The crystal structure of the Thermus aquaticus DnaB helicase monomer (original) (raw)
Abstract
The ring-shaped hexameric DnaB helicase unwinds duplex DNA at the replication fork of eubacteria. We have solved the crystal structure of the full-length Thermus aquaticus DnaB monomer, or possibly dimer, at 2.9 Å resolution. DnaB is a highly flexible two domain protein. The C-terminal domain exhibits a RecA-like core fold and contains all the conserved sequence motifs that are characteristic of the DnaB helicase family. The N-terminal domain contains an additional helical hairpin that makes it larger than previously appreciated. Several DnaB mutations that modulate its interaction with primase are found in this hairpin. The similarity in the fold of the DnaB N-terminal domain with that of the C-terminal helicase-binding domain (HBD) of the DnaG primase also includes this hairpin. Comparison of hexameric homology models of DnaB with the structure of the papillomavirus E1 helicase suggests the two helicases may function through different mechanisms despite their sharing a common ancestor.
INTRODUCTION
The DnaB helicase is the main replicative helicase of eubacteria (1). The enzyme sits on the 5′ single-stranded DNA (ssDNA) arm of the replication fork (1) and utilizes the hydrolysis of ATP to fuel the unwinding of double-stranded DNA (dsDNA). Its movement along the DNA proceeds in the 5′–3′ direction (1) and at 25°C it has an intrinsic rate of ∼ 291 bp per second (2). At the replication fork, DnaB forms a critical interaction with the DnaG-primase that stimulates the activities of both the helicase and the primase (3).
Helicases assemble into a variety of oligomeric forms, ranging from monomers to heptamers and have been classified into several families based on analysis of their sequences (4). All helicases share the Walker A and B motifs, whose most conserved residues are implicated in nucleotide binding and hydrolysis (4). Specific helicase families are defined by the presence of additional specific motifs. Structural studies of helicases from many families have shown that all these proteins contain a core fold that was first visualized in the crystal structure of RecA (5).
DnaB belongs to a small family of hexameric helicases whose members function in replication. In addition to DnaB, this family includes the replicative helicases of the T4 and T7 bacteriophages as well as the RepA helicase of plasmid RSF1010. All members of this family contain by definition five conserved motifs (H1, H1a, H2, H3 and H4) that are located in the C-terminal region of the protein (6). Motifs H1 and H2 are equivalent to the Walker A and B motifs. The N-terminal region of these helicases is less well conserved. Indeed, in the T7 helicase a domain related to DnaG primase is fused to the N-terminal portion of the helicase.
Electron microscopy (EM) has revealed that DnaB family helicases form hexameric ring-shaped structures that exists in equilibrium among these different quaternary states (7,8), exhibiting either 6-fold rotational symmetry (C6), 3-fold rotational symmetry (C3) or an intermediate state between the two (C3C6). While these EM reconstructions predict a movement of the C-terminal region of DnaB, the transition from the C3 to the C6 particle is believed to be principally determined by the N-terminal region (7,8). Fluorescence transfer experiments have suggested that DnaB is positioned at the replication fork with the 5′ ssDNA arm inside the ring while the 3′ ssDNA arm is occluded from the ring, hence providing a mechanism for duplex separation (9).
Several crystal structures of the T7 gp4 helicase have been determined (10–12). The presence of the same sequence motifs in the helicase domain of the T7 protein and the C-terminal region of DnaB would suggest that the structure of the DnaB C-terminal region resembles that of the T7 helicase domain. To date, the only structural information available for DnaB are the crystal (13) and NMR (14) structures of an N-terminal fragment. Here we present the 2.9 Å resolution crystal structure of the full-length DnaB helicase from the thermophile Thermus aquaticus (Taq DnaB) that shows a larger, complete N-terminal domain exhibiting four different orientations relative to the C-terminal domain.
MATERIALS AND METHODS
Protein expression and purification
The expression plasmid for Taq DnaB (15) was transformed into BL21(DE3) cells (Novagen). The transformed cells were grown in LB medium containing 200 μg/ml ampicillin at 37°C. When the cells reached an A600 of between 0.4 and 0.8, gene expression was induced by addition of isopropyl-β-d-thiogalactopyranoside (Sigma) to a final concentration of 1 mM. After 3 h, the cells were harvested by centrifugation and quickly flash frozen in liquid nitrogen.
The cell paste containing Taq DnaB was resuspended in Buffer B1 containing 20 mM Tris pH 8.0, 50 mM NaCl, 5 mM MgCl2 and 10% (w/v) sucrose supplemented with complete EDTA-free protease inhibitors (Roche). The cells were lysed in a microfluidizer and then heated at 65°C for 20 min. Following centrifugation, the protein was run over a Q-sepharose column (GE Healthcare) that had been equilibrated with buffer B1. Taq DnaB was eluted using a linear gradient from 50 mM to 1 M NaCl. The pooled fractions were then applied to a Hi-Trap Heparin HP column (GE Healthcare) and eluted in a linear gradient from 0.1 to 1.0 M NaCl. Finally, the protein was gel filtered over a 26/60 Superdex 200 prep grade column (GE Healthcare) that had been equilibrated in B2 buffer containing 10 mM Tris pH 8.0, 10 mM NaCl, 1 mM MgCl2 and 25 mM Maltose. Taq DnaB factions were pooled and spin concentrated before being flash frozen in liquid nitrogen. The final protein was over 99% pure as judged by SDS-PAGE and coomasie blue staining. The Seleno-methionine (SeMet) substituted Taq DnaB was expressed in B834(DE3) (Novagen) and purified using the same methods that had been used as the native protein with the exception that the reducing agent, dithiothreitol (Sigma), was added at all buffers.
