break an extremely favorable interaction between Cys ligands and Cu1+ ion (soft–soft interactions). Detailed investigation in the chemical mechanism of SOD1 activation by CCS is now underway in our laboratory.
FIG. 3.
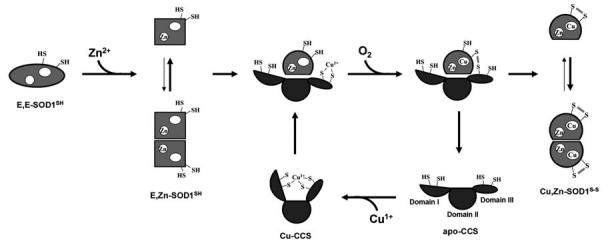
Our proposed model for the SOD1 activation cycle by Cu-CCS in yeast. Cu-CCS will interact with the Zn-bound and disulfide-reduced form of SOD1, E,Zn-SOD1SH. Yeast SOD1 favors monomeric state when the disulfide bond is reduced. Oxygen attacks the SOD1-CCS heterodimer to trigger the copper transfer, resulting in the formation of the intermolecular disulfide bond. Then the disulfide isomerization results in the intramolecular disulfide bond in SOD1 to form the dimeric holo-SOD1.
There are several precedents for O2-dependent disulfide formation in proteins; one of those is sulfhydryl oxidase (SOX). SOX is a flavin-containing enzyme and is comprised of two distinct domains intervened by a long amino acid stretch (175). N-terminal domain has a thioredoxin-like structure with CXXC motif, and C-terminal domain is related to Erv1 protein (vide infra), where another CXXC motif and a flavin-binding site are found. Oxidizing equivalents for disulfide formation are relayed from O2 via FAD, CXXC in C-terminal domain, CXXC in N-terminal domain and finally to substrate proteins (175). A relaying mechanism of oxidizing equivalents in disulfide formation is also found in pathways for protein oxidative folding. In eukaryotes, Ero1p transfers oxidizing equivalents directly to PDI protein, which oxidizes the substrate proteins through a disulfide-exchange mechanism (52). Ero1p, a single domain protein, contains two disulfide bonds and FAD (67). Erv protein, another protein responsible for PDI oxidation, also contains FAD, CXXC and CXC motifs at its C-terminus, where oxidizing equivalents from O2 are transferred to form a disulfide bond in the substrate (68). Given that CCS has a copper ion, CXXC and CXC motifs, CCS can be paralleled with these enzymes for O2-dependent disulfide formation to the extent that a redox active cofactor with disulfide bond(s) seems a general architecture for disulfide formation. CCS is nonetheless unique in that not only oxidizing equivalents but also the redox active cofactor itself (i.e., copper ion) are transferred to the substrate, SOD1. While CCS has been defined as one of metallochaperone proteins, its assist of SOD1 folding through the disulfide formation suggests its role as a real “chaperone” protein.
In one proposed mechanism of CCS-dependent SOD1 activation (Fig. 3), the first step is incorporation of a Zn2+ ion into the disulfide-reduced protein, given that fully demetallated SOD1 was not activated with Cu-CCS (94). Our gel filtration studies also reveal that the apo and disulfide-reduced form of yeast SOD1 tends to unfold/aggregate much more readily than more mature forms, but incorporation of a Zn2+ ion in the disulfide-reduced SOD1 can lead to the protein folding (55). In the next step, the reduced (but not the oxidized) state of E,Zn-SOD1 forms a heterodimeric complex with the copper chaperone, which would be consistent with the fact that disulfide-reduced yeast SOD1 favors monomeric state (55). The subsequent reaction of CCS-SOD1 heterodimer with dissolved oxygen forms an intermolecular disulfide bond between CCS and SOD1. Following the copper insertion in the heterodimeric complex, Cu binding in SOD1 could induce the conformational changes especially around the Cys residues, promoting the protein disulfide isomerase-like reaction from intermolecular to intramolecular SOD1 disulfide (vide infra). Disulfide exchange within the heterodimeric intermediate further drives dissociation of the docked complex to the individual proteins, Cu,Zn-SOD1S-S and apo-CCS, due to the thermodynamic preference of the disulfide-bonded SOD1 protein for the dimeric state. This reaction cycle predicts that CCS will be able to rapidly catalyze formation of the active SOD1 with uptake of the substrates: Cu1+, E,Zn-SOD1SH, and O2.
As reviewed above, copper insertion and disulfide formation in apo-SOD1 are performed by CCS; however, despite important roles of Zn(II) ion in the SOD1 folding and structure, little is known on how this protein acquires a Zn(II) ion in the cell. Given that human CCS can bind a Zn(II) ion in Domain II and possibly Atx1-like Domain I, CCS may also function as a “Zn chaperone” protein. Interestingly, our recent studies have shown that apo and disulfide-reduced human SOD1 becomes dimeric upon binding of Zn(II) ion, which is in apparent contrast to the observations in yeast SOD1. Human CCS may thus favorably interact with a monomeric state of human SOD1 (i.e., E,E-hSOD1SH); further in vitro and in vivo studies will prove how and when Zn ion is incorporated in SOD1 during the activation process. Posttranslational activation of SOD1 including Cu/Zn binding and disulfide formation is regulated in a well-concerted manner, but if there are deficits in the posttranslational activation pathway, SOD1 protein itself turns to be toxic to the cell.
TOXICITY INDUCED BY MUTATIONS IN SOD1– RELEVANCE TO ALS
Aberrant copper chemistry
In 1993, several point mutations in SOD1 were reported as a cause of the familial form of amyotrophic lateral sclerosis (ALS), which has shed light on a dark side of this antioxidant enzyme (146). Given that significant decline of SOD1 activity is found in several mutant proteins (e.g., A4V), it had been initially considered that inability to remove superoxide anion would lead to increase of intracellular oxidative stress and damage to neuronal cells (142, 178). The other ALS-associated mutant SOD1 proteins, however, retain partial or even full enzymatic activity (e.g., G37R, G93A) (22, 192). Point mutations in SOD1 thus do not seem to induce neurodegeneration based on the “loss-of-function” mechanism, which has been further supported by the following animal models. Overexpression of wild-type SOD1 does not reduce or even deteriorates ALS symptoms in the transgenic mice expressing mutant SOD1 proteins (25, 81), and an SOD1-knockout mouse exhibits no overt signs of the ALS pathology in spinal cord (140). A consensus has thus been established that SOD1 protein gains new toxic properties upon introduction of ALS-associated mutations.
One of the proposed functions acquired by mutations is the increased peroxidase activity; H2O2, normally a product of the superoxide disproportionation, is catalytically converted into highly reactive and toxic hydroxyl radical at Cu1+ ion bound in SOD1 mutant proteins (189). In fact, increased levels of hydroxyl radical have been reported in the transgenic mouse expressing human SOD1 with G93A mutation (20). Another aberrant copper chemistry proposed in SOD1 mutant proteins involves a strong oxidant, peroxynitrite (ONOO−). ONOO− forms with reaction between nitric oxide (NO) and superoxide anion at a diffusion-controlled rate. When SOD1 has no Zn ion bound, a Cu1+ ion in SOD1 is proposed to undergo one-electron reduction of molecular oxygen (O2) to form superoxide anion, which further reacts with NO and leads to the peroxynitrite production (47). Given that many ALS-associated mutant SOD1 proteins have reduced affinity for Zn ion (34), an acquired toxic function of SOD1 by mutations may arise from the peroxynitrite chemistry including the tyrosine nitration (34). This is supported by the result that amounts of free 3-nitrotyrosine are increased in the transgenic mice and the ALS patients (14, 24). Mishandling of a copper ion may thus lead to oxidative damage in neuronal cells; consistent with this, administration of copper chelators such as trientine (116) and _d_-penicillamine (76) exhibits protective effects on the disease onset and progression in mouse models.
Recently, however, aberrant copper chemistry has been questioned as a cause of the SOD1-linked familial ALS; symptoms have been found in the transgenic mouse overexpressing the copper-binding-site-null mutant SOD1, in which all four copper ligands (His 46, 48, 63, and 120) are mutated (185). Human SOD1 protein with a mutation in one of these copper binding residues, H46R, has been shown to bind a copper ion at a site where copper is not typically seen (i.e., at a surface Cys residue, Cys 111) (103). This has led to the proposal that binding of copper ion at region(s) other than a canonical binding site may be associated with the toxicity of the ALS-variants. This proposal does not, however, explain the fact that the presence of ALS-associated mutations in mouse SOD1 can cause ALS-like symptoms (141), even though mouse SOD1 has no Cys residue at position 111. Besides, there is no in vivo evidence that copper ions bind at the SOD1 protein surface with significant affinity under physiological conditions. While increased oxidative stress is a major pathology found in the ALS-model mice and ALS patients, the bulk of the evidence to date does not strongly support the idea that aberrant copper chemistry originating with mutant SOD1 proteins is responsible for the disease.
Aggregation of SOD1: defects in posttranslational modifications
As an alternative mechanism of this disease, protein aggregation has been proposed on the basis of the fact that Bunina body and Lewy body-like hyaline inclusion are found in motor neurons of sporadic and familial ALS patients, respectively (153). Accumulation of protein aggregates in motor neuronal cells will interfere with axonal transport, protein degradation and mitochondrial function, resulting in neuronal cell death (26). The protein aggregates found in SOD1-related familial ALS patients and transgenic mouse models are strongly immunoreactive to SOD1, whereas involvement of SOD1 in protein aggregates has been controversial in sporadic cases (154). Aggregation generally occurs through unfolding/misfolding of protein molecules; therefore, the intracellular regulation of SOD1 stability will be an important factor in the molecular mechanism of familial ALS pathology.
Holo-form of SOD1 (Cu,Zn-SOD1) is a very stable protein to the extent that the melting temperature, _T_m, is around 90°C (51); however, removing a copper ion from holo-SOD1 (E,Zn-SOD1) decreases _T_m to ∼70°C, and further removal of Zn ion from E,Zn-SOD1 (E,E-SOD1) results in ∼60°C of _T_m (145). Destabilization of SOD1 protein upon demetallation has also been confirmed in experiments using a chemical denaturant, guanidium hydrochloride (Gdn-HCl) (100). The metal-binding process thus maintains structural integrity as a folded SOD1 molecule and seems to protect the protein from aggregation; in fact, a Cu-bound but Zn-deficient SOD1 molecule tends to aggregate through oxidation of the histidine residues into 2-oxohistidine (136), although there is no in vivo evidence of this type of SOD1 aggregates.
Roles of the metal binding processes in keeping the protein integrity are further supported by the crystal structure of metal-deficient human SOD1 protein, in which an extended arrangement of β-barrel shaped apo-SOD1 is reminiscent of the common structure associated with amyloid-like fibrils (165). Besides, demetallation-induced conformational changes of the SOD1 proteins with pathogenic mutations (H46R, S134N) can create a new protein–protein interaction site (gain-of-interaction), which tolerates formation of higher-order amyloid-like assemblies of SOD1 molecules (42). While the pathogenic SOD1 aggregates do not seem to have an amyloid structure (based on Congo Red staining) (123), mutant SOD1 in the absence of bound metal ions can adopt alternative conformations that facilitate protein aggregate formation. X-ray scattering data on the mutant SOD1 (A4V, I113T) have indeed predicted significant alterations of the subunit orientation in solution; structural destabilization of the dimer interface would trigger the protein aggregation (77). An aberrant increase in the hydrophobicity of the disulfide-reduced metal-deficient SOD1 mutant proteins has been reported in the presence of SDS, and is consistent with conformational differences in these forms of mutant and WT SOD1 (177). Monomerization induced by demetallation has also been proposed as a step leading to misfolded intermediates in the SOD1 aggregation pathway (135). Given that many ALS-associated mutations in SOD1 are found to provoke significant destabilization and conformational changes, especially in the metal-deficient state (100, 144), instability of apo mutant SOD1 may be a “common denominator” for the increased aggregation propensity among over 100 ALS-linked mutant proteins (100).
