Cervical keratinocytes containing stably replicating extrachromosomal HPV-16 are refractory to transformation by oncogenic H-Ras (original) (raw)
. Author manuscript; available in PMC: 2007 Sep 25.
Abstract
Ras expression in human epithelial cells with integrated HPV genomes has been shown to cause tumorigenic transformation. The effects of Ras in cells representing early stage HPV-associated disease (i.e., when HPV is extrachromosomal and the oncogenes are under control of native promoters) have not been examined. Here, we used human cervical keratinocyte cell lines containing stably replicating extrachromosomal HPV-16 and present the novel finding that these cells resist transformation by oncogenic H-Ras. Ras expression consistently diminished anchorageindependent growth (AI), reduced E6 and E7 expression, and caused p53 induction in these cells. Conversely, AI was enhanced or maintained in Ras-transduced cervical cells that were immortalized with a 16E6/E7 retrovirus, and minimal effects on E6 and E7 expression were observed. Ras expression with either episomal HPV-16 or LXSN-E6/E7 was insufficient for tumorigenic growth suggesting that other events are needed for tumorigenic transformation. In conclusion, our results indicate that Ras-mediated transformation depends on the context of HPV oncogene expression and that this is an important point to address when developing HPV tumor models.
Keywords: Ras, Episome, Extrachromosomal, Episomal HPV, Papillomavirus, HPV, Transformation, Anchorage independence, Cervical keratinocyte
Introduction
High-risk human papillomavirus (HPV) infection is a causative agent of cervical cancer, with nearly all malignancies containing HPV DNA (Walboomers et al., 1999). During the natural course of infection, HPV is first established as an extrachromosomally replicating genome (often referred to as episomal replication) in the basal cells of stratifying epidermis. The association of the E6 and E7 oncogenes with several cellular factors, like the tumor suppressor p53, the hTERT catalytic component of telomerase, and the retinoblastoma (Rb) protein, promotes cellular immortalization and transformation (Mantovani and Banks, 2001 ;Munger et al., 2001).
Progression to malignancy is commonly associated with transcriptional upregulation of E6/E7 as a result of integration of the viral genome with preferential deletion of the E1 and E2 ORFs (Andersson et al., 2005 ;Romanczuk and Howley, 1992). Many studies have relied on the use of cloned HPV E6 and E7 genes artificially expressed from a retroviral or CMV promoter to mimic HPV-infected cells and the use of extensively cultured cell lines that contain integrated HPV to model advanced cervical disease. Models of HPV transformation based on these systems, however, do not consider that a significant number of cervical lesions, even some high grade, continue to maintain extrachromosomal copies of the viral genome. Arias-Pulido et al. showed that integration most commonly occurs in the presence of a multitude of HPV episomes (Arias-Pulido et al., 2006), and in light of a recent report, viral genes expressed episomally, namely E2, likely influence expression from integrated HPV DNA in a natural infection (Pett et al., 2006). The maintenance of extrachromosomal genomes and its effect on oncogene regulation, both in cis and trans, are important considerations for understanding how high-risk HPV malignancies develop.
Examination of the early stages of HPV infection and transformation has relied on the establishment of viral replication in vitro. These studies have been hindered by the tendency of viral genomes to integrate during cell culture, although a handful of reports have demonstrated stable episomal replication of HPV-11, -16, -18, -31, and -45 in foreskin keratinocytes (Flores et al., 1999; Frattini et al., 1996 , 1997 ; McLaughlin-Drubin et al., 2003 ; Thomas et al., 2001 ) and HPV-18 in cervical keratinocytes ( Meyers et al., 1997 ). Study of episomal HPV-16 specifically in cervical keratinocytes has relied on the use of W12 cells and its subclonal derivations. W12 cells were isolated from a pre-existing CIN I lesion (Stanley et al., 1989), and early passage cells from the parental cell line maintain episomal copies of HPV-16 which are susceptible to integration over time (Jeon et al., 1995). We previously reported the in vitro establishment of episomally replicating HPV-16 in isogenic cell lines derived from primary human cervical keratinocytes by transfection of HPV-16 genomes isolated from W12 clone E or by a novel adenoviral vector (Lee et al., 2004; Sprague et al., 2002). The cell lines were isolated from independently derived clones soon after introduction of HPV into primary cells. Extrachromosomal replication of HPV-16 in these cells is quite stable when cultured with irradiated feeder fibroblasts (Sprague et al., 2002). Thus, the HPV-positive cell lines generated in vitro provide a model of early stage cervical disease to study malignant progression of HPV-16-infected cells by factors such as oncogenic Ras.
