rVista for Comparative Sequence-Based Discovery of Functional Transcription Factor Binding Sites (original) (raw)
Abstract
Identifying transcriptional regulatory elements represents a significant challenge in annotating the genomes of higher vertebrates. We have developed a computational tool, rVISTA, for high-throughput discovery of _cis_-regulatory elements that combines clustering of predicted transcription factor binding sites (TFBSs) and the analysis of interspecies sequence conservation to maximize the identification of functional sites. To assess the ability of rVISTA to discover true positive TFBSs while minimizing the prediction of false positives, we analyzed the distribution of several TFBSs across 1 Mb of the well-annotated cytokine gene cluster (Hs5q31; Mm11). Because a large number of AP-1, NFAT, and GATA-3 sites have been experimentally identified in this interval, we focused our analysis on the distribution of all binding sites specific for these transcription factors. The exploitation of the orthologous human–mouse dataset resulted in the elimination of >95% of the ∼58,000 binding sites predicted on analysis of the human sequence alone, whereas it identified 88% of the experimentally verified binding sites in this region.
A major challenge of the postgenome-sequencing era is decoding the regulatory networks underlining gene expression. In eukaryotes, modulation of gene expression is achieved through the complex interaction of regulatory proteins (_trans_-factors) with specific DNA regions (_cis_-acting regulatory sequences). Intensive efforts over several decades have identified numerous regulatory proteins, transcription factors (TF), whose sequence-specific DNA binding activity is central to transcriptional regulation. Traditionally, DNA binding specificity of many TFs has been experimentally determined primarily with in vitro techniques such as DNase I footprinting and electromobility shift assay (EMSA) (Rooney et al. 1995). Recently, alternative techniques such as expression DNA microarrays, in silico oligonucleotide binding, and phylogenetic footprinting have been adopted to identify DNA targets for TFs (Fickett and Wasserman 2000).
Unfortunately, despite the fact that the binding sites of many TF have been experimentally defined, most TFs bind to short (6–12 base pairs [bp]), degenerate sequence motifs that occur very frequently in the human genome. The binding specificities of these factors can be summarized as position weight matrices (PWM) (Heinemeyer et al. 1998) that are compiled in various databases such as the TRANSFAC database (http://www.biobase.de) (Wingender et al. 2001). Pattern-recognition programs such as MATCH or MatInspector (Quandt et al. 1995) use these libraries of TF-PWMs to identify significant matches in DNA sequences. A major confounding factor in the use of PWMs to identify transcription factor binding sites (TFBSs) is that only a very small fraction of predicted binding sites are functionally significant. Accordingly, the use of PWMs has proved to be a poor resource for sequence-based discovery of biologically relevant regulatory elements (Fickett and Wasserman 2000).
In complex organisms, gene expression results from the cooperative action of many different proteins exerting different effects in time and space. Multiple TFs are simultaneously required to cooperatively activate and modulate eukaryotic gene expression (Berman et al. 2002). One potential avenue for improving the discovery of functional regulatory elements is to identify multiple TFBSs that are specifically clustered together (Wagner 1999). This strategy has been successfully implemented in the analysis of regulatory regions involved in muscle (Wasserman and Fickett 1998) and liver-specific gene expression (Krivan and Wasserman 2001).
An additional powerful strategy that has been shown to counter the large numbers of false positives derived from the analysis of sequences from a single organism is the use of multispecies comparative sequence alignments or phylogenetic footprinting (Gumucio et al. 1996; Hardison et al. 1997b; Duret and Bucher 1997; Levy et al. 2001). Several recent studies have shown that noncoding regulatory sequences tend to be evolutionarily conserved and support the use of comparative genomics as an extremely effective tool for the discovery of biologically active gene regulatory elements (Hardison et al. 1997a; Oeltjen et al. 1997; Hardison et al. 2000; Loots et al. 2000; Wasserman et al. 2000). The computational algorithms developed to perform comparative sequence analysis are based either on local alignments (BLAST [Altschul et al. 1990]; PIPMaker [Schwartz et al. 2000]) or on global alignments (AVID; VISTA [Mayor et al., 2000;]), both of which have proved very efficient in detecting regions of high DNA conservation.
To facilitate the efficient and accurate identification of regulatory sequences in large genomic intervals from complex organisms, we have developed a computational tool, Regulatory VISTA (rVISTA: http://pga.lbl.gov/rvista.html), that enriches for evolutionarily conserved TFBSs. rVISTA uses orthologous sequence analysis and clustering to overcome some of the limitations associated with TFBS predictions of sequences derived from a single organism. Here we introduce the rVISTA program and illustrate its ability to identify functional TFBSs as it dramatically reduces the total number of AP-1, NFAT, and GATA-3 sites predicted in a ∼1-Mb genomic interval of the well-annotated cytokine gene cluster (Hs5q31; Mm11) (Frazer et al. 1997; Wenderfer et al. 2000).
RESULTS
Computational Design of the rVISTA Program
To take advantage of combining sequence motif recognition and multiple sequence alignment of orthologous regions in an unbiased manner, rVISTA analysis proceeds in four major steps: (1) identification of TFBS matches in the individual sequences, (2) identification of globally aligned noncoding TFBSs, (3) calculation of local conservation extending upstream and downstream from each orthologous TFBS, and (4) visualization of individual or clustered noncoding TFBSs (Fig. 1). The program uses available PWMs in the TRANSFAC database and independently locates all TFBS matches in each sequence with the MATCH program. A global alignment generated by the AVID program (http://bio.math.berkeley.edu/avid/) and the corresponding sequence annotations are used to identify aligned TFBS matches in noncoding genomic intervals.