The stability of the apo-state of SOD1 is increased by several types of mutations [H46R and D124V (143), which incidentally are associated with relatively milder symptoms (4, 86)], but most of the mutant apo-proteins (21 out of 25 mutant proteins) are still significantly destabilized (143). Besides, stability of apo-SOD1 mutant proteins exhibits apparent inverse correlation with the mean survival time after disease onset (100), indicating that the protein stability plays an important role in the SOD1-linked ALS pathogenesis. We have, nonetheless, noted that metal-deficient form of mutant SOD1 still has a higher/comparable melting temperature than physiological body temperature, ∼37°C; for example, apo-form of A4V, G93A, and E100G hSOD1 has 50.7, 49.1, and 51.8°C, respectively (163). Significant fractions of mutant holo-forms of SOD1 are thus expected to be folded under the physiological conditions; these may not be predisposed to the protein aggregation mechanism. This suggests that other states in the posttranslational SOD1 activation pathway may be more important in the aggregation process.
As described in a previous section, our proposed mechanism of the SOD1 activation consists of three major posttranslational modifications; copper/zinc binding, and disulfide formation (Fig. 3). Depending upon these modifications plus monomer-dimer equilibrium, 44 canonical microstates are possible in SOD1 protein (Fig. 4). This only takes into account SOD1 proteins where the zinc is bound in the zinc site and copper in its site. Given that these modifications generally increase the protein structural stability, it is well expected that the most immature form [i.e., apo- and disulfide-reduced SOD1 (E,E-SOD1SH], is susceptible to aggregation. In fact, we have confirmed that disulfide reduction in wild-type apo-SOD1 drops the melting temperature to 46°C (54). This melting temperature is about 45°C lower than that of holo-SOD1 and close to body temperature (∼37°C), implying that a small fraction of even wild-type protein may be unfolded when any posttranslational modifications are absent. Structural instability of unmodified wild-type SOD1 may relate to the fact that spinal cord tissues of some sporadic ALS cases contain protein aggregates immunoreactive toward SOD1 (154). When SOD1 has an ALS-associated mutation, the melting temperature of the E,E-SOD1SH state further drops to 35 (A4V) or 31 (G93A)°C even in the presence of the stabilizing reagent, glycerol (54). These values are >10°C lower than that of the wild-type protein and also lower than body temperature (∼37°C); apo and disulfide-reduced SOD1 with ALS-associated mutations would be unfolded in physiological conditions. Interestingly, in the absence of glycerol, E,E-hSOD1SH of A4V and G93A exhibit no endothermic transition between 5° and 100°C, implying the protein unfolding/aggregation even below 5°C. This is consistent with the observation that, using gel filtration chromatography at 4°C, an oligomeric state larger than dimer has been detected in the apo and disulfide-reduced SOD1 with ALS-associated mutations (54). Our recent findings have thus suggested that E,E-hSOD1SH state is most prone to protein aggregation at physiological temperature.
FIG. 4.
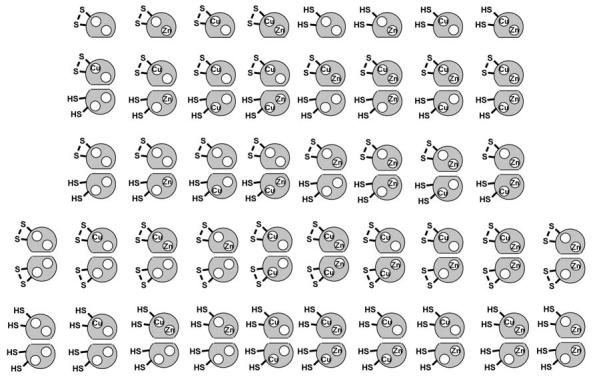
44 canonical microstates in SOD1. SOD1 microstates are dependent upon copper and zinc binding, intramolecular disulfide bond, and dimerization.
Furthermore, ALS-associated mutations increase a chance to restore SOD1 protein to the most immature state by decreasing both disulfide stability and metal affinity. While the intramolecular disulfide bond in wild-type SOD1 is kinetically robust against reducing agents (55), the disulfide in ALS-mutant SOD1 would not persist in reducing environment of the cytosol (176). SOD1 has extremely high Cu/Zn affinity to the extent that it functions as an intracellular Cu/Zn sink (35, 187), but significantly reduced affinity and/or altered binding geometry of Zn ion in some mutant SOD1 proteins has been shown by spectrophotometric techniques (34, 107). We have confirmed that the disulfide-reduced form of mutant SOD1 also exhibits decreased association with zinc ion by using a cobalt substitution method (Furukawa Y. and O'Halloran T.V., unpublished) (108). Addition of an equimolar amount of CoCl2 to E,E-hSOD1SH gives the absorption peaks at 535, 570, and 585 nm (Fig. 5A). This spectral shape is virtually the same with that previously reported in E,Co-hSOD1S-S(WT) (107), suggesting little perturbation of Zn binding upon disulfide reduction. Using apo form of mutant SOD1, in which all four Cys residues (Cys 6, 57, 111, and 146) are mutated to Ser, a similar spectrum is obtained after addition of equimolar CoCl2, excluding possibility of the Co(Zn) ion ligation through Cys residues (data not shown). Addition of a strong metal chelator, EDTA (_K_a Co(II) = 1013 M_−1 at pH 7), to E,Co-hSOD1SH(WT) does not change the spectrum (Fig. 5A) at least within an hour. Co2+ ion at the Zn-binding site is hence kinetically inert or thermodynamically stable in the reduced wild-type protein. By comparison, the disulfide-reduced mutant proteins (A4V, G93A) with equimolar CoCl2 show relatively broadened absorption between 500 and 600 nm with its reduced intensity (Figs. 5B and C), although the detailed binding geometries around Zn ion are still elusive in mutant proteins. Addition of 100 μ_M EDTA to the Co-substituted mutant proteins immediately decreases the absorption intensity (Figs. 5B and C), consistent with a lower affinity for zinc ion in the disulfide-reduced state. Given high capacity for chelation of zinc ion in reducing environment of the cytosol (125), we suggest that the intracellular milieu facilitates formation of the aggregation-prone state, E,E-hSOD1SH, in the ALS-associated mutant SOD1 proteins.
FIG. 5.
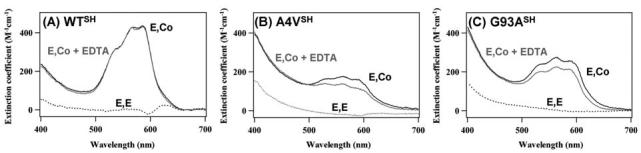
ALS-associated mutations in SOD1 cause the reduction of affinity for a Zn ion. Electronic absorption spectra of E,E-hSOD1SH (A: wild-type, B: A4V, C: G93A) in 50 m_M_ HEPES, 1 m_M_ tris-(2-carboxyethyl)phosphine, pH 7.4, with an equimolar CoCl2 at 20°C (solid black curve). 0.1 m_M_ EDTA was further added to the E,Co-hSOD1SH samples, and the spectrum was obtained after 1 h incubation at 20°C (solid gray curve). Spectra of E,E-hSOD1SH alone were also shown as dotted curves. Molar extinction coefficient shown in the figure was calculated based upon the absorption at 280 nm, and the following protein concentration was used for the measurements: 67.3 μ_M_ wild-type, 74.5 μ_M_ A4V, 78.2 μ_M_ G93A hSOD1.
Oxidative damage on SOD1—disulfide-linked multimers
Reduction of the disulfide bond not only decreases protein stability but also affords vulnerability toward oxidative stress to SOD1 protein; oxidative injuries in the cell have been reported to augment in both sporadic and familial ALS patients (48). A thiol group (−SH) of a Cys residue is susceptible to oxidation under physiological conditions, resulting in the formation of sulfenic (−SOH), sulfinic (−SO2H) and sulfonic acid (−SO3H). As found in the brains of patients with Alzheimer and Parkinson diseases, Cys 146 in SOD1 can be irreversibly oxidized to cysteine sulfonic acid (31). Given that Cys 146 constitutes one of the thiol groups for formation of the intramolecular disulfide bond (Fig. 1), stability of the SOD1 protein oxidized at Cys 146 would be as much decreased as that of the disulfide-reduced form, which may explain the SOD1-immunoreactive senile plaques and neurofibrillary tangles in Alzheimer disease patients. As an alternative oxidized state of SOD1, we have found that E,EhSOD1SH molecules are cross-linked with each other by forming intermolecular disulfide bonds upon addition of either hydrogen peroxide or even milder oxidant, oxidized glutathione, resulting in the insoluble SOD1 multimers (54). While the covalent aggregation of SOD1 has been reported to occur in the presence of carbonate radical anion through the tryptophan oxidation in vitro (194), involvement of disulfide formation in the protein aggregation has been recently noticed in several protein conformation diseases such as transmissible spongiform encephalopathy (prion) (99) and familial amyloid polyneuropathy (transthyretin) (195). Random crosslinks between Cys residues sometimes lead to formation of insoluble protein inclusions during purification of recombinant proteins in E. coli, but disulfide crosslinks of SOD1 is not completely random; the conserved Cys residues (Cys 57 and 146) exhibit higher reactivity for formation of the disulfide-linked multimers than the nonconserved Cys residues (Cys 6 and 111). Covalent addition of persulfide at the thiol group of Cys 111 makes SOD1 more resistant to the oxidation-induced aggregation (40), suggesting that selective quench of the thiol reactivity by chemical modifications and/or genetic engineering reduce the disulfide-linked multimerization and is a potential therapeutics for SOD1-related familial ALS disease. In vivo evidence of the disulfide-linked multimerization of SOD1 would be invoked in the observations using the transgenic C. elegans co-expressing the mutant SOD1 proteins (A4V, G37R, and G93A) (118); mutant SOD1 is degraded more rapidly than the wild-type protein, but oxidative stress by paraquat retards the degradation of mutant SOD1 and induces formation of discrete aggregates. It needs to be established whether the disulfide-linked multimer is a toxic species causing neurodegeneration or an end product of the oxidative modifications of SOD1 molecules. It is also well expected that SOD1 exhibits intrinsically high propensity for aggregate formation, since significant truncations from the C-terminus of SOD1 (as much as 117 amino acids) still tolerate the protein aggregation (58). Our recent findings have, nonetheless, suggested that oxidative crosslinks at Cys residues play an important role in the physiological SOD1 aggregate formation.
Cytotoxicity of ALS–SOD1 aggregates?
