Interferon γ Eliminates Responding Cd4 T Cells during Mycobacterial Infection by Inducing Apoptosis of Activated Cd4 T Cells (original) (raw)
Abstract
In Mycobacterium bovis Bacille Calmette-Guérin (BCG)-infected wild-type mice, there was a large expansion of an activated (CD44hi) splenic CD4 T cell population followed by a rapid contraction of this population to normal numbers. Contraction of the activated CD4 T cell population in wild-type mice was associated with increased apoptosis of activated CD4 T cells. In BCG-infected interferon (IFN)-γ knockout (KO) mice, the activated CD4 T cell population did not undergo apoptosis. These mice accumulated large numbers of CD4+CD44hi T cells that were responsive to mycobacterial antigens. Addition of IFN-γ to cultured splenocytes from BCG-infected IFN-γ KO mice induced apoptosis of activated CD4 T cells. IFN-γ–mediated apoptosis was abolished by depleting adherent cells or Mac-1+ spleen cells or by inhibiting nitric oxide synthase. Thus, IFN-γ is essential to a regulatory mechanism that eliminates activated CD4 T cells and maintains CD4 T cell homeostasis during an immune response.
Keywords: T lymphocytes, homeostasis, cell death, knockout mice, nitric oxide
Introduction
During immune responses, large numbers of activated T cell effectors are generated. The majority of these cells are eliminated by apoptosis to restore the T cell compartment to homeostasis 1. Three major pathways for terminating immune responses have been identified (for review see reference 2). Fas–Fas ligand (FasL) interactions mediate activation-induced cell death. CTLA-4 expression on T cells inhibits the proliferation of activated T cells. IL-2 promotes activation and then apoptosis of T cells. Mice lacking Fas, FasL, CTLA-4, IL-2, or IL-2R accumulate activated T cells and are susceptible to autoimmune disease 2 3. However, it is not clear whether these molecules play crucial roles in terminating normal immune responses, as the T cell population in Fas- and CTLA-4–deficient mice shows normal expansion and contraction after in vivo antigen stimulation 4 5. It is important to fully understand the mechanisms by which effector cells are eliminated after an immune response. Mechanisms for terminating immune responses may affect the generation of protective memory T cells and may eliminate potentially harmful activated T cells.
In an earlier study, IFN-γ was postulated to play a critical role in death of effector CD4 T cells as a possible mechanism for self-tolerance 6. However, this work was done entirely in vitro with a cloned T cell line. No in vivo evidence for such a role for IFN-γ was presented. IFN-γ has an essential role in stimulating macrophages to produce nitric oxide (NO) 7. Macrophages restrict the expansion of splenocytes in vitro to various stimuli by making NO 8. NO is an inducer of apoptosis in a variety of cell types, including T cell clones 9 10 11. However, it is evident from numerous recent reviews of T cell downregulation that IFN-γ and NO are not widely considered to have a role in T cell downregulation and homeostasis 2 12 13 14 15. This may be because there is no clear in vivo evidence that activated CD4 T cells fail to undergo apoptosis and therefore accumulate in mice lacking IFN-γ.
We observed that during Bacille Calmette-Guérin (BCG) infection of IFN-γ knockout (KO) mice, the CD4 T cell population expanded markedly to comprise 30–50% of total lymphocytes in the spleen and liver. Our investigation of this phenomenon led to novel evidence that IFN-γ KO mice fail to induce apoptosis of activated CD4 T cells during BCG infection, resulting in accumulation of activated CD4 T cells. We also found that IFN-γ induces apoptosis of activated CD4 T cells in vitro during BCG infection and that activated macrophages and NO are a part of the mechanism. Thus, IFN-γ is an essential component of a mechanism for terminating immune responses.
Materials and Methods
Infection of Mice.
Homozygous IFN-γ KO breeding pairs, backcrossed for 10 generations onto C57BL/6, were set up to generate IFN-γ KO mice 16. C57BL/6 mice (8–10 wk of age) were used as wild-type controls. 7-wk-old B6.MRL-Faslpr (lpr/lpr on C57BL/6) were from The Jackson Laboratory. Mycobacterium bovis BCG was from The Trudeau Institute Mycobacterial Collection (TM 1011), stored as a live, frozen suspension. Mice were injected intravenously through the lateral tail vein with 2 × 106 CFU of BCG in a volume of 0.2 ml. All investigations involving mice were approved by The Trudeau Institute Institutional Animal Care and Use Committee.