Figure 1.

rVISTA data flow. The user submits a global alignment file (generated by the AVID program) and optional annotation files for the two orthologous sequences. The imported TRANSFAC matrix library and the MATCH program are consequently used to identify all transcription factor binding site (TFBS) matches in each individual sequence and to generate a file with all TFBS matches in the reference sequence (used as baseline for visualization). Next, the global alignment and the sequence annotations provided are used to identify all aligned TFBSs present in the noncoding DNA (in the absence of annotation, the program will identify all aligned sites across the entire alignment). A second file is generated containing aligned noncoding TFBSs. DNA sequence conservation is determined by the hula-hoop module, which identifies TFBSs surrounded by conserved sequences and generates a data table with detailed statistics. The final data processing step includes a user-interactive visualization module. The user customizes the data by choosing which TF sites to visualize (we are giving an example for choosing GATA-3 sites), what TRANSFAC parameters to use for all TF matches (rVISTA default 0.75/0.8), and by selectively clustering individual or combinatorial sites. The user can customize the clustering of any of the three data sets (all matches in the reference sequence are depicted as blue tick marks, aligned TFBS matches are in red, and conserved TFBS matches are in green).
An aligned TFBS represents a region in the global alignment that corresponds to identical TFBS matches in each orthologous sequence. Orthologous regions correspond to similar DNA sequences from different species that arose from a common ancestral gene during speciation and are likely to be involved in similar biological functions. Because the global alignment forces two closely related sequences to generate the best possible pairwise alignment by introducing gaps, an aligned TFBS site can be present in a region of poor DNA conservation that is below 80% ID. To identify TFBSs present in regions of high DNA conservation, the “hula hoop” component of the algorithm calculates DNA conservation for each aligned TFBS as percent identity (% ID) over a dynamically shifting window of 21 bp that centers on a nucleotide inside the TFBS with the maximum % ID. This process identifies TFBSs located at the edges of highly conserved sequences that would falsely fall below the established conservation criteria threshold if the DNA conservation was determined by a static DNA window perfectly centered on the TFBS alignment.
By use of the same principle, rVISTA calculates the maximum DNA conservation over larger DNA segments (up to 201 bp) facilitating the identification of sites present in larger, highly conserved regions. The rVISTA algorithm generates two types of outputs: (1) a static data table with detailed statistics for all aligned TFBSs and (2) a dynamic web-interactive module that allows the user to customize the data for unfiltered, aligned, or conserved TFBS sites and graphically visualize them as colored tick marks. Visualized conserved binding sites fit the criteria of ≥80% ID over a 21-bp region.
Combinatorial Analysis of TFBSs with Multiclustering
Detailed molecular analyses addressing the architecture of complex regulatory regions in higher eukaryotes have established that the majority of transcriptional control elements such as enhancers and repressors represent a conglomerate of multiple TFBSs that act in concordance to directly modulate the expression patterns of the linked genes (Pilpel et al. 2001). In addition, it has been observed that regulatory elements involved in similar physiological functions, such as the enhancement of liver-specific genes (Krivan and Wasserman 2001), are associated with distinct patterns of coordinate TF binding. These regulatory regions are frequently present in clusters of two or more repeated sites for the same TF or in combinatorial clusters of two or more adjacent sites belonging to unique regulatory proteins that act together to modulate gene expression (Fickett and Wasserman 2000; Pilpel et al. 2001; Berman et al. 2002).
To analyze combinations of multiple TFBSs and identify TF binding patterns that control gene expression in novel sequences, rVISTA calculates the distance between all neighboring TFBSs and allows the user to perform customized clustering of individual or multiple unique transcription factors. One clustering module allows the user to selectively cluster two or more sites of the same TF present in regions of user-defined lengths (Fig 2A, B), facilitating the identification of evolutionarily conserved elements that harbor multiple clusters of various unrelated TFBSs. A second clustering module allows the user to identify groups of multiple TFBSs present in DNA segments of user-specified length (Fig 2C).
Figure 2.

Visualizing rVISTA cluster analysis for a 25-kb region across the GM-CSF and IL-3 genomic interval. (A) Ikaros-2 TFBS clusters (two sites over 60-bp region). Ikaros-2 matches fitting the clustering criteria for the human sequence alone are depicted in blue, aligned clusters in red, and conserved clusters in green. (B) Multiclustering of individual sites can be performed by independently choosing the clustering criteria for each TF. AP-1 (blue), NFAT (red), and GATA-3 (green) clusters (two sites over 100 bp) of conserved TFBSs are illustrated. (C) Combinatorial clustering of TFBS. By use of the clustering criteria of 1 NFAT and 1 AP-1 across a 60-bp DNA fragment, the rVISTA program identifies all the AP-1 (blue) and NFAT (red) paired and displays them as tickmarks. This clustering module can be applied to the three data sets allowing the visualization of clusters in the reference sequence, among the aligned sites, and the conserved sites.
Collection of Experimental Data and Validation of rVISTA
To evaluate the biological significance of TFBS data generated by the rVISTA algorithm, we analyzed ∼1 Mb of a well-annotated cytokine gene cluster (Hs5q31; Mm11) (IL-3; IL-4; IL-5; IL-13; IRF-1; GM-CSF) (Frazer et al. 1997; Wenderfer et al. 2000) plus the intensively characterized cytokine 2 (IL-2) promoter region (Hs4q26; Mm3) (Rooney et al. 1995). Cytokines are of particular biomedical importance because they augment the growth and differentiation of T helper cell subsets and have been directly implicated in having a major role in determining susceptibilities to asthma phenotypes and inflammatory disorders (Lacy et al. 2000; O'Garra and Arai 2000). As such, much interest has focused on the regulatory mechanisms by which naive helper CD4+ T cells establish their cytokine repertoires, events that are predominantly regulated at the transcriptional level.