The toxicity of ALS–SOD1 aggregates in cell culture studies has been controversial. Lee and coworkers concluded that aggregate formation does not correlate with neuronal death in differentiated PC12 cells (98); however, a very recent time-lapse study following the fate of individual differentiated PC12 cells reveals that cell death correlates with formation of the aggregates and occurs within 6–24 h after aggregate appearance (109). While neither study addresses the status of the intrinsic disulfide, we note that inappropriate disulfide formation in the mutant SOD1 would promote and stabilize aggregate formation. These new results from Morimoto and coworkers are thus consistent with mechanisms that invoke SOD1 protein aggregation as a trigger for neurodegeneration (26). One of those is that the essential chaperone proteins are entangled in the SOD1 aggregates (180). Increased interaction between SOD1 and Hsp70 has been reported upon treatment of SOD1 with DTT (182), which corroborates reported susceptibility to the disulfide reduction in mutant SOD1 (176). Hsp70, Hsp40, and αβ-crystallin are coimmunoprecipitated with mutant SOD1 (156), suggesting deactivation of protein chaperones in the aggregate formation. Another proposed mechanism involves inhibition of the ubiquitin-proteasome protein degradation system by protein aggregates (182, 183). The aggregates containing SOD1 are intensely immunoreactive with antibodies to ubiquitin (155), and mutant SOD1 is selectively ubiquitinated by the RING finger-type E3 ubiquitin ligase, dorfin (117). Addition of proteasomal inhibitors facilitates the aggregation of mutant SOD1 in cultured cells (84). Given that intermolecular disulfide crosslinks inhibit complete unfolding of SOD1 into the polypeptide thread state, it is interesting to speculate that the disulfide-linked SOD1 aggregates are stuck in the proteasome and lead to its dysfunction. Due to its insolubility and bulkiness, SOD1 aggregates have been also proposed to obstruct the axonal transport of essential components including neurotrophic factors (26). In fact, intracerebroventicular delivery of vascular endothelial growth factor, VEGF, has been recently found to significantly reduce the ALS-like symptoms in the rat model (164). SOD1 itself is delivered to the nerve termini in a slow axonal transport system (190), the rate of which is on the order of 1–2 mm/day (74). For the meter-long axon, it will take one to two years for SOD1 to arrive at the nerve termini (32). Such a long time will lead to an increased chance of oxidative damage for SOD1, and, therefore, neuronal cell may provide an oxidative environment facilitating the formation of disulfide-linked SOD1 aggregates.
With regards to oxidative stress, SOD1 protein will experience significant oxidative environment when incorporated into mitochondrial IMS, where reactive oxygen species are found as byproducts of the consumption of molecular oxygen in the respiratory chain. We have found that SOD1 must be in a disulfide-reduced state for its import into the intermembrane space of mitochondria (Fig. 7, inset) (49). Reduction of the conserved disulfide leads to the protein monomerization (6), which downsizes SOD1 protein and possibly facilitates its crossing over the outer membrane of mitochondria. Disulfide-linkage in a polypeptide may also be a three-dimensional obstruct for crossing the membrane. Upon mitochondrial import of SOD1, therefore, the reduced Cys residues will be exposed to a relatively high oxidative environment of mitochondrial IMS. Although CCS and/or the disulfide relay system, Mia40/Erv1 (113), in mitochondrial IMS correctly introduce the intramolecular disulfide in SOD1, oxidative modifications of thiol groups including the disulfide-linked multimerization would occur in the IMS, which may further lead to the apoptotic cell death cascade. Interestingly, high molecular-weight SDS-resistant SOD1 aggregates are associated with the spinal cord mitochondria of transgenic mice expressing ALS-mutant SOD1 (104) and can interact with antiapoptotic protein, Bcl-2 (128).
FIG. 7.
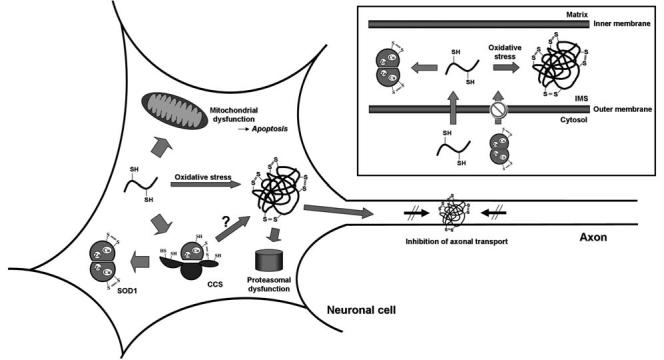
Proposed processing of SOD1 protein in neuronal cells. Unmodified SOD1 polypeptide acquires a copper ion and a disulfide bond by forming a heterodimer with CCS, resulting in an enzymatic activation of SOD1. The free thiol groups on the SOD1 polypeptide can also be oxidized to form the disulfide-linked protein aggregates, which would lead to dysfunctions of proteasome and/or axonal transport. (Inset) The disulfide-reduced form of SOD1 can cross the outer membrane of mitochondria, but the disulfide form cannot. Due to the highly oxidative environment in the mitochondrial IMS, the imported SOD1 polypeptide is susceptible to disulfide crosslinks and aggregation in the absence of any posttranslational modifications.
Possible therapeutics—facilitating posttranslational modifications on SOD1
Based on our proposed aggregation mechanism, investigation of several therapeutic avenues are warranted, including a search for agents that stimulate maturation of the reduced and apo-forms of the mutant proteins, as well as agents that inhibit SOD1 oligomerization and disulfide-linked multimerization. Protein stabilization has been shown as one of treatment options for other aggregate-associated neurodegenerative diseases (171, 172) and amyloidosis (70); for example, in a transgenic mouse model for Huntington disease, where a protein with polyglutamine expansion is overexpressed, oral administration of trehalose decreases protein aggregates and improves motor dysfunction through stabilization of partially unfolded polyglutamine-containing protein (171). In the transgenic ALS-model mouse overexpressing G93A human SOD1, administration of trehalose (5,000 mg/kg/day) resulted in little effect on the disease onset and duration (http://www.als.net/). Our experiments have, nonetheless, shown that glycerol is another candidate to stabilize mutant SOD1, given that addition of glycerol increases the protein thermal stability and inhibits the oligomerization of E,EhSOD1SH state of the ALS-associated mutant proteins (A4V and G93A) (54). Dietary zinc administration also seems to be a promising treatment because of its role in increasing the protein thermal stability; in fact, moderate supplementation of zinc (12 mg/kg/day) delayed death in the G93A SOD1 transgenic mice by 11 days compared to mice on a zinc-deficient diet (46). Dietary copper supplementation should be also evaluated in light of the recent progress of Multhaup and co-workers in mouse models of other neurodegenerative diseases (13). Stabilization of the dimeric state has been attempted as an effective approach to reduce/inhibit SOD1 aggregation; insertion of an engineered intersubunit disulfide bond into mutant SOD1 (A4V) (139), although this approach does not seem potent in vivo (58). Using in silico screening, small molecules have been searched that can bind at the subunit interface and inhibit the monomerization/aggregation (138). Some reducing reagents as well as the physiological antioxidant systems are another effective choice to lessen the intracellular oxidative stress (3, 85, 119) and possibly inhibit formation of the disulfide-linked SOD1 aggregates. In the light of the aggregate suppression by stabilizing a protein molecule, we have shown that CCS is responsible for at least two posttranslational modifications of SOD1, copper insertion and disulfide formation; therefore, stimulation of CCS activity or protein levels is expected to be another good therapeutic approach for SOD1-associated familial ALS.
Effects of CCS on SOD1 aggregation pathway
CCS has been excluded as a candidate gene for ALS (157), but protein aggregation occurs when Domain II of CCS has a mutation homologous to an ALS-associated SOD1 mutation (158). Using immunohistochemistry, CCS has been reported to co-aggregate with SOD1 in neuronal hyaline inclusions found in familial ALS patients with an SOD1 mutation of either A4V or a frame-shift mutation at 126 (88) and in transgenic mice expressing G85R or G93A mutant SOD1 (186); however, possible involvement of CCS in the SOD1 aggregation pathway is still unclear. To prove roles of CCS in SOD1-linked familial ALS disease, the ccs gene knock-out has been examined in transgenic mice overexpressing human SOD1 mutant proteins (G37R, G85R, and G93A); both onset and progression of the ALS-like symptoms have been little affected (167), implying that CCS does not play a primary role in causing the ALS disease. Small fractions of SOD1 are, however, active even in the CCS-knockout mice (15), which has left some ambiguities on the involvement of CCS in the ALS disease pathology. Indeed, it has been later shown that, unlike yeast, mammalian cells are equipped with CCS-independent pathway(s) for SOD1 activation, in which reduced glutathione plays an important role (28). CCS-dependent activation pathway can function in the ALS-associated mutant SOD1 (33), which has led us to the expectation that changes in the CCS expression in transgenic ALS-model mice will perturb the disulfide formation and/or the copper insertion in SOD1 and reduce the cellular fractions of the aggregation-prone E,E-hSOD1SH state.
As reported in the yeast SOD1-yeast CCS system (55), Cu,Zn-hCCS interacts with E,Zn-hSOD1SH(WT) and introduces the disulfide bond in hSOD1 within 10 min under aerobic conditions (Furukawa Y and O'Halloran TV, unpublished). Likewise, the intramolecular disulfide bond is introduced in the ALS-mutant proteins by aerobic mixing with an equimolar Cu,Zn-hCCS. Notable in mutant SOD1 proteins is formation of a high molecular-weight (MW) species, which is more significant in H46R and G85R compared to A4V and G93A mutant proteins (Furukawa Y and O'Halloran TV, unpublished). The higher MW species is also found in the reaction between Cu,Zn-hCCS and the hSOD1 protein with Cys → Ser mutations at positions 6, 111, and 146. Analysis using MALDI–TOF mass spectrometry has confirmed this higher MW species as a disulfide-linked SOD1-CCS heterodimer. Stabilization of the disulfide-linked heterodimer may relate to reduced binding of a copper ion, because it is one of shared properties between H46R and G85R (73). This is also supported by the fact that the disulfide-linked heterodimer has been successfully characterized in yeast SOD1/CCS by a mutation at a copper-binding ligand of SOD1 (H48F) (94, 95). In a subset of the SOD1 mutant proteins, therefore, it is expected that CCS cannot exercise its role in facilitating the posttranslational maturation of SOD1, given that metal binding process influences the CCS-dependent pathway of SOD1 activation.
Binding of a copper ion affects the SOD1 structure (9); the arrangement of Loops IV and VII is significantly altered with a minimal change in the overall folding of SOD1 protein (Fig. 6) (8-10). This is because Loops IV and VII contain the ligands for copper and zinc ions. Given that one of the conserved Cys residues (Cys 57) is also a component of Loop IV, it is plausible that binding of metal ions facilitates the intramolecular disulfide formation by exerting conformational changes of Loop IV. Comparison of the structures among E,E-,E,Zn- and Cu,Zn-hSOD1S-S indeed shows orientational changes of the disulfide bond (Fig. 6), which may present a mechanistic implication of how the intramolecular disulfide bond is favored upon binding of metal ions over the intermolecular disulfide bond with CCS. In the copper-binding mutant SOD1 such as H46R and G85R, therefore, the structural changes required to drive the disulfide isomerization step may not be exerted, resulting in stabilization of the disulfide-linked heterodimer.
FIG. 6.
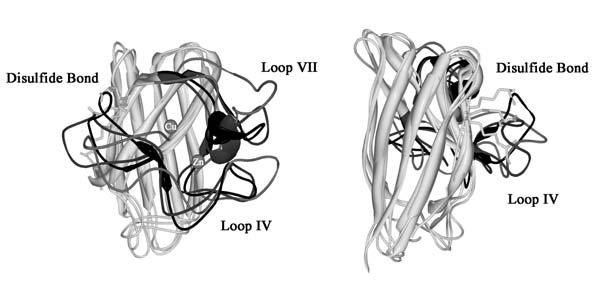
Superimposed images of E,E-, E,Znand Cu,Zn-hSOD1 S-S structures (PDB ID: 1RK7, 1KMG, and 1BA9, respectively). An image from a different angle is also shown in the right panel. The conserved disulfide bond is shown in stick, and the Loops IV and VII are also highlighted by color; light gray, dark gray, and black in E,E-, E,Zn-, and Cu,Zn-hSOD1S-S, respectively.