Flow Cytometric Analysis.
Cells from the spleens were stained with antibodies, collected on a FACSCalibur™ instrument (Becton Dickinson) and analyzed using CELLQuest™ software. The following conjugated antibodies were obtained from PharMingen or Caltag Labs.: anti-CD4–allophycocyanin (RM4-5), anti-CD44–PE (IM7), anti-CD45RB–PE (16A), anti-CD25–PE (3C7), and anti-CD62L–PE (MEL-14). Propidium iodide (PI; 0.25 μg) was added to each tube just before collection of 5,000–20,000 live cells.
Proliferation of CD4 T Cells to Antigen.
LPS-stimulated B cell blasts, 95% B220+ cells, mitomycin C treated and pulsed with antigen, were prepared as described 17. 80–90% pure CD4 T cells were enriched from the spleens of BCG-infected wild-type and IFN-γ KO mice by depleting CD8+, B220+, Mac-1+, Gr-1+, and MHC class II+ cells from the spleens with magnetic beads (Miltenyi Biotec). APCs and CD4 T cells, each at 2 × 106 cells/ml, were cultured in the presence of antigen at 10 μg/ml, Con A (3 μg/ml), or medium only, without IL-2. [3H]thymidine was added to the wells between 24 and 40 h of culture. Purified protein derivative of M. tuberculosis (PPD) was a gift of Dr. John Griffen (Mycos, Ft. Collins, CO). Antigen 85 protein complex (Ag 85; lot #97.Rv.2.4.2.8.Ag85) of M. tuberculosis was obtained from Colorado State University, produced under the National Institute of Allergy and Infectious Diseases contract N01 AI-75320. OVA and Con A were from Sigma-Aldrich.
Apoptosis Assay.
Cells (105) were surface stained with anti-CD4–allophycocyanin and anti-CD44–PE, washed, and then stained with FITC-labeled annexin V and PI (R & D Systems). At least 1,000 CD4+CD44hi events were collected for annexin/PI analysis. CD4+CD44hi cells in early apoptosis (annexin+PI−) were in the lower right quadrant of Fig. 3 A. Live cells (annexin−PI−) were in the lower left quadrant. Dead cells (annexin+PI+) were in the upper right quadrant.
Figure 3.

Measurement of apoptosis of CD4+CD44hi T cells ex vivo during BCG infection. (A) Typical dot plots of annexin V and PI staining of gated CD4+CD44hi T cells in spleens of wild-type and IFN-γ KO mice at 4 wk of BCG infection. (B) Kinetics of apoptosis of gated CD4+CD44hi T cells from the spleens of BCG-infected wild-type and IFN-γ KO mice. A gate was set on CD4+CD44hi T cells, and the percentage of apoptotic CD4+CD44hi T cells was determined by annexin V and PI staining. Numbers shown are the percentages of cells that are in the early stages of apoptosis. Each point is the average and SD of at least six to eight mice done in two to three or more separate experiments. (C) Apoptosis of gated CD4+CD44hi T cells from the spleens of BCG-infected wild-type, lpr/lpr (on C57BL/6), and IFN-γ KO mice; all three groups were compared in the same experiment each time. Numbers shown are percentages of CD4+CD44hi T cells in early apoptosis. Each point is the average and SD of six mice done in two separate experiments.
Induction of Apoptosis In Vitro with IFN-γ.
Splenocytes were cultured at a 5 × 106 cells/ml in 0.32-cm2 wells in T cell medium, which is RPMI 1640 with penicillin, streptomycin, glutamine, 2-ME, and 7.5% FBS (Hyclone), with recombinant murine IL-2 at 80 U/ml (R & D Systems). Purified recombinant mouse IFN-γ at 10 ng/ml (R & D Systems) was added to induce apoptosis. Induction of apoptosis was monitored over a 65-h time course using the annexin/PI assay. NG-methyl-l-arginine (LMNA) was obtained from Sigma-Aldrich and used at a concentration of 1 mM.
