Tracking Single Kinesin Molecules in the Cytoplasm of Mammalian Cells (original) (raw)
Abstract
Understanding dynamic cellular processes requires precise knowledge of the distribution, transport, and interactions of individual molecules in living cells. Despite recent progress in in vivo imaging, it has not been possible to express and directly track single molecules in the cytoplasm of live cells. Here, we overcome these limitations by combining fluorescent protein-labeling with high resolution total internal reflection fluorescence microcopy, using the molecular motor Kinesin-1 as model system. First, we engineered a three-tandem monomeric Citrine tag for genetic labeling of individual molecules and expressed this motor in COS cells. Detailed analysis of the quantized photobleaching behavior of individual fluorescent spots demonstrates that we are indeed detecting single proteins in the cytoplasm of live cells. Tracking the movement of individual cytoplasmic molecules reveals that individual Kinesin-1 motors in vivo move with an average speed of 0.78 ± 0.11 _μ_m/s and display an average run length of 1.17 ± 0.38 _μ_m, which agrees well with in vitro measurements. Thus, Kinesin-1's speed and processivity are not upregulated or hindered by macromolecular crowding. Second, we demonstrate that standard deviation maps of the fluorescence intensity computed from single molecule image sequences can be used to reveal important physiological information about infrequent cellular events in the noisy fluorescence background of live cells. Finally, we show that tandem fluorescent protein tags enable single-molecule, in vitro analyses of extracted, mammalian-expressed proteins. Thus, by combining direct genetic labeling and single molecule imaging in vivo, our work establishes an important new biophysical method for observing single molecules expressed and localized in the mammalian cytoplasm.
INTRODUCTION
During recent years, new quantitative and mechanistic insights into biomolecule function have frequently been derived from single-molecule, in vitro assays using purified molecules. While the benefit to such analysis is to avoid many of the uncertainties associated with ensemble-averaging experiments, in vitro approaches eliminate many physiologically relevant components of the system such as interactions with other molecules, localization of molecules or macromolecular complexes to cellular subdomains, and specific physiological conditions. In addition, many processes in biology (including gene expression, cell signaling, and cellular trafficking) are controlled by a single or very few protein molecules. Consequently, a major objective of modern life sciences research is the development of single-molecule, in vivo imaging techniques and assays to study the movements, interactions, and biochemical activity of biomolecules in intact cells (see (1,2)).
Current live cell, single-molecule imaging is most readily possible with events occurring on the plasma membranes (3–6). Analysis of cytoplasmic events has been limited to specialized labeling techniques (for example (7–9)) or bright semiconductor quantum dots (10–13) to overcome cellular autofluorescence. Harnessing the full potential of in vivo, single-molecule imaging will, however, require the ability to follow any individual molecule in the cytoplasm by direct, fluorescent protein-based labeling to maintain biomolecule functionality and avoid artifacts associated with the uptake of external probes. Here we develop a three-tandem monomeric Citrine tag (3×mCit) for labeling and demonstrate, using the motor molecule Kinesin-1 as model system, that it is indeed possible to track the movement of single, genetically labeled, fully functional protein molecules in the cytoplasm of live cells with high temporal and spatial resolution. Kinesin is an extremely interesting model for such studies, because 1), its rapid movement along microtubules is challenging to track; and 2), the in vivo structural and motile properties enabling kinesin motors to power cellular transport processes remain largely unknown.
EXPERIMENTAL METHODS AND MATERIALS
Plasmids
Myc-tagged rat kinesin heavy chain (KHC)(1-891) has been described previously (14). KHC(1-891) and KHC(1-339) fluorescent protein (FP)-fusion proteins were created by PCR and verified by DNA sequencing. A 12-amino-acid linker (TVPRARDPPVAT) was inserted between the KHC coding sequence and fluorescent protein tags. Vectors containing three tandem copies of mCit (p3×mCit-N1 and p3×mCit-C1) were created by PCR using the EGFP-N1 and EGFP-C1 vectors (Clontech, Palo Alto, CA) as a backbone. Ten amino-acid linkers (GAPGGSPVAT and GAPGTSGASG) connect the three copies of mCit.