Because of the vast interest in understanding the regulation of cytokines, we focused our analysis on transcription factors known to transcriptionally activate these genes. One of the best known examples of cooperative binding is the NFAT : AP-1 TF complex that has been described for genes involved in various immune responses. NFAT and AP-1 synergistically form stable complexes with DNA sequences that contain composite elements of adjacent NFAT and AP-1 TFBSs to induce the expression of genes (Macian et al. 2001). We have compiled a representative collection of AP-1 and NFAT experimentally defined TFBSs (Table 1) from the published data on this ∼1-Mb interval and used it to examine the ability of rVISTA to identify true TFBSs. By analyzing ∼925 kb noncoding human sequence independent of the mouse sequence, the MATCH program predicted 23,457 AP-1 and 14,900 NFAT sites with the PWMs available in the TRANSFAC database for these transcription factors (parameters: 0.75/0.8). A comparable number of sites were independently predicted for the orthologous mouse sequence. Among the large number of predicted AP-1 and NFAT sites for the human sequence were also included 17 of the 19 functional AP-1 sites and 19 of the 21 functional NFAT sites (Fig. 3A). The omitted AP-1 and NFAT functional sites failed to meet the TRANSFAC default parameters.
Table 1.
Localization of Functionally Characterized Binding Sites to Conserved Blocks
| Location | Ref | Element sequence | NFAT (%ID) | AP-1 (%ID) | ||
|---|---|---|---|---|---|---|
| IL-2 | E1 | 280 | (15) | AGGAAAATTTGTTTCATA | 90.48 | 90.48 |
| E2 | 160 | (15) | AGAAATTCCAGAGAGTCA | 90.48 | 95.24 | |
| E3 | 135 | (15) | AGGAAAAACAAAGGTAAT | 95.24 | 80.95 | |
| E4 | 90 | (15) | TTGAAAATATGTGTAATA | 95.25 | 100 | |
| E5 | 45 | (15) | TGGAAAAAT | 95.25 | n/a | |
| IL-3 | E6 | 306 | (11) | TGAGCTGAGTCAGGCTTCCCCTT | 76.2a | 71.43 |
| IL-4 | P0 | −106 | (1, 2) | GTAAACTCATTTTCCCTTGGTTTC | 95.25 | 100 |
| P1 | −121 | (1, 2) | GTAATAAAATTTTCCAATGTAAAC | 95.25 | 100 | |
| P2 | −124 | (1, 2) | ACAGGTAAATTTTCCTGTGAAATC | 95.25 | 100 | |
| P3 | −238 | (1, 2, 3) | GGTGTTTCATTTTCCAATTTGTCT | 95.25 | 95.24a | |
| P4 | −287 | (1, 2) | TATGGTGTAATTTCCTATGCTTGA | 100 | 100a | |
| P5 | −406 | (16) | GCAGTCCTCCTGGGGAAAGATAGAGTAATATCA | 95.25 | 95.24 | |
| IL-5 | E7 | 163 | (4, 6, 7) | GCATTGGAAACATTTAGTTTCACGAT | 80.95 | 80.95 |
| E8 | 104 | (4, 5) | GAAATTATTCATTTCCTCAAAG | 90.48 | 95.24 | |
| IL-13 | E9 | 135 | (12) | CTGGATTTTCCA | 85.71 | n/a |
| E10 | 154 | (13) | CATGAGAAATCAAATCTTTCCTTTA | 90.48 | 80.95 | |
| GMCSF | E11 | −67 | (9, 10) | CACCATTAATCATTTCCTCTG | 80.95 | 100 |
| E12 | −101 | (14) | AGGAGATTCCACAGTTCAGGTAGTTCCCCCGCCTCC | 95.1 | 90.48 | |
| E13 | −163 | (13) | CCTAGGGAAAAGGCTCACCGT | 80.95 | 90.48 | |
| E14 | −2.8kb | (8) | GCCCTGATGTCATCTTTCCATGA | 90.48 | 90.48 | |
| E15 | −2.9kb | (8) | CCATCGGAGCCCCTGAGTCAGCAT | 85.71a | 85.71 | |
| GATA-3 (%ID) | Distance (bp) | |||||
| IL-4 | E16 | −87 | (17) | ATTACACCAGATTGTCAGTTA- | 95.25 | 20 |
| TTCTGGGCCAATCAGCACC | 100 | |||||
| IL-5 | E17 | −75 | (18) | CTCTATCTGATTGTTA | 95.24 | 5 |
| 95.24 |
Figure 3.

rVISTA analysis algorithm identifies experimentally characterized TFBS. (A) Two functionally characterized NFAT/AP-1 clusters indicated by black vertical arrows ([two sites/60 bp] [Table 1: E7 and E8]) are identified by rVISTA and are the only two clusters of conserved TFBSs present in the IL-5 promoter. The VISTA alignment highlights exons in blue, UTRs in yellow, and conserved noncoding in red. (B) A GATA-3 pair in the IL-5 promoter indicated by black vertical arrow is highly conserved and represents the only functional GATA-3 cluster ([2 GATA/60 bp] [Table 1]) in the proximal promoter (500 bp upstream of the 5′UTR) of this cytokine.