In some experiments, 99% of splenic Mac-1+ cells were removed by magnetic bead depletion using CD11b Microbeads (Miltenyi Biotec). CD4 T cells (80–95% pure) were enriched with CD4 Microbeads and positive selection columns (Miltenyi Biotec). Adherent cells were depleted by culture of whole spleen in tissue culture flasks for 2 h at 37°C, and then nonadherent cells were removed to another plate. Adherent PECs, 80–90% macrophages, were prepared from BCG-infected wild-type mice as described 18.
Results
Accumulation of Activated Splenic CD4 T Cells in IFN-γ KO Mice.
Wild-type and IFN-γ KO mice were infected intravenously with 2 × 106 CFU of BCG. This resulted in 3 × 105 CFU in the spleens at the time of infection. At 3 wk of BCG infection, wild-type spleens had an average of 3.9 × 106 CFU, and IFN-γ KO spleens had an average of 2.4 × 107 CFU. These organisms were not rapidly cleared in the wild-type mice, persisting at 1–3 × 105 CFU in the spleen for 8 wk. In IFN-γ KO mice, the BCG organisms grew in an uncontrolled manner due to an impairment of macrophage activation in the absence of IFN-γ 16.
We monitored the number of activated CD4 T cells in the spleens of mice during BCG infection using CD44hi as a marker for activation. When naive cells encounter antigen and are activated to become effectors, there is a stable upregulation of CD44 on the cell surface of effectors 19. After week 3 of BCG infection, the CD4 T cell population in the spleens of IFN-γ KO mice was highly skewed toward high levels of expression of CD44 (Fig. 1 A). The CD4+CD44hi T cells expressed low levels of CD62L and CD45RB, consistent with an activated phenotype 20.
Figure 1.

Kinetics of expansion and contraction of activated CD4 T cells during BCG infection. (A) Typical FACS® histogram plots of CD44 expression on live CD4 T cells of uninfected mice or BCG-infected wild-type and IFN-γ KO mice at 4.5 wk of BCG infection. The CD44 marker was always set on uninfected mice as shown. The CD4 T cell population of IFN-γ KO mice is extremely skewed toward high levels of expression of CD44. (B) Total number of CD4+CD44hi T cells in the spleens of wild-type and IFN-γ KO mice during BCG infection. Cells were stained and analyzed by flow cytometry to measure CD4 and CD44 expression. Shown is the total number of CD4+CD44hi T cells, calculated as: percentage of live cells that are CD4+CD44hi × total number of live splenocytes. Numbers shown are average and SD of six to eight mice per time point, done in two to three experiments per time point. (C) Kinetics of expression of high levels of CD44 on splenic CD4 T cells during BCG infection. Numbers shown are percentage of live CD4 T cells with CD44hi. This is the average and SD of six to eight mice per group, done in replicate experiments.
Fig. 1 B shows the kinetics of accumulation of splenic CD4+CD44hi T cells during BCG infection. The wild-type mice showed an ∼10-fold expansion of the activated CD4 T cell population at 3.5 wk of infection. This was followed by a rapid contraction of the population 1 wk later. In the IFN-γ KO mice, the activated CD4 T cell population expanded by 27-fold at 3.5 wk of infection. This population did not contract but accumulated to large numbers in the spleen for several weeks. A sharp decline in the number of activated CD4 T cells occurred at 8 wk of BCG infection in IFN-γ KO mice; this coincided with cell death in the necrotic spleen. Thus, the apparent deletion of activated CD4 T cells in IFN-γ KO mice was most likely a consequence of nonspecific cell death. Furthermore, the CD4 T cell population became increasingly skewed toward high levels of CD44 expression during the course of infection rather than returning to the homeostasis level of 25% CD44hi, as observed in BCG-infected wild-type mice (Fig. 1 C). There was no change in the number of naive CD4 T cells or CD8 T cells in the spleens of IFN-γ KO mice during BCG infection.
Increased CD4 T Cell Responsiveness to Mycobacterial Antigens in IFN-γ KO Mice.
