Developmental programming of health and disease (original) (raw)
. Author manuscript; available in PMC: 2007 Jun 4.
Published in final edited form as: Proc Nutr Soc. 2006 Feb;65(1):97–105. doi: 10.1079/pns2005478
Abstract
The environment encountered in fetal and neonatal life exerts a profound influence on physiological function and risk of disease in adult life. Epidemiological evidence suggests that impaired fetal growth followed by rapid catch-up in infancy is a strong predictor of obesity, hypertension, non-insulin-dependent diabetes and CHD. Whilst these associations have been widely accepted to be the product of nutritional factors operating in pregnancy, evidence from human populations to support this assertion is scarce. Animal studies clearly demonstrate that there is a direct association between nutrient imbalance in fetal life and later disease states, including hypertension, diabetes, obesity and renal disease. These associations are independent of changes in fetal growth rates. Experimental studies examining the impact of micro- or macronutrient restriction and excess in rodent pregnancy provide clues to the mechanisms that link fetal nutrition to permanent physiological changes that promote disease. Exposure to glucocorticoids in early life appears to be an important consequence of nutrient imbalance and may lead to alterations in gene expression that have major effects on tissue development and function. Epigenetic mechanisms, including DNA methylation, may also be important processes in early-life programming.
Keywords: Fetal programming, Pregnancy, CHD, Hypertension
The early-life origins of health and disease
The environment encountered during fetal life and infancy appears to be strongly related to risk of non-communicable diseases in adult life (Barker, 2004). In order to explain these apparently causal relationships it is proposed that adaptations during critical phases of growth and development may ensure the maintenance of homeostasis, and hence survival, when the environment is compromised (Gluckman & Hanson, 2004). Variation in nutrient supply during early development appears to be a strong signal initiating these adaptive processes. The means through which events in early life trigger permanent responses have been described as nutritional or metabolic programming (Lucas, 1991). These terms describe the process through which a stimulus or insult during a critical window of fetal or infant development elicits permanent responses that produce long-term changes in tissue structure or function. Programming is the consequence of the innate capacity of developing tissues to adapt to the conditions that prevail during early life, which for almost all cell types in all organs is an ability that is present for only a short period before the time of birth.
The epidemiological studies that first indicated that disease could be programmed by intrauterine influences formed the basis of what became known as the ‘fetal origins of adult disease hypothesis’, or the ‘Barker hypothesis’ (see Langley-Evans, 2004b). This constantly evolving concept is now described as the developmental origins of health and disease hypothesis. The developmental origins of health and disease hypothesis was originally developed to explain associations between patterns of fetal and infant growth and major disease states in human populations, but has received strong support from experimental studies in animals. These studies will be the main focus of the present review.
Clues from epidemiology
The first clues that the environment encountered in early life could determine risk of disease in adulthood came from ecological studies evaluating the causes of the north-south divide in disease patterns in England and Wales (Barker & Osmond, 1986). These studies identified the period around the time of birth as playing a critical role in the development of CHD (Osmond et al. 1990). Studies of large retrospective cohorts in Hertfordshire (Barker et al. 1989; Hales et al. 1991), Preston, Lancs. (Barker et al. 1990) and Sheffield, South Yorkshire (Barker et al. 1993b) that included men and women who had been born in the first third of the twentieth century have confirmed these observations. These studies have shown that anthropometry at birth is predictive of later disease. Low weight at birth is associated with increased risk of CHD mortality (Fall et al. 1995), raised adult blood pressure (Law et al. 1991), non-insulin-dependent diabetes (Phillips et al. 1994) and risk of the metabolic syndrome (Barker et al. 1993a).
The birth weight-disease associations have been confirmed in a large number of independent cohorts all around the developed and developing world. Based on fairly limited evidence the association has been attributed to the impact of a poor plane of nutrition before and during pregnancy (see Fig. 1). This simplistic model has been updated to allow for observations that measurements such as thinness at birth, reduced abdominal circumference and a large head circumference in proportion to truncal length are also predictive of later disease (Godfrey, 1998). The main basis of the Barker hypothesis is that undernutrition in pregnancy impairs fetal growth or promotes disproportionate fetal growth, and as a trade-off these adaptations that promote survival in adverse conditions lead to limited physiological function and disease in the long term.
Fig. 1.
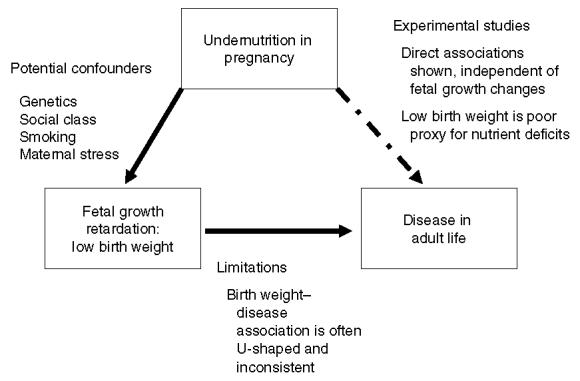
Schematic representation of the Barker hypothesis (Langley-Evans, 2004_b_). The simplest form of the hypothesis is that undernutrition impairs fetal growth. The association between fetal growth and long-term disease outcomes is likely to be confounded by a direct association between undernutrition and disease.
The best evidence relating periods of undernutrition in pregnancy with health and disease in the resulting offspring comes from follow-ups of famines associated with military action in the Second World War. The best studied settings are the Leningrad siege (Stanner & Yudkin, 2001) and the Dutch Hunger Winter (Roseboom et al. 2001). These wartime famines subjected large populations to periods of severe undernutrition. However, children continued to be conceived and born under these harsh conditions. Follow-ups of these individuals, now in their sixth decade, provide important clues about the impact of prenatal stressors on human development and long-term health. In the case of the Dutch Hunger Winter it is clear that prenatal undernutrition had only a small impact on fetal growth, but in the long-term it programmed greater risk of CHD (Roseboom et al. 2000), obesity (Ravelli et al. 1999), renal dysfunction (Painter et al. 2005) and non-insulin-dependent diabetes (Ravelli et al. 2000). With this population it has been possible to distinguish the effects of famine during discrete periods of development, and it appears that exposure to undernutrition in the first trimester of pregnancy, which interestingly increased birth weight, is the strongest predictor of CHD and adult obesity.