The disulfide-linked heterodimer is on the physiological pathway to activate SOD1 by CCS (Fig. 3) (55). Given that CCS is about 30-fold less abundant than SOD1 in mammals (147), stabilization of the disulfide-linked heterodimer in H46R and G85R SOD1 would result in sequestration of available CCS proteins. Interestingly, SOD1 homologues in poxviruses, which do not encode a cysteine at position 146, act as the catalytically inert “decoy” by forming the disulfide-linked heterodimer with CCS of the host cell and interfering in the proper metallation of SOD1 of the host cell (173). The metal-binding mutant SOD1 can thus be also a catalytically inert decoy by forming and stabilizing the disulfide-linked heterodimer with CCS, possibly causing significant shortage of intracellular CCS and increase of the cytosolic fractions of aggregation-prone E,E-hSOD1SH state. In this respect, it is interesting to note that the familial ALS is linked to the C146R mutation in SOD1 (80), which can stabilize the disulfide-linked heterodimer with CCS (Furukawa Y and O'Halloran TV, unpublished observations). Besides, stabilization of SOD1 through CCS-independent activation pathway will not be operated in metal-binding mutant SOD1. In this respect, it is interesting to note that the metal-binding ALS mutant SOD1 leads to different pathologies relative to other mutants; more protein inclusions are found in the transgenic mice with the Cu binding mutants, H46R and G85R, relative to the mutants with normal Cu binding such as G37R and G93A (115, 186).
Interestingly, the latter mutations are associated with more aggressive forms of the disease, thus propensity for aggregation may not correlate with disease severity. Although Subramaniam et al. have concluded that knockout of ccs gene exhibits no effects on the disease onset/progression (167), it is interesting to note that G85R but not G37R and G93A transgenic mice exhibit slightly longer life span in the absence of ccs gene (see Fig. 5 in ref. 167). This result, if supported by further studies, suggests that effects of CCS on the ALS pathology are dependent upon the type of mutations in SOD1; CCS-dependent/independent posttranslational modifications of SOD1 may help to inhibit protein aggregation in those mutant SOD1s that can bind a copper ion, while overexpression of CCS may be incompetent or even toxic in the familial ALS with the SOD1 mutations showing reduced affinity for a copper ion. To explore roles of CCS in the pathology of SOD1-linked familial ALS disease and pursue a possibility of CCS as a therapeutic target, in vivo characterization of the CCS-dependent pathway of SOD1 posttranslational activation is now underway in our laboratory.
CONCLUSION
As summarized in Fig. 7, we depict the intracellular fate of SOD1 protein from peptide synthesis at mRNA-ribosome complex, through folding, zinc and copper ion acquisition, and formation of the essential intrasubunit disulfide with the latter two steps mediated by CCS (also see Fig. 3). As long as this activation pathway functions correctly, SOD1 will protect the cell from various oxidative injuries as an enzymatic antioxidant. Mutations in SOD1, however, increase the lifetime of incorrectly folded states, and, depending on the concentration of apoSOD1, allows for reversible formation of SOD1 oligomers. If these oligomers are exposed to even mild oxidative stress, incorrect disulfide links form and stabilize larger aggregates that may be resistant to the degradation by the quality control machinery of the cell. Thus, two events may be necessary to explain how mutations in SOD1 can lead to toxic aggregates: first the mutations must destabilize the folded state of immature SOD1 polypeptide or significantly slow down the rate at which the peptide folds into the proper conformation; secondly, an oxidative stress event can lead to crosslinking of and thus stabilization of the aggregated state. As some of the mutant SOD1 is sequestered in inactive aggregates, some cell may respond to further oxidative stress by induction of SOD1 transcription and translation. This would lead to a snow-ball effect in the stressed cells, perhaps explaining why aggregates are not observed in all cell types. As discussed above, aggregates have been observed in the mitochondria (104), which we note is an oxidative compartment. This then is a possible site for adventitious disulfide-oxidation and formation of incorrect disulfides that would further stabilize mutant SOD1 aggregates (Fig. 7, Inset). The disulfide-linked SOD1 aggregate at this site would be particularly dangerous if it compromised mitochondrial membrane functions: this could trigger apoptosis and may be involved in the neuronal cell death (Fig. 7). The SOD1 protein can be described as a “double-edged sword” to the extent that it can function as either a beneficial antioxidant or, upon mutation of a single residue, form an intractable protein aggregate that, depending upon the quality control machinery and the amount of excess apoSOD1 protein produced, may prove toxic to the cell. If this model is born out by additional studies, it would place SOD1-linked ALS squarely as a disease of protein folding. Although exact path from mutations in SOD1 to neurodegeneration remains unclear, agents that stimulate productive posttranslational modifications in SOD1 may be one of effective therapeutic approaches to cure ALS disease.
ABBREVIATIONS
ALS
Amyotrophic lateral sclerosis
CCS
copper chaperone for SOD1
IMS
intermembrane space of mitochondria
SOD1
Cu,Zn-superoxide dismutase
Footnotes
We have recently found the disulfide-linked SOD1 aggregates in the spinal cord of the transgenic ALS model mice (Proc Natl Acad Sci USA 103: 7148–7153, 2006). Involvement of the wild-type protein in the disulfide-linked aggregates also explains aggravation of ALS phenotype in mutant SOD1 transgenic mice with overexpressed wild-type protein (Proc Natl Acad Sci USA 103: 7142–7147, 2006).
REFERENCES
- 1.Abernethy JL, Steinman HM, Hill RL. Bovine erythrocyte superoxide dismutase. Subunit structure and sequence location of the intrasubunit disulfide bond. J Biol Chem. 1974;249:7339–7347. [PubMed] [Google Scholar]
- 2.Ahmad S, Gromiha M, Fawareh H, Sarai A. ASAView: database and tool for solvent accessibility representation in proteins. BMC Bioinformatics. 2004;5:51. doi: 10.1186/1471-2105-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andreassen OA, Dedeoglu A, Klivenyi P, Beal MF, Bush AI. N-acetyl-l-cysteine improves survival and preserves motor performance in an animal model of familial amyotrophic lateral sclerosis. Neuroreport. 2000;11:2491–2493. doi: 10.1097/00001756-200008030-00029. [DOI] [PubMed] [Google Scholar]
- 4.Aoki M, Ogasawara M, Matsubara Y, Narisawa K, Nakamura S, Itoyama Y, Abe K. Mild ALS in Japan associated with novel SOD mutation. Nat Genet. 1993;5:323–324. doi: 10.1038/ng1293-323. [DOI] [PubMed] [Google Scholar]
- 5.Arai K, Iizuka S, Tada Y, Oikawa K, Taniguchi N. Increase in the glucosylated form of erythrocyte Cu-Zn-superoxide dismutase in diabetes and close association of the nonenzymatic glucosylation with the enzyme activity. Biochim Biophys Acta. 1987;924:292–296. doi: 10.1016/0304-4165(87)90025-0. [DOI] [PubMed] [Google Scholar]
- 6.Arnesano F, Banci L, Bertini I, Martinelli M, Furukawa Y, O'Halloran TV. The unusually stable quaternary structure of human SOD1 is controlled by both metal occupancy and disulfide status. J Biol Chem. 2004;279:47998–48003. doi: 10.1074/jbc.M406021200. [DOI] [PubMed] [Google Scholar]
- 7.Asayama K, Burr IM. Rat superoxide dismutases. Purification, labeling, immunoassay, and tissue concentration. J Biol Chem. 1985;260:2212–2217. [PubMed] [Google Scholar]
- 8.Banci L, Benedetto M, Bertini I, Del Conte R, Piccioli M, Viezzoli MS. Solution structure of reduced monomeric Q133M2 copper, zinc superoxide dismutase (SOD). Why is SOD a dimeric enzyme? Biochemistry. 1998;37:11780–11791. doi: 10.1021/bi9803473. [DOI] [PubMed] [Google Scholar]
- 9.Banci L, Bertini I, Cantini F, D'Onofrio M, Viezzoli MS. Structure and dynamics of copper-free SOD: The protein before binding copper. Protein Sci. 2002;11:2479–2492. doi: 10.1110/ps.0210802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banci L, Bertini I, Cramaro F, Del Conte R, Viezzoli MS. Solution structure of Apo Cu,Zn superoxide dismutase: role of metal ions in protein folding. Biochemistry. 2003;42:9543–9553. doi: 10.1021/bi034324m. [DOI] [PubMed] [Google Scholar]
- 11.Battistoni A, Mazzetti AP, Rotilio G. In vivo formation of Cu,Zn superoxide dismutase disulfide bond in Escherichia coli. FEBS Lett. 1999;443:313–316. doi: 10.1016/s0014-5793(98)01725-6. [DOI] [PubMed] [Google Scholar]
- 12.Battistoni A, Rotilio G. Isolation of an active and heat-stable monomeric form of Cu,Zn superoxide dismutase from the periplasmic space of Escherichia coli. FEBS Lett. 1995;374:199–202. doi: 10.1016/0014-5793(95)01106-o. [DOI] [PubMed] [Google Scholar]
- 13.Bayer TA, Schafer S, Simons A, Kemmling A, Kamer T, Tepest R, Eckert A, Schussel K, Eikenberg O, Sturchler–Pierrat C, Abramowski D, Staufenbiel M, Multhaup G. Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice. Proc Natl Acad Sci USA. 2003;100:14187–14192. doi: 10.1073/pnas.2332818100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beal MF, Ferrante RJ, Browne SE, Matthews RT, Kowall NW, Brown RH., Jr. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol. 1997;42:644–654. doi: 10.1002/ana.410420416. [DOI] [PubMed] [Google Scholar]
- 15.Beckman JS, Esetvez AG, Barbeito L, Crow JP. CCS knockout mice establish an alternative source of copper for SOD in ALS. Free Radic Biol Med. 2002;33:1433–1435. doi: 10.1016/s0891-5849(02)01092-4. [DOI] [PubMed] [Google Scholar]
- 16.Benov L, Sage H, Fridovich I. The copper- and zinc-containing superoxide dismutase from Escherichia coli: molecular weight and stability. Arch Biochem Biophys. 1997;340:305–310. doi: 10.1006/abbi.1997.9940. [DOI] [PubMed] [Google Scholar]
- 17.Bertinato J, Iskandar M, L'Abbe MR. Copper deficiency induces the upregulation of the copper chaperone for Cu/Zn superoxide dismutase in weanling male rats. J Nutr. 2003;133:28–31. doi: 10.1093/jn/133.1.28. [DOI] [PubMed] [Google Scholar]