Crystallization and data collection
Taq DnaB crystals were prepared using the sitting drop method of vapor diffusion, mixing equal volumes of protein sample and well solution. Well solutions contained 1.3 M ammonium sulfate, 10 mM magnesium sulfate, 5% 1,6-hexanediol and 100 mM MES pH 6.5. To provide cryo-protection, the crystals were transferred in 0.5 M increments into a solution of 3.0 M ammonium sulfate, 10 mM magnesium sulfate, 2.5% 1,6-hexanediol, 100 mM MES pH 6.5 and 10% ethylene glycol. Native diffraction data were collected at the National Synchrotron Light Source (NSLS) station X25. SeMet substituted crystals were used to collect multi-wavelength anomalous dispersion (MAD) data at the Argonne Photon Source (APS) station 19-ID. The HKL suite of programs was used for all data integration and scaling (16). The crystals belong to space group P3121 with unit cell dimensions a = 104.7 Å, b = 104.7 Å, c = 363.3 Å and have a diffraction limit of 2.9 Å resolution where its I/σ drops to 1.3. Data collection statistics are shown in Table 1.
Table 1.
Data collection, phasing and refinement statistics
| Se inflection | Se peak | Se remote | Native | |
|---|---|---|---|---|
| Data collection | ||||
| Wavelength (Å) | 0.97949 | 0.97935 | 0.96411 | 1.1404 |
| Resolution (Å) | 50 − 3.1 (3.21−3.1) | 50 − 3.1 (3.21−3.1) | 50 − 3.2 (3.31−3.2) | 50 − 2.9 (2.974−2.9) |
| _R_merge (%) | 9.1 (>100)a | 8.0 (>100)a | 9.1 (>100)a | 9.8 (>100)a |
| I/σI | 11.7 (1.4)a | 12.9 (1.2)a | 11.8 (1.1)a | 13.5 (1.9)a |
| Completeness (%) | 99.9 (100.0)a | 98.7 (100.0)a | 99.9 (100.0)a | 98.3 (99.9)a |
| Redundancy | 6.3 | 6.5 | 6.1 | 5.4 |
| Phasing | ||||
| Resolution (Å) | 50 − 3.5 | 50 − 3.5 | 50 − 3.5 | |
| Solve Z-score | 161.6 | |||
| Solve figure of merit | 0.56 | |||
| Refinement | ||||
| _R_work (%) | 24.2 (36.6) | |||
| _R_free (%) | 28.5 (38.9) | |||
| rmsd bond length (Å) | 0.007 | |||
| rmsd bond angle (°) | 1.136 | |||
| Ramachandran | ||||
| Most favored (%) | 92% | |||
| Additionally allowed (%) | 8% |
Structure determination and refinement
The positions of 47 Se atoms were determined using the direct methods procedure as implemented in the program SnB (17). After MAD phasing using the program SOLVE (18), the electron density was solvent flattened and each of the four N-terminal and C-terminal domains were 4-fold averaged with the program RESOLVE (19). The maps were further improved by sharpening the data by applying a B factor of 50 to the experimental amplitudes. Cycles of model building in the programs O (20) and COOT (21) and refinement in REFMAC (22) resulted in a structure with an _R_cryst of 24.2% and _R_free of 28.5%. The current model contains 1641 out of 1776 amino acids across four molecules of DnaB. Residues 1, 175–182, 326–332 and 441–444 where disordered in the electron density maps and therefore are not included in any of the DnaB chains of the current model. Phasing and refinement statistics are given in Table 1 and a portion of a sigma-A weighted 2_F_o − _F_c electron density omit map can be seen in Figure 1A.
Figure 1.

(A) Portion of a sigma-A weighted 2_F_o − _F_c electron density omit map calculated at 2.9 Å resolution. The diffraction amplitudes were sharpened by a temperature factor of 50. (B) Ribbon representation of the backbone structure of the DnaB monomer. The N-terminal domain (NTD), C-terminal domain (CTD) and linker region are colored blue, red and yellow, respectively. (C) Similar ribbon representations of the three additional copies of the DnaB monomer found in the asymmetric unit. Each of the monomer structures in panels B and C were orientated by pairwise superposition of their respective CTDs. (D) A schematic diagram of the domain structure of the DnaB helicase. Residues found at the domain boundaries are labeled.
RESULTS
Overall structure
The full-length DnaB structure consists of two domains that are connected by a long flexible linker (Figure 1A and B). The N-terminal domain or NTD (residues 1–150, _Eco_1-168; here and throughout the manuscript residue numbering in E. coli will have the prefix Eco) is an all α-helical structure containing a largely spherical bundle of helices terminated by an extended helical hairpin (Figures 1 and 2A). The C-terminal domain or CTD (residues 186–444; _Eco_207-471) consists of a predominantly parallel β-sheet flanked by α-helices (Figures 1 and 3A). The flexible linker of 34 residues (residues 151–185; _Eco_169-206) has an extended structure composed of a single α-helix flanked by two loops that connect the helix to the NTD and the CTD.