A large expansion of the CD4+ CD44hi T cell population was never seen in uninfected IFN-γ KO mice but occurred only in response to BCG infection. We investigated whether this large population of activated CD4 T cells in IFN-γ KO mice was responsive to mycobacterial antigens. Enriched CD4 T cells from IFN-γ KO mice showed greatly increased proliferation, incorporating four to six times more [3H]thymidine in response to both PPD and Ag 85 compared with CD4 T cells from wild-type mice (Fig. 2). Wild-type and IFN-γ KO CD4 T cells proliferated equally well to the T cell mitogen Con A (not shown), and both failed to proliferate to an irrelevant antigen, OVA. These data and the increased CD4+CD44hi T cell numbers suggest that there were increased numbers of mycobacterial antigen–specific CD4 T cells in the spleens of BCG-infected IFN-γ KO mice compared with BCG-infected wild-type mice. This is consistent with a failure to delete antigen-specific CD4 T cells during the immune response to BCG in IFN-γ KO mice.
Figure 2.

Comparison of the proliferative response of enriched CD4 T cells to mycobacterial antigens. Shown is the Δ cpm: cpm of CD4 T cells cultured with APCs prepulsed with 10 μg/ml antigen − cpm of CD4 T cells incubated with APCs and no antigens.
Activated CD4 T Cells Underwent Increased Apoptosis in Wild-Type but not IFN-γ KO Mice.
We considered the possibility that the failure of the activated CD4 T cell population to contract in IFN-γ KO mice might reflect a failure of these cells to undergo apoptosis. We measured apoptosis of activated splenic CD4 T cells during BCG infection with a quantitative flow cytometry assay that uses annexin V–FITC 21. The activated CD4 T cell population in BCG-infected wild-type mice typically had more than twice as many cells in early apoptosis as the same population in BCG-infected IFN-γ KO mice (Fig. 3 A).
We measured the kinetics of apoptosis of activated CD4 T cells during BCG infection (Fig. 3 B). On day 17 of BCG infection, apoptosis of the activated CD4 T cell population in wild-type mice increased dramatically to 45–50%, remaining high for at least 8 wk. The activated CD4 T cell population of IFN-γ KO mice did not show any increase in apoptosis over baseline levels during BCG infection. These data indicate that IFN-γ is required, either directly or indirectly, for upregulating apoptosis of activated CD4 T cells during BCG infection.
Fas Was Not Required for Upregulation of Apoptosis of Activated CD4 T Cells.
We investigated whether Fas was involved in this IFN-γ–dependent pathway of apoptosis. We infected wild-type, IFN-γ KO, and Fas-deficient lpr/lpr mice with BCG and monitored the kinetics of apoptosis of activated CD4 T cells. Interestingly, the lpr/lpr mice behaved identically to wild-type mice, exhibiting a surge of apoptosis of activated CD4 T cells on day 17 (Fig. 3 C). Apoptosis of activated CD4 T cells in IFN-γ KO mice was significantly lower than that observed in lpr/lpr and wild-type mice on days 17 and 28 after BCG infection. Thus, increased apoptosis of activated CD4 T cells during BCG infection required IFN-γ but did not require Fas.
IFN-γ Induced Apoptosis of Activated CD4 T Cells In Vitro.
We investigated whether adding exogenous IFN-γ to spleen cells from BCG-infected IFN-γ KO mice could induce apoptosis of CD4+CD44hi T cells (Fig. 4). Addition of IFN-γ to cultures of BCG-infected IFN-γ KO mice induced significantly increased apoptosis of CD4+CD44hi T cells as early as 22 h, increased at 45 h, but was most dramatic at 65 h (Fig. 4). We obtained similar results measuring apoptosis of CD4+CD44hi T cells by decreased forward and increased side light scatter 22 and by using the TUNEL (TdT-mediated dUTP-biotin nick-end labeling) assay for apoptosis (data not shown).
Figure 4.

Induction of apoptosis of CD4+CD44hi T cells in vitro by IFN-γ. Spleen cells from BCG-infected wild-type and IFN-γ KO mice, week 3 or 4 after infection, were cultured in medium with IL-2, with or without IFN-γ. A gate was set on CD4+CD44hi T cells, and the percentage of cells that were live, apoptotic, and dead at 65 h of culture was determined from the annexin V/PI dot plots. Each bar is the average and SD of six mice per group, done in two different experiments. Statistical significance (t test) between groups is shown.
During the 65 h of culture, wild-type activated CD4 T cells showed greater apoptosis and death than the same cells in IFN-γ KO cultures (compare wild-type versus IFN-γ KO cultures in medium, Fig. 4). In wild-type cultures, addition of IFN-γ did not significantly increase apoptosis of CD4+CD44hi T cells (Fig. 4). These wild-type CD4 T cells may be programmed to die during the culture period by signals they received in vivo.