Ras is a cellular GTPase that signals through MAPK, PI3K, and Ral-GEF pathways (Katz and McCormick, 1997). Ras expression in primary fibroblasts induces growth arrest and a flattened, enlarged phenotype characteristic of premature senescence (Mason et al., 2004;Serrano et al., 1997). Murine keratinocytes (Lin et al., 1998), human ovarian surface epithelial cells (Nicke et al., 2005), and human esophageal keratinocytes (Takaoka et al., 2004) react similarly. However, it has been reported that human thyroid epithelial cells proliferate upon introduction of Ras (Skinner et al., 2004), suggesting that the effects of Ras on proliferation are cell type specific. In cells where Ras mediates senescence, it is generally accepted that senescence is caused by activation of the ARF-p53 and p16INK4a pathways which trigger cell cycle arrest in G1 (Ferbeyre et al., 2002;Lin et al., 1998;Serrano et al., 1997). This response is considered a tumor suppressing mechanism to prevent aberrant cell signaling and proliferation. Unlike rodent cells, human diploid fibroblasts require disruption of both p53 and p16INK4a pathways to overcome senescence (Serrano et al., 1997). Esophageal keratinocytes lacking a functional p53 pathway (Takaoka et al., 2004) or hTERT-immortalized human adenoid epithelial cells lacking p14ARF ( Farwell et al., 2000 ) undergo Ras-induced senescence, further supporting that disruption of the ARF-p53 pathway alone is insufficient for human cells to escape growth arrest. The contribution of p16INK4a in Ras-induced senescence in epithelial cells is less clear. In one study, overexpression of Cdk4 (which counteracts p16INK4a) in human epidermal keratinocytes was sufficient for Ras-mediated transformation ( Lazarov et al., 2002) while hTERT-immortalized human foreskin keratinocytes deficient for p16INK4a remained sensitive to Ras oncogene expression ( Farwell et al., 2000).
Constitutive activation of the Ras signaling pathway is an important component of malignant progression for a number of different cancers. Overexpression or mutation of Ras has been identified to varying degrees in cervical cancers depending on the experimental design (reviewed in Mammas et al., 2005). Several studies indicate an association of Ras with high-grade cervical lesions (Alonio et al., 2003; Bernard et al., 1994; Riou et al., 1988;Sagae et al., 1990). Alonio et al. detected mutated H-Ras in 20% of CIN III cases and 41% of in situ carcinomas (Alonio et al., 2003). In another study, elevated Ras expression was detected in 17.9% of CIN I lesions as compared to 53.9% of CIN III (Sagae et al., 1990). There are also reports that Ras alterations rarely occur in cervical cancer, however, and these studies conclude that there is no correlation between disease severity and Ras expression (Ngan et al., 1999;O’Leary et al., 1998; Van Le et al., 1993). Thus, it is unclear at what stage Ras overexpression or mutation occurs during malignant transformation and whether HPV episome-containing cells tolerate constitutive activation of the Ras pathway, since there was no distinction between episomal or integrated HPV in the studies mentioned.
It is understood that HPV infection alone is not sufficient for tumor formation and that carcinogenesis is a multistep process (reviewed in Hanahan and Weinberg, 2000). In rodent models, Ras has been shown to cooperate with oncogenes from HPV in vivo by inducing tumor growth in HPV-16 E6/E7 transgenic mice (Schreiber et al., 2004). In human cells, understanding the contribution of Ras to cellular transformation in the context of the full HPV genome has been limited to studies using constructs which integrate. Integrated HPV constructs have been shown to cooperate with H-Ras or K-Ras in cervical keratinocytes (DiPaolo et al., 1989), foreskin keratinocytes (Durst et al., 1989,1991), and prostate epithelial cells (Rhim et al., 1994) by inducing tumor growth in mice. Retroviral transductions using only the 16-E6/E7 oncogenes, however, along with H-Ras and hTERT in fibroblasts and HEK cells did not result in anchorage independence or tumor growth (Hahn et al., 2002; Morales et al., 1999), indicating other events or the full HPV genome are necessary for malignant transformation in those tumor models.