- 18.Bertini I, Mangani S, Viezzoli MS. Structure and properties of copper-zinc superoxide dismutase. Adv Inorg Chem. 1998;45:127–250. doi: 10.1007/s007750050353. [DOI] [PubMed] [Google Scholar]
- 19.Beyer WF, Jr., Fridovich I, Mullenbach GT, Hallewell R. Examination of the role of arginine-143 in the human copper and zinc superoxide dismutase by site-specific mutagenesis. J Biol Chem. 1987;262:11182–11187. [PubMed] [Google Scholar]
- 20.Bogdanov MB, Ramos LE, Xu Z, Beal MF. Elevated “hydroxyl radical” generation in vivo in an animal model of amyotrophic lateral sclerosis. J Neurochem. 1998;71:1321–1324. doi: 10.1046/j.1471-4159.1998.71031321.x. [DOI] [PubMed] [Google Scholar]
- 21.Bordo D, Djinovic K, Bolognesi M. Conserved patterns in the Cu,Zn superoxide dismutase family. J Mol Biol. 1994;238:366–386. doi: 10.1006/jmbi.1994.1298. [DOI] [PubMed] [Google Scholar]
- 22.Bowling AC, Barkowski EE, McKenna–Yasek D, Sapp P, Horvitz HR, Beal MF, Brown RH., Jr. Superoxide dismutase concentration and activity in familial amyotrophic lateral sclerosis. J Neurochem. 1995;64:2366–2369. doi: 10.1046/j.1471-4159.1995.64052366.x. [DOI] [PubMed] [Google Scholar]
- 23.Brown NM, Torres AS, Doan PE, O'Halloran TV. Oxygen and the copper chaperone CCS regulate posttranslational activation of Cu,Zn superoxide dismutase. Proc Natl Acad Sci USA. 2004;101:5518–5523. doi: 10.1073/pnas.0401175101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruijn LI, Beal MF, Becher MW, Schulz JB, Wong PC, Price DL, Cleveland DW. Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci USA. 1997;94:7606–7611. doi: 10.1073/pnas.94.14.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 26.Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 27.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 28.Carroll MC, Girouard JB, Ulloa JL, Subramaniam JR, Wong PC, Valentine JS, Culotta VC. Mechanisms for activating Cu- and Zn-containing superoxide dismutase in the absence of the CCS Cu chaperone. Proc Natl Acad Sci USA. 2004;101:5964–5969. doi: 10.1073/pnas.0308298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casareno RLB, Waggoner D, Gitlin JD. The copper chaperone CCS directly interacts with copper/zinc superoxide dismutase. J Biol Chem. 1998;273:23625–23628. doi: 10.1074/jbc.273.37.23625. [DOI] [PubMed] [Google Scholar]
- 30.Chang LY, Slot JW, Geuze HJ, Crapo JD. Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J Cell Biol. 1988;107:2169–2179. doi: 10.1083/jcb.107.6.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi J, Rees HD, Weintraub ST, Levey AI, Chin LS, Li L. Oxidative modifications and aggregation of Cu,Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J Biol Chem. 2005;280:11648–11655. doi: 10.1074/jbc.M414327200. [DOI] [PubMed] [Google Scholar]
- 32.Cleveland DW. Neuronal growth and death: order and disorder in the axoplasm. Cell. 1996;84:663–666. doi: 10.1016/s0092-8674(00)81044-2. [DOI] [PubMed] [Google Scholar]
- 33.Corson LB, Strain JJ, Culotta VC, Cleveland DW. Chaperone-facilitated copper binding is a property common to several classes of familial amyotrophic lateral sclerosis-linked superoxide dismutase mutants. Proc Natl Acad Sci USA. 1998;95:6361–6366. doi: 10.1073/pnas.95.11.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- 35.Culotta VC, Joh HD, Lin SJ, Slekar KH, Strain J. A physiological role for Saccharomyces cerevisiae copper/zinc superoxide dismutase in copper buffering. J Biol Chem. 1995;270:29991–29997. doi: 10.1074/jbc.270.50.29991. [DOI] [PubMed] [Google Scholar]
- 36.Culotta VC, Klomp LWJ, Strain J, Casareno RLB, Krems B, Gitlin JD. The copper chaperone for superoxide dismutase. J Biol Chem. 1997;272:23469–23472. doi: 10.1074/jbc.272.38.23469. [DOI] [PubMed] [Google Scholar]
- 37.Dahlgren C, Karlsson A. Respiratory burst in human neutrophils. J Immunol Methods. 1999;232:3–14. doi: 10.1016/s0022-1759(99)00146-5. [DOI] [PubMed] [Google Scholar]
- 38.Dameron CT, Harris ED. Regulation of aortic CuZn-superoxide dismutase with copper. Caeruloplasmin and albumin re-activate and transfer copper to the enzyme in culture. Biochem J. 1987;248:669–675. doi: 10.1042/bj2480669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davies KJ. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life. 2000;50:279–289. doi: 10.1080/713803728. [DOI] [PubMed] [Google Scholar]
- 40.de Beus MD, Chung J, Colon W. Modification of cysteine 111 in Cu/Zn superoxide dismutase results in altered spectroscopic and biophysical properties. Protein Sci. 2004;13:1347–1355. doi: 10.1110/ps.03576904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eisses JF, Stasser JP, Ralle M, Kaplan JH, Blackburn NJ. Domains I and III of the human copper chaperone for superoxide dismutase interact via a cysteine-bridged dicopper(I) cluster. Biochemistry. 2000;39:7337–7342. doi: 10.1021/bi000690j. [DOI] [PubMed] [Google Scholar]
- 42.Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, Hasnain SS, Hayward LJ, Valentine JS, Yeates TO, Hart PJ. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol. 2003;10:461–467. doi: 10.1038/nsb935. [DOI] [PubMed] [Google Scholar]
- 43.Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L, Van Remmen H, Epstein CJ, Huang TT. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 44.Endo T, Fujii T, Sato K, Taniguchi N, Fujii J. A pivotal role of Zn-binding residues in the function of the copper chaperone for SOD1. Biochem Biophys Res Commun. 2000;276:999–1004. doi: 10.1006/bbrc.2000.3581. [DOI] [PubMed] [Google Scholar]
- 45.Engidawork E, Lubec G. Protein expression in Down syndrome brain. Amino Acids. 2001;21:331–361. doi: 10.1007/s007260170001. [DOI] [PubMed] [Google Scholar]
- 46.Ermilova IP, Ermilov VB, Levy M, Ho E, Pereira C, Beckman JS. Protection by dietary zinc in ALS mutant G93A SOD transgenic mice. Neurosci Lett. 2005;379:42–46. doi: 10.1016/j.neulet.2004.12.045. [DOI] [PubMed] [Google Scholar]
- 47.Estevez AG, Crow JP, Sampson JB, Reiter C, Zhuang YX, Richardson GJ, Tarpey MM, Barbeito L, Beckman JS. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- 48.Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, Kowall NW, Brown RH, Jr, Beal MF. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69:2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- 49.Field LS, Furukawa Y, O'Halloran TV, Culotta VC. Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J Biol Chem. 2003;278:28052–28059. doi: 10.1074/jbc.M304296200. [DOI] [PubMed] [Google Scholar]
- 50.Fisher CL, Cabelli DE, Tainer JA, Hallewell RA, Getzoff ED. The role of arginine 143 in the electrostatics and mechanism of Cu,Zn superoxide dismutase: computational and experimental evaluation by mutational analysis. Proteins. 1994;19:24–34. doi: 10.1002/prot.340190105. [DOI] [PubMed] [Google Scholar]
- 51.Forman HJ, Fridovich I. On the stability of bovine superoxide dismutase. The effects of metals. J Biol Chem. 1973;248:2645–2649. [PubMed] [Google Scholar]
- 52.Frand AR, Kaiser CA. Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol Cell. 1999;4:469–477. doi: 10.1016/s1097-2765(00)80198-7. [DOI] [PubMed] [Google Scholar]
- 53.Fridovich I. Superoxide dismutases. Annu Rev Biochem. 1975;44:147–159. doi: 10.1146/annurev.bi.44.070175.001051. [DOI] [PubMed] [Google Scholar]
- 54.Furukawa Y, O'Halloran TV. ALS mutations have the greatest destabilizing effect on the apo, reduced form of SOD1, leading to unfolding and oxidative aggregation. J Biol Chem. 2005;280:17266–17274. doi: 10.1074/jbc.M500482200. [DOI] [PubMed] [Google Scholar]
- 55.Furukawa Y, Torres AS, O'Halloran TV. Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J. 2004;23:2872–2881. doi: 10.1038/sj.emboj.7600276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galiazzo F, Ciriolo MR, Carri MT, Civitareale P, Marcocci L, Marmocchi F, Rotilio G. Activation and induction by copper of Cu/Zn superoxide dismutase in Saccharomyces cerevisiae. Presence of an inactive proenzyme in anaerobic yeast. Eur J Biochem. 1991;196:545–549. doi: 10.1111/j.1432-1033.1991.tb15848.x. [DOI] [PubMed] [Google Scholar]
- 57.Galiazzo F, Labbe–Bois R. Regulation of Cu,Zn- and Mn-superoxide dismutase transcription in Saccharomyces cerevisiae. FEBS Lett. 1993;315:197–200. doi: 10.1016/0014-5793(93)81162-s. [DOI] [PubMed] [Google Scholar]
- 58.Ghadge GD, Wang L, Sharma K, Monti AL, Bindokas V, Stevens FJ, Roos RP. Truncated wild-type SOD1 and FALS-linked mutant SOD1 cause neural cell death in the chick embryo spinal cord. Neurobiol Dis. 2005;21:194–205. doi: 10.1016/j.nbd.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 59.Gilbert HF. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 60.Goto Y, Hamaguchi K. The role of the intrachain disulfide bond in the conformation and stability of the constant fragment of the immunoglobulin light chain. J Biochem (Tokyo) 1979;86:1433–1441. doi: 10.1093/oxfordjournals.jbchem.a132661. [DOI] [PubMed] [Google Scholar]
- 61.Goto Y, Hamaguchi K. Formation of the intrachain disulfide bond in the constant fragment of the immunoglobulin light chain. J Mol Biol. 1981;146:321–340. doi: 10.1016/0022-2836(81)90391-0. [DOI] [PubMed] [Google Scholar]
- 62.Goto Y, Hamaguchi K. Unfolding and refolding of the reduced constant fragment of the immunoglobulin light chain. Kinetic role of the intrachain disulfide bond. J Mol Biol. 1982;156:911–926. doi: 10.1016/0022-2836(82)90147-4. [DOI] [PubMed] [Google Scholar]
- 63.Gralla EB, Thiele DJ, Silar P, Valentine JS. ACE1, a copper-dependent transcription factor, activates expression of the yeast copper, zinc superoxide dismutase gene. Proc Natl Acad Sci USA. 1991;88:8558–8562. doi: 10.1073/pnas.88.19.8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Graumann J, Lilie H, Tang XL, Tucker KA, Hoffmann JT, Vijayalakshmi J, Saper M, Bardwell JCA, Jakob U. Activation of the redox-regulated molecular chaperone Hsp33–A two-step mechanism. Structure. 2001;9:377–387. doi: 10.1016/s0969-2126(01)00599-8. [DOI] [PubMed] [Google Scholar]
- 65.Gregory EM, Fridovic I. Oxygen toxicity and superoxide dismutase. J Bacteriol. 1973;114:1193–1197. doi: 10.1128/jb.114.3.1193-1197.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gregory EM, Goscin SA, Fridovich I. Superoxide dismutase and oxygen toxicity in a eukaryote. J Bacteriol. 1974;117:456–460. doi: 10.1128/jb.117.2.456-460.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gross E, Kastner DB, Kaiser CA, Fass D. Structure of Ero1p, source of disulfide bonds for oxidative protein folding in the cell. Cell. 2004;117:601–610. doi: 10.1016/s0092-8674(04)00418-0. [DOI] [PubMed] [Google Scholar]