The epidemiological evidence suggesting that CHD and the metabolic syndrome may be programmed by factors operating in utero has been criticised on a number of fronts (Kramer & Joseph, 1996; Huxley et al. 2002). Issues with cohort selection, reproducibility of findings and problems with correction for confounders are an inevitable weakness of this sort of work, as the study of nutritional programming of diseases that afflict individuals in their middle age inevitably relies on historical data and retrospective cohorts. Some critical issues are difficult to dismiss. Huxley and co-workers (Huxley et al. 2002; Huxley & Neil, 2004) have demonstrated through meta-analyses that the effect size for associations between birth weight and vascular disease indicators actually decreases as cohort size increases, leading to the suggestion that the fetal origins hypothesis is a product of publication bias and measurement error. Moreover, the explanation that maternal nutritional factors underpin any association between impaired fetal growth and later disease risk is weak, as human fetal growth is remarkably resistant to variation in quantity and quality of nutrient intake, particularly within the normal range of intakes (Mathews et al. 1999; Langley-Evans & Langley-Evans, 2003). Birth weight is a poor proxy for nutritional events during gestation. Any individual baby of lower weight at birth may have arrived at that particular weight via a number of routes, with restraint resulting from undernutrition being just one explanation (Law, 2002). The developmental origins of health and disease hypothesis therefore requires demonstration of biological plausibility before any major conclusions can be drawn regarding its importance in terms of public health.
Biological plausibility
The ideal demonstration of the biological plausibility of nutritional programming requires cohorts of human subjects with high-quality data relating to nutrient intake in pregnancy, or cohorts such as those collected following the Dutch Hunger Winter from which the longer-term impact of deficit can be examined. However, whilst the Dutch famine data generally supports the developmental origins of health and disease hypothesis, there are potential problems with this data. First the level of food restriction during this famine may have been highly variable between women and difficult to evaluate. Moreover, wartime is by definition stressful and it is known that psychological stressors impact on fetal development (Stein, 2004).
There are very few studies that have been able to consider physiology or disease states in relation to maternal diet in pregnancy, but those studies that do exist are supportive of the developmental origins of health and disease hypothesis. Godfrey et al. (1994) have reported that the blood pressures of pre-pubescent Jamaican boys are inversely related to the triceps skinfold thicknesses and Hb status of their mothers pre-pregnancy. It has also been reported that the blood pressures of middle-aged men in Aberdeen are inversely related to maternal intake of animal protein, but only when carbohydrate intakes are high (Campbell et al. 1996). Both these studies are suggestive of a role for nutritional programming at least in the determination of blood pressure. They do not, however, provide unequivocal support for the concept of nutritional programming in a wider disease context.
The use of appropriate animal models has been of greatest use in providing the critical data that supports the developmental origins of health and disease hypothesis and enables an understanding of the mechanisms that may link nutritional factors in early life to the functional capacity of organs and systems in the mature state. Animal models allow the examination of the specific effects of a simple dietary change, independently of the confounding factors that are inevitably associated with long-term epidemiological studies. Many different approaches to the study of nutritional programming have been adopted using a variety of species, including rat, mouse, guinea-pig, sheep and pigs (Langley-Evans, 2004a). All these approaches have demonstrated beyond any doubt that variation in the quality or quantity of nutrient provision in pregnancy and/or lactation have a major impact on tissue development and function and can promote both disease susceptibility and disease resistance. These effects are often independent of major changes in fetal growth.
Life-course perspectives on health and disease
The aetiology of non-communicable disease is invariably complex, and is now regarded as involving influences at all stages of the life-course, a concept best considered using the example of CHD. It is long-established that CHD is causally related to adult environmental and lifestyle factors, including a high-fat diet, poor dietary antioxidant status and smoking. These environmental factors are clearly not the only determinants of risk, as the individual genotype determines the impact of dietary risk factors. In other words, the adult risk of disease is related to a ‘phenotype’ that is defined by interaction between the genotype and the environment. This adult phenotype is, however, also shaped by nutrient-gene interactions in adolescence, in childhood, infancy and in fetal life (Langley-Evans, 2004b). Thus, early-life programming is just one facet of the way in which adaptations to insults or stimuli at different life stages determine the adult physiology and metabolic profile and the responsiveness of the individual to metabolic or endocrine signals.
The interaction with prenatal and postnatal factors is well-illustrated by two examples in both man and animals. Eriksson et al. (2003a,b) have extensively reported on a unique cohort from Helsinki, Finland for which data is available detailing birth anthropometry, with follow-ups in childhood and adulthood. It is clear for this cohort that risk of type 2 diabetes is related to both prenatal and postnatal factors, with low birth weight having the greatest impact when coupled to catch-up growth in infancy and rapid weight gain in adolescence (Eriksson et al. 2003b). In rats severe restriction of maternal food intake in pregnancy (feeding 30% ad libitum intake) produces growth-retarded offspring (Woodall et al. 1996). When these offspring are fed a high-fat diet from weaning, massive central obesity and metabolic sequelae are noted (Vickers et al. 2000). This outcome is a perfect example of the thrifty phenotype hypothesis originally proposed by Hales & Barker (2001; Fig. 2) and subsequently revisited in the predictive adaptive response hypothesis of Gluckman & Hanson (2004).
Fig. 2.
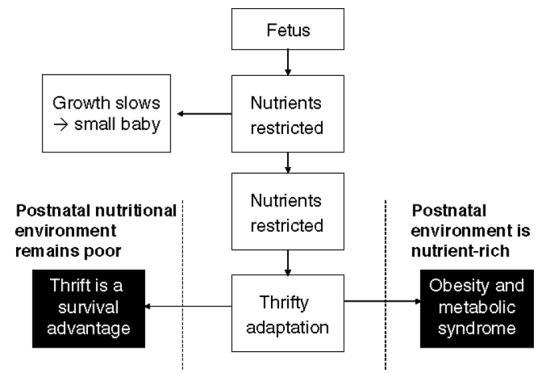
The thrifty phenotype hypothesis (Hales & Barker, 2001; Gluckman & Hanson, 2004). Exposure of the developing organism to a low plane of nutrition promotes metabolic thrift in order to ensure survival. In a postnatal environment in which nutrients are in short supply this metabolic thrift continues to be a survival trait, but if nutrients are present in excess the thrifty trait will promote the metabolic syndrome.