Subjecting the orthologous human and mouse sequences to rVISTA analysis reduced the total number of predicted AP-1 and NFAT sites by >95%, identifying 1114 conserved AP-1 and 734 conserved NFAT sites. rVISTA also identified 16 of the 19 AP-1 and 19 of the 21 functionally characterized NFAT sites. Whereas only 4.5% of the total NFAT and AP-1 predicted sites for the human sequence were conserved in the orthologous mouse sequence, in sharp contrast, 88% of the experimentally defined TFBSs were present in highly conserved DNA blocks. This data establishes a strong correlation between the presence of TFBSs in regions of high DNA conservation and biological function (Table 2). However, only a small percentage of the total identified conserved sites correspond to functional sites that have been experimentally verified. Some of the other conserved TFBSs may also be functional but remain to be experimentally confirmed.
Table 2.
Enrichment of Functional TFBS Using Clustering and DNA Conservation
| AP-1 | NFAT | AP-1/NFAT clusters | GATA-3 | GATA clusters | |
|---|---|---|---|---|---|
| Total (∼1 Mb) | 23,079 | 14,900 | 12,015 | 21,362 | 19,986 |
| Experimental | 17/19a (89%) | 19/21a (90%) | 17/19a (89%) | 4/4 (100%) | 4/4 (100%) |
| Aligned | 2598 (11%) | 1617 (11%) | 787 (6.5%) | 2445 (11.4%) | 1289 (6.4%) |
| Experimental | 17 (89%) | 19 (90%) | 17 (89%) | 4 (100%) | 4 (100%) |
| Conserved | 1045 (4.5%) | 717 (4.8%) | 324 (2.7%) | 946 (4.4%) | 459 (2.3%) |
| Experimental | 16 (84%) | 19 (90%) | 16 (84%) | 4 (100%) | 4 (100%) |
Cytokine Promoter Analysis to Assess rVISTA Predictions
In addition to AP-1 and NFAT, the GATA-3 TF has also been implicated in the transcriptional control of the large number of Th2-specific cytokines present in this interval (IL-4, IL-13, IL-5, GMCSF, IL-3) (Ranganath and Murphy 2001). GATA-3's direct involvement in gene activation has been extensively shown for the IL-4 and IL-5 promoters (Zheng and Flavell 1997; Lee et al. 1998) and has been postulated for the activation (or repression) of all the cytokine genes present in this interval (Zheng and Flavell 1997). On the basis of GATA-3's predicted binding to upstream regions of cytokine genes, we hypothesized that there should be an increased distribution of GATA-3 sites across the six cytokine promoters compared with the promoters of the 16 non-Th1/Th2 expressing genes in this region.
To test this hypothesis, we determined the GATA-3 site distribution for the 2-kb promoter region of all 22 annotated genes in this interval. Because of the highly degenerate nature of the GATA binding profile that is recognized by all members of the GATA-family (Merika and Orkin 1993), TRANFAC predicted an average of 50 GATA-3 sites per promoter that were evenly distributed across both cytokine and noncytokine gene promoters. In contrast, the rVISTA analysis dramatically reduced the total number of GATA-3 sites per promoter and, most importantly, resulted in an increased representation of GATA-3 sites in cytokine promoters (Fig. 4A). On average, rVISTA detected eight conserved GATA-3 sites per cytokine promoter while yielding only two conserved GATA-3 sites per noncytokine promoter. In addition, the experimentally characterized GATA-3 sites in both the IL-4 and IL-5 promoters (Zheng and Flavell 1997; Lee et al. 1998) were among the highly conserved sites identified by rVISTA (Fig. 3B).
Figure 4.

Distribution of conserved GATA-3 binding sites across the 22 promoter regions (2 kb upstream of 5′UTR) of all annotated genes from the 1-Mb cytokine gene cluster (Hs5q31; Mm11). Cytokine genes are labeled by arrows, gray bars indicate observed GATA-3 sites, and open bars represent predicted GATA-3 sites as a result of random distribution. Random distribution was estimated on the basis of the frequency of GATA-3 sites across the 1-Mb human sequence and the DNA conservation of each promoter. (A), conserved individual GATA-3 sites. (B), conserved GATA-3 present in clusters (two or more conserved sites enclosed in a 60-bp DNA fragment).
Because functional GATA-3 sites are present in pairs (Table 1), we next analyzed the distribution of GATA-3 sites clustered (two or more sites present within 60-bp regions). By clustering the conserved GATA-3 sites, we observed a further enrichment of GATA-3 sites in the cytokine promoters. In each cytokine promoter there were an average of six GATA-3 clustered sites, whereas no such clustered sites were noted in the promoters of noncytokine genes. These GATA-3 clustered sites, although not yet experimentally verified, were exclusively found in the promoters of genes predicted to be GATA-3 responsive. rVISTA's ability to recognize what are likely true TFBSs in the promoters of cytokine genes supports the hypothesis that GATA-3 plays an important role in the regulation of all the cytokine genes present on human 5q31 (Fig. 4B).
DISCUSSION
Annotating the noncoding portion of the human genome remains among the greatest challenges of the post-sequencing era. Clues for identifying sequences involved in the complex regulatory networks of eukaryotic genes are provided by the presence of TFBS motifs, the clustering of such binding site motifs, and the conservation of these sites between species. rVISTA takes advantage of all these established strategies to enhance the detection of functional transcriptional regulatory sequences controlling gene expression through its ability to identify evolutionarily conserved and clustered TFBSs.