Because there is a lack of understanding of cancer progression based on Ras and cervical keratinocytes maintaining HPV-16 DNA extrachromosomally, we utilized clonal cell lines generated in our laboratory to examine whether oncogenic Ras cooperates with stable episomal HPV-16, specifically during the early stages of transformation. Since we also generated clonal cell lines derived from the same genetic background as the episomal cells using LXSN-16E6/E7 retrovirus, we were able to compare the effects of Ras between two different contexts of oncogene expression. While Ras enhanced or maintained anchorage independence of E6/E7-transduced cells, our results demonstrate that Ras actually caused a loss in transformation potential of cells containing HPV-16 episomes. Our findings suggest that HPV-infected cells are refractory to Ras-induced transformation commensurate with reduction of E6 and E7 and induction of p53.
Results
Ras expression does not alter episome copy number
Since no in-depth study has analyzed whether Ras and episomal HPV-16 cooperate in transformation of human cervical cells, we grew clonal cervical keratinocyte cell linescontaining HPV-16 episomes, called HPVEP cells, and trans-duced them with H-RasG12V. We also used isogenically derived clonal cell lines transduced with LXSN-16E6/E7, called E6/E7LX cells, as well as primary human cervical keratinocytes (HCKs). Ras protein expression was confirmed by Western blot (Fig. 1A). The H-RasG12V construct used in our studies produces a functional protein capable of stimulating the Ras pathway in both HPVEP and E6/E7LX cells, as evidenced by increased phosphorylation of MEK and MAPK effector proteins (Fig. 1B). The reason for higher basal levels of hosphorylated p44/42 MAPK in the E6/E7LX cells is unknown but may have to do with higher expression levels of E6 in these cells ( Chakrabarti et al., 2004). To verify that Ras did not disrupt episome copy number control or cause integration, Southern analysis was performed (Fig. 1C). Even in the presence of high Ras expression, HPVEP cells maintained stable episomal DNA for at least 20 population doublings after Ras transduction with equivalent copies between Ras-transduced cells and vector controls. We have also observed episome maintenance with no evidence of integration in untransduced HPVEP cells even when grown for ˜30 population doublings on plastic without irradiated feeders, thought necessary for episome stability, in serum-free keratinocyte media (data not shown). This finding along with Fig. 1C demonstrates the notable stability of episomes in the HPVEP cells created from primary cervical keratinocytes.
Fig. 1.
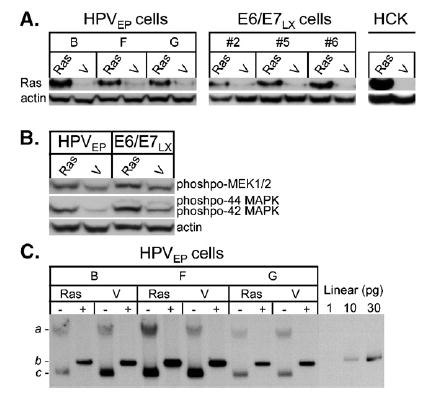
(A) Western blot verifying Ras protein expression one passage after retroviral transduction of pBabe-puro H-RasG12V (Ras) or pBabe-puro vector (V). Cell lines used are HPVEP clones (B, F, and G), E6/E7LX clones (#2, #5, and #6), or primary HCKs. (B) Western blot verifying functionality of the H-RasG12V construct by activation of downstream effectors via phosphorylation of MEK1/2 and 44/42 MAPK proteins in HPVEP and E6/E7LX cells. (C) Southern blot displaying viral DNA present in HPVEP clonal populations expressing Ras or vector. Cells were cultured for 10 passages (˜20 population doublings) after Ras-retrovirus infection and DNA was digested either with _Bg_ιII (− lanes) or _Bam_HI (+ lanes). _Bg_ιII cleaves cellular genomic DNA, leaving the HPV genome as an intact supercoiled episome (c) or nicked-circular plasmid (a). _Bam_HI linearizes the HPV genome (b). Picogram quantities of linearized HPV-16 plasmid were used to estimate copy number as previously described (see Materials and methods). The 30 pg lane is equivalent to 5 copies per cell.