Activated Macrophages Were Required for IFN-γ to Induce Apoptosis of Activated CD4 T Cells.
IFN-γ did not kill highly enriched CD4 T cells but did kill CD4+CD44hi T cells in whole spleen cultures (Fig. 5 A), suggesting that the effect of IFN-γ was indirect. Mac-1 or CD11b is expressed at high levels on differentiated macrophages 23. In Mac-1–depleted cultures, addition of IFN-γ did not kill activated CD4 T cells. Similarly, depletion of adherent cells greatly decreased the cytotoxic effects of IFN-γ (Fig. 5 A). Adherent peritoneal exudate cells (PECs) from wild-type BCG-infected mice are highly enriched in activated macrophages and have been shown to have cytotoxic effects in vitro on tumor cells 24. We cultured wild-type PECs with purified IFN-γ KO CD4 T cells (Fig. 5 A). As the wild-type PECs had most likely been exposed to IFN-γ in vivo, they reduced the viability of the CD4+CD44hi T cells even without addition of IFN-γ. However, wild-type PECs killed significantly more IFN-γ KO CD4+CD44hi T cells with exogenously added IFN-γ (Fig. 5 A).
Figure 5.
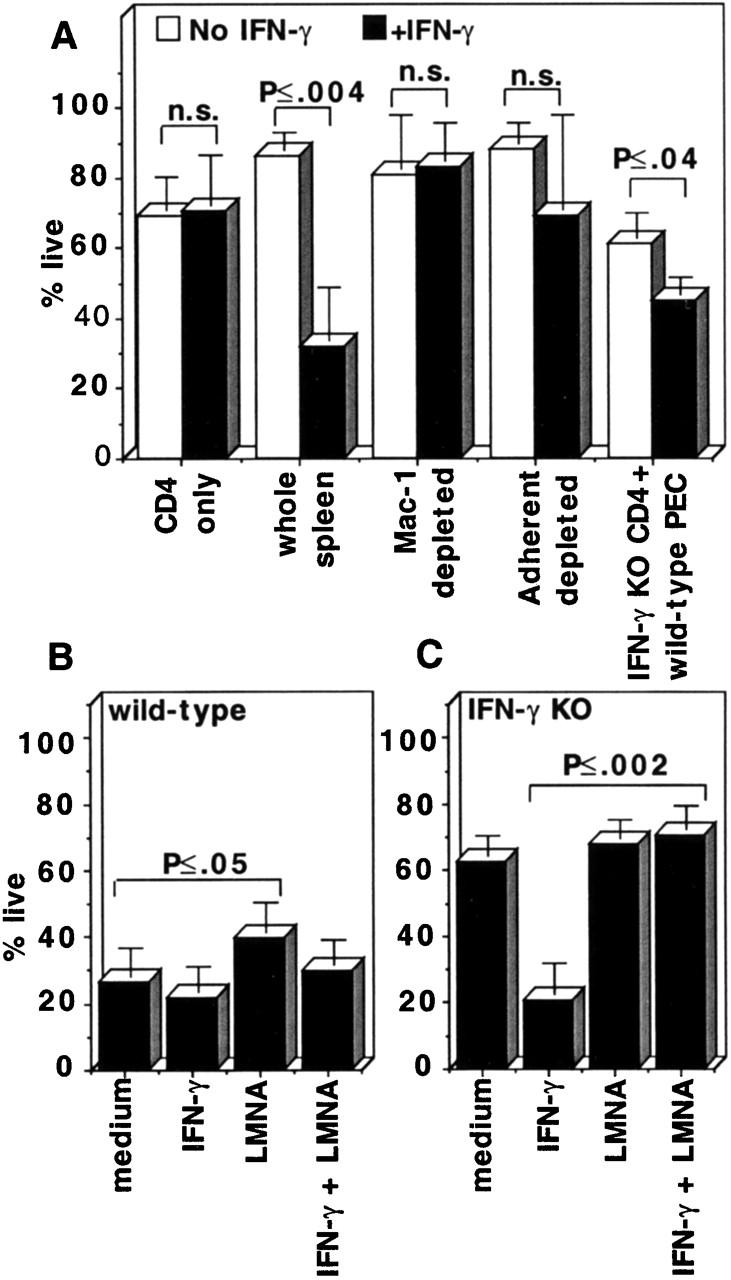
Activated macrophages and NO were required for IFN-γ to induce apoptosis of activated CD4 T cells. (A) The cells indicated, from BCG-infected IFN-γ KO mice or IFN-γ KO CD4 T cells with wild-type PECs, all at week 3–4 of BCG infection, were cultured in the presence or absence of IFN-γ. The percentage of live CD4+CD44hi T cells at 65 h of culture was measured using the annexin/PI assay. Average and SD of three to nine mice done in multiple experiments. (B) Wild-type and (C) IFN-γ KO spleen cells, used at week 3–4 of BCG infection, were cultured in the indicated conditions. Percent live CD4+CD44hi cells at 65 h is shown. Statistical significance (t test) between groups is shown.