COS cell culture, transfection, cell lysis, and Western blotting
105 COS cells (14) were added to 35-mm cell culture dishes containing a 25 mm × 25 mm, #1.5 cover glass (Corning Glass, Corning, NY). Cells were allowed to settle and then transiently transfected with 1 _μ_g of plasmid DNA and 3 _μ_l TransIT-LTI (Takara Mirus Bio, Madison, WI). After 4–10 h, COS cells were used for live cell or in vitro single molecule experiments. Cells were lysed in 50 _μ_l of SLB lysis buffer (40 mM HEPES/KOH, 120 mM NaCl, 1 mM EDTA, 10 mM pyrophosphate, 10 mM _β_-glycerophosphate, 50 mM NaF, pH 7.5) containing 0.5% Triton X-100, protease inhibitors (1 mM PMSF, 10 _μ_g/ml leupeptin, 5 _μ_g/ml chymostatin, 3 _μ_g/ml eastatinal, 1 mg/ml pepstatin A) and 1 mM ATP. Cell lysates were cleared by centrifugation at 14,000 rpm for 10 min at 4°C in a table-top centrifuge. The supernatant was used immediately or flash-frozen by immersion in liquid nitrogen and stored at −80°C. After SDS-PAGE and transfer to nitrocellulose, proteins were immunoblotted with polyclonal antibodies to KHC (#13 (14)) or GFP (Molecular Probes, Eugene, OR).
Live cell single molecule total internal reflection fluorescence microscopy (TIRFM)
After 4–8 h expression, transfected COS cells on a cover glass were carefully rinsed with Ringers buffer (10 mM HEPES/KOH, 155 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, 10 mM glucose, pH 7.2). The cover glass was assembled into a flow chamber with double-sided tape and a microscopy slide. After sealing with candle wax, the cells could be maintained in Ringers buffer for several hours at 24°C. Objective-type total internal reflection fluorescence microscopy (TIRFM) was performed on a custom-modified Zeiss Axiovert 135TV microscope (Carl Zeiss, Göttingen, Germany), equipped with a 1.45 NA _α_-Plan Fluor objective, 2.5× optovar, 505DCXR dichroic and HQ540/70M emission filters (Chroma Technology, Rockingham, VT) and a back-illuminated EMCCD camera (Cascade 512B, Roper Scientific, Trenton, NJ). The 488 nm line of a tunable, single-mode, fiber-coupled Argon Ion Laser with Littrow prism (Schäfter und Kirchhoff, Melles Griot, Carlsbad, CA) at incident powers of 0.26 mW or 0.55 mW was used to illuminate a circular region of ∼30 _μ_m in diameter for capturing video sequences at 30 Hz or 100 Hz, respectively.
In vitro single molecule TIRFM
In vitro single molecule motility assays were performed in a flow chamber made from a 25 mm × 25 mm #1.5 cover glass and a microscopy slide spaced by double-sided tape (chamber volume ∼30 _μ_l). Plus end-labeled Cy5 microtubules were diluted 10-fold in P12 buffer (12 mM PIPES/KOH, 1 mM EGTA, 2 mM MgCl2, pH 6.8) with 10 _μ_M taxol, flowed into the chamber, and incubated at room temperature for 2 min. Fifteen milligrams per milliliter of BSA (in P12 buffer with 10 _μ_M taxol) was then flowed in and incubated for 10 min at room temperature. Fifty microliters of oxygen scavenger buffer (1 mM DTT, 1 mM MgCl2, 2 mM ATP, 10 mM glucose, 0.1 mg/ml glucose oxidase, 0.08 mg/ml catalase, 5 mg/ml BSA, and 10 _μ_M taxol in P12) containing 1 _μ_l of COS lysate was flowed in and the chamber was sealed with wax. The sealed chamber was then observed under the same conditions as live cell single molecule TIRFM experiments.
Off-line imaging processing
Movies and images were prepared with ImageJ (National Institutes of Health, Bethesda, MD), Photoshop and Illustrator (Adobe Systems, San Jose, CA). To determine the location of microtubule tracks frequently utilized by kinesin motors, standard deviation maps were generated by calculating the statistical intensity variation of each pixel location from the raw images of a video sequence (typically referred to as image stack) and plotting them in form of an image. In brief, for an image stack containing Z slices of images with M × N pixels, the intensity (I) of each pixel in the standard deviation map was calculated with ImageJ (ZProjector_StandardDeviation) as
 |
(1) |
|---|
For low expression COS cells (Supplementary Material, Movie 5), the autofluorescence background of cells was determined by computing an average map (ZProjector_Average in ImageJ) of the raw images using
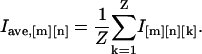 |
(2) |
|---|
The KHC(1-891)-3×mCit fluorescence signal was then extracted by subtracting the autofluorescence background (average map) from the raw images. Background autofluorescence was pseudo-colored in blue, single motor fluorescence was pseudo-colored in green, and the standard deviation map was pseudo-colored in pink or red by ImageJ to generate the movies.