Effects of Ras on cell growth
Because Ras is capable of inducing senescence in some cells and stimulating growth in others (Skinner et al., 2004), we first examined whether expression of mutated Ras induced morphological changes indicative of senescence in the HPVEP and E6/E7LX cells. Functionality of the H-RasG12V retroviral construct used in our study was further confirmed in primary HCKs as indicated by the presence of enlarged, highly vacuolated cells typical of senescence-associated morphology (Fig. 2A). Ras-HPVEP cells did not exhibit as dramatic of an effect, although there were sporadic populations of enlarged, senescent-like cells (Fig. 2A). Differences between Ras and vector transduced E6/E7LX were not readily apparent (Fig. 2A). We stained for senescence-associated beta-galactosidase (SA-β-gal) activity, a marker that is commonly observed in senescent fibroblasts, but detected only weak SA-β-gal staining in late passage senescent primary keratinocytes and Ras-expressing clones (data not shown), suggesting that this is not a reliable marker for senescence in keratinocytes. To quantify the senescent-like cells, a blinded count of cells with an enlarged morphology was performed (Fig. 2B). Senescent-like cells were more frequently found in the Ras-HPVEP cultures than vector control. To a lesser extent, a small fraction of senescent-like cells was also counted in the Ras-E6/E7LX cultures as compared to vector. We also examined the cells for changes in cell cycle profile and did not see dramatic differences between Ras and vector controls in either cell type (data not shown). We next examined the effect of Ras on population doubling (PD) time based on growth assays in monolayer culture (Fig. 3). Ras lengthened the PD time of two HPVEP clones by 4% in clone B and 8% in clone F, yet had no effect in clone G. Two of three E6/E7LX clones had decreased PD time upon Ras expression (i.e., a 3% decrease in PD time for Ras-E6/E7LX clone #6 and only a 1% decrease for Ras-E6/E7LX clone #5). No difference in PD time was observed for E6/E7LX clone #2. In summary, Ras expression had a negative but small effect on growth of HPVEP cells in monolayer culture, and growth of E6/E7LX clones was either enhanced or unchanged.
Fig. 2.
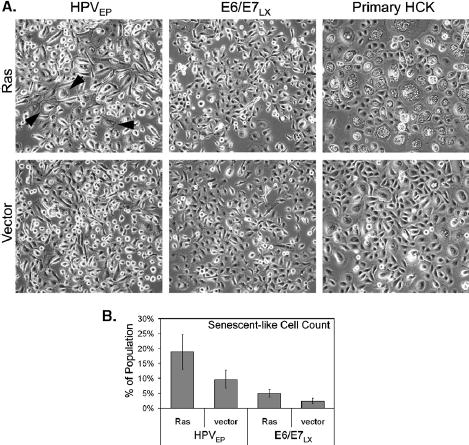
(A) Microphotographs of morphological changes induced by H-RasG12V. Photographed cultures of HPVEP clone G, HPVEP clone #2, and HCKs were grown without irradiated feeders. Black arrows point to enlarged cells in Ras-expressing HPVEP cells, which are rarely found in E6/E7LX cultures. HCKs demonstrate typical morphology induced by oncogenic Ras in primary epithelial cells. (B) Blinded counts of enlarged and flattened cells, indicative of senescence, from triplicate plates of either HPVEP clone G or HPVEP clone #2 are expressed as a percentage of the total number of cells counted. Data are expressed as the mean±SD.
Fig. 3.