By performing an unbiased analysis of the distribution of NFAT and AP-1 binding sites across ∼1 Mb of human/mouse orthologous region, we were able to show that although rVISTA reduces more than 95% of the predicted TFBSs derived from the sequence analysis of a single organism, it still recognizes 88% of the biologically characterized AP-1 and NFAT in this region. The PWM compiled from experimentally determined TFBSs available in the TRANSFAC database pose a major limitation in the rVISTA analysis, because the computational approach described relies on the available DNA binding profiles of known transcription factors (Table 1). Of the total 19 AP-1 and 21 NFAT experimentally described sites, 17 AP-1 and 19 NFAT sites had TRANSFAC values greater than 0.75/ 0.8, two AP-1 and one NFAT site had values of 0.7/0.7, and one NFAT site had a value of 0.6/0.7 (Table 1). Of the 36 experimentally defined AP-1 and NFAT sites recognized by the PWMs available in the TRANSFAC database (with the 0.75/0.8 parameters), only one aligned AP-1 site (71%) was below our established conservation threshold (≥80%) and failed to be identified by the rVISTA program. Our data indicates that the rVISTA program dramatically eliminates a large number of false-positive TFBSs while it enriches for functional TFBSs.
Although the identification of conserved TFBSs on a small genomic interval can be achieved by phylogenetic footprinting (Hardison et al. 1997; Krivan and Wasserman 2001), a great strength of the rVISTA algorithm is its ability to efficiently analyze large genomic intervals and potentially whole genomes. The clustering modules and the user-defined customization of visualized sites makes this a further useful tool for the investigation of TFBSs. Through the use of a global alignment, rVISTA takes into account the linear structure of sequence conservation across a large DNA segment. By allowing small gaps and DNA shifts in the aligned TFBSs, we are maximizing the identification of functional TFBSs that have diverged slightly yet are present in highly conserved regions; ∼25–35% of all aligned and ∼15–20% of all conserved TFBSs identified have one gap in their alignment (data not shown). In addition, ∼25% of the aligned and ∼18% of the conserved NFAT and AP-1 sites have shifts (1–6 bp) across their alignments (data not shown). The presence of gaps in the alignments of experimentally characterized TFBSs further supports the use of a global alignment for rVISTA analysis and the need for loose parameters for the identification of aligned sites, as well as stringent percent identity criteria for detecting highly conserved TFBSs.
Properties related to protein–protein interaction and chromatin structure, as well as clusters of multiple unique sites that have been reshuffled in one of the human or the mouse genome and have lost their positional linearity, are not addressed. Also, clustering does not take into account the spacing between sites but rather counts the number of adjacent sites of a given TF spanning DNA segments of specified length. Although TFBS clustering has been suggested for identifying regulatory sequence, no data to date has proved the effectiveness of this approach (Wagner 1997; Wagner 1999). Our clustering analysis results indicate that this approach has the potential to efficiently prioritize functionally relevant noncoding sequences. rVISTA represents the only publicly available program that allows the user to identify customized clusters of multiple TFBSs in large genomic intervals.
Our analysis of the AP-1 and NFAT TFBS in the cytokine gene cluster illustrates the effectiveness of the rVISTA algorithm in eliminating many false positives while retaining the majority of experimentally verified sites. In our analysis of GATA-3 sites in the putative promoters of the 23 genes from human 5q31 we were able to prioritize, exclusively on the basis of sequence analysis, a limited number of GATA-3 sites with a high likelihood of being functional that can be used for further biological investigation. With the increasing availability of sequence data for multiple organisms, rVISTA's ability to use comparative data and clustering options in a user-friendly manner makes it particularly suited to assist investigators focused on biologically defined genomic intervals, as well as those interested in performing whole genome analyses to identify functional TFBSs and regulatory elements.
METHODS
rVISTA is implemented as a publicly available web-based tool (http://pga.lbl.gov/rvista.html) that requires a sequence alignment file and optional gene annotation files as user input. The rVISTA analysis tool consists of four major modules: (1) motif recognition, (2) identification of aligned TFBSs, (3) conservation analysis, and (4) visualization of TFBSs. The system units are implemented with the C++ computer language equipped with Web user-interactive interface written in Perl. For the conservation analysis rVISTA uses an alignment file in the AVID format obtained using the AVID program from the AVID (http://bio.math.berkeley.edu/avid/) or VISTA (http://www-gsd.lbl.gov/VISTA/) servers. The following methodological scheme was implemented as a core for the rVISTA tool. Initially, the user chooses a set of TFs and the PWM parameters to be used. Next, rVISTA extracts all TFBS coordinates independently in the two orthologous sequences before the analysis of the alignment. The locally installed TRANFAC 5.2 library and the MATCH program from Biobase, Inc. (http://www.biobase.de) are used at this step.
Subsequently, the global alignment is scanned for pairs of neighboring human and mouse TFBSs that are aligned and match identically in both sequences. An aligned TFBS is allowed to have a maximum 6-bp shift (majority of TF matrices have core sequences of 4–6 bp) in the alignment of the TFBS core and a single gap present across the entire local alignment of the TFBS. The conservation analysis module contains one major unit, the hula hoop, which is designed to analyze the local DNA conservation of each aligned TFBS to eliminate aligned sites present in regions of weak DNA conservation. A fixed-size DNA window (21 bp) is being shifted through all the positions of an aligned TFBS, whereas the entire sequence spanning the TFBS is permanently enclosed by the shifting DNA window. The percent identity is calculated at every base pair across the aligned TFBS and extending 10 nucleotides upstream and downstream from it, similar to a hula hoop. The position with the highest percent identity is used to assign the conservation level of that particular TFBS. This process allows the identification of the maximum percent identity for the local alignment of a conserved TFBS. The program calculates % ID for each binding site with dynamically shifting windows of up to 200 bp. These data are provided in a table format and allow the identification of TFBSs present in large regions of high conservation.