Programming in fetal life: animal studies
Manipulations of diet in pregnancy
Many approaches have been taken to study nutritional programming in animals. A commonly-favoured approach is to limit the food intake of pregnant or lactating animals (global nutrient-restriction models), which is known to impair fetal and neonatal growth (Woodall et al. 1996). More specific manipulations of the maternal diet include overfeeding of macronutrients (saturated fat, Khan et al. 2003; protein, Daenzer et al. 2002), restriction of macronutrient intake and restriction of the intake of specific micronutrients (Fe; Gambling et al. 2003, Zn; Beach et al. 1982, Ca; Bergel & Belizan, 2002). It is consistently noted in rats, mice and guinea-pigs that fetal exposure to anyform of undernutrition produces elevated blood pressure (Langley-Evans, 2004a). Similar observations in large animal species such as the sheep suggest that programming of cardiovascular function occurs in all mammals (Gopalakrishnan et al. 2004). Similar findings are noted when considering glucose intolerance and insulin resistance (Reusens et al. 2004), and a common finding in studies of animals subject to intrauterine nutritional restriction is that central adiposity is increased (Langley-Evans et al. 2005).
The largest programming effects of nutritional manipulations on the development of hypertension are observed when the maternal diet is modified to produce an imbalance in the nutrient supply. For example, the feeding of low-protein diets in rat pregnancy generates offspring that have a blood pressure that is 15-30 mmHg above that of control animals by the age of weaning (Langley-Evans et al. 1996d). Similarly, low-Fe diets (Gambling et al. 2003) or high-saturated-fat diets (Khan et al. 2003) in rat pregnancy produce large effects on the blood pressure of the offspring. Where the diet is manipulated in such a way that the intakes of all nutrients are reduced in a balanced manner during pregnancy the impact on the blood pressure of the subsequent offspring is still observed, but tends to be of a lower magnitude (Woodall et al. 1996; Kind et al. 2002). Despite the variation in the magnitude of the effects produced by different dietary protocols, there is a high extent of consistency in the type of physiological changes that stem from very different manipulations of the diet during pregnancy. This commonality of response suggests that there may be a relatively small number of common mechanisms that link the programming stimulus (maternal diet) to the associated disease outcomes.
Maternal low-protein diets
Experiments in the author’s laboratory have focused on the feeding of a maternal low-protein (MLP) diet in rat pregnancy. This approach, using a very mild nutritional intervention, has now been extensively characterised and it is known that the offspring of rats fed MLP will undergo a late-gestation retardation of growth, which particularly affects the development of the truncal organs such as the lungs and kidneys (Langley-Evans et al. 1996a). Although of low to normal birth weight, rats exposed to a MLP diet in fetal life develop raised blood pressure by 3-4 weeks of age and this relative hypertension persists into adult life (Langley-Evans et al. 1994; Langley-Evans & Jackson, 1995). In the postnatal period these animals have an accelerated progression towards renal failure (Nwagwu et al. 2000) and their lifespan is shorter than that of rats exposed to a protein-replete diet in fetal life (Sayer et al. 2001). Programming of lifespan may occur through an increased susceptibility to oxidative injury that has been noted in a number of tissues (Langley-Evans et al. 1997; Langley-Evans & Sculley, 2005).
The offspring of rats fed the MLP diet also display behavioural differences that have the capacity to promote positive energy balance and hence obesity. When fed a low-fat standard laboratory chow diet MLP offspring are hypophagic yet still maintain a normal body weight. When allowed to self-select from a range of foods rich in either fat, protein or carbohydrate the MLP offspring increase food intake markedly and show a preference for high-fat foods (Bellinger et al. 2004). As with other features of this model this behaviour differs between males and females, with females showing the greater fat preference. As has also been reported by Vickers et al. (2003) following global nutrient restriction, MLP-exposed offspring are less physically active and are prone to increased fat deposition with ageing. These effects are to some extent dependent on the timing of the protein restriction in utero.
Mechanisms of nutritional programming
Tissue remodelling
The simplest process through which developmental insults could exert permanent effects on physiology, metabolism and health is through alteration of tissue morphology. Changes to the numbers of cells or the type of cells present within a tissue could have profound effects on organ function. For example, within the kidney the principal functional unit is the nephron. The number of nephrons in man is determined before birth, whilst in rats nephrogenesis continues until postnatal day 10 (Mackenzie et al. 1996). Factors that limit nephron formation will impair renal function, raise local and systemic blood pressure and ultimately promote renal failure. In the MLP rat model of nutritional programming kidney size is largely unaffected by prenatal nutritional insult but nephron number is reduced by as much as 30% (Marchand & Langley-Evans, 2001). The decrease in functional units alongside normal tissue mass indicates that specialised cell types comprising the nephron have been replaced by non-specialised lineages.
This form of remodelling could occur as a result of disruption of cell proliferation or differentiation at key developmental stages. All tissues and organs are essentially derived from small populations of embryonic progenitor cell lines. These cell lines proliferate in the embryonic and fetal periods and differentiate into specialised forms as organs mature. It is easy to envisage that a lack of nutrients or key signals during these developmental stages can have irreversible consequences.
The consequences of tissue remodelling can be far-reaching. Changing the numbers and types of cells present within a tissue will impact not only on specialised functions, such as in the case of the kidney, but will also change the profile of genes expressed within a tissue, will alter the cell-cell signalling pathways, modify hormone production and the capacity of cells to respond to hormone signals (for example, by changing expression of receptors such as the glucocorticoid receptor; Bertram et al. 2001). This impact is well illustrated by the endocrine pancreas in which, in the rat, development is sensitive to the level of protein in the diet. Low-protein diets result in a pancreas with reduced numbers of islets, which are smaller and less effectively vascularised than in control animals (Snoeck et al. 1990). This outcome has a major impact on insulin production and glucose homeostasis (Dahri et al. 1990).