- 68.Gross E, Sevier CS, Vala A, Kaiser CA, Fass D. A new FAD-binding fold and intersubunit disulfide shuttle in the thiol oxidase Erv2p. Nat Struct Biol. 2002;9:61–67. doi: 10.1038/nsb740. [DOI] [PubMed] [Google Scholar]
- 69.Hall LT, Sanchez RJ, Holloway SP, Zhu HN, Stine JE, Lyons TJ, Demeler B, Schirf V, Hansen JC, Nersissian AM, Valentine JS, Hart PJ. X-ray crystallographic and analytical ultracentrifugation analyses of truncated and full-length yeast copper chaperones for SOD (LYS7): A dimerdimer model of LYS7-SOD association and copper delivery. Biochemistry. 2000;39:3611–3623. doi: 10.1021/bi992716g. [DOI] [PubMed] [Google Scholar]
- 70.Hammarstrom P, Schneider F, Kelly JW. Transsuppression of misfolding in an amyloid disease. Science. 2001;293:2459–2462. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- 71.Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J. 2001;353:411–416. doi: 10.1042/0264-6021:3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haskins K, Kench J, Powers K, Bradley B, Pugazhenthi S, Reusch J, McDuffie M. Role for oxidative stress in the regeneration of islet beta cells? J Investig Med. 2004;52:45–49. doi: 10.1136/jim-52-01-25. [DOI] [PubMed] [Google Scholar]
- 73.Hayward LJ, Rodriguez JA, Kim JW, Tiwari A, Goto JJ, Cabelli DE, Valentine JS, Brown RH., Jr. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2002;277:15923–15931. doi: 10.1074/jbc.M112087200. [DOI] [PubMed] [Google Scholar]
- 74.Hoffman PN, Lasek RJ. The slow component of axonal transport. Identification of major structural polypeptides of the axon and their generality among mammalian neurons. J Cell Biol. 1975;66:351–366. doi: 10.1083/jcb.66.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Holmgren A. Thioredoxin and glutaredoxin systems. J Biol Chem. 1989;264:13963–13966. [PubMed] [Google Scholar]
- 76.Hottinger AF, Fine EG, Gurney ME, Zurn AD, Aebischer P. The copper chelator d-penicillamine delays onset of disease and extends survival in a transgenic mouse model of familial amyotrophic lateral sclerosis. Eur J Neurosci. 1997;9:1548–1551. doi: 10.1111/j.1460-9568.1997.tb01511.x. [DOI] [PubMed] [Google Scholar]
- 77.Hough MA, Grossmann JG, Antonyuk SV, Strange RW, Doucette PA, Rodriguez JA, Whitson LJ, Hart PJ, Hayward LJ, Valentine JS, Hasnain SS. Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc Natl Acad Sci USA. 2004;101:5976–5981. doi: 10.1073/pnas.0305143101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huie RE, Padmaja S. The reaction of NO with superoxide. Free Radic Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 79.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 80.Ito K, Uchiyama T, Fukutake T, Arai K, Kanesaka T, Hattori T. Different clinical phenotypes of siblings with familial amyotrophic lateral sclerosis showing Cys146Arg point mutation of superoxide dismutase 1 gene. Clinica Neurol. 2002;42:175–177. [PubMed] [Google Scholar]
- 81.Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, Verspaget HW, London J, Holstege JC. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis. 2000;7:623–643. doi: 10.1006/nbdi.2000.0299. [DOI] [PubMed] [Google Scholar]
- 82.Jakob U, Muse W, Eser M, Bardwell JC. Chaperone activity with a redox switch. Cell. 1999;96:341–352. doi: 10.1016/s0092-8674(00)80547-4. [DOI] [PubMed] [Google Scholar]
- 83.Jensen LT, Culotta VC. Activation of Cu/Zn superoxide dismutases from C. elegans does not require the copper chaperone CCS. J Biol Chem. 2005;280:41373–41379. doi: 10.1074/jbc.M509142200. [DOI] [PubMed] [Google Scholar]
- 84.Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu,Zn- superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jokic N, Di Scala F, Dupuis L, Rene F, Muller A, De Aguilar JL, Loeffler JP. Early activation of antioxidant mechanisms in muscle of mutant Cu/Zn-superoxide dismutase-linked amyotrophic lateral sclerosis mice. Ann NY Acad Sci. 2003;1010:552–556. doi: 10.1196/annals.1299.102. [DOI] [PubMed] [Google Scholar]
- 86.Juneja T, Pericak-Vance MA, Laing NG, Dave S, Siddique T. Prognosis in familial amyotrophic lateral sclerosis: progression and survival in patients with glu100gly and ala4val mutations in Cu,Zn superoxide dismutase. Neurology. 1997;48:55–57. doi: 10.1212/wnl.48.1.55. [DOI] [PubMed] [Google Scholar]
- 87.Kagi JH, Schaffer A. Biochemistry of metallothionein. Biochemistry. 1988;27:8509–8515. doi: 10.1021/bi00423a001. [DOI] [PubMed] [Google Scholar]
- 88.Kato S, Sumi-Akamaru H, Fujimura H, Sakoda S, Kato M, Hirano A, Takikawa M, Ohama E. Copper chaperone for superoxide dismutase co-aggregates with superoxide dismutase 1 (SOD1) in neuronal Lewy body-like hyaline inclusions: an immunohistochemical study on familial amyotrophic lateral sclerosis with SOD1 gene mutation. Acta Neuropathol. 2001;102:233–238. doi: 10.1007/s004010000355. [DOI] [PubMed] [Google Scholar]
- 89.Kessova IG, Ho YS, Thung S, Cederbaum AI. Alcohol-induced liver injury in mice lacking Cu, Zn-superoxide dismutase. Hepatology. 2003;38:1136–1145. doi: 10.1053/jhep.2003.50450. [DOI] [PubMed] [Google Scholar]
- 90.Khare SD, Caplow M, Dokholyan NV. The rate and equilibrium constants for a multistep reaction sequence for the aggregation of superoxide dismutase in amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2004;101:15094–15099. doi: 10.1073/pnas.0406650101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kroll JS, Langford PR, Wilks KE, Keil AD. Bacterial [Cu,Zn]-superoxide dismutase: phylogenetically distinct from the eukaryotic enzyme, and not so rare after all! Microbiology. 1995;141:2271–2279. doi: 10.1099/13500872-141-9-2271. [DOI] [PubMed] [Google Scholar]
- 92.Kurobe N, Suzuki F, Okajima K, Kato K. Sensitive enzyme immunoassay for human Cu/Zn superoxide dismutase. Clin Chim Acta. 1990;187:11–20. doi: 10.1016/0009-8981(90)90257-s. [DOI] [PubMed] [Google Scholar]
- 93.Laliberte J, Whitson LJ, Beaudoin J, Holloway SP, Hart PJ, Labbe S. The Schizosaccharomyces pombe Pccs protein functions in both copper trafficking and metal detoxification pathways. J Biol Chem. 2004;279:28744–28755. doi: 10.1074/jbc.M403426200. [DOI] [PubMed] [Google Scholar]
- 94.Lamb AL, Torres AS, O'Halloran TV, Rosenzweig AC. Heterodimer formation between superoxide dismutase and its copper chaperone. Biochemistry. 2000;39:14720–14727. doi: 10.1021/bi002207a. [DOI] [PubMed] [Google Scholar]
- 95.Lamb AL, Torres AS, O'Halloran TV, Rosenzweig AC. Heterodimeric structure of superoxide dismutase in complex with its metallochaperone. Nat Struct Biol. 2001;8:751–755. doi: 10.1038/nsb0901-751. [DOI] [PubMed] [Google Scholar]
- 96.Lamb AL, Wernimont AK, Pufahl RA, Culotta VC, O'Halloran TV, Rosenzweig AC. Crystal structure of the copper chaperone for superoxide dismutase. Nat Struct Biol. 1999;6:724–729. doi: 10.1038/11489. [DOI] [PubMed] [Google Scholar]
- 97.Lamb AL, Wernimont AK, Pufahl RA, O'Halloran TV, Rosenzweig AC. Crystal structure of the second domain of the human copper chaperone for superoxide dismutase. Biochemistry. 2000;39:1589–1595. doi: 10.1021/bi992822i. [DOI] [PubMed] [Google Scholar]
- 98.Lee JP, Gerin C, Bindokas VP, Miller R, Ghadge G, Roos RP. No correlation between aggregates of Cu/Zn su-peroxide dismutase and cell death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;82:1229–1238. doi: 10.1046/j.1471-4159.2002.01056.x. [DOI] [PubMed] [Google Scholar]
- 99.Lee S, Eisenberg D. Seeded conversion of recombinant prion protein to a disulfide-bonded oligomer by a reduction-oxidation process. Nat Struct Biol. 2003;10:725–730. doi: 10.1038/nsb961. [DOI] [PubMed] [Google Scholar]
- 100.Lindberg MJ, Tibell L, Oliveberg M. Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: decreased stability of the apo state. Proc Natl Acad Sci USA. 2002;99:16607–16612. doi: 10.1073/pnas.262527099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lindenau J, Noack H, Possel H, Asayama K, Wolf G. Cellular distribution of superoxide dismutases in the rat CNS. Glia. 2000;29:25–34. doi: 10.1002/(sici)1098-1136(20000101)29:1<25::aid-glia3>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 102.Liochev SI, Fridovich I. The role of O2.- in the production of HO.: in vitro and in vivo. Free Radic Biol Med. 1994;16:29–33. doi: 10.1016/0891-5849(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 103.Liu H, Zhu H, Eggers DK, Nersissian AM, Faull KF, Goto JJ, Ai J, Sanders-Loehr J, Gralla EB, Valentine JS. Copper(2+) binding to the surface residue cysteine 111 of His46Arg human copper-zinc superoxide dismutase, a familial amyotrophic lateral sclerosis mutant. Biochemistry. 2000;39:8125–8132. doi: 10.1021/bi000846f. [DOI] [PubMed] [Google Scholar]
- 104.Liu J, Lillo C, Jonsson PA, Velde CV, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brannstrom T, Gredal O, Wong PC, Williams DS, Cleveland DW. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 105.Liu SX, Fabisiak JP, Tyurin VA, Borisenko GG, Pitt BR, Lazo JS, Kagan VE. Reconstitution of apo-superoxide dismutase by nitric oxide-induced copper transfer from metallothioneins. Chem Res Toxicol. 2000;13:922–931. doi: 10.1021/tx0000623. [DOI] [PubMed] [Google Scholar]
- 106.Locker JK, Griffiths G. An unconventional role for cytoplasmic disulfide bonds in vaccinia virus proteins. J Cell Biol. 1999;144:267–279. doi: 10.1083/jcb.144.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lyons TJ, Liu HB, Goto JJ, Nersissian A, Roe JA, Graden JA, Cafe C, Ellerby LM, Bredesen DE, Gralla EB, Valentine JS. Mutations in copper-zinc superoxide dismutase that cause amyotrophic lateral sclerosis alter the zinc binding site and the redox behavior of the protein. Proc Natl Acad Sci USA. 1996;93:12240–12244. doi: 10.1073/pnas.93.22.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maret W, Vallee BL. Cobalt as probe and label of proteins. Methods Enzymol. 1993;226:52–71. doi: 10.1016/0076-6879(93)26005-t. [DOI] [PubMed] [Google Scholar]