The visualization module is a web-based tool that post-processes the rVISTA output. One unit of the program eliminates redundancy. Overlapping TFBS matches (within 3 bp from each other) belonging to the same family of regulatory proteins are considered to be an identical match. A second unit of the program measures the distance between adjacent matches belonging to the same TF family and allows the user to selectively cluster TFBSs into groups of x number of sites over y base pair length. The clustering parameters are user-defined and are assigned independently for every family of TF. Any combination of unfiltered, aligned, or conserved TFBSs with customized clustering for the selected set of TFs are interactively visualized as a ‘tick-plot’ track overlaid on the conservation VISTA-type track and the gene annotation track. All conserved binding sites displayed fit the criteria of ≥80% over a 21-bp alignment.
Evaluation of True TF Enrichment with GATA Sites in Promoters
To quantitatively measure the enrichment of GATA predictions for functional sites, we performed a statistical simulation for the expected number of conserved sites and compared it with the observed number of conserved sites (≥80% ID; 21 bp) present in promoter regions (2 kb upstream of the 5′UTR). Redundant GATA sites (defined to be overlapping sites) were excluded before the analysis. GATA site clustering was performed for two or more neighboring GATA sites present over a ≤60-bp region. The upper bound for the expected number of conserved GATA sites in a promoter under consideration, i, was calculated as follows:
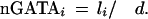
(d = 41 bp; d is the average distance between two nonredundant GATA sites in the human sequence; 1/d is the probability of a given nucleotide to be at the starting position for a GATA site; and li is the number of nucleotides inside a promoter region conserved ≥80%.)
To obtain a more accurate value for the number of expected conserved and aligned GATA sites, we also considered the fact that a human GATA site present in a region of high DNA conservation (≥80%) will not always have an aligned match in the mouse sequence. Any given conserved GATA site could either have no corresponding aligned binding site in the second sequence or the binding site alignment could exceed the gap and shift requirements. We approached this problem by introducing a scaling parameter σ. (σ is the probability of a conserved site to be aligned and is approximated to be a constant for all the promoter regions.) The estimation for the σ value was calculated on the basis of the number of conserved sites that were also aligned in noncytokine promoters. We estimated the expected distribution of conserved GATA sites across the promoter sequences as follows:
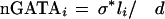
Thus, we were able to compare the distribution of observed conserved and aligned GATA sites in cytokine promoters with the estimated distribution of such sites extrapolated from noncytokine promoters (Fig. 4A).
Similarly, we estimated the number of expected conserved, aligned, and clustered GATA sites. The length of the conserved and clustered segment of each promoter was obtained by checking all the possible paired coordinates in the conserved regions of all promoters and obtaining the ratio of sites closer than 60 bp but greater than 4 bp apart from each other. The σ values for conserved and for conserved and clustered sites were found to be 0.23 and 0.19, respectively. Close σ values for these two types of sites (conserved; conserved and clustered) indicate that the probability of a conserved site to be aligned is independent from the probability of the same site to be clustered (Fig. 4B).
WEB SITE REFERENCES
http://bio.math.berkeley.edu/avid/; AVID program.
http://pga.lbl.gov/rvista.html; rVISTA program.
http://www.biobase.de; TRANSFAC database.
http://www.biobase.de; MATCH program, Biobase, Inc.
Acknowledgments
We are grateful to Moshe Malkin, Jody Schwartz, Alexander Fabrikant, and Michael Brudno for technical assistance. We thank the Rubin Laboratory for insightful comments on the manuscript. This work was supported by the Program for Genomic Applications (PGAs) funded by the National Heart, Lung, and Blood Institute (NHLBI/NIH); G.G. Loots was supported by the Department of Energy Alexander Hollaender Fellowship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL ggloots@lbl.gov; ildubchak@lbl.gov; FAX (510) 486-6746.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.225502\. Article published online before print in April 2002.