Disruption of homeostasis will be most profound if prenatal undernutrition impacts on the hypothalamus, which is the interface between sensory inputs and the adjusting endocrine and neuronal outputs. There is evidence that the hypothalamus can be programmed at a gross tissue remodelling level. Exposure of rats to low-protein diets in both the fetal and suckling period alters the volume of key hypothalamic centres and changes the neuronal density (Plagemann et al. 2000). Accordingly, there are shifts in the profile of neuropeptides produced at these centres, which have been proposed to impact on feeding behaviour. Programming of homeostatic mechanisms in this way fits well with the life-course model of disease risk outlined earlier, as clearly this mechanism will influence the capacity of the individual to respond to the environment subsequent to the early developmental insult.
Materno-fetal endocrine exchange
The placenta is more than a conduit for the exchange of nutrients, gases and waste products between mother and fetus. Endocrine signals between placenta and fetus and between mother and placenta play a critical role in the regulation of fetal development and nutrient partitioning (Godfrey, 2002). Some hormonal exchanges require tight regulation, however, in order to avoid in appropriate fetal responses.
Glucocorticoids are steroid hormones and therefore have the capacity to move freely across the placenta through simple diffusion. Glucocorticoids are powerful modulators of gene expression, and experimental studies have shown that they accelerate fetal organ maturation, a property exploited clinically where premature delivery is likely. In all species there is a massive gradient of glucocorticoid concentrations across the placenta, with maternal concentrations being 100-1000 times greater than those seen in the fetal circulation. This gradient is maintained, and the independence of the developing fetal hypothalamic-pituitary-adrenal axis assured, by the presence of the enzyme 11β-hydroxysteroid dehydrogenase type 2 in the placenta (Edwards et al. 1993). This gatekeeper enzyme is believed to be critical in protecting the fetal tissues from inappropriately high concentrations of the active corticosteroids.
A role for overexposure of the fetus to glucocorticoids of maternal origin has been proposed as a key step in nutritional programming. The administration of synthetic glucocorticoids that are poor substrates of 11β-hydroxysteroid dehydrogenase type 2 programmes hypertension and renal defects in animals (Benediktsson et al. 1993; Dodic et al. 2002). Studies with the MLP rat model, however, show that down-regulation of 11β-hydroxysteroid dehydrogenase type 2 by undernutrition may be the common pathway through which a broad range of nutritional insults produce a narrow and similar range of programmed responses. The feeding of low-protein diets in rat pregnancy reduces both the activity and mRNA expression of placental 11β-hydroxysteroid dehydrogenase type 2 (Langley-Evans et al 1996c; Bertram et al. 2001). Moreover, blockade of maternal glucocorticoid synthesis through pharmacological adrenalectomy prevents the programming of hypertension in the offspring of MLP-fed rats, demonstrating the glucocorticoid-dependence of the nutritional effect (Langley-Evans et al. 1996b; Langley-Evans, 1997a; McMullen & Langley-Evans, 2005). 11β-hydroxysteroid dehydrogenase type 2 inhibition programmes high blood pressure in the offspring of protein-replete animals (Langley-Evans, 1997b).
Epigenetic mechanisms
One particularly favoured explanation for the association between nutritional variation in early life and long-term physiological functions lies with the epigenetic modification of gene expression (Razin, 1998). Mechanisms such as DNA methylation or histone acetylation effectively silence gene expression, and recent animal studies have shown that the effects of nutrition can be quite selective, with variation in the provision of potential methyl donors in the diet during pregnancy impacting on the expression of transposable elements in the Agouti locus (Waterland & Jirtle, 2003). Furthermore, the window of sensitivity for this epigenetic modification of gene expression appears to extend far beyond the embryonic period, which was originally proposed as the only time at which this form of gene silencing might be sensitive to the environment. Stress-induced behaviours of rat mothers during lactation have been shown to alter DNA methylation in their suckling young (Weaver et al. 2004).
The overall level and pattern of DNA methylation in the developing embryo or fetus is governed by flux through the methionine-homocysteine pathway, suggesting that it is sensitive to nutritional factors such as the supply of amino acids, folic acid, vitamin B12 and vitamin B6 (Young et al. 2004). The low-protein diet protocol established in the author’s laboratory has been suggested to provide methionine at a higher concentration than that required by the pregnant animal (Rees, 2002), which may disturb the balance of the methionine-homocysteine cycle and, therefore, produce programmed effects through changes in DNA methylation status (Petrie et al. 2002). However, there is no strong evidence to support this assertion and it has recently been shown that maternal and fetal plasma homocysteine concentrations are not markedly altered by the low-protein feeding (Lilley & Langley-Evans, 2005). This finding does not exclude the possibility of epigenetic change, as recently demonstrated by Lillycrop et al. (2005).
Gene expression
There are now many reports of changes in gene expression following a prenatal or early postnatal nutritional insult. For example, it has been reported that expression of the angiotensin II AT2 receptor is modified by feeding a low-protein diet in utero (McMullen et al. 2004), and the processes that bring about this change and its downstream consequences for blood pressure control and renal function are being explored (McMullen & Langley-Evans, 2005). Furthermore, DNA microarray studies have shown that the expression of 102 genes in the hypothalamus and thirty-six genes in the kidney are modified by intrauterine protein restriction (Langley-Evans et al. 2005).
The fundamental problem with these findings is that it is almost impossible to assess whether the changes in gene expression are a cause of abnormal physiology and disease or whether they are a consequence. Furthermore, is it not clear whether the changes in gene expression are related to tissue remodelling, effects of glucocorticoids or even epigenetic silencing. In the case of the AT2 receptor it is known that expression is controlled by glucocorticoids (McMullen & Langley-Evans, 2005) and levels of expression both determine the development of specialised structures within the kidney and are regulated by the presence of those specialised structures. The situation is extremely complex and detailed studies are required that consider factors such as the timing of nutritional insult and the ontological stage at which tissues are sampled for the expression studies.