Figure 2.

Ribbon representations of (A) Taq NTD, (B) E. coli P12 fragment and (C) E. coli helicase-binding domain (HBD) of primase. The Taq NTD is colored as in Figure 2. The E. coli P12 fragment and HBD are colored orange and green, respectively. Escherichia coli P12 and HBD were orientated by superposition of their structures on that of the Taq NTD. The C-terminal residue of the E. coli P12 fragment and the equivalent residue in the intact Taq NTD are labeled. (D) Stereo view of a ribbon representation of the Taq NTD dimer observed in the crystal lattice.
Figure 3.
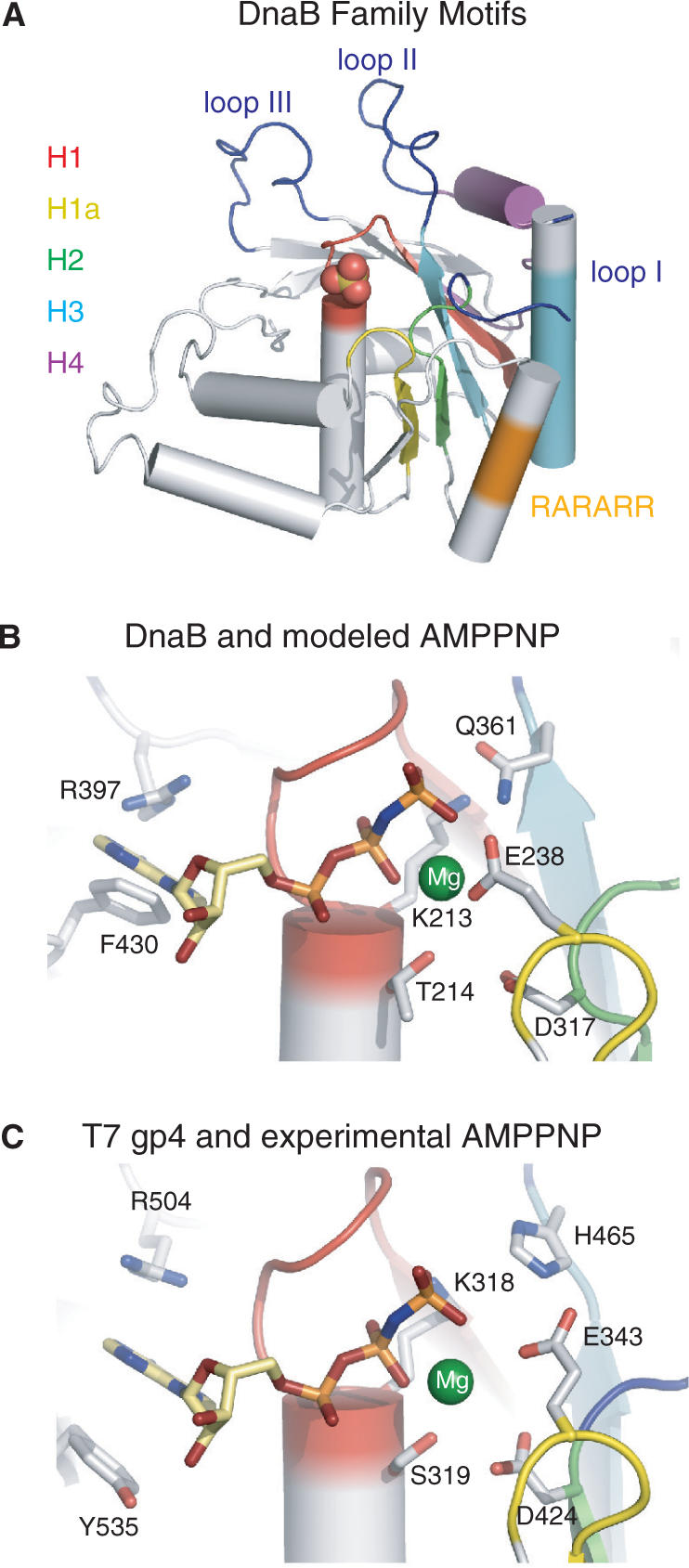
(A) Ribbon representation of the Taq CTD colored according to the conserved DnaB family motifs: H1 red, H1a yellow, H2 green, H4 cyan and H4 purple. The RARARR basic region (see text) and the three proposed DNA-binding loops are colored orange and blue, respectively. (B) DnaB nucleotide-binding site with the homology modeled AMPPNP (stick model) and magnesium ion (green sphere). Side chains of key residues are displayed. Secondary structure elements are colored as in panel A. (C) T7 gp4 helicase nucleotide-binding pocket (PDB code: 1E0J). Representations and coloring are the same as panel B.