Plus end-labeled Cy5 microtubules
Tubulin was purified from pig brain and nonspecifically labeled with Cy5-succinimidyl-esters (Amersham Bioscience, Piscataway, NJ (15)) Microtubules were first polymerized with low ratio of Cy5-tubulin in BRB80 buffer (80 mM PIPES/KOH, 1 mM EGTA, 2 mM MgCl2, pH 6.9) containing 1 mM GTP and 2 mM MgCl2 at 37°C for 15 min, and stabilized by 10 _μ_M taxol (Paclitaxel, Calbiochem, San Diego, CA). The plus-ends were then labeled by mixing with 10-fold excess of Cy5-tubulin at 37°C for 5 min. The microtubules, dimly labeled overall and heavily labeled at the plus ends, were used within three days. Imaging of Cy5-microtubule was performed by epifluorescence simultaneously with TIRFM (see above) using an HQ602/13M exciter, 51008BS/ dichroic mirror, and 51008M emission filter (Chroma Technology).
Speed and processivity analysis
Single molecule tracking of the KHC(1-891)-3×mCit was performed on diffraction-limited fluorescence spots (5 × 5 pixels) that were clearly separated from the neighboring fluorescence. For in vivo data, only those fluorescence spots moving on stable, stationary microtubule tracks identified in standard deviation maps were selected. Comparison of microtubule standard deviation maps over time periods of many minutes was used to confirm that the selected microtubule tracks did not undergo significant movement as compared to the speed of kinesin. ImageJ's SpotTracker plug-in (16) was modified and used for measuring the speed and run length of single motors.
Photobleaching analysis
Analysis of the photobleaching behavior of kinesins in vivo and in vitro was performed with TIRFM using conditions identical to those used for motility assays. Incident laser power was 0.26 mW or 0.55 mW, illuminating a circular region of ∼30 _μ_m in diameter for image capture at 30 Hz. For analyzing fluorophore properties in vivo, mCit-labeled motors were forced to remain in a microtubule-bound state by addition of the nonhydrolyzable ATP analog AMPPNP to ensure that the entire photobleaching event was captured. Transfected COS cells in a live cell chamber were permeabilized for 30 s with 30 _μ_l of 0.1 _μ_g/_μ_l Streptolysin O in Permeabilization Buffer I (25 mM HEPES/KOH, 5 mM MgCl2, 115 mM KOAc, 5 mM NaOAc, 0.5 mM EGTA, pH 7.2) with 10 mg/ml of BSA. After washing three times with 50 _μ_l of Buffer I, 2 mM of AMPPNP was flowed in and incubated for 10 min. For analyzing fluorophore properties in vitro, a flow chamber was first incubated with 10 mg/ml of BSA (in oxygen scavenger buffer) for 10 min. Fifty microliters of 1 mg/ml of BSA (in oxygen scavenger buffer) containing 1 _μ_l of COS cell lysate was then flowed in and incubated for another 10 min. In each case, stationary fluorescent spots located within the light diffraction areas and separated from neighboring spots were chosen for analysis. Background-subtracted fluorescence intensity over time was plotted using an ImageJ plug-in developed in our lab. Briefly, a 5 × 5 pixel (320 nm × 320 nm) area covering a fluorescence spot was manually selected. A 9 × 9 pixel area was then automatically generated, centered by the selected 5 × 5 pixel area. The background-subtracted fluorescence intensity was then measured as the average intensity of the central 25 pixels minus the average intensity of the surrounding 56 pixels. Fluorescence bleaching steps and the single molecule initial photobleaching time (the earliest time when fluorescence intensity reaches background) were determined from the plots.
RESULTS
To track the motility of single Kinesin-1 motors in the cytoplasm of live cells, we utilized a constitutively active version of the kinesin heavy chain (KHC) subunit. Truncation of the KHC tail (KHC(1-891), Fig. 1 a) removes both the regulatory tail domain required for autoinhibition of motor activity and the ATP-independent cryptic microtubule binding site that can obscure ATP-dependent microtubule-based motility (17). KHC(1-891) was genetically tagged with a monomeric version of the fluorescent protein (FP) Citrine (mCit), a brighter and more photostable version of EYFP (18). COS cells were chosen for these studies due to their flat morphology (Fig. 1 b) and their low levels of endogenous Kinesin-1 expression (Supplementary Material, Fig. S1). When observed by epi-fluorescence microscopy, KHC(1-891)-mCit expressed in COS cells showed diffuse cytoplasmic localization with some accumulation on microtubules (Fig. 1 b). Imaging of individual Kinesin-1 molecules was not feasible due to cellular autofluorescence and freely diffusing KHC(1-891)-mCit molecules. However, the shallow evanescent wave-excitation of total internal reflection fluorescence microscopy (TIRFM) dramatically reduced the background and made it possible to image individual fluorescent spots in the cell periphery (Fig. 2). Analysis of KHC(1-891)-mCit transfected COS cells by TIRFM revealed clearly detectable, diffraction-limited fluorescent spots that underwent kinesin-like processive motility (Fig. 2, a and b; and see Supplementary Material, Movie 1). Rapid diffusion of the soluble fraction could also be seen. However, quantitative observations of individual KHC(1-891)-mCit molecules were hindered by 1), the low signal level relative to cellular autofluorescence; and 2), rapid bleaching and blinking (reversible photobleaching) (19).