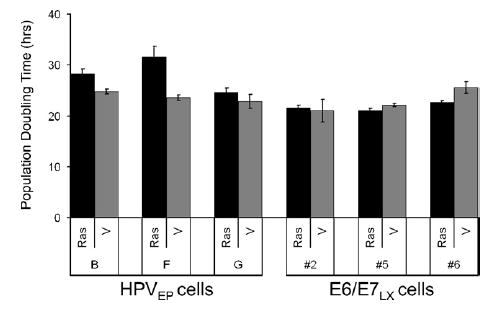
Population doubling (PD) time was determined over a 3-day period from absorbance data generated from crystal violet staining of cultures (see Materials and methods). H-RasG12V increased PD time in two of three HPVEP clones. No change in PD time was observed for two E6/E7LX clones, #2 and #5, while Ras slightly decreased PD time in one cell line, clone #6. Data are expressed as the mean±SD of triplicate data points.
Ras reduces transformation potential of HPVEP but not E6/E7LX cells
Growing cells in soft agar is a common in vitro transformation assay that measures anchorage-independent (AI) growth (i.e., the ability to grow without substrate attachment). To test if Ras altered anchorage independence of the HPVEP and E6/E7LX cervical cell lines, we assayed cell growth in a modified soft agar assay. Two experiments were performed ( Table 1 ) using cell lines generated from two independent transductions with Ras and vector. For visualization purposes, the results of the first experiment are shown graphically (Fig. 4). AI growth of both the HPVEP and E6/E7LX clones was heterogenous, with vector controls ranging from 1 to 16% CFE (Fig. 4). More striking than the growth changes induced by Ras in monolayer culture (Fig. 3), we observed profound effects on growth in soft agar. Oncogenic Ras significantly limited AI growth of all three HPVEP cell lines (p<0.001) (Fig. 4). In contrast, two E6/E7LX clones (#2 and #6) showed significant growth enhancement upon Ras expression (p<0.001) while one cell line (#5) showed no difference (Fig. 4). The lack of enhanced anchorage independence by Ras in E6/E7LX clone #5 likely reflects heterogeneity in E6/E7LX cell clones. Overall, our results indicate that Ras can cause different effects on AI growth depending on whether cells contain episomal HPV-16 or the 16E6/E7 retroviral construct.
Table 1.
The effect of H-RasG12V on colony-forming efficiency in soft agar and tumor growth
| Cell line | Clone construct | Anchorage independence (Experiment #1)a | Anchorageindependence (Experiment #2) | No. of mice with tumors |
|---|---|---|---|---|
| % CFE±SD | % CFE±SD | |||
| HPVEP | B-Ras | 9.5±1.8 | 5.2±1.0 | 0/3 |
| B-vector | 14.6±0.6 | 12.0±1.0 | 0/3 | |
| F-Ras | 3.4±0.9 | 1.0±0.7 | 0/3 | |
| F-vector | 6.3±0.8 | 9.6±1.4 | 0/3 | |
| G-Ras | 2.3±0.3 | 2.1±0.1 | 0/3 | |
| G-vector | 15.8±3.4 | 6.8±2.8 | 0/3 | |
| E6/E7LX | #2-Ras | 7.1±0.6 | 2.5±0.5 | 0/3 |
| #2-vector | 3.7±0.5 | 0.9±0.2 | 0/3 | |
| #5-Ras | 8.1±0.8 | 7.0±0.7 | 0/3 | |
| #5-vector | 8.4±1.1 | 7.8±0.7 | 0/3 | |
| #6-Ras | 22.1±2.2 | 12.8±1.0 | 0/3 | |
| #6-vector | 11.8±1.3 | 3.7±0.6 | 0/3 | |
| Control | HPV-18 tumor b | N/D±N/D | N/D±N/D | 3/3 |
| C33A | 7.1±0.5 | N/D±N/D | N/D | |
| HCK | 0±0 | 0±0 | 0/3 |
Fig. 4.