DnaB does not form a closed hexameric ring quaternary structure in this crystal lattice as does the isolated molecule visualized by EM (7,8) nor does DnaB form a 6-fold filament, as observed in the original crystal structure of the T7 gp4 helicase domain (10). Instead, the four copies of DnaB observed in the crystal asymmetric unit appear to be arranged as individual monomers or dimers (see below). Size-exclusion chromatography of Taq DnaB indicates the protein is hexameric in the absence of nucleotide, even under high salt (1 M NaCl) conditions (Supplementary Figure S1). To determine whether the conditions used for crystallization perturbed the equilibrium between hexamer and monomer, we performed size-exclusion chromatography experiments under conditions mimicking, as closely as possible, those of crystallization. Thus, Taq DnaB was gel filtered in a buffer containing 50 mM MES pH 6.0, 5% 1,6-hexanediol, 10 mM magnesium sulfate and 200 mM ammonium sulfate (Supplementary Figure S1). Using this approach, we were unable to assay higher ammonium sulfate concentrations as the helicase began to interact with the column matrix (data not shown). This gel filtration experiment clearly showed that, within the experimental detection limits, the helicase remains hexameric (Supplementary Figure S1). We therefore assume that crystals of the monomeric or dimeric helicase are formed from the equilibrium with the hexamer, which may or may not favor the monomer at high ammonium sulfate concentrations.
Comparison of the positions of the NTD relative to the CTD in each of the four monomers reveals a highly flexible monomer structure. Alignment of the four DnaB molecules found in the asymmetric unit by superposition of their CTDs reveals that each of the linkers takes a different trajectory from the CTD and in turn each NTD adopts a different orientation with respect to the linker (Figure 1). The flexible joints generated by the two linker region loops result in almost unrestrained positioning of the two domains with respect to one another. The organization of the NTD, linker region and CTD are highly reminiscent of the helicase domain, linker region and primase domain of the gp4 helicase-primase subunit structure (12). This flexibility is consistent with previous hydrodynamic studies that predict an extended structure for the Escherichia coli DnaB monomer (23).
The NTD of DnaB
Identification of the boundaries of the two DnaB domains has previously been based on its digestion with trypsin (24), which cuts the protein into an N-terminal 12 kDa fragment (P12) and a C-terminal 33 kDa fragment (P33). Superposition of the coordinates of the Taq DnaB NTD onto those of the E. coli P12 fragment reveals that the structures are highly similar with a root mean squared deviation (rmsd) between them of 1.2 Å over 97 Cα atoms. However, comparison of the two structures reveals that the NTD of intact DnaB is 40 residues larger than the P12 fragment. These additional residues of the DnaB NTD extend the C-terminal helix of P12, which then interacts with a further C-terminal helix to form a helical hairpin subdomain (Figure 2A). This helical hairpin includes residues that have been shown by mutagenesis experiments to modulate the interaction of the helicase with the DnaG-primase (25). Previously these residues had been ascribed to be part of the linker region.
The four copies of the DnaB NTD in the asymmetric unit appear to form two independent dimers. Each dimer interface is formed by the helical hairpin subdomains that pack together to produce a four helix bundle (Figure 2D). Short chain hydrophobic residues dominate the interface that buries 1250 Å2 of surface area per monomer. The two dimers are highly similar as their superposition results in an rmsd of 1.5 Å over 302 Cα atoms. A recent NMR structure determination of an E. coli DnaB fragment protein that included residues C-terminal to 171 (150 in Taq), which we now know corresponds very well to the NTD of the intact protein, revealed that the C-terminus of the isolated NTD does not form a helical hairpin as is observed in the full-length structure (26). Instead this region was found to be unstructured. Although this could be due to inherent differences between the two species, this seems unlikely as secondary structure predictions indicate that this region within the E. coli protein should also be helical and form a hairpin (26). More likely, the hairpin may be stabilized in the context of the hexamer assembly, possibly in the C3 symmetric particles by the formation of the NTD dimer observed here (Figure 2D).
Recent structures of the C-terminal helicase-binding domain (HBD) of the DnaG-primase have revealed that the fold of this domain is related to that of the P12 fragment of DnaB (26–28). Comparison of the structure of the NTD in the intact DnaB with that of the NMR structure of the E. coli HBD (26) shows that this similarity extends to the additional helical hairpin (Figure 2). Thus, the C-terminal HBD of DnaG primase and the NTD of DnaB helicase have related folds.
The CTD of DnaB
The five motifs that define the dnaB family are all located within its CTD. Four of the motifs (H1, H1a, H2 and H3) form the core β-sheet and provide the residues that line the nucleotide-binding pocket (Figure 3A). The fifth motif (H4) forms a helix, the N-terminus of a core β-strand as well as their connecting loop (Figure 3A) and has been proposed to contain residues that are involved in DNA binding (11). As expected the structure of the CTD of DnaB closely resembles that of the helicase domain of T7 gp4 (rmsd of 1.5 Å over 172 Cα atoms) and that of the RecA core (rmsd of 1.9 Å over 151 Cα atoms) (Supplementary Figure S2). Structural similarity is also observed between DnaB, a 5′–3′ helicase and the papillomavirus 3′–5′ E1 helicase. The CTDs of these two helicases superimpose with an rmsd of 3.0 Å over 102 Cα atoms.