FIGURE 1.

COS expression system for tracking single kinesin-1 motors in live cells. (a) Schematic of KHC fusion proteins. One or three monomeric Citrine (mCit) fluorescent protein tags were fused to the C-terminus of homodimeric KHC(1-891) or monomeric KHC(1-339). (b) Epi-fluorescence microscopy of fixed COS cells expressing KHC(1-891)-mCit (right panels) or untransfected (left panels). The flat morphology of the COS cell periphery can be seen by DIC microscopy. The microtubule tracks were stained with an anti-tubulin antibody. The expressed KHC(1-891)-mCit can be seen in the mCit channel. (Bottom panels) Merged anti-tubulin and mCit images. Scale bar = 10 _μ_m.
FIGURE 2.

Single molecule tracking of mCit-labeled KHC motors in live COS cells. TIRFM of COS cells expressing panels a_–_d, KHC(1-891)-mCit; panels e_–_h, KHC(1-891)-3×mCit; and panels i_–_l, KHC(1-339)-3×mCit. (a,e,i) Field of view of TIRFM near the periphery of the cells. The edges of the cells are outlined. (b,f,j) Kymographs of the boxed areas in panels a, e, and i, respectively. Scale bars, 2 _μ_m and, 0.2 s. The homodimeric kinesin-1 motors KHC(1-891)-mCit (b) and KHC(1-891)-3×mCit (f) move processively, whereas the monomeric motor KHC(1-339)-3×mCit (j) does not. The presence of two mCit FPs in KHC(1-891)-mCit (a) results in loss of signal due to blinking and photobleaching whereas the six mCit FPs in KHC(1-891)-3×mCit (f) are brighter and unlikely to blink to background before complete photobleaching. (c,g,k) Representative traces of quantized photobleaching. mCit-labeled KHC molecules were locked on microtubules by the addition of AMPPNP to prevent diffusion or motility. Stepwise photobleaching and the number of bleaching steps indicate the present of single KHC motors in the light diffraction-limited areas. (d,h,l) Standard deviation maps of the image series shown in panels a, e, and i, respectively. By deemphasizing the fluorescence background and emphasizing the motility events, the standard deviation maps highlight the microtubule tracks utilized for motility. Processive, linear motility can be seen for the homodimeric kinesin-1 motors KHC(1-891)-mCit (d) and KHC(1-891)-3×mCit (h) but not the monomeric motor KHC(1-339)-3×mCit (l).
To improve the fluorescence signal for in vivo imaging, we tagged KHC(1-891) with three tandem copies of mCit (KHC(1-891)-3×mCit, Fig. 1 a). When KHC(1-891)-3×mCit was expressed in COS cells, we observed significantly brighter diffraction-limited fluorescent spots (Fig. 2, e and f; and see Supplementary Material, Movie 2). Using a sensitive frame-transfer CCD camera with on-chip multiplication gain, the average fluorescence excitation power was limited to ∼0.3 _μ_W/_μ_m2, which resulted in a global time constant of 3×mCit photobleaching of ∼2.2 s (Supplementary Material, Fig. S2). A higher excitation level of ∼0.6 _μ_W/_μ_m2 was sufficient to record movies at up to 100 frames/s (Supplementary Material, Movie 7), although slightly longer exposure times (33 ms) and lower laser power (0.3 _μ_W/_μ_m2) represented the best compromise between image quality and resolving the movement of kinesin. Under these conditions, characterization of the processive run length of motors in vivo was not limited by premature fluorophore bleaching.
To demonstrate that the recorded fluorescent spots are single proteins and not aggregates, we characterized the fluorescence properties of the spots. The maximum fluorescence intensity for KHC(1-891)-3×mCit fluorescent spots was approximately three times that of KHC(1-891)-mCit spots (Fig. 2, g and c, respectively). Individual fluorescent spots showed abrupt bleaching in unitary steps (photodestruction of mCit), characteristic of single molecules. Multiple steps indicate the presence of multiple FPs, with one to two steps for dimeric KHC(1-891)-mCit (Fig. 2 c), four to six steps for dimeric KHC(1-891)-3×mCit (Fig. 2 g; and see Supplementary Material, Fig. S3) and two to three steps for monomeric KHC(1-339)-3×mCit (Fig. 2 k). Deviations from an ideal bleaching response are most pronounced when multiple FPs are present in a single molecule (e.g., KHC(1-891)-3×mCit, Fig. 2 g) and are likely due to 1), frequent blinking of individual FPs; 2), fluorescence resonance energy transfer between fluorophores (homo-FRET); 3), partial bleaching of FPs before microtubule binding; and 4), incomplete FP maturation. Overall, the fluorescent properties of the spots, including the maximum fluorescence, the number of steps to a nonfluorescent dark state, and the rate of bleaching, are consistent with the number of FPs per constructs (Fig. 1 a, Supplementary Material, Fig. S3) and the global bleaching properties of 3×mCit (Supplementary Material, Fig. S2). Thus, the 3×mCit tag provided a better signal/noise ratio, blinked less frequently to a nonfluorescent dark state, and emitted more photons than mCit.