Future directions
Epidemiological studies clearly show that early-life events determine risk of CHD, with robust associations between CHD mortality and birth anthropometry. Whilst animal studies have reproducibly demonstrated that blood pressure and vascular reactivity are subject to nutritional programming, these end points are relatively crude markers of CVD and in man only represent simple risk factors for CHD. One approach to the study of CHD programming in animals has been to study the response of the isolated heart to ischaemic-reperfusion injury, essentially modelling the impact of a myocardial infarction (Sutherland & Hearse, 2000). Although at an early stage, the author’s studies using the Langendorff isolated heart system indicate that exposure to a low-protein diet in utero, impairs the ability of the hearts of male rats to recover function following 30 min of ischaemia (Elmes et al. 2005).
More convincing evidence of nutritional programming and the relevance of any identified mechanisms to human health could be generated if models could be developed in which atherosclerosis could be programmed by undernutrition in pregnancy. Wild-type rats, mice and hamsters are all relatively resistant to the development of atherosclerosis, which has in the past limited the scope of programming studies. However, there are now a variety of GM strains of mouse that are much more prone to the development of aortic atherosclerotic lesions. Many of these strains, e.g. the LDL receptor knock-out mouse (Fazio & Linton, 2001), will develop lesions without the need for any nutritional stimulus, which limits their usefulness in studies that consider the interaction between prenatal programming influences and the postnatal diet. The generation of strains in which development of atherosclerosis is conditional on diet, such as the apo E*3 Leiden mouse that develops atherosclerotic lesions only in response to high-fat high-cholesterol feeding (Groot et al. 1996) provides a unique opportunity to improve models of nutritional programming of atherosclerosis.
As described earlier, nutritional remodelling of tissue morphology and the associated changes to specialised cell types and associated physiological function may be an important mediator of programming effects on disease susceptibility. Vulnerability to CVD may be programmed in utero through disturbance of normal vascular development. The vascular system develops at a very early stage in embryonic organogenesis. As exposure to low-protein diets for very short periods in early pregnancy has been shown to induce hypertension in the offspring (Kwong et al. 2000), it is possible that the very early formation of vascular lineages is compromised by undernutrition. Transgenic mouse strains are available that allow this aspect to be easily explored and these strains may provide a productive approach for future research. An example is the Tie2 GFP mouse, which expresses green fluorescent protein in vascular endothelial cells (Motoike et al. 2000).
Conclusion
Nutritional factors operating in early life exert strong effects on physiology and metabolism in adult life. Experimental studies that have the capacity to examine nutrient-gene interactions during this period of developmental plasticity are of fundamental importance in expanding the understanding of how human disease may be programmed in fetal life. It is likely that over the coming decade these studies will indicate that the importance of nutritional programming is greater than predicted from earlier epidemiological studies and identify novel therapies and preventive strategies that will be important in a public health context.
Acknowledgements
I am proud to accept the 2005 Nutrition Society Silver Medal and the recognition of the Society for my work in the field described in the present review. Of course, none of this work would have been possible without the help and support of others, and I would like to express my gratitude to a number of individuals. The contributions made by all those who have worked in my laboratory over the last 10 years have been invaluable, as has been the support of all our collaborators. Special thanks go to Professor Alan Jackson for all his ideas, support and encouragement, which fuelled my development as an independent researcher. I am grateful to Professor Peter Buttery for his helpful input and encouragement, which has helped me to broaden the scope and strategic importance of my work.
Abbreviation
MLP
maternal low-protein
References
- Barker DJ. The developmental origins of chronic adult disease. Acta Paediatrica. 2004;93(Suppl):26–33. doi: 10.1111/j.1651-2227.2004.tb00236.x. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Bull AR, Osmond C, Simmonds SJ. Fetal and placental size and risk of hypertension in adult life. British Medical Journal. 1990;301:259–262. doi: 10.1136/bmj.301.6746.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993a;36:62–67. doi: 10.1007/BF00399095. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;i:1077–1081. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Simmonds SJ, Wield GA. The relation of small head circumference and thinness at birth to death from cardiovascular disease in adult life. British Medical Journal. 1993b;306:422–426. doi: 10.1136/bmj.306.6875.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;ii:577–580. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- Beach RS, Gershwin ME, Hurley LS. Gestational zinc deprivation in mice: persistence of immunodeficiency for three generations. Science. 1982;218:469–471. doi: 10.1126/science.7123244. [DOI] [PubMed] [Google Scholar]
- Bellinger L, Lilley CL, Langley-Evans SC. Prenatal exposure to a maternal low-protein diet programmes a preference for high-fat foods in the young adult rat. British Journal of Nutrition. 2004;92:513–520. doi: 10.1079/bjn20041224. [DOI] [PubMed] [Google Scholar]
- Benediktsson R, Lindsay RS, Noble J, Seckl JR, Edwards CR. Glucocorticoid exposure in utero: new model for adult hypertension. Lancet. 1993;341:339–341. doi: 10.1016/0140-6736(93)90138-7. [DOI] [PubMed] [Google Scholar]