NO Production Was Required for IFN-γ to Induce Apoptosis of Activated CD4 T Cells.
We tested the effect of LMNA, an NO synthase enzyme inhibitor, on IFN-γ–induced apoptosis of CD4+CD44hi T cells. Addition of LMNA to wild-type cultures resulted in significantly increased percentages of live CD4+CD44hi CD4 T cells compared with cultures with medium only (Fig. 5 B). Addition of LMNA along with IFN-γ to IFN-γ KO cultures abolished IFN-γ–induced cell death (Fig. 5 C). Similar results were obtained using another NO inhibitor, N 6-(1-iminoethyl)-dihydrochloride (data not shown). These data suggest that at least part of the killing of CD4+CD44hi T cells in IFN-γ KO cultures by IFN-γ is mediated by NO.
Discussion
We propose that a negative feedback loop maintains CD4 T cell homeostasis. During BCG infection, CD4 T cells secrete IFN-γ and activate macrophages to increased microbicidal activity. Activated macrophages generate NO and other effector molecules, such as superoxide, that enable them to kill the BCG organisms. In the presence of activated macrophages, the CD4 T cells begin to undergo increased apoptosis, returning the number of activated CD4 T cells to normal levels. Therefore, activated CD4 T cells secreting IFN-γ indirectly trigger their own apoptosis by activating macrophages. As IFN-γ is uniquely required for macrophage activation, IFN-γ KO mice are expected to be lacking other macrophage effector molecules besides NO. Whether other macrophage effector mechanisms are involved is currently under investigation.
Naive 6–10-wk-old IFN-γ KO mice do not spontaneously accumulate activated CD4 T cells in the absence of BCG infection or an antigenic challenge. This suggests that IFN-γ is involved in regulating CD4 T cell homeostasis only during immune responses. In contrast, mice lacking CTLA-4, IL-2, and the common cytokine γ chain receptor spontaneously accumulate activated CD4 T cells without apparent infection 3 25 26. Thus, the IFN-γ KO mouse has a novel phenotype that provides insight into the regulation of the immune response.
Several recent studies provide supporting evidence for a role of IFN-γ in apoptosis of immune cells during other infectious diseases. During infection of mice with Toxoplasma gondii, neutralization of IFN-γ with antibody resulted in decreased apoptosis in tissue sections of Peyer's patch lymphocytes 27. This was associated with decreased Fas expression. In contrast, we showed that mice lacking Fas upregulated apoptosis of activated CD4 T cells during BCG infection identically to wild-type mice. In another study, IFN-γ KO mice infected with Trypanosome cruzi showed decreased apoptosis of whole splenocytes 28. However, neither study specifically addressed the role of IFN-γ in apoptosis of CD4 T cells, nor did they demonstrate increased accumulation of activated CD4 T cells in the absence of IFN-γ. Mice lacking inducible NO synthase showed increased numbers of CD4 T cells after infection with Trypanosome brucei. However, the level of apoptosis of these CD4 T cells was not measured 29.