To verify that these fluorescent spots are single Kinesin-1 motor proteins, we analyzed the motile behavior of the spots. Importantly, both KHC(1-891)-mCit and KHC(1-891)-3×mCit fluorescent spots displayed linear processive movement (Fig. 2, b and f, respectively), characteristic of kinesin motors. In contrast, KHC(1-339)-3×mCit, a monomeric Kinesin-1 that cannot move processively (20), showed only static binding to microtubules and fast diffusion in the cytoplasm (Fig. 2, i and j; see Supplementary Material, Movie 3). Clearly, this movement suggests that the recorded spots are kinesins, not some other autofluorescent proteins or small structures.
Like many cellular events, tracking the motility of Kinesin-1 along microtubules requires fast imaging of infrequent events. Thus, to reveal this structural interaction and gather physiological data from multiple individual KHC(1-891)-3×mCit motility events, we computed standard deviation maps from the image series (see Experimental Methods and Materials). Such maps provide a statistical representation of the intensity fluctuations in each pixel such that background fluorescence is deemphasized and large changes in fluorescence intensity are enhanced. Clearly, the binding and movement of fluorescently-labeled motors along microtubules should lead to the largest intensity changes. Indeed, the computed maps from KHC(1-891)-mCit (Fig. 2 d) and KHC(1-891)-3×mCit (Fig. 2 h) reveal linear tracks that closely resemble the microtubule network in the periphery of COS cells (Fig. 1 b). In contrast, the standard deviation map from monomeric KHC(1-339)-3×mCit shows only static binding to the microtubules (Fig. 2 l). Such image processing can be applied to the analysis of many other cellular events, like DNA replication and transcription as well as other transport mechanisms, signaling events or catalytic processes that are associated with structural components of the cell.
Since the tandem FP tags enable high resolution tracking in vivo, we tested whether they could be used to study mammalian-expressed proteins in typical in vitro assays. Such an approach would avoid the drawbacks of bacterial expression, allow the analysis of multiprotein complexes and molecules containing the correct post-translational modifications, and make possible direct correlations between in vivo and in vitro properties. We extracted mCit-tagged KHC proteins from transfected COS cells for low background in vitro analysis (Fig. 3). Analysis of the bleaching properties of the mCit-tagged motors in vitro was consistent with the in vivo observations. KHC(1-891)-mCit, KHC(1-891)-3×mCit, and KHC(1-339)-3×mCit bleached in distinct steps (Fig. 3, a, d, and g, respectively). The maximum number of photobleaching steps (Fig. 3, c, f, and i) and the rate of initial photobleaching (Fig. 3, b, e, and h) are directly related to the number of FPs for each fusion protein. The bleaching behavior is in agreement with Western blot analysis that the majority of KHC motors are mCit-tagged with a minority incorporating endogenous KHC (Supplementary Material, Fig. S1). Taken together, these results support the conclusion that we are tracking the movement of single Kinesin-1 molecules on microtubule tracks.
FIGURE 3.

Fluorescence properties of mCit-labeled KHC motors in vitro. Extracts of COS cells expressing panels a_–_c, KHC(1-891)-mCit; panels d_–_f, KHC(1-891)-3×mCit; or panels g_–_i, KHC(1-339)-3×mCit were absorbed onto the cover glass and imaged under the same TIRFM conditions as live cell experiments (Fig. 2). (a,d,g) Representative examples of quantized photobleaching of mCit-labeled KHC motors within the light-diffraction-limited fluorescent spots. (b,e,h) Fitting of single exponential decay fitting (solid lines) of the fluorescence bleaching time of mCit-labeled KHC motors showed that the decay time constants are 1.1 ± 0.2 s for KHC(1-891)-mCit (N = 77, b), 2.2 ± 0.3 s for KHC(1-891)-3×mCit (N = 86, e), and 1.8 ± 0.2 s for KHC(1-339)-3×mCit (N = 89, h). (c,f,i) Histograms of the photobleaching events show primarily two steps for KHC(1-891)-mCit (c), four or five bleaching steps for KHC(1-891)-3×mCit (f), and two or three steps for KHC(1-339)-3×mCit (i).