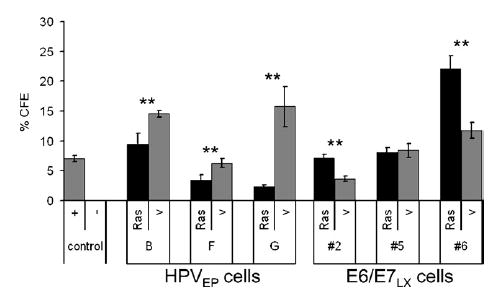
H-RasG12V significantly reduced anchorage-independent (AI) colony formation of all HPVEP clones (**p<0.001) and stimulated growth of two E6/E7LX clones (**p<0.001). Percent colony forming efficiency (%CFE) is reported as the percentage of colonies that grew in soft agar per number of cells initially seeded. HPV-negative C33A cells (+) control; primary HCKs (−) control. Data are expressed as the mean±SD of triplicate data points.
Since AI growth does not necessarily correlate to an ability to form tumors in vivo, full transformation of human cells is typically determined by injecting cells into immuno-compromised mice. Therefore, we tested whether Ras could cooperate with episomal HPV or LXSN-E6/E7 to form tumors in athymic nude mice after subcutaneous injection. After 100 days postinjection, no tumorigenic growth was observed for HPVEP or E6/E7LX cells expressing Ras or vector ( Table 1 ), indicating that Ras and expression of E6/E7 are insufficient for causing tumorigenic transformation.
Effects of Ras on HPV oncogene expression
To determine whether differential oncogene expression is responsible for the observed differences between the HPVEP and E6/E7LX cells, we used real-time RT-PCR to measure the abundance of E6 and E7 mRNA. For reference, mRNA levels were calculated relative to W12 cells. Vector control HPVEP cells, which contain W12-E genomes, had comparable amounts of E6 and E7 to the parental W12 cell line (Figs. 5A and C). Clearly, vector control E6/E7LX cells expressed more E6 than the HPVEP vector control cells (13-fold more on average) (Figs. 5A and B; note different scales). Interestingly, the HPVEP and E6/ E7LX vector control cell lines had comparable amounts of E7 mRNA with E6/E7LX vector controls expressing only 1.6-fold more on average (Figs. 5C and D). In agreement with previous reports, this supports the finding that the dual LXSN-E6/E7 retroviral construct expresses E7 inefficiently ( Halbert et al., 1991). These results indicate that E6 expression is one key distinction between HPVEP and E6/E7LX cell types.
Fig. 5.
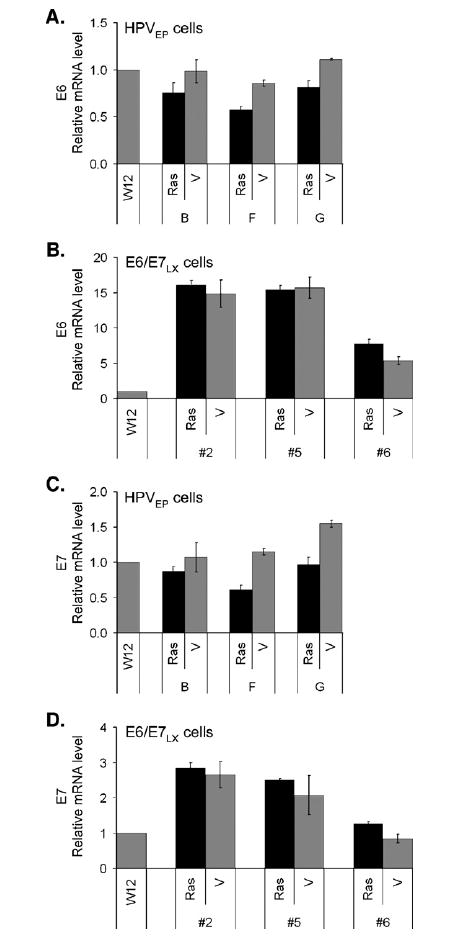
Real-time RT-PCR for E6 transcripts in Ras or vector-transduced HPVEP (A) or E6/E7LX cells (B) and E7 transcripts in HPVEP (C) or E6/E7LX cells (D) all standardized to 18S mRNA levels. HPVEP cells contain the full genome derived from W12-E cells. E6/E7LX cells dually express E6 and E7 from a single retroviral promoter. For reference, RNA levels were calculated relative to W12 cervical cells, the cell line containing episomal HPV-16 from which the W12-E clone was derived. Data are expressed as the mean±SD of triplicate data points.