REFERENCES
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Berman BP, Nibu Y, Pfeiffer BD, Tomancak P, Celniker SE, Levine M, Rubin GM, Eisen MB. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. PNAS. 2002;99:757–762. doi: 10.1073/pnas.231608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke TF, Casolaro V, Georas SN. Characterization of P5, a novel NFAT/AP-1 site in the human IL-4 promoter. Biochem Biophys Res Commun. 2000;270:1016–1023. doi: 10.1006/bbrc.2000.2508. [DOI] [PubMed] [Google Scholar]
- Cakouros D, Cockerill PN, Bert AG, Mital R, Roberts DC, Shannon MF. A NF-kappa B/Sp1 region is essential for chromatin remodeling and correct transcription of a human granulocyte-macrophage colony-stimulating factor transgene. J Immunol. 2001;167:302–310. doi: 10.4049/jimmunol.167.1.302. [DOI] [PubMed] [Google Scholar]
- Cockerill GW, Bert AG, Ryan GR, Gamble JR, Vadas MA, Cockerill PN. Regulation of granulocyte-macrophage colony-stimulating factor and E-selectin expression in endothelial cells by cyclosporin A and the T-cell transcription factor NFAT. Blood. 1995;86:2689–2698. [PubMed] [Google Scholar]
- De Boer ML, Mordvinov VA, Thomas MA, Sanderson CJ. Role of nuclear factor of activated T cells (NFAT) in the expression of cytokine-5 and other cytokines involved in the regulation of hemopoietic cells. Int J Biochem Cell Biol. 1999;31:1221–1236. doi: 10.1016/s1357-2725(99)00069-2. [DOI] [PubMed] [Google Scholar]
- Duret L, Bucher P. Searching for regulatory elements in human noncoding sequences. Curr Opin Struct Biol. 1997;7:399–406. doi: 10.1016/s0959-440x(97)80058-9. [DOI] [PubMed] [Google Scholar]
- Ficket JW, Wasserman WW. Discovery and modeling of transcriptional regulatory regions. Curr Opin Biotechnol. 2000;11:19–24. doi: 10.1016/s0958-1669(99)00049-x. [DOI] [PubMed] [Google Scholar]
- Frazer KA, Ueda Y, Zhu Y, Gifford VR, Garofalo MR, Mohandas N, Martin CH, Palazzolo MJ, Cheng JF, Rubin EM. Computational and biological analysis of 680 kb of DNA sequence from the human 5q31 cytokine gene cluster region. Genome Res. 1997;7:495–512. doi: 10.1101/gr.7.5.495. [DOI] [PubMed] [Google Scholar]
- Gottschalk LR, Giannola DM, Emerson SG. Molecular regulation of the human IL-3 gene: Inducible T cell-restricted expression requires intact AP-1 and Elf-1 nuclear protein binding sites. J Exp Med. 1993;178:1681–1692. doi: 10.1084/jem.178.5.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumucio DL, Shelton DA, Zhu W, Millinoff D, Gray T, Bock JH, Slightom JL, Goodman M. Evolutionary strategies for the elucidation of cis and trans factors that regulate the developmental switching programs of the beta-like globin genes. Mol Phylogenet Evol. 1996;5:18–32. doi: 10.1006/mpev.1996.0004. [DOI] [PubMed] [Google Scholar]
- Hardison R, Slightom JL, Gumucio DL, Goodman M, Stojanovic N, Miller W. Locus control regions of mammalian beta-globin gene clusters: Combining phylogenetic analyses and experimental results to gain functional insights. Gene. 1997b;205(1–2):73–94. doi: 10.1016/s0378-1119(97)00474-5. ; 12. [DOI] [PubMed] [Google Scholar]
- Hardison RC, Oeltjen J, Miller W. Long human-mouse sequence alignments reveal novel regulatory elements: A reason to sequence the mouse genome. Genome Res. 1997a;7:959–966. doi: 10.1101/gr.7.10.959. [DOI] [PubMed] [Google Scholar]
- Hardison RC. Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet. 2000;16:369–372. doi: 10.1016/s0168-9525(00)02081-3. [DOI] [PubMed] [Google Scholar]
- Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, et al. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26:362–367. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins F, Cockerill PN, Bohmann D, Shannon MF. Multiple signals are required for function of the human granulocyte-macrophage colony-stimulating factor gene promoter in T cells. J Immunol. 1995;155:1240–1251. [PubMed] [Google Scholar]
- Kel A, Kel-Margoulis O, Babenko V, Wingender E. Recognition of NFATp/AP-1 composite elements within genes induced upon the activation of immune cells. J Mol Biol. 1999;288:353–376. doi: 10.1006/jmbi.1999.2684. [DOI] [PubMed] [Google Scholar]
- Krivan W, Wasserman WW. A predictive model for regulatory sequences directing liver-specific transcription. Genome Res. 2001;11:1559–1566. doi: 10.1101/gr.180601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy DA, Wang ZE, Symula DJ, McArthur CJ, Rubin EM, Frazer KA, Locksley RM. Faithful expression of the human 5q31 cytokine cluster in transgenic mice. J Immunol. 2000;164:4569–4574. doi: 10.4049/jimmunol.164.9.4569. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Masuda ES, Arai N, Arai K, Yokota T. Definition of cis-regulatory elements of the mouse cytokine-5 gene promoter. Involvement of nuclear factor of activated T cell-related factors in cytokine-5 expression. J Biol Chem. 1995;270:17541–17550. doi: 10.1074/jbc.270.29.17541. [DOI] [PubMed] [Google Scholar]
- Lee HJ, O'Garra A, Arai K, Arai N. Characterization of cis-regulatory elements and nuclear factors conferring Th2-specific expression of the IL-5 gene: A role for a GATA-binding protein. J Immunol. 1998;160:2343–2352. [PubMed] [Google Scholar]
- Levy S, Hannenhalli S, Workman C. Enrichment of regulatory signals in conserved non-coding genomic sequence. Bioinformatics. 2001;17:871–877. doi: 10.1093/bioinformatics/17.10.871. [DOI] [PubMed] [Google Scholar]
- Li-Weber M, Davydov IV, Krafft H, Krammer PH. The role of NF-Y and IRF-2 in the regulation of human IL-4 gene expression. J Immunol. 1994;153:4122–4133. [PubMed] [Google Scholar]