Finally, we set out to compare the in vivo motile behavior of Kinesin-1 with its in vitro properties. When extracted from COS cells, dimeric KHC(1-891)-3×mCit molecules moved processively toward the plus-end of polarity-marked, taxol-stabilized microtubules (Fig. 4, a and b; see Supplementary Material, Movie 4) at an average speed of 0.77 ± 0.14 _μ_m/s (Fig. 4 c) and an average run-length of 0.83 ± 0.29 _μ_m (Fig. 4 d). Thus, the in vitro motile properties of our mammalian-expressed dimeric KHC constructs are directly comparable to those of recombinant KHC motors (21). In vivo, single KHC(1-891)-3×mCit motors showed unidirectional processive motility along linear tracks (Fig. 4, e_–_h; and see Supplementary Material, Movies 5–7). Histograms of the speed and run length demonstrated that Kinesin-1 moved at an average speed of 0.78 ± 0.11 _μ_m/s in cells (Fig. 4 i) and an average run-length of 1.17 ± 0.38 _μ_m (Fig. 4 j). The similarity in motile properties between Kinesin-1 in vitro and in vivo demonstrates that Kinesin-1 activity is neither upregulated in cells nor hindered by macromolecular crowding along the microtubule track.
FIGURE 4.

Motile properties of single KHC(1-891)-3×mCit molecules in vitro and in vivo. (a_–_d) Motile properties of KHC(1-891)-3×mCit in vitro. (a) Taxol-stabilized microtubules, heavily labeled with Cy5-tubulin (red) at their plus ends, were incubated in a BSA-coated flow chamber with KHC(1-891)-3×mCit COS lysate and 1 mM ATP. Kymograph shows one representative fluorescence spot moving processively on a Cy5-labeled microtubule. (b) Graph of displacement (red) and fluorescence intensity (black) over time for the same fluorescence spot shown in panel a. Stepwise photobleaching behavior indicates the presence of one KHC(1-891)-3×mCit in the light-diffraction-limited area. (c) Gaussian fitting of the speed histogram shows that KHC(1-891)-3×mCit motors move at 0.77 ± 0.14 _μ_m/s (N = 54). (d) Single exponential decay fitting of the run length histogram shows the same motors move processively for 0.83 ± 0.29 _μ_m/run. (e_–_j) Motile properties of KHC(1-891)-3×mCit in vivo. (e,f) Merge of final TIRFM image and standard deviation map of COS cells expressing low (e) or moderate (f) levels of KHC(1-891)-3×mCit (Supplementary Material, Movies 5 and 6, respectively). KHC(1-891)-3×mCit (green); standard deviation map of image series (pink or red); cellular autofluorescence background (blue). (g) Kymograph of boxed region of panel e shows KHC(1-891)-3×mCit moving processively. (h) Graph of displacement (red) and fluorescence intensity (black) over time of the same fluorescent spot shown in panel g. (i) Gaussian fitting of the speed histogram shows that KHC(1-891)-3×mCit motors move at 0.78 ± 0.11 _μ_m/s (N = 54). (j) Single exponential decay fitting of the run length histogram shows the same motors move processively for 1.17 ± 0.38 _μ_m/run. White scale bars, 2 _μ_m; yellow scale bars, 0.1 s.
DISCUSSION
We developed and validated a new, powerful method for in vivo single molecule imaging. Our work clearly proves that it is possible to track individual, genetically labeled molecules in the cytoplasm of mammalian cells with high spatial and temporal resolution. For the experimental conditions used in this report, we achieve single molecule recordings in vivo with frame rates up to 100 Hz. Fitting of Gaussian functions to diffraction-limited fluorescent spots yields a spatial resolution of ∼20 nm at frame rates of 30 Hz. Our data also suggest that spatial and temporal resolutions are not limited by the in vivo background fluorescence, but are determined by the tradeoff between the rate of photobleaching and fluorescence emission intensity at various excitation levels.
As a biological model system, we characterized the movement of single Kinesin-1 motor molecules in COS cells. Past research on the biochemical and structural properties of Kinesin-1, together with various single molecule assays using purified motors in vitro, has revealed detailed mechanistic insights into Kinesin-1 function (21), yet these biophysical advances contributed little to understanding kinesin's role in intracellular transport. Our work is aimed at closing the gap between the single molecule biophysical properties of Kinesin-1 in vitro and the cellular functions of Kinesin-1 in vivo. Toward this end, we provide the first direct observations and analysis of the motile properties of a single molecular motor in vivo. We reveal a remarkable consistency between the motility of Kinesin-1 in living cells and in reconstituted in vitro assays using purified components. This result has two important implications for the study of molecular motors in intracellular transport. First, our work suggests that neither cellular factors present in vivo nor macromolecular crowding on the microtubule track hinder the motility of Kinesin-1. This conclusion is broadly in agreement with in vitro experiments on the processive movement of individual kinesins on crowded microtubules (22). Second, this work provides an important step beyond previous live cell studies where entire organelles with uncertain motor composition were tracked as indirect reporters of motor activity (1,11,13,23) or where semiconductor quantum dots sparsely labeled with recombinant Kinesin-1 were introduced into cells (10). The latter experiments, in particular, could confirm neither the tracking of single kinesin molecules, the precise labeling stoichiometry, nor the activity of the recombinant motors.