Interestingly, we found that Ras reduced E6 mRNA levels in the HPVEP clones (Fig. 5A). However, Ras expression affected E6 transcripts in only one of three E6/E7LX clones (#6), where it actually increased levels (Fig. 5B). Similar results were attained for E7. Ras reduced E7 transcripts in the HPVEP clones (Fig. 5C) but did not have a significant effect on E7 transcript levels in E6/E7LX clones #2 and #5 and marginally increased levels in E6/E7LX clone #6 (Fig. 5D). Since we observed changes in E7 transcripts in Ras-expressing cells, we analyzed E7 protein levels (Fig. 6). In HPVEP cells, Ras reduced E7 protein and this corresponds to the reduction of E7 mRNA shown in Fig. 5B. With the E6/E7LX clones, some reduction of E7 protein was observed upon Ras expression but, overall, this was not as great as that observed for the HPVEP cells. The reason for the slight differences between E7 transcript (Figs. 5B and D) and protein levels (Fig. 6) is unknown. Since reliable antibodies for E6 are not available, we were unable to assay for E6 protein in our cells. Overall, however, our real-time RT-PCR and Western data provide evidence that Ras represses expression of E6 and E7 in HPVEP cells but has minimal effects in E6/E7LX cells.
Fig. 6.
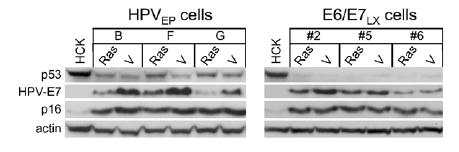
Western blot of p53, HPV16-E7, and p16INK4a proteins in HPVEP and E6/E7LX clonal populations. Primary HCKs were used for reference. Lanes consist of lysates collected one passage after transduction and selection for H-RasG12V or vector (V).
Oncogenic Ras induces p53 but not p16INK4a in cells with episomal HPV-16
Mutant Ras has been shown to upregulate p53 and p16INK4a in normal human fibroblasts, resulting in growth inhibition and senescence ( Serrano et al., 1997). Since we observed senescent-like cells, reduction in AI growth, and changes in E6 and E7 transcript levels upon Ras transduction we examined expression of p53 and p16INK4a in our different cell lines. Western analysis was performed using protein lysates from early passage cells (i.e., within 1 to 2 passages after transduction of vector or Ras). As expected from basal levels of E6 mRNA (Figs. 5 A and B), HPVEP cells have higher p53 basal expression than E6/E7LX cells (Fig. 6). An increase in p53 protein was detected in Ras-HPVEP cells (Fig. 6), corresponding to a decrease in E6 mRNA. Changes in p53 were not evident between Ras-E6/E7LX and vector controls and this is likely related to higher levels of E6 mRNA in these cells. Since disruption of the p16INK4a pathway is sufficient to overcome Ras-induced senescence in some epithelial cell types ( Lazarov et al., 2002) but not in others (Farwell et al., 2000), we also examined p16INK4a protein levels. p16INK4a in either cell type was unchanged upon Ras expression, indicating that induction of p16INK4a was not associated with inhibition of AI growth by Ras in HPVEP cells.
Discussion
The use of cervical cell lines with stably replicating HPV-16 extrachromosomal genomes allowed us to examine the effects of Ras in cells that more closely resemble early HPV infection. The finding that episomal HPV-16 does not support Rasmediated transformation in human keratinocytes indicates that HPV genome status has a significant impact on transformation capacity. HPV E6 and E7 oncogenes are essential for transformation and the downregulation of both E6 and E7 by Ras may partially explain why HPVEP cells have reduced AI growth when Ras is introduced. Because p53 closely regulates cell growth and can induce senescence, the significant inhibition of AI growth is likely related to p53 upregulation by Ras. Together, increased p53 and decreased E6 and E7 may limit the growth of HPVEP cells in tissue culture and in soft agar. The results with HPVEP cells contrast with those obtained from cells expressing E6/E7 from a retroviral construct. The E6/E7LX cells were able to maintain or enhance anchorage independence likely because Ras expression did not profoundly affect p53, E6, and E7 levels. Because of the skewed oncogene expression favoring E6 transcription from the heterologous promoter in the E6/E7LX cells, we speculate that it was artificial overexpression of E6 and not E7 that specifically contributed to maintenance of the transformed phenotype in the presence of Ras. Therefore, we conclude that the context of oncogene expression may be a determinant for transformation by oncogenic Ras.