The proposal that the CTD of DnaB contains a leucine zipper dimerization motif is not supported by the crystal structure. Analysis of sequence of residues 361–390 in E. coli (338–368 in Taq), that lie upstream of a region rich in basic residues (Eco324-329, residues 299–304 in Taq), suggested that it forms a leucine zipper reminiscent of the eukaryotic transcription factors, such as cFOS and cJUN (29). This sequence overlaps with the H3 motif sequence in the DnaB family. The basic region is highly conserved across DnaB proteins, although it is not found in the T7 gp4 helicase. Site-directed mutagenesis of the conserved basic residues of this region results in significant attenuation of DNA binding, ATPase activity and consequently the helicase activity of DnaB (29). Inspection of the CTD fold reveals that the basic sequence RARARR (in Taq) is located on a helix that lies on the opposite face of the β-sheet from the nucleotide-binding pocket (Figure 3A). However, the residues implicated in forming a leucine zipper, which is an all α-helical structure, in fact form a helix-loop-strand motif in the core of the RecA fold of the CTD (Figure 3A).
Despite attempts to prepare a crystalline complex of DnaB with its nucleotide substrate, either by soaking crystals or co-crystallizing in solutions containing ATP, ADP or non-hydrolyzable analogs of ATP, no binding of nucleotide was observed. We presume that nucleotide binding was inhibited by the high concentration of sulfate ions (at least 1.0 M) in the crystallization buffers. Indeed, a sulfate ion is bound to the Walker A motif of each monomer in the crystal, presumably mimicking a phosphate of ATP (Figure 3A). Although the structure of a nucleotide complex has not been achieved as yet, the similarity between the structure of the CTD of DnaB and that of the T7 gp4 helicase domain allows us to use the structure of the nucleotide complex of T7 gp4 helicase (Figure 3B) to homology model the ATP onto the DnaB structure (Figure 3C). The resulting homology model is in excellent agreement with that of the T7 gp4 structure. Coordination of the modeled magnesium ion is provided by the side chains of residues T214 (Eco T238) and D317 (Eco D343). Residues R397 (Eco R420) and F430 (Eco F452) are ideally positioned to stack on the adenine ring of the modeled nucleotide and the γ-phosphate is in contact distance with residues K213 (Eco K237) and Q361 (Eco Q384).
The T7 gp4 helicase ring is stabilized through interactions between its linker helix in one subunit and a small α-helical pocket, found on the periphery of the RecA core, in the adjacent subunit (11,12). Although it varies slightly in secondary structure (Supplementary Figure S3), DnaB also contains this hexamerization pocket (residues 240–283, _Eco_264-308). Within the lattice of the monomer crystal each linker helix is bound in the hexamerization pocket of an adjacent CTD but in such a way that does not form a closed hexameric ring. However, comparison of DnaB with the T7 gp4 ring structures (11,12) suggest that the linker region helix interface observed here will be similar in a closed hexameric assembly of DnaB (Supplementary Figure S3).
Possible hexamer assemblies
A homology model of the hexameric arrangement of the DnaB CTD can be built by superimposing its CTD onto the hexameric ring structure of the T7 gp4 helicase (Figure 4). The resulting model is free of any main chain steric clashes and the dimensions and shape are consistent with the EM reconstructions of DnaB from E. coli and the bacteriophage SPP1 (7,8). Structural and mutagenesis experiments with the T7 gp4 helicase have suggested residues within motif H4 and loops I, II and III are likely candidates for DNA binding (11). The homology model of the DnaB CTD likewise positions the equivalent elements in DnaB at the center of the modeled ring, and this arrangement is consistent with fluorescence energy transfer experiments done using E. coli DnaB (9). The highly basic region, whose mutagenesis has been shown to helicase activity (29), is found on the N-terminal surface of the modeled hexamer. At the replication fork, the duplex DNA would be located at the C-terminal surface of the helicase ring and the ssDNA would emanate from the N-terminal surface (30). The N-terminal surface would also be the location of the NTD in the intact protein.
Figure 4.

Three perpendicular views of a surface representation of the Taq DnaB CTD hexamer that has been constructed by homology modeling using the T7 gp4 helicase structure (PDB code: 1E0J). The loops implicated in DNA binding are colored blue, motif H4 magenta and the RARARR sequence orange.
In order to model the locations of the NTDs within a hexamer, we attempted to manually fit the Taq DnaB NTD coordinates into the published EM reconstructions. The reconstructions used where of the C3 and C6 symmetrized particles from E. coli DnaB (7) and the C3, C6, C3C6 symmetrized particles from bacteriophage SPP1 G40P DnaB homolog (8). However, no satisfactory placement of the NTD or the observed NTD dimer into any these reconstructions could be found. In each case, the NTD could not be fit within the EM volume without severe steric clashes. Difficulties in fitting the NTD into the low resolution EM reconstructions could arise from several sources. The NTD subdomains could adopt a different conformation with respect to one another in the context of a hexameric assembly than observed. Alternatively, the EM reconstructions could be distorted, either by the inherent difficulties in generating reconstructions of highly polymorphic molecules (such as DnaB) or from negative staining techniques which are known to cause compression of the specimen (7,8).