It is interesting to note that previous live cell studies have demonstrated that various organelles and vesicles move with speeds in vivo that are often significantly higher than the in vitro velocities of kinesin motors (13,24,25). Our work rules out the possibility that the fast organelle and vesicle speeds observed in vivo are due to increased speed of Kinesin-1 motors in vivo. An alternative hypothesis (13), that multiple motors cooperate to produce increased speeds for intracellular transport, cannot be ruled out by our data. However, since single motors and multiple motors show identical speeds in vitro (26), the fact that single motors behave identically in vivo as in vitro leads to the logical hypothesis that multiple motors will also move with identical speed in vivo. Indeed, existing experimental and theoretical work (27–31) provides strong evidence that it is the forces of cooperatively interacting Kinesin-1 motors that sum. We currently favor the hypothesis that the fast velocities of organelles and vesicles are due to the presence of different kinesin family members with distinct kinetic properties and/or cooperativity between multiple motors, including different actin and microtubule-based motors, on the same vesicle (13,23,32). Since it is still unknown for most organelles which motor(s) is (are) responsible for the observed cellular motility, identifying the species and numbers of motors associated with a specific cargo is an important goal for future experiments.
In conclusion, our results demonstrate for the first time the direct tracking of single molecules in the cytoplasm of living cells with high temporal and spatial resolutions (Figs. 2 and 4; and see Supplementary Material, Movies 2, 5, 6, and 7). By using genetically engineered tandem FP tags with improved fluorescence properties, this work has broad applications to the analysis of a wide variety of cytoplasmic events without the requirements of plasma membrane localization (1,2) or special labeling methods (8) and creates unique opportunities to investigate how single molecules work in living cells. Continual improvements in the photophysical properties (like quantum yield, stability, and bleaching) of fluorescent proteins are likely to make it possible to track multiple proteins simultaneously. In addition, our work shows that the tandem FP tags enable in vitro analysis of mammalian-expressed proteins. Furthermore, the standard deviation maps provide a novel way to characterize multiple infrequent events that occur within a specific subcellular locale. By combining single molecule biophysical and cell biological approaches, our work opens now unique opportunities to address how individual molecules work and interact in living cells.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank A. Hoppe, T. Rapoport, and J Swanson for stimulating discussions and reading of the article, and Y. Goldman for his generous release of an ImageJ plug-in for Gaussian function fitting.
This work was supported in part by National Institutes of Health, National Science Foundation, and Defense Advanced Research Projects Agency grants to E.M. and K.J.V.
References
- 1.Xie, X. S., J. Yu, and W. Y. Yang. 2006. Living cells as test tubes. Science. 312:228–230. [DOI] [PubMed] [Google Scholar]
- 2.Sako, Y., and T. Yanagida. 2003. Single-molecule visualization in cell biology. Nat. Rev. Mol. Cell Biol. Suppl:SS1–5. [PubMed]
- 3.Schutz, G. J., G. Kada, V. P. Pastushenko, and H. Schindler. 2000. Properties of lipid microdomains in a muscle cell membrane visualized by single molecule microscopy. EMBO J. 19:892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harms, G. S., L. Cognet, P. H. Lommerse, G. A. Blab, H. Kahr, R. Gamsjager, H. P. Spaink, N. M. Soldatov, C. Romanin, and T. Schmidt. 2001. Single-molecule imaging of l-type Ca2+ channels in live cells. Biophys. J. 81:2639–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iino, R., I. Koyama, and A. Kusumi. 2001. Single molecule imaging of green fluorescent proteins in living cells: E-cadherin forms oligomers on the free cell surface. Biophys. J. 80:2667–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Douglass, A. D., and R. D. Vale. 2005. Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell. 121:937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang, W., J. Gelles, and S. M. Musser. 2004. Imaging of single-molecule translocation through nuclear pore complexes. Proc. Natl. Acad. Sci. USA. 101:12887–12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shav-Tal, Y., X. Darzacq, S. M. Shenoy, D. Fusco, S. M. Janicki, D. L. Spector, and R. H. Singer. 2004. Dynamics of single mRNPs in nuclei of living cells. Science. 304:1797–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watanabe, N., and T. J. Mitchison. 2002. Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science. 295:1083–1086. [DOI] [PubMed] [Google Scholar]