We considered the possibility that the IFN-γ KO mice accumulated activated CD4 T cells only because of the large difference in bacterial burden between wild-type and IFN-γ KO mice. However, for several reasons we think it is more likely that the principal cause for accumulation of activated CD4 T cells was the failure to induce apoptosis of these cells in the absence of IFN-γ. First, BCG proved to be a very persistent pathogen in both wild-type and IFN-γ KO mice. The bacterial burden in wild-type spleens did not fall below the initiating number of ∼105 CFU during the entire 8 wk of BCG infection. However, elimination of the activated CD4 T cells in wild-type spleens occurred well before the bacteria were cleared. Second, the peak numbers of activated CD4 T cells were at 3 wk of infection in IFN-γ KO mice; at this time, the bacterial burden was only sixfold greater than in wild-type mice. Third, in the an accompanying article in this issue by Chu et al., we show that activated CD4 T cells fail to undergo apoptosis and accumulate in large numbers in the spleen and central nervous system of IFN-γ KO mice during experimental autoimmune encephalomyelitis 30. This is an immune response to a persisting, but noninfectious, antigen. Taken together, these data strongly argue that IFN-γ is required to eliminate activated CD4 T cells by apoptosis during both infectious and autoimmune disease.
Acknowledgments
This work was supported by National Institutes of Health grant R29 AI41258-02 and by Trudeau Institute grant IHP-85.
References
- Murali-Krishna K., Altman J.D., Suresh M., Sourdive D.J., Zajac A.J., Miller J.D., Slansky J., Ahmed R. Counting antigen-specific CD8 T cellsa reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- Van Parijs L., Abbas A.K. Homeostasis and self-tolerance in the immune systemturning lymphocytes off. Science. 1998;280:243–248. doi: 10.1126/science.280.5361.243. [DOI] [PubMed] [Google Scholar]
- Chambers C.A., Krummel M.F., Boitel B., Hurwitz A., Sullivan T.J., Fournier S., Cassell D., Brunner M., Allison J.P. The role of CTLA-4 in the regulation and initiation of T-cell responses. Immunol. Rev. 1996;153:27–46. doi: 10.1111/j.1600-065x.1996.tb00919.x. [DOI] [PubMed] [Google Scholar]
- Van Parijs L., Peterson D.A., Abbas A.K. The Fas/Fas ligand pathway and Bcl-2 regulate T cell responses to model self and foreign antigens. Immunity. 1998;8:265–274. doi: 10.1016/s1074-7613(00)80478-1. [DOI] [PubMed] [Google Scholar]
- Bachmann M.F., Waterhouse P., Speiser D.E., McKall-Faienza K., Mak T.W., Ohashi P.S. Normal responsiveness of CTLA-4-deficient anti-viral cytotoxic T cells. J. Immunol. 1998;160:95–100. [PubMed] [Google Scholar]
- Liu Y., Janeway C., Jr. Interferon gamma plays a critical role in induced cell death of effector T cella possible third mechanism of self-tolerance. J. Exp. Med. 1990;172:1735–1739. doi: 10.1084/jem.172.6.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding A.H., Nathan C.F., Stuehr D.J. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J. Immunol. 1988;141:2407–2412. [PubMed] [Google Scholar]
- MacMicking J., Xie Q.W., Nathan C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- Williams M.S., Noguchi S., Henkart P.A., Osawa Y. Nitric oxide synthase plays a signaling role in TCR-triggered apoptotic death. J. Immunol. 1998;161:6526–6531. [PubMed] [Google Scholar]
- Murphy M.P. Nitric oxide and cell death. Biochim. Biophys. Acta. 1999;1411:401–414. doi: 10.1016/s0005-2728(99)00029-8. [DOI] [PubMed] [Google Scholar]
- Haendeler J., Zeiher A.M., Dimmeler S. Nitric oxide and apoptosis. Vitam. Horm. 1999;57:49–77. doi: 10.1016/s0083-6729(08)60640-8. [DOI] [PubMed] [Google Scholar]
- Lenardo M., Chan K.M., Hornung F., McFarland H., Siegel R., Wang J., Zheng L. Mature T lymphocyte apoptosis—immune regulation in a dynamic and unpredictable antigenic environment. Annu. Rev. Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- Winoto A. Cell death in the regulation of immune responses. Curr. Opin. Immunol. 1997;9:365–370. doi: 10.1016/s0952-7915(97)80083-0. [DOI] [PubMed] [Google Scholar]