- Bergel E, Belizan JM. A deficient maternal calcium intake during pregnancy increases blood pressure of the offspring in adult rats. British Journal of Obstetrics and Gynaecology. 2002;109:540–545. [PubMed] [Google Scholar]
- Bertram C, Trowern AR, Copin N, Jackson AA, Whorwood CB. The maternal diet during pregnancy programs altered expression of the glucocorticoid receptor and type 2 11beta-hydroxysteroid dehydrogenase: potential molecular mechanisms underlying the programming of hypertension in utero. Endocrinology. 2001;142:2841–2853. doi: 10.1210/endo.142.7.8238. [DOI] [PubMed] [Google Scholar]
- Campbell DM, Hall MH, Barker DJ, Cross J, Shiell AW, Godfrey KM. Diet in pregnancy and the offspring’s blood pressure 40 years later. British Journal of Obstetrics and Gynaecology. 1996;103:273–280. doi: 10.1111/j.1471-0528.1996.tb09718.x. [DOI] [PubMed] [Google Scholar]
- Daenzer M, Ortmann S, Klaus S, Metges CC. Prenatal high protein exposure decreases energy expenditure and increases adiposity in young rats. Journal of Nutrition. 2002;132:142–144. doi: 10.1093/jn/132.2.142. [DOI] [PubMed] [Google Scholar]
- Dahri S, Snoeck A, Reusens-Billen B, Remacle C, Hoet JJ. Islet function in offspring of mothers on low protein diet during gestation. Diabetes. 1990;40:115–120. doi: 10.2337/diab.40.2.s115. [DOI] [PubMed] [Google Scholar]
- Dodic M, Abouantoun T, O’Connor A, Wintour EM, Moritz KM. Programming effects of short prenatal exposure to dexamethasone in sheep. Hypertension. 2002;40:729–734. doi: 10.1161/01.hyp.0000036455.62159.7e. [DOI] [PubMed] [Google Scholar]
- Edwards CR, Benediktsson R, Lindsay RS, Seckl JR. Dysfunction of placental glucocorticoid barrier: link between fetal environment and adult hypertension. Lancet. 1993;341:355–357. doi: 10.1016/0140-6736(93)90148-a. [DOI] [PubMed] [Google Scholar]
- Elmes MJ, Gardner DS, Langley-Evans SC. Prenatally programmed hypertension and its effects on the left ventricular pressure (LVP) function of the rat heart following ischemia reperfusion. Proceedings of the Nutrition Society. 2005;64:82A. [Google Scholar]
- Eriksson J, Forsen T, Osmond C, Barker D. Obesity from cradle to grave. International Journal of Obesity. 2003a;27:722–727. doi: 10.1038/sj.ijo.0802278. [DOI] [PubMed] [Google Scholar]
- Eriksson JG, Forsen T, Tuomilehto J, Osmond C, Barker DJ. Early adiposity rebound in childhood and risk of Type 2 diabetes in adult life. Diabetologia. 2003b;46:190–194. doi: 10.1007/s00125-002-1012-5. [DOI] [PubMed] [Google Scholar]
- Fall CH, Osmond C, Barker DJ, Clark PM, Hales CN, Stirling Y, Meade TW. Fetal and infant growth and cardio-vascular risk factors in women. British Medical Journal. 1995;310:428–432. doi: 10.1136/bmj.310.6977.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio S, Linton MF. Mouse models of hyperlipidemia and atherosclerosis. Frontiers in Biosciences. 2001;6:D515–D525. doi: 10.2741/fazio. [DOI] [PubMed] [Google Scholar]
- Gambling L, Dunford S, Wallace DI, Zuur G, Solanky N, Srai SK, McArdle H. Iron deficiency during pregnancy affects postnatal blood pressure in the rat. Journal of Physiology. 2003;552:603–610. doi: 10.1113/jphysiol.2003.051383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. The developmental origins of the metabolic syndrome. Trends in Endocrinology and Metabolism. 2004;15:183–187. doi: 10.1016/j.tem.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Godfrey KM. Maternal regulation of fetal development and health in adult life. European Journal of Obstetrics Gynecology and Reproductive Biology. 1998;78:141–150. doi: 10.1016/s0301-2115(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Godfrey KM. The role of the placenta in fetal programming - a review. Placenta. 2002;23(Suppl A):S20–S27. doi: 10.1053/plac.2002.0773. [DOI] [PubMed] [Google Scholar]
- Godfrey KM, Forrester T, Barker DJ, Jackson AA, Landman JP, Hall JS, Cox V, Osmond C. Maternal nutritional status in pregnancy and blood pressure in childhood. British Journal of Obstetrics and Gynaecology. 1994;101:398–403. doi: 10.1111/j.1471-0528.1994.tb11911.x. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan GS, Gardner DS, Rhind SM, Rae MT, Kyle CE, Brooks AN, Walker RM, Ramsay MM, Keisler DH, Stephenson T, Symonds ME. Programming of adult cardiovascular function after early maternal undernutrition in sheep. American Journal of Physiology. 2004;287:R12–R20. doi: 10.1152/ajpregu.00687.2003. [DOI] [PubMed] [Google Scholar]
- Groot PH, van Vlijmen BJ, Benson GM, Hofker MH, Schiffelers R, Vidgeon-Hart M, Havekes LM. Quantitative assessment of aortic atherosclerosis in APOE*3 Leiden transgenic mice and its relationship to serum cholesterol exposure. Arteriosclerosis, Thrombosis, and Vascular Biology. 1996;16:926–933. doi: 10.1161/01.atv.16.8.926. [DOI] [PubMed] [Google Scholar]
- Hales CN, Barker DJP. The thrifty phenotype hypothesis. British Medical Bulletin. 2001;60:5–20. doi: 10.1093/bmb/60.1.5. [DOI] [PubMed] [Google Scholar]
- Hales CN, Barker DJP, Clark PMS, Cox LJ, Fall C, Osmond C, Winter PD. Fetal and infant growth and impaired glucose-tolerance at age 64. British Medical Journal. 1991;303:1019–1022. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley R, Neil A, Collins R. Unravelling the fetal origins hypothesis: is there really an inverse association between birthweight and subsequent blood pressure. Lancet. 2002;360:659–665. doi: 10.1016/S0140-6736(02)09834-3. [DOI] [PubMed] [Google Scholar]
- Huxley RR, Neil NA. Does maternal nutrition in pregnancy and birth weight influence levels of CHD risk factors in adult life. British Journal of Nutrition. 2004;91:459–468. doi: 10.1079/BJN20031052. [DOI] [PubMed] [Google Scholar]
- Khan IY, Taylor PD, Dekou V, Seed PT, Lakasing L, Graham D, Dominiczak AF, Hanson MA, Poston L. Gender-linked hypertension in offspring of lard-fed pregnant rats. Hypertension. 2003;41:168–175. doi: 10.1161/01.hyp.0000047511.97879.fc. [DOI] [PubMed] [Google Scholar]