- 109.Matsumoto G, Stojanovic A, Holmberg CI, Kim S, Morimoto RI. Structural properties and neuronal toxicity of amyotrophic lateral sclerosis-associated Cu/Zn super-oxide dismutase 1 aggregates. J Cell Biol. 2005;171:75–85. doi: 10.1083/jcb.200504050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Matzuk MM, Dionne L, Guo Q, Kumar TR, Lebovitz RM. Ovarian function in superoxide dismutase 1 and 2 knockout mice. Endocrinology. 1998;139:4008–4011. doi: 10.1210/endo.139.9.6289. [DOI] [PubMed] [Google Scholar]
- 111.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 112.McFadden SL, Ding D, Reaume AG, Flood DG, Salvi RJ. Age-related cochlear hair cell loss is enhanced in mice lacking copper/zinc superoxide dismutase. Neurobiol Aging. 1999;20:1–8. doi: 10.1016/s0197-4580(99)00018-4. [DOI] [PubMed] [Google Scholar]
- 113.Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, Herrmann JM. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–1069. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 114.Moriwaki Y, Yamamoto T, Suda M, Nasako Y, Takahashi S, Agbedana OE, Hada T, Higashino K. Purification and immunohistochemical tissue localization of human xanthine oxidase. Biochim Biophys Acta. 1993;1164:327–330. doi: 10.1016/0167-4838(93)90266-t. [DOI] [PubMed] [Google Scholar]
- 115.Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, Kasai N, Brown RH, Jr, Itoyama Y. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nagano S, Ogawa Y, Yanagihara T, Sakoda S. Benefit of a combined treatment with trientine and ascorbate in familial amyotrophic lateral sclerosis model mice. Neurosci Lett. 1999;265:159–162. doi: 10.1016/s0304-3940(99)00227-x. [DOI] [PubMed] [Google Scholar]
- 117.Niwa J, Ishigaki S, Hishikawa N, Yamamoto M, Doyu M, Murata S, Tanaka K, Taniguchi N, Sobue G. Dorfin ubiquitylates mutant SOD1 and prevents mutant SOD1-mediated neurotoxicity. J Biol Chem. 2002;277:36793–36798. doi: 10.1074/jbc.M206559200. [DOI] [PubMed] [Google Scholar]
- 118.Oeda T, Shimohama S, Kitagawa N, Kohno R, Imura T, Shibasaki H, Ishii N. Oxidative stress causes abnormal accumulation of familial amyotrophic lateral sclerosis-related mutant SOD1 in transgenic Caenorhabditis elegans. Hum Mol Genet. 2001;10:2013–2023. doi: 10.1093/hmg/10.19.2013. [DOI] [PubMed] [Google Scholar]
- 119.Ogawa Y, Kosaka H, Nakanishi T, Shimizu A, Ohoi N, Shouji H, Yanagihara T, Sakoda S. Stability of mutant superoxide dismutase-1 associated with familiar amyotrophic lateral sclerosis determines the manner of copper release and induction of thioredoxin in erythrocytes. Biochem Biophys Res Commun. 1997;241:251–257. doi: 10.1006/bbrc.1997.7804. [DOI] [PubMed] [Google Scholar]
- 120.O'Halloran TV, Culotta VC. Metallochaperones, an intracellular shuttle service for metal ions. J Biol Chem. 2000;275:25057–25060. doi: 10.1074/jbc.R000006200. [DOI] [PubMed] [Google Scholar]
- 121.Oh-ishi S, Kizaki T, Ookawara T, Sakurai T, Izawa T, Nagata N, Ohno H. Endurance training improves the resistance of rat diaphragm to exercise-induced oxidative stress. Am J Respir Crit Care Med. 1997;156:1579–1585. doi: 10.1164/ajrccm.156.5.96-11035. [DOI] [PubMed] [Google Scholar]
- 122.Okado–Matsumoto A, Fridovich I. Amyotrophic lateral sclerosis: A proposed mechanism. Proc Natl Acad Sci USA. 2002;99:9010–9014. doi: 10.1073/pnas.132260399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Okamoto K, Hirai S, Yamazaki T, Sun XY, Nakazato Y. New ubiquitin-positive intraneuronal inclusions in the extra-motor cortices in patients with amyotrophic lateral sclerosis. Neurosci Lett. 1991;129:233–236. doi: 10.1016/0304-3940(91)90469-a. [DOI] [PubMed] [Google Scholar]
- 124.Ookawara T, Kawamura N, Kitagawa Y, Taniguchi N. Site-specific and random fragmentation of Cu,Zn-superoxide dismutase by glycation reaction. Implication of reactive oxygen species. J Biol Chem. 1992;267:18505–18510. [PubMed] [Google Scholar]
- 125.Outten CE, O'Halloran TV. Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science. 2001;292:2488–2492. doi: 10.1126/science.1060331. [DOI] [PubMed] [Google Scholar]
- 126.Pacello F, Langford PR, Kroll JS, Indiani C, Smulevich G, Desideri A, Rotilio G, Battistoni A. A novel heme protein, the Cu,Zn-superoxide dismutase from Haemophilus ducreyi. J Biol Chem. 2001;276:30326–30334. doi: 10.1074/jbc.M010488200. [DOI] [PubMed] [Google Scholar]
- 127.Pardo CA, Xu Z, Borchelt DR, Price DL, Sisodia SS, Cleveland DW. Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc Natl Acad Sci USA. 1995;92:954–958. doi: 10.1073/pnas.92.4.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 129.Polavarapu R, Spitz DR, Sim JE, Follansbee MH, Oberley LW, Rahemtulla A, Nanji AA. Increased lipid peroxidation and impaired antioxidant enzyme function is associated with pathological liver injury in experimental alcoholic liver disease in rats fed diets high in corn oil and fish oil. Hepatology. 1998;27:1317–1323. doi: 10.1002/hep.510270518. [DOI] [PubMed] [Google Scholar]
- 130.Prohaska JR, Brokate B. Lower copper, zinc-superoxide dismutase protein but not mRNA in organs of copper-deficient rats. Arch Biochem Biophys. 2001;393:170–176. doi: 10.1006/abbi.2001.2470. [DOI] [PubMed] [Google Scholar]
- 131.Prohaska JR, Geissler J, Brokate B, Broderius M. Copper, zinc-superoxide dismutase protein but not mRNA is lower in copper-deficient mice and mice lacking the copper chaperone for superoxide dismutase. Exp Biol Med (Maywood) 2003;228:959–966. doi: 10.1177/153537020322800812. [DOI] [PubMed] [Google Scholar]
- 132.Pufahl RA, Singer CP, Peariso KL, Lin SJ, Schmidt PJ, Fahrni CJ, Culotta VC, PennerHahn JE, O'Halloran TV. Metal ion chaperone function of the soluble Cu(I) receptor Atx1. Science. 1997;278:853–856. doi: 10.1126/science.278.5339.853. [DOI] [PubMed] [Google Scholar]
- 133.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O'Halloran TV. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 134.Rae TD, Torres AS, Pufahl RA, O'Halloran TV. Mechanism of Cu,Zn-superoxide dismutase activation by the human metallochaperone hCCS. J Biol Chem. 2001;276:5166–5176. doi: 10.1074/jbc.M008005200. [DOI] [PubMed] [Google Scholar]
- 135.Rakhit R, Crow JP, Lepock JR, Kondejewski LH, Cashman NR, Chakrabartty A. Monomeric Cu,Znsuperoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J Biol Chem. 2004;279:15499–15504. doi: 10.1074/jbc.M313295200. [DOI] [PubMed] [Google Scholar]
- 136.Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi XF, Crow JP, Cashman NR, Kondejewski LH, Chakrabartty A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem. 2002;277:47551–47556. doi: 10.1074/jbc.M207356200. [DOI] [PubMed] [Google Scholar]
- 137.Ralle M, Lutsenko S, Blackburn NJ. X-ray absorption spectroscopy of the copper chaperone HAH1 reveals a linear two-coordinate Cu(I) center capable of adduct formation with exogenous thiols and phosphines. J Biol Chem. 2003;278:23163–23170. doi: 10.1074/jbc.M303474200. [DOI] [PubMed] [Google Scholar]
- 138.Ray SS, Nowak RJ, Brown RH, Jr, Lansbury PT., Jr Small-molecule-mediated stabilization of familial amyotrophic lateral sclerosis-linked superoxide dismutase mutants against unfolding and aggregation. Proc Natl Acad Sci USA. 2005;102:3639–3644. doi: 10.1073/pnas.0408277102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ray SS, Nowak RJ, Strolovich K, Brown RH, Jr., Walz T, Lansbury PT., Jr An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amyotrophic lateral sclerosis. Biochemistry. 2004;43:4899–4905. doi: 10.1021/bi030246r. [DOI] [PubMed] [Google Scholar]
- 140.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr., Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 141.Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Robberecht W, Sapp P, Viaene MK, Rosen D, McKenna-Yasek D, Haines J, Horvitz R, Theys P, Brown R., Jr. Cu/Zn superoxide dismutase activity in familial and sporadic amyotrophic lateral sclerosis. J Neurochem. 1994;62:384–387. doi: 10.1046/j.1471-4159.1994.62010384.x. [DOI] [PubMed] [Google Scholar]
- 143.Rodriguez JA, Shaw BF, Durazo A, Sohn SH, Doucette PA, Nersissian AM, Faull KF, Eggers DK, Tiwari A, Hayward LJ, Valentine JS. Destabilization of apoprotein is insufficient to explain Cu,Zn-superoxide dismutase-linked ALS pathogenesis. Proc Natl Acad Sci USA. 2005;102:10516–10521. doi: 10.1073/pnas.0502515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rodriguez JA, Valentine OS, Eggers DK, Roe JA, Tiwari A, Brown RH, Hayward LJ. Familial amyotrophic lateral sclerosis-associated mutations decrease the thermal stability of distinctly metallated species of human copper/zinc superoxide dismutase. J Biol Chem. 2002;277:15932–15937. doi: 10.1074/jbc.M112088200. [DOI] [PubMed] [Google Scholar]
- 145.Roe JA, Butler A, Scholler DM, Valentine JS, Marky L, Breslauer KJ. Differential scanning calorimetry of Cu,Zn-superoxide dismutase, the apoprotein, and its zinc-substituted derivatives. Biochemistry. 1988;27:950–958. doi: 10.1021/bi00403a017. [DOI] [PubMed] [Google Scholar]
- 146.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 147.Rothstein JD, Dykes–Hoberg M, Corson LB, Becker M, Cleveland DW, Price DL, Culotta VC, Wong PC. The copper chaperone CCS is abundant in neurons and astrocytes in human and rodent brain. J Neurochem. 1999;72:422–429. doi: 10.1046/j.1471-4159.1999.0720422.x. [DOI] [PubMed] [Google Scholar]
- 148.Rush JW, Laughlin MH, Woodman CR, Price EM. SOD-1 expression in pig coronary arterioles is increased by exercise training. Am J Physiol Heart Circ Physiol. 2000;279:H2068–H2076. doi: 10.1152/ajpheart.2000.279.5.H2068. [DOI] [PubMed] [Google Scholar]
- 149.Rush JW, Turk JR, Laughlin MH. Exercise training regulates SOD-1 and oxidative stress in porcine aortic endothelium. Am J Physiol Heart Circ Physiol. 2003;284:H1378–H1387. doi: 10.1152/ajpheart.00190.2002. [DOI] [PubMed] [Google Scholar]
- 150.Schmidt PJ, Kunst C, Culotta VC. Copper activation of superoxide dismutase 1 (SOD1) in vivo—Role for protein-protein interactions with the copper chaperone for SOD1. J Biol Chem. 2000;275:33771–33776. doi: 10.1074/jbc.M006254200. [DOI] [PubMed] [Google Scholar]
- 151.Schmidt PJ, Rae TD, Pufahl RA, Hamma T, Strain J, O'Halloran TV, Culotta VC. Multiple protein domains contribute to the action of the copper chaperone for superoxide dismutase. J Biol Chem. 1999;274:23719–23725. doi: 10.1074/jbc.274.34.23719. [DOI] [PubMed] [Google Scholar]