- Wong B., Choi Y. Pathways leading to cell death in T cells. Curr. Opin. Immunol. 1997;9:358–364. doi: 10.1016/s0952-7915(97)80082-9. [DOI] [PubMed] [Google Scholar]
- Rathmell J.C., Thompson C.B. The central effectors of cell death in the immune system. Annu. Rev. Immunol. 1999;17:781–828. doi: 10.1146/annurev.immunol.17.1.781. [DOI] [PubMed] [Google Scholar]
- Dalton D.K., Pitts-Meek S., Keshav S., Figari I.S., Bradley A., Stewart T.A. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- Croft M., Carter L., Swain S.L., Dutton R.W. Generation of polarized antigen-specific CD8 effector populationsreciprocal action of interleukin (IL)-4 and IL-12 in promoting type 2 versus type 1 cytokine profiles. J. Exp. Med. 1994;180:1715–1728. doi: 10.1084/jem.180.5.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drapier J.C., Hibbs J., Jr. Differentiation of murine macrophages to express nonspecific cytotoxicity for tumor cells results in L-arginine-dependent inhibition of mitochondrial iron-sulfur enzymes in the macrophage effector cells. J. Immunol. 1988;140:2829–2838. [PubMed] [Google Scholar]
- Budd R.C., Cerottini J.C., Horvath C., Bron C., Pedrazzini T., Howe R.C., MacDonald H.R. Distinction of virgin and memory T lymphocytes. Stable acquisition of the Pgp-1 glycoprotein concomitant with antigenic stimulation. J. Immunol. 1987;138:3120–3129. [PubMed] [Google Scholar]
- Bell E.B., Sparshott S.M., Bunce C. CD4+ T-cell memory, CD45R subsets and the persistence of antigen—a unifying concept. Immunol. Today. 1998;19:60–64. doi: 10.1016/s0167-5699(97)01211-5. [DOI] [PubMed] [Google Scholar]
- Vermes I., Haanen C., Steffens-Nakken H., Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J. Immunol. Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- Chrest F.J., Buchholz M.A., Kim Y.H., Kwon T.K., Nordin A.A. Identification and quantitation of apoptotic cells following anti-CD3 activation of murine G0 T cells. Cytometry. 1993;14:883–890. doi: 10.1002/cyto.990140806. [DOI] [PubMed] [Google Scholar]
- Springer T., Galfre G., Secher D.S., Milstein C. Mac-1a macrophage differentiation antigen identified by monoclonal antibody. Eur. J. Immunol. 1979;9:301–306. doi: 10.1002/eji.1830090410. [DOI] [PubMed] [Google Scholar]
- Drapier J.C., Wietzerbin J., Hibbs J.B., Jr. Interferon-gamma and tumor necrosis factor induce the L-arginine-dependent cytotoxic effector mechanism in murine macrophages. Eur. J. Immunol. 1988;18:1587–1592. doi: 10.1002/eji.1830181018. [DOI] [PubMed] [Google Scholar]
- Sadlack B., Merz H., Schorle H., Schimpl A., Feller A.C., Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–261. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- Nakajima H., Shores E.W., Noguchi M., Leonard W.J. The common cytokine receptor gamma chain plays an essential role in regulating lymphoid homeostasis. J. Exp. Med. 1997;185:189–195. doi: 10.1084/jem.185.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesenfeld O., Kosek J.C., Suzuki Y. Gamma interferon induces Fas-dependent apoptosis of Peyer's patch T cells in mice following peroral infection with Toxoplasma gondii . Infect. Immun. 1997;65:4682–4689. doi: 10.1128/iai.65.11.4682-4689.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins G.A., Vieira L.Q., Cunha F.Q., Silva J.S. Gamma interferon modulates CD95 (Fas) and CD95 ligand (Fas-L) expression and nitric oxide-induced apoptosis during the acute phase of Trypanosoma cruzi infectiona possible role in immune response control. Infect. Immun. 1999;67:3864–3871. doi: 10.1128/iai.67.8.3864-3871.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar A.E., Sternberg J., McSharry C., Wei X.Q., Liew F.Y., Turner C.M. T-cell responses during Trypanosoma brucei infections in mice deficient in inducible nitric oxide synthase. Infect. Immun. 1999;67:3334–3338. doi: 10.1128/iai.67.7.3334-3338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C.-Q., Wittmer S., Dalton D.K. Failure to suppress the expansion of the activated CD4 T cell population in interferon γ–deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J. Exp. Med. 2000;191:123–128. doi: 10.1084/jem.192.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]