- Kind KL, Simonetta G, Clifton PM, Robinson JS, Owens JA. Effect of maternal feed restriction on blood pressure in the adult guinea pig. Experimental Physiology. 2002;87:469–477. doi: 10.1111/j.1469-445x.2002.tb00060.x. [DOI] [PubMed] [Google Scholar]
- Kramer MS, Joseph KS. Enigma of fetal/infant-origins hypothesis. Lancet. 1996;348:1254–1255. doi: 10.1016/s0140-6736(05)65750-9. [DOI] [PubMed] [Google Scholar]
- Kwong WY, Wild AE, Roberts P, Willis AC, Fleming TP. Maternal undernutrition during the preimplantation period of rat development causes blastocyst abnormalities and programming of postnatal hypertension. Development. 2000;127:4195–4202. doi: 10.1242/dev.127.19.4195. [DOI] [PubMed] [Google Scholar]
- Langley-Evans AJ, Langley-Evans SC. Relationship between maternal nutrient intakes in early and late pregnancy and infants weight and proportions at birth. Journal of the Royal Society for the Promotion of Health. 2003;123:210–216. doi: 10.1177/146642400312300409. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC. Hypertension induced by foetal exposure to a maternal low-protein diet, in the rat, is prevented by pharmacological blockade of maternal glucocorticoid synthesis. Journal of Hypertension. 1997a;15:537–544. doi: 10.1097/00004872-199715050-00010. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC. Maternal carbenoxolone treatment lowers birthweight and induces hypertension in the offspring of rats fed a protein-replete diet. Clinical Science. 1997b;93:423–429. doi: 10.1042/cs0930423. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, editor. Fetal Nutrition and Adult Disease: Programming of Chronic Disease through Fetal Exposure to Undernutrition. CABI; Wallingford, Oxon.: 2004a. Experimental models of hypertension and cardiovascular disease; pp. 129–156. [Google Scholar]
- Langley-Evans SC, editor. Fetal Nutrition and Adult Disease: Programming of Chronic Disease through Fetal Exposure to Undernutrition. CABI; Wallingford, Oxon.: 2004b. Fetal programming of adult disease: an overview; pp. 1–20. [Google Scholar]
- Langley-Evans SC, Bellinger L, McMullen S. Animal models of programming: Early life influences on appetite and feeding behaviour. Maternal and Child Nutrition. 2005 doi: 10.1111/j.1740-8709.2005.00015.x. (In the Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley-Evans SC, Gardner DS, Jackson AA. Association of disproportionate growth of fetal rats in late gestation with raised systolic blood pressure in later life. Journal of Reproduction and Fertility. 1996a;106:307–312. doi: 10.1530/jrf.0.1060307. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Jackson AA. Captopril normalises systolic blood pressure in rats with hypertension induced by fetal exposure to maternal low protein diets. Comparative Biochemistry and Physiology. 1995;110A:223–228. doi: 10.1016/0300-9629(94)00177-u. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Phillips GJ, Jackson AA. In utero exposure to maternal low protein diets induces hypertension in weanling rats, independently of maternal blood pressure changes. Clinical Nutrition. 1994;13:319–324. doi: 10.1016/0261-5614(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Phillips GJ, Jackson AA. Fetal exposure to low protein maternal diet alters the susceptibility of young adult rats to sulfur dioxide-induced lung injury. Journal of Nutrition. 1997;127:202–209. doi: 10.1093/jn/127.2.202. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Phillips GJ, Gardner DS, Jackson AA. Role of glucocorticoids in programming of maternal-diet-induced hypertension. Journal of Nutritional Biochemistry. 1996b;7:173–178. [Google Scholar]
- Langley-Evans SC, Phillips GJ, Benediktsson R, Gardner DS, Edwards CR, Jackson AA, Seckl JR. Protein intake in pregnancy, placental glucocorticoid metabolism and the programming of hypertension in the rat. Placenta. 1996c;17:169–172. doi: 10.1016/s0143-4004(96)80010-5. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Sculley DV. Programming of hepatic antioxidant capacity and oxidative injury in the ageing rat. Mechanisms of Ageing and Development. 2005;126:804–812. doi: 10.1016/j.mad.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Welham SJM, Sherman RC, Jackson AA. Weanling rats exposed to maternal low protein diets during discrete periods of gestation exhibit differing severity of hypertension. Clinical Science. 1996d;91:607–615. doi: 10.1042/cs0910607. [DOI] [PubMed] [Google Scholar]
- Law CM. Significance of birth weight for the future. Archives of Disease in Childhood. 2002;86:F7–F8. doi: 10.1136/fn.86.1.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CM, Barker DJ, Bull AR, Osmond C. Maternal and fetal influences on blood pressure. Archives of Disease in Childhood. 1991;66:1291–1295. doi: 10.1136/adc.66.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley C, Langley-Evans SC. Fetal programming of adult disease may be induced by oxidative damage and alterations in antioxidant activity. Proceedings of the Nutrition Society. 2005;64:81A. [Google Scholar]
- Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. Journal of Nutrition. 2005;135:1382–1386. doi: 10.1093/jn/135.6.1382. [DOI] [PubMed] [Google Scholar]
- Lucas A. Programming by early nutrition in man. Ciba Foundation Symposium. 1991;156:38–50. [PubMed] [Google Scholar]
- McMullen S, Langley-Evans SC. Maternal low-protein diet in rat pregnancy programs blood pressure through sex-specific mechanisms. American Journal of Physiology. 2005;288:R85–R90. doi: 10.1152/ajpregu.00435.2004. [DOI] [PubMed] [Google Scholar]
- McMullen S, Gardner DS, Langley-Evans SC. Prenatal programming of angiotensin II type 2 receptor expression in the rat. British Journal of Nutrition. 2004;91:133–140. doi: 10.1079/bjn20031029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie HS, Lawler EV, Brenner BM. Congenital oligonephropathy: The fetal flaw in essential hypertension. Kidney International. 1996;55(Suppl):S30–S34. [PubMed] [Google Scholar]