- 152.Senkevich TG, White CL, Koonin EV, Moss B. A viral member of the ERV1/ALR protein family participates in a cytoplasmic pathway of disulfide bond formation. Proc Natl Acad Sci USA. 2000;97:12068–12073. doi: 10.1073/pnas.210397997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shibata N, Asayama K, Hirano A, Kobayashi M. Immunohistochemical study on superoxide dismutases in spinal cords from autopsied patients with amyotrophic lateral sclerosis. Dev Neurosci. 1996;18:492–498. doi: 10.1159/000111445. [DOI] [PubMed] [Google Scholar]
- 154.Shibata N, Hirano A, Kobayashi M, Sasaki S, Kato T, Matsumoto S, Shiozawa Z, Komori T, Ikemoto A, Umahara T, et al. Cu/Zn superoxide dismutase-like immunore-activity in Lewy body-like inclusions of sporadic amyotrophic lateral sclerosis. Neurosci Lett. 1994;179:149–152. doi: 10.1016/0304-3940(94)90956-3. [DOI] [PubMed] [Google Scholar]
- 155.Shibata N, Hirano A, Kobayashi M, Siddique T, Deng HX, Hung WY, Kato T, Asayama K. Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol. 1996;55:481–490. doi: 10.1097/00005072-199604000-00011. [DOI] [PubMed] [Google Scholar]
- 156.Shinder GA, Lacourse MC, Minotti S, Durham HD. Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis. J Biol Chem. 2001;276:12791–12796. doi: 10.1074/jbc.M010759200. [DOI] [PubMed] [Google Scholar]
- 157.Silahtaroglu AN, Brondum-Nielsen K, Gredal O, Werdelin L, Panas M, Petersen MB, Tommerup N, Tumer Z. Human CCS gene: genomic organization and exclusion as a candidate for amyotrophic lateral sclerosis (ALS) BMC Genet. 2002;3:5. doi: 10.1186/1471-2156-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Son M, Cloyd CD, Rothstein JD, Rajendran B, Elliott JL. Aggregate formation in Cu,Zn superoxide dismutase-related proteins. J Biol Chem. 2003;278:14331–14336. doi: 10.1074/jbc.M211698200. [DOI] [PubMed] [Google Scholar]
- 159.Southon A, Burke R, Norgate M, Batterham P, Camakaris J. Copper homoeostasis in Drosophila melanogaster S2 cells. Biochem J. 2004;383:303–309. doi: 10.1042/BJ20040745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Spagnolo L, Toro I, D'Orazio M, O'Neill P, Pedersen JZ, Carugo O, Rotilio G, Battistoni A, Djinovic–Carugo K. Unique features of the sodC-encoded super-oxide dismutase from Mycobacterium tuberculosis, a fully functional copper-containing enzyme lacking zinc in the active site. J Biol Chem. 2004;279:33447–33455. doi: 10.1074/jbc.M404699200. [DOI] [PubMed] [Google Scholar]
- 161.St John G, Steinman HM. Periplasmic copper-zinc superoxide dismutase of Legionella pneumophila: role in stationary-phase survival. J Bacteriol. 1996;178:1578–1584. doi: 10.1128/jb.178.6.1578-1584.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Stasser JP, Eisses JF, Barry AN, Kaplan JH, Blackburn NJ. Cysteine-to-serine mutants of the human copper chaperone for superoxide dismutase reveal a copper cluster at a domain III dimer interface. Biochemistry. 2005;44:3143–3152. doi: 10.1021/bi0478392. [DOI] [PubMed] [Google Scholar]
- 163.Stathopulos PB, Rumfeldt JA, Scholz GA, Irani RA, Frey HE, Hallewell RA, Lepock JR, Meiering EM. Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc Natl Acad Sci USA. 2003;100:7021–7026. doi: 10.1073/pnas.1237797100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Storkebaum E, Lambrechts D, Dewerchin M, Moreno–Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, Carmeliet P. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- 165.Strange RW, Antonyuk S, Hough MA, Doucette PA, Rodriguez JA, Hart PJ, Hayward LJ, Valentine JS, Hasnain SS. The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J Mol Biol. 2003;328:877–891. doi: 10.1016/s0022-2836(03)00355-3. [DOI] [PubMed] [Google Scholar]
- 166.Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem. 2001;276:38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- 167.Subramaniam JR, Lyons WE, Liu J, Bartnikas TB, Rothstein J, Price DL, Cleveland DW, Gitlin JD, Wong PC. Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat Neurosci. 2002;5:301–307. doi: 10.1038/nn823. [DOI] [PubMed] [Google Scholar]
- 168.Suzuki KT, Kuroda T. Direct transfer of copper from metallothionein to superoxide dismutase: a possible mechanism for differential supply of Cu to SOD and ceruloplasmin in LEC rats. Res Commun Mol Pathol Pharmacol. 1994;86:15–23. [PubMed] [Google Scholar]
- 169.Suzuki KT, Kuroda T. Transfer of copper and zinc from ionic and metallothionein-bound forms to Cu, Zn–superoxide dismutase. Res Commun Mol Pathol Pharmacol. 1995;87:287–296. [PubMed] [Google Scholar]
- 170.Tainer JA, Getzoff ED, Richardson JS, Richardson DC. Structure and mechanism of copper, zinc superoxide dismutase. Nature. 1983;306:284–287. doi: 10.1038/306284a0. [DOI] [PubMed] [Google Scholar]
- 171.Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. 2004;10:148–154. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- 172.Tatzelt J, Prusiner SB, Welch WJ. Chemical chaperones interfere with the formation of scrapie prion protein. EMBO J. 1996;15:6363–6373. [PMC free article] [PubMed] [Google Scholar]
- 173.Teoh ML, Walasek PJ, Evans DH. Leporipoxvirus Cu,Zn-superoxide dismutase (SOD) homologs are catalytically inert decoy proteins that bind copper chaperone for SOD. J Biol Chem. 2003;278:33175–33184. doi: 10.1074/jbc.M300644200. [DOI] [PubMed] [Google Scholar]
- 174.Thornton JM. Disulphide bridges in globular proteins. J Mol Biol. 1981;151:261–287. doi: 10.1016/0022-2836(81)90515-5. [DOI] [PubMed] [Google Scholar]
- 175.Thorpe C, Hoober KL, Raje S, Glynn NM, Burnside J, Turi GK, Coppock DL. Sulfhydryl oxidases: emerging catalysts of protein disulfide bond formation in eukaryotes. Arch Biochem Biophys. 2002;405:1–12. doi: 10.1016/s0003-9861(02)00337-5. [DOI] [PubMed] [Google Scholar]
- 176.Tiwari A, Hayward LJ. Familial ALS mutants of Cu/Zn superoxide dismutase are susceptible to disulfide reduction. J Biol Chem. 2003;278:5984–5992. doi: 10.1074/jbc.M210419200. [DOI] [PubMed] [Google Scholar]
- 177.Tiwari A, Xu Z, Hayward LJ. Aberrantly increased hydrophobicity shared by mutants of Cu/Zn superoxide dismutase in familial amyotrophic lateral sclerosis. J Biol Chem. 2005;280:29771–29779. doi: 10.1074/jbc.M504039200. [DOI] [PubMed] [Google Scholar]
- 178.Tsuda T, Munthasser S, Fraser PE, Percy ME, Rainero I, Vaula G, Pinessi L, Bergamini L, Vignocchi G, McLachlan DR, et al. Analysis of the functional effects of a mutation in SOD1 associated with familial amyotrophic lateral sclerosis. Neuron. 1994;13:727–736. doi: 10.1016/0896-6273(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 179.Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol. 2004;164:341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Tummala H, Jung C, Tiwari A, Higgins CM, Hayward LJ, Xu Z. Inhibition of chaperone activity is a shared property of several Cu,Zn-superoxide dismutase mutants that cause amyotrophic lateral sclerosis. J Biol Chem. 2005;280:17725–17731. doi: 10.1074/jbc.M501705200. [DOI] [PubMed] [Google Scholar]
- 181.Tyler DD. Polarographic assay and intracellular distribution of superoxide dismutase in rat liver. Biochem J. 1975;147:493–504. doi: 10.1042/bj1470493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Urushitani M, Kurisu J, Tateno M, Hatakeyama S, Nakayama K, Kato S, Takahashi R. CHIP promotes proteasomal degradation of familial ALS-linked mutant SOD1 by ubiquitinating Hsp/Hsc70. J Neurochem. 2004;90:231–244. doi: 10.1111/j.1471-4159.2004.02486.x. [DOI] [PubMed] [Google Scholar]
- 183.Urushitani M, Kurisu J, Tsukita K, Takahashi R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;83:1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x. [DOI] [PubMed] [Google Scholar]
- 184.Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25:2463–2470. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR. Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet. 2003;12:2753–2764. doi: 10.1093/hmg/ddg312. [DOI] [PubMed] [Google Scholar]
- 186.Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis. 2001;8:933–941. doi: 10.1006/nbdi.2001.0443. [DOI] [PubMed] [Google Scholar]
- 187.Wei JPJ, Srinivasan C, Han H, Valentine JS, Gralla EB. Evidence for a novel role of copper-zinc superoxide dismutase in zinc metabolism. J Biol Chem. 2001;276:44798–44803. doi: 10.1074/jbc.M104708200. [DOI] [PubMed] [Google Scholar]
- 188.West EC, Prohaska JR. Cu,Zn-superoxide dismutase is lower and copper chaperone CCS is higher in erythrocytes of copper-deficient rats and mice. Exp Biol Med (Maywood) 2004;229:756–764. doi: 10.1177/153537020422900807. [DOI] [PubMed] [Google Scholar]
- 189.Wiedau–Pazos M, Goto JJ, Rabizadeh S, Gralla EB, Roe JA, Lee MK, Valentine JS, Bredesen DE. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- 190.Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- 191.Wintz H, Vulpe C. Plant copper chaperones. Biochem Soc Trans. 2002;30:732–735. doi: 10.1042/bst0300732. [DOI] [PubMed] [Google Scholar]
- 192.Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 193.Wong PC, Waggoner D, Subramaniam JR, Tessarollo L, Bartnikas TB, Culotta VC, Price DL, Rothstein J, Gitlin JD. Copper chaperone for superoxide dismutase is essential to activate mammalian Cu/Zn superoxide dismutase. Proc Natl Acad Sci USA. 2000;97:2886–2891. doi: 10.1073/pnas.040461197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang H, Andrekopoulos C, Joseph J, Crow J, Kalyanaraman B. The carbonate radical anion-induced covalent aggregation of human copper, zinc superoxide dismutase, and alpha-synuclein: intermediacy of tryptophan- and tyrosine-derived oxidation products. Free Radic Biol Med. 2004;36:1355–1365. doi: 10.1016/j.freeradbiomed.2004.02.038. [DOI] [PubMed] [Google Scholar]
- 195.Zhang Q, Kelly JW. Cys10 mixed disulfides make transthyretin more amyloidogenic under mildly acidic conditions. Biochemistry. 2003;42:8756–8761. doi: 10.1021/bi030077a. [DOI] [PubMed] [Google Scholar]
- 196.Zhu HN, Shipp E, Sanchez RJ, Liba A, Stine JE, Hart PJ, Gralla EB, Nersissian AM, Valentine JS. Cobalt(2+) binding to human and tomato copper chaperone for su-peroxide dismutase: implications for the metal ion transfer mechanism. Biochemistry. 2000;39:5413–5421. doi: 10.1021/bi992727+. [DOI] [PubMed] [Google Scholar]