- 10.Courty, S., C. Luccardini, Y. Bellaiche, G. Cappello, and M. Dahan. 2006. Tracking individual kinesin motors in living cells using single quantum-dot imaging. Nano Lett. 6:1491–1495. [DOI] [PubMed] [Google Scholar]
- 11.Nan, X., P. A. Sims, P. Chen, and X. S. Xie. 2005. Observation of individual microtubule motor steps in living cells with endocytosed quantum dots. J. Phys. Chem. B Condens. Matter Mater. Surf. Interfaces Biophys. 109:24220–24224. [DOI] [PubMed] [Google Scholar]
- 12.Michalet, X., F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir, and S. Weiss. 2005. Quantum dots for live cells, in vivo imaging, and diagnostics. Science. 307:538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kural, C., H. Kim, S. Syed, G. Goshima, V. I. Gelfand, and P. R. Selvin. 2005. Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement? Science. 308:1469–1472. [DOI] [PubMed] [Google Scholar]
- 14.Verhey, K. J., D. L. Lizotte, T. Abramson, L. Barenboim, B. J. Schnapp, and T. A. Rapoport. 1998. Light chain-dependent regulation of Kinesin's interaction with microtubules. J. Cell Biol. 143:1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lakamper, S., and E. Meyhofer. 2005. The E-hook of tubulin interacts with kinesin's head to increase processivity and speed. Biophys. J. 89:3223–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sage, D., F. R. Neumann, F. Hediger, S. M. Gasser, and M. Unser. 2005. Automatic tracking of individual fluorescence particles: application to the study of chromosome dynamics. IEEE Trans. Image Process. 14:1372–1383. [DOI] [PubMed] [Google Scholar]
- 17.Cai, D., A. D. Hoppe, J. A. Swanson, and K. J. Verhey. 2007. Kinesin-1 structural organization and conformational changes revealed by FRET stoichiometry in live cells. J. Cell Biol. 176:51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griesbeck, O., G. S. Baird, R. E. Campbell, D. A. Zacharias, and R. Y. Tsien. 2001. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 276:29188–29194. [DOI] [PubMed] [Google Scholar]
- 19.Dickson, R. M., A. B. Cubitt, R. Y. Tsien, and W. E. Moerner. 1997. On/off blinking and switching behavior of single molecules of green fluorescent protein. Nature. 388:355–358. [DOI] [PubMed] [Google Scholar]
- 20.Hancock, W. O., and J. Howard. 1998. Processivity of the motor protein kinesin requires two heads. J. Cell Biol. 140:1395–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schliwa, M. 2003. Molecular Motors. Wiley-VCH, Wenheim.
- 22.Seitz, A., and T. Surrey. 2006. Processive movement of single kinesins on crowded microtubules visualized using quantum dots. EMBO J. 25:267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levi, V., A. S. Serpinskaya, E. Gratton, and V. Gelfand. 2006. Organelle transport along microtubules in Xenopus melanophores: evidence for cooperation between multiple motors. Biophys. J. 90:318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaether, C., P. Skehel, and C. G. Dotti. 2000. Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol. Biol. Cell. 11:1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill, D. B., M. J. Plaza, K. Bonin, and G. Holzwarth. 2004. Fast vesicle transport in PC12 neurites: velocities and forces. Eur. Biophys. J. 33:623–632. [DOI] [PubMed] [Google Scholar]
- 26.Howard, J., A. J. Hudspeth, and R. D. Vale. 1989. Movement of microtubules by single kinesin molecules. Nature. 342:154–158. [DOI] [PubMed] [Google Scholar]
- 27.Leibler, S., and D. A. Huse. 1993. Porters versus rowers: a unified stochastic model of motor proteins. J. Cell Biol. 121:1357–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hunt, A. J., F. Gittes, and J. Howard. 1994. The force exerted by a single kinesin molecule against a viscous load. Biophys. J. 67:766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Svoboda, K., C. F. Schmidt, B. J. Schnapp, and S. M. Block. 1993. Direct observation of kinesin stepping by optical trapping interferometry. Nature. 365:721–727. [DOI] [PubMed] [Google Scholar]
- 30.Meyhofer, E., and J. Howard. 1995. The force generated by a single kinesin molecule against an elastic load. Proc. Natl. Acad. Sci. USA. 92:574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inoue, Y., Y. Y. Toyoshima, A. H. Iwane, S. Morimoto, H. Higuchi, and T. Yanagida. 1997. Movements of truncated kinesin fragments with a short or an artificial flexible neck. Proc. Natl. Acad. Sci. USA. 94:7275–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogers, S. L., and V. I. Gelfand. 1998. Myosin cooperates with microtubule motors during organelle transport in melanophores. Curr. Biol. 8:161–164. [DOI] [PubMed] [Google Scholar]