DISCUSSION
The crystal structure of the full-length helicase reveals that DnaB is a highly flexible two domain molecule in the absence of the restraints imposed by the formation of the hexameric quaternary structure. We observe a potential dimer interface within the crystal lattice that is mediated by the helicase NTDs. Although dimers of E. coli DnaB have not been reported previously, both monomers and trimers have been detected by analytic ultracentrifugation in the absence of magnesium (23). Models have also been proposed that implicate the dimerization of the NTD in the transition of the DnaB ring from 6-fold to 3-fold symmetry (13,14). However, more recent EM reconstructions have discounted dimerization of the NTD within the 3-fold symmetric ring (7,8). Therefore the significance, if any, of the dimer interface observed here awaits further study.
The optimal activity of DnaB during replication is achieved through its interaction with the DnaG-primase. Mutagenesis studies suggest that the interaction interface between the two proteins is extensive and involves residues from the NTD and the CTD as well as their flexible linker (3,25). However, the structure of the intact protein reveals that the residues that had previously been assigned to the linker region form the C-terminal helical hairpin of the NTD. Several of the DnaB mutations that affect its interaction with primase lie in this hairpin structure (25) and to our knowledge none of the mutations studied previously lie within the reassigned linker region of the intact protein. Hence, the structure of DnaB in conjunction with the mutagenesis studies suggest that the primase interaction interface of the helicase is formed largely by the redefined NTD.
The structure of the NTD of DnaB has a fold that is highly similar to the fold of the DnaG-primase C-terminal HBD. The HBD has been shown to be the only domain of the DnaG-primase that is required for interaction with and stimulation of the helicase (3) and the NTD of DnaB forms at least a large part of the primase interaction surface. In light of this, it is tempting to speculate that the DnaB NTD and the DnaG HBD could utilize their respective hairpins to interact in a manner similar to that of the NTD dimer observed in the crystal. However, this would require the energetically unfavorable burying of several charged residues from the HBD hairpin with hydrophobic residues from the DnaB NTD hairpin. Therefore, it seems highly unlikely that HBD will interact with DnaB in this manner.
Interrogation of the protein structure database reveals that no other known protein structure shares the fold observed in the DnaB NTD and DnaG HBD. These results have lead to the suggestion that the last common ancestor of the DnaB family helicase was a bi-functional helicase-primase molecule. In this scenario the N-terminal domain of primase and C-terminal domain of helicase where separated by a linker, whose fold was related to that of the modern day NTD of DnaB. Duplication of the linker region followed by gene separation has presumably resulted in the current helicase and primase (31).
Very little is understood about the mechanism by which DnaB unwinds duplex DNA. Homology modeling of the DnaB CTD onto the structure of the T7 gp4 hexamer suggests that DnaB could in principle utilize the ‘binding change’ model of sequential nucleotide hydrolysis put forward on the basis of the T7 gp4 hexamer structure (11). However, several differences exist between the DnaB and the T7 gp4 helicases that may imply differences in their respective mechanisms. DnaB contains an NTD that is not present in the T7 gp4 helicase and the helical bundle moiety of this domain has recently been shown to modulate the polarity of the DnaB helicase. DnaB truncation mutants that lack this helical bundle have been shown to poses 3′–5′ helicase activity (32). Kinetic studies also suggest that DnaB has three high affinity binding sites for ATP (33), which is consistent with the 3-fold symmetry observed by EM (8,9). However, the model based on the T7 helicase crystal structure predicts four ATP-binding sites and 3-fold symmetric molecules of the T7 helicase have not been detected by EM.
The DnaB helicase and the papillomavirus E1 helicase share several common features; both are ring-shaped hexameric helicases that unwind genomic DNA at replication forks and both are formed by an all α-helical NTD (although the structures of the two NTD folds are unrelated) and a CTD that contains a core RecA-like fold (34). However, these proteins have distinct biochemical properties as DnaB unwinds DNA in the 5′–3′ direction where as the E1 helicase unwinds DNA in the 3′–5′ direction (34) and furthermore, these two proteins are structurally distinct. Comparisons of their respective RecA-like folds shows that although the Walker A and Walker B motifs (motifs H1 and H2 in the DnaB family) are similarly positioned, the DNA-binding elements are located in different regions of the fold, shown schematically in Figure 5A. But, in the quaternary structures, the DNA-binding elements are still found at the center of the ring of each helicase. This is possible because both helicases form hexamers in distinct manners; the plane of the core β-sheet of the E1 helicase are found approximately parallel to the ring axis whereas in DnaB-family helicases, the plane of the core β-sheet is approximately perpendicular to the ring axis (Figure 5B). A mechanism for ssDNA translocation by the E1 helicase has been proposed based on the crystal structure of the E1 helicase bound to ssDNA (34). In this structure, the DNA-binding loops from each subunit of the hexamer form a spiral, with each loop bound to one nucleotide of the ssDNA. The mechanism by which the helicase translocates along the DNA has been proposed to involve each DNA-binding loop carrying a nucleotide of ssDNA across the ring in response to ATP binding and hydrolysis (34). A similar spiral method of translocation is attractive for DnaB, and indeed the T7 gp4 helicase structure also displays a spiral conformation of its subunits, suggesting this spiraling mechanism may be common to all hexameric helicases. However, our analysis of the structure of the E1 hexamer and that of DnaB implies that the way in which DnaB and the E1 helicase utilize ATP binding and hydrolysis to move the DNA-binding elements are very likely to differ.
Figure 5.

Comparison of the ternary and quaternary structures of DnaB and the E1 helicase. (A) Topology diagrams of the CTD of DnaB (left) and the E1 helicase (right). The core RecA fold is colored green. Unique structural elements within each domain are colored gray. The Walker A and Walker B motifs are colored red and orange, respectively. Arginine fingers are highlighted as magenta circles and DNA-binding loops are in blue. The N and C-termini are indicated as black circles with white text. (B) Arrangement of essential helicase elements within (left) the DnaB CTD hexamer modeled by superposition (see text) onto the T7 gp4 structure and right the E1 CTD assembly. Key elements are colored as in panel A. For clarity only the β-strand preceding and the α-helix following the Walker A motif are shown. Shown in white are the molecular surfaces of the two structures with the nucleotide (yellow) displayed in a space-filling representation.
The high degree of structural similarity observed between DnaB and RecA supports the previous hypothesis that DnaB originated from a gene duplication of a RecA-like ancestor (35). Sequence analysis and the structural comparisons of DnaB and the E1 helicase suggest this event occurred after the divergence of eubacteria from archaea and eukaryotes. The replicative DnaB helicase of bacteria and the replicative helicases of eukaryotes e.g. the E1 helicase and related MCM proteins (34), are clearly distinct and thus appear to have evolved separately from a RecA-like ancestor, which was most likely not a helicase.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank the staff at ALS beamline 19-ID and NSLS beamline X25. We would also like to thank Dr Edward Egelman for the E. coli DnaB EM reconstructions. This work was supported by NIH grant GM57510 to T.A.S. Atomic coordinates and structure factor amplitudes have been deposited to the RCS PDB with the accession codes 2Q6T. Funding to pay the Open Access publication charges for this article was provided by Howard Hughes Medical Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1.LeBowitz JM, McMacken R. The Escherichia coli dnaB protein is a DNA helicase. J. Biol. Chem. 1986;261:4738–4748. [PubMed] [Google Scholar]
- 2.Galletto R, Jezewska MJ, Bujalowski W. Unzipping mechanism of the double-stranded DNA unwinding by a hexameric helicase: quantitative analysis of the rate of the dsDNA unwinding, processivity and kinetic step-size of the Escherichia coli DnaB helicase using rapid quench-flow methods. J. Mol. Biol. 2004;343:83–99. doi: 10.1016/j.jmb.2004.07.055. [DOI] [PubMed] [Google Scholar]
- 3.Bird LE, Pan H, Soultanas P, Wigley DB. Mapping protein-protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry. 2000;39:171–182. doi: 10.1021/bi9918801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorbalenya AE, Koonin EV. Helicases: amino acid sequence comparisons and structure-function relationships. Curr. Opin. Struct. Biol. 1993;3:419–429. [Google Scholar]
- 5.Story RM, Weber IT, Steitz TA. The structure of the E. coli recA protein monomer and polymer. Nature. 1992;355:318–325. doi: 10.1038/355318a0. [DOI] [PubMed] [Google Scholar]
- 6.Ilyina TV, Gorbalenya AE, Koonin EV. Organization and evolution of bacterial and bacteriophage primase-helicase systems. J. Mol. Evol. 1992;34:351–357. doi: 10.1007/BF00160243. [DOI] [PubMed] [Google Scholar]
- 7.Yang S, Yu X, VanLoock MS, Jezewska MJ, Bujalowski W, Egelman EH. Flexibility of the rings: structural asymmetry in the DnaB hexameric helicase. J. Mol. Biol. 2002;321:839–849. doi: 10.1016/s0022-2836(02)00711-8. [DOI] [PubMed] [Google Scholar]
- 8.Ramirez RN, Robledo Y, Mesa P, Ayora S, Alonso JC, Carazo JM, Donate E. Quaternary polymorphism of replicative helicase G40P: structural mapping and domain rearrangements. J. Mol. Biol. 2006;357:1063–1076. doi: 10.1016/j.jmb.2006.01.091. [DOI] [PubMed] [Google Scholar]
- 9.Jezewska MJ, Rajendran S, Bujalowski D, Bujalowski W. Does the single-stranded DNA pass through the inner channel of the protein hexamer in the complex with the Escherichia coli DnaB helicase? J. Biol. Chem. 1998;273:10515–10529. doi: 10.1074/jbc.273.17.10515. [DOI] [PubMed] [Google Scholar]
- 10.Sawaya MR, Guo S, Tabor S, Richardson CC, Ellenberger T. Crystal structure if the helicase domain from the replicative helicase-primase of bacteriophage T7. Cell. 1999;99:167–177. doi: 10.1016/s0092-8674(00)81648-7. [DOI] [PubMed] [Google Scholar]
- 11.Singleton MR, Sawaya MR, Ellenberger T, Wigley DB. Crystal structure of T7 gene ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell. 2000;101:589–600. doi: 10.1016/s0092-8674(00)80871-5. [DOI] [PubMed] [Google Scholar]
- 12.Toth EA, Li Y, Sawaya MR, Cheng Y, Ellenberger T. The crystal structure of the bifunctional primase-helicase of bacteriophage T7. Mol. Cell. 2003;12:1113–1123. doi: 10.1016/s1097-2765(03)00442-8. [DOI] [PubMed] [Google Scholar]
- 13.Fass D, Bogden CE, Berger JM. Crystal structure of the N-terminal domain of the DnaB hexameric helicase. Structure. 1999;7:691–698. doi: 10.1016/s0969-2126(99)80090-2. [DOI] [PubMed] [Google Scholar]
- 14.Weigelt J, Brown SE, Miles CS, Dixon NE, Otting G. NMR structure of the N-terminal domain of E. coli DnaB helicase: implications for structure rearrangements in the helicase hexamer. Structure. 1999;7:681–690. doi: 10.1016/s0969-2126(99)80089-6. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan DL, Steitz TA. DnaB from Thermus aquaticus unwinds forked duplex DNA with an asymmetric tail length dependence. J. Biol. Chem. 1999;274:6889–6897. doi: 10.1074/jbc.274.11.6889. [DOI] [PubMed] [Google Scholar]
- 16.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter JCW, Sweet RM, editors. Macromolecular Crystallography, Part A. Academic Press: New York; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 17.Weeks CM, Miller R. The design and implementation of SnB v2.0. J. Appl. Cryst. 1999;32:120–124. [Google Scholar]
- 18.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr. 1999;D55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terwilliger TC. Statistical density modification with non-crystallographic symmetry. Acta Crystallogr. 2002;D58:2082–2086. doi: 10.1107/S0907444902016360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 21.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 22.Murchudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta. Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 23.Bujalowski W, Klonowska MM, Jezewska MJ. Oligomeric structure of Escherichia coli primary replicative helicase DnaB protein. J. Biol. Chem. 1994;269:31350–31358. [PubMed] [Google Scholar]
- 24.Nakayama N, Arai N, Kaziro Y, Arai KI. Structural and functional studies of the dnaB protein using limited proteolysis. Characterization of domains for DNA-dependent ATP hydrolysis and for protein association in the primosome. J. Biol. Chem. 1984;259:88–96. [PubMed] [Google Scholar]
- 25.Thirlway J, Soultanas P. In the Bacillus stearothermophilus DnaB-DnaG complex, the activities of the two proteins are modulated by distinct but overlapping networks of residues. J. Bacteriol. 2006;188:1534–1539. doi: 10.1128/JB.188.4.1534-1539.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su XC, Schaeffer PM, Loscha KV, Gan PHP, Dixon NE, Otting G. Monomeric solution structure of the helicase-binding domain of Escherichia coli DnaG primase. FEBS J. 2006;273:4997–5009. doi: 10.1111/j.1742-4658.2006.05495.x. [DOI] [PubMed] [Google Scholar]
- 27.Syson K, Thirlway J, Hounslow AM, Soultanas P, Waltho JP. Solution structure of the helicase-interaction domain of the primase DnaG: a model for helicase activation. Structure. 2005;13:609–616. doi: 10.1016/j.str.2005.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oakley AJ, Loscha KV, Schaeffer PM, Liepinsh E, Pintacuda G, Wilce MCJ, Otting G, Dixon NE. Crystal and solution structures of the helicase-binding domain of Escherichia coli primase. J. Biol. Chem. 2005;280:11495–11504. doi: 10.1074/jbc.M412645200. [DOI] [PubMed] [Google Scholar]
- 29.Biswas EE, Biswas SB. Mechanism of DNA binding by the DnaB helicase of Escherichia coli: analysis of the roles of domain γ in DNA binding. Biochemistry. 1999;38:10929–10939. doi: 10.1021/bi990049l. [DOI] [PubMed] [Google Scholar]
- 30.Jezewska MJ, Rajendran S, Bujalowski W. Complex of Escherichia coli primary replicative helicase DnaB protein with a replication fork: recognition and structure. Biochemistry. 1998;37:3116–3136. doi: 10.1021/bi972564u. [DOI] [PubMed] [Google Scholar]
- 31.Soultanas P. The bacterial helicase-primase interaction: a common structural/functional module. Structure. 2005;13:839–844. doi: 10.1016/j.str.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mesa P, Alonso JC, Ayora S. Bacillus subtilis bacteriophage SPP1 G40P helicase lacking the n-terminal domain unwinds DNA bidirectionally. J. Mol. Biol. 2006;357:1077–1088. doi: 10.1016/j.jmb.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 33.Bujalowski W, Jezewska MJ. Kinetic mechanism of nucleotide cofactor binding to Escherichia coli replicative helicase DnaB protein. Stopped-flow kinetic studies using fluorescent, ribose-, and base-modified nucleotide analogues. Biochemistry. 2000;39:2106–2122. doi: 10.1021/bi992413m. [DOI] [PubMed] [Google Scholar]
- 34.Enemark EJ, Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442:270–275. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 35.Leipe DD, Aravind L, Grishin NV, Koonin EV. The bacterial replicative helicase DnaB evolved from a RecA duplication. Genome Res. 2000;10:5–16. [PubMed] [Google Scholar]