- Marchand MC, Langley-Evans SC. Intrauterine programming of nephron number: the fetal flaw revisited. Journal of Nephrology. 2001;14:327–331. [PubMed] [Google Scholar]
- Mathews F, Yudkin P, Neil A. Influence of maternal nutrition on outcome of pregnancy: prospective cohort study. British Medical Journal. 1999;319:339–343. doi: 10.1136/bmj.319.7206.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoike T, Loughna S, Perens E, Roman BL, Liao W, Chau TC, et al. Universal GFP reporter for the study of vascular development. Genesis. 2000;28:75–81. doi: 10.1002/1526-968x(200010)28:2<75::aid-gene50>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Nwagwu MO, Cook A, Langley-Evans SC. Evidence of progressive deterioration of renal function in rats exposed to a maternal low protein diet in utero. British Journal of Nutrition. 2000;83:79–85. [PubMed] [Google Scholar]
- Osmond C, Barker DJ, Slattery JM. Risk of death from cardiovascular disease and chronic bronchitis determined by place of birth in England and Wales. Journal of Epidemiology and Community Health. 1990;44:139–141. doi: 10.1136/jech.44.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter RC, Roseboom TJ, van Montfrans GA, Bossuyt PM, Krediet RT, Osmond C, Barker DJ, Bleker OP. Microalbuminuria in adults after prenatal exposure to the Dutch famine. Journal of the American Society for Nephrology. 2005;16:189–194. doi: 10.1681/ASN.2004060474. [DOI] [PubMed] [Google Scholar]
- Petrie L, Duthie SJ, Rees WD, McConnell JM. Serum concentrations of homocysteine are elevated during early pregnancy in rodent models of fetal programming. British Journal of Nutrition. 2002;88:471–477. doi: 10.1079/BJN2002695. [DOI] [PubMed] [Google Scholar]
- Phillips DIW, Barker DJP, Hales CN, Hirst S, Osmond C. Thinness at birth and insulin resistance in adult life. Diabetologia. 1994;37:150–154. doi: 10.1007/s001250050086. [DOI] [PubMed] [Google Scholar]
- Plagemann A, Harder T, Rake A, Melchior K, Rohde W, Dorner G. Hypothalamic nuclei are malformed in weanling offspring of low protein malnourished rat dams. Journal of Nutrition. 2000;130:2582–2589. doi: 10.1093/jn/130.10.2582. [DOI] [PubMed] [Google Scholar]
- Ravelli AC, van der Meulen JH, Osmond C, Barker DJ, Bleker OP. Obesity at the age of 50 y in men and women exposed to famine prenatally. American Journal of Clinical Nutrition. 1999;70:811–816. doi: 10.1093/ajcn/70.5.811. [DOI] [PubMed] [Google Scholar]
- Ravelli AC, van der Meulen JH, Osmond C, Barker DJ, Bleker OP. Infant feeding and adult glucose tolerance, lipid profile, blood pressure, and obesity. Archives of Disease in Childhood. 2000;82:248–252. doi: 10.1136/adc.82.3.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin A. CpG methylation, chromatin structure and gene silencing - a three-way connection. EMBO Journal. 1998;17:4905–4908. doi: 10.1093/emboj/17.17.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees WD. Manipulating the sulfur amino acid content of the early diet and its implications for long-term health. Proceedings of the Nutrition Society. 2002;61:71–77. doi: 10.1079/pns2001137. [DOI] [PubMed] [Google Scholar]
- Reusens B, Kalbe L, Remacle C. Programming of diabetes: experimental models. In: Langley-Evans SC, editor. Fetal Nutrition and Adult Disease: Programming of Chronic Disease through Fetal Exposure to Undernutrition. CABI; Wallingford, Oxon.: 2004. pp. 171–194. [Google Scholar]
- Roseboom TJ, van der Meulen JH, Osmond C, Barker DJ, Ravelli AC, Schroeder-Tanka JM, van Montfrans GA, Michels RP, Bleker OP. Coronary heart disease after prenatal exposure to the Dutch famine, 1944-45. Heart. 2000;84:595–598. doi: 10.1136/heart.84.6.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Twin Research. 2001;4:293–298. doi: 10.1375/1369052012605. [DOI] [PubMed] [Google Scholar]
- Sayer AA, Dunn RL, Langley-Evans SC, Cooper C. Prenatal exposure to a maternal low protein diet shortens life span in rats. Gerontology. 2001;47:9–14. doi: 10.1159/000052764. [DOI] [PubMed] [Google Scholar]
- Snoeck A, Remacle C, Reusens B, Hoet JJ. Effect of a low protein diet during pregnancy on the fetal rat endocrine pancreas. Biology of the Neonate. 1990;57:107–118. doi: 10.1159/000243170. [DOI] [PubMed] [Google Scholar]
- Stanner SA, Yudkin JS. Fetal programming and the Leningrad Siege study. Twin Research. 2001;4:287–292. doi: 10.1375/1369052012498. [DOI] [PubMed] [Google Scholar]
- Stein A. Birthweight and the development of over-weight and obesity. In: Langley-Evans SC, editor. Fetal Nutrition and Adult Disease: Programming of Chronic Disease through Fetal Exposure to Undernutrition. CABI; Wallingford, Oxon.: 2004. pp. 195–210. [Google Scholar]
- Sutherland FJ, Hearse DS. The isolated blood and perfusion fluid perfused heart. Pharmacological Research. 2000;41:613–627. doi: 10.1006/phrs.1999.0653. [DOI] [PubMed] [Google Scholar]
- Vickers MH, Breier BH, Cutfield WS, Hofman PL, Gluckman PD. Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. American Journal of Physiology. 2000;279:E83–E87. doi: 10.1152/ajpendo.2000.279.1.E83. [DOI] [PubMed] [Google Scholar]
- Vickers MH, Breier BH, McCarthy D, Gluckman PD. Sedentary behavior during postnatal life is determined by the prenatal environment and exacerbated by postnatal hypercaloric nutrition. American Journal of Physiology. 2003;285:R271–R273. doi: 10.1152/ajpregu.00051.2003. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Molecular and Cellular Biology. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nature Neuroscience. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Woodall SM, Johnston BM, Breier BH, Gluckman PD. Chronic maternal undernutrition in the rat leads to delayed postnatal growth and elevated blood pressure of offspring. Pediatric Research. 1996;40:438–443. doi: 10.1203/00006450-199609000-00012. [DOI] [PubMed] [Google Scholar]
- Young L, Rees WD, Sinclair KD. Programming in the pre-implantation embryo. In: Langley-Evans SC, editor. Fetal Nutrition and Adult Disease: Programming of Chronic Disease through Fetal Exposure to Undernutrition. CABI; Wallingford, Oxon.: 2004. pp. 333–352. [Google Scholar]