Not uncommon for some transformed cells (Bihani et al., 2004), our cell lines may retain properties of primary cells which trigger tumor suppressing mechanisms, as in p53 induction in Ras-HPVEP cells. It has been previously shown that oncogenic Ras synergizes with integrated HPV-18 in HeLa cells by stimulating cell cycle progression ( Cordova-Alarcon et al., 2005), but it was also noted in a related paper that a subpopulation of Ras-transduced HeLa cells have aberrant, giant cell morphology and underwent “mitotic death” (Miranda et al., 1996). Thus, even HeLa cells may still retain some normal responses to Ras. Interestingly, it has been noted, using reporter constructs and HeLa cells, that Ras can mediate activation of the HPV-18 viral upstream regulatory region (URR) (Medina-Martinez et al., 1997). Regarding integrated HPV-16, one report showed increased E6/E7 RNA expression in CX16-2 cells with a human H-Ras oncogene (Gaiotti et al., 2000) while another study reported no change in E6/E7 protein levels in the same CX16-2 cells using a viral H-Ras homolog (DiPaolo et al., 1989). We conclude that the effects of Ras on E6 and E7 RNA and protein levels vary depending on HPV type, source of oncogenic Ras, and most importantly, the context of oncogene expression either from integrated HPV, integrated HPV constructs containing non-HPV DNA sequences, or extrachromosomal genomes as shown here. A mechanism of how Ras regulates E6/E7 is not fully understood but may involve regulation of AP-1 transcription factors such as c-Jun and c-Fos or other factors (Medina-Martinez et al., 1997).
As with p53, p16INK4a is another protein associated with senescence ( Serrano et al., 1997). Although we have observed weak p16INK4a induction by Ras in primary cervical keratinocytes (unpublished data), Ras did not alter p16INK4a expression in the HPVEP or E6/E7LX cells (Fig. 6). This is likely due to the fact that Rb inactivation by E7 causes misregulation of the p16INK4a pathway leading to high accumulation of p16INK4a protein ( Giarre et al., 2001), thus overshadowing any effect by Ras. Therefore, while growth inhibition of HPVEP cells may be in part due to p53 induction, it does not appear to be due to Ras induction of p16INK4a. It should be noted that other functions of E6 and E7, besides inactivation of p53 and pRb, may play a role in Ras-mediated effects on AI growth. Both E6 and E7 bind to a plethora of cellular proteins and these interactions may be important for maintaining or even promoting cell growth upon Ras activation (Mantovani and Banks, 2001; Munger et al., 2001). Further work is necessary to determine what functions of E6 and/or E7 are important for allowing transformation by Ras.
The answer to why Ras reduces transformation of cells with episomal HPV-16 may also not be as simple as changes in expression levels of E6 and E7. In our studies, E6 and E7 mRNA levels do not strictly correlate to %CFE in the AI growth assays. For example, there is a notable increase in AI of E6/E7LX clone #6 by Ras (Fig. 4) yet this clone expresses lower amounts of oncogenes than the other E6/E7LX clones (Fig. 5). There may be a heterogeneous response to Ras in E6/E7LX cells or any number of other unknown cellular defects that could have occurred in this cell line. In addition to expression levels, whether Ras inhibits or enhances AI growth may also depend on spliced E6 mRNA species. Transcription of the E6 ORF produces alternatively spliced E6* in addition to full-length E6. The function of E6* is unclear but may regulate E6/E7 expression ( Sedman et al., 1991) and inhibit degradation of p53 by unspliced E6 (