- Loots GG, Locksley RM, Blankespoor CM, Wang ZE, Miller W, Rubin EM, Frazer KA. Identification of a coordinate regulator of cytokines 4, 13, and 5 by cross-species sequence comparisons. Science. 2000;288:136–140. doi: 10.1126/science.288.5463.136. [DOI] [PubMed] [Google Scholar]
- Macian F, Lopez-Rodriguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476–2489. doi: 10.1038/sj.onc.1204386. [DOI] [PubMed] [Google Scholar]
- Masuda ES, Tokumitsu H, Tsuboi A, Shlomai J, Hung P, Arai K, Arai N. The granulocyte-macrophage colony-stimulating factor promoter cis-acting element CLE0 mediates induction signals in T cells and is recognized by factors related to AP1 and NFAT. Mol Cell Biol. 1993;13:7399–7407. doi: 10.1128/mcb.13.12.7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, Pachter LS, Dubchak I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics. 2000;16:1046–1047. doi: 10.1093/bioinformatics/16.11.1046. [DOI] [PubMed] [Google Scholar]
- Merika M, Orkin SH. DNA-binding specificity of GATA family transcription factors. Mol Cell Biol. 1993;13:3999–4010. doi: 10.1128/mcb.13.7.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeltjen JC, Malley TM, Muzny DM, Miller W, Gibbs RA, Belmont JW. Large-scale comparative sequence analysis of the human and murine Bruton's tyrosine kinase loci reveals conserved regulatory domains. Genome Res. 1997;7:315–329. doi: 10.1101/gr.7.4.315. [DOI] [PubMed] [Google Scholar]
- O'Garra A, Arai N. The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends Cell Biol. 2000;10:542–550. doi: 10.1016/s0962-8924(00)01856-0. [DOI] [PubMed] [Google Scholar]
- Pilpel Y, Sudarsanam P, Church GM. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat Genet. 2001;29:153–159. doi: 10.1038/ng724. [DOI] [PubMed] [Google Scholar]
- Prieschl EE, Gouilleux-Gruart V, Walker C, Harrer NE, Baumruker T. A nuclear factor of activated T cell-like transcription factor in mast cells is involved in IL-5 gene regulation after IgE plus antigen stimulation. J Immunol. 1995;154:6112–6119. [PubMed] [Google Scholar]
- Quandt K, Frech K, Karas H, Wingender E, Werner T. MatInd and MatInspector: New fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganath S, Murphy KM. Structure and specificity of GATA proteins in Th2 development. Mol Cell Biol. 2001;21:2716–2725. doi: 10.1128/MCB.21.8.2716-2725.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JW, Sun YL, Glimcher LH, Hoey T. Novel NFAT sites that mediate activation of the cytokine-2 promoter in response to T-cell receptor stimulation. Mol Cell Biol. 1995;15:6299–6310. doi: 10.1128/mcb.15.11.6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Zhang Z, Frazer KA, Smit A, Riemer C, Bouck J, Gibbs R, Hardison R, Miller W. PipMaker—a web server for aligning two genomic DNA sequences. Genome Res. 2000;10:577–586. doi: 10.1101/gr.10.4.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranick KS, Zambas DN, Uss AS, Egan RW, Billah MM, Umland SP. Identification of transcription factor binding sites important in the regulation of the human cytokine-5 gene. J Biol Chem. 1997;272:16453–16465. doi: 10.1074/jbc.272.26.16453. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Gold JS, Murphy TL, Murphy KM. Identification of cis-acting regulatory elements controlling cytokine-4 gene expression in T cells: Roles for NF-Y and NF-ATc. Mol Cell Biol. 1993;13:4793–4805. doi: 10.1128/mcb.13.8.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto N, Koyano-Nakagawa N, Arai N, Arai K, Yokota T. Four P-like elements are required for optimal transcription of the mouse IL-4 gene: Involvement of a distinct set of nuclear factor of activated T cells and activator protein-1 family proteins. Int Immunol. 1997;9:1329–1338. doi: 10.1093/intimm/9.9.1329. [DOI] [PubMed] [Google Scholar]
- Thomas MA, Mordvinov VA, Sanderson CJ. The activity of the human cytokine-5 conserved lymphokine element 0 is regulated by octamer factors in human cells. Eur J Biochem. 1999;265:300–307. doi: 10.1046/j.1432-1327.1999.00732.x. [DOI] [PubMed] [Google Scholar]
- Wagner A. A computational genomics approach to the identification of gene networks. Nucleic Acids Res. 1997;25:3594–3604. doi: 10.1093/nar/25.18.3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. Genes regulated cooperatively by one or more transcription factors and their identification in whole eukaryotic genomes. Bioinformatics. 1999;10:776–784. doi: 10.1093/bioinformatics/15.10.776. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Fickett JW. Identification of regulatory regions which confer muscle-specific gene expression. J Mol Biol. 1998;278:167–181. doi: 10.1006/jmbi.1998.1700. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Palumbo M, Thompson W, Fickett JW, Lawrence CE. Human-mouse genome comparisons to locate regulatory sites. Nat Genet. 2000;2:225–228. doi: 10.1038/79965. [DOI] [PubMed] [Google Scholar]
- Wenderfer SE, Slack JP, McCluskey TS, Monaco JJ. Identification of 40 genes on a 1-Mb contig around the IL-4 cytokine family gene cluster on mouse chromosome 11. Genomics. 2000;63:354–373. doi: 10.1006/geno.1999.6100. [DOI] [PubMed] [Google Scholar]
- Wingender E, Chen X, Fricke E, Geffers R, Hehl R, Liebich I, Krull M, Matys V, Michael H, Ohnhauser R, et al. The TRANSFAC system on gene expression regulation. Nucleic Acids Res. 2001;29:281–283. doi: 10.1093/nar/29.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W-P, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for TH2 cytokine gene expression in CD4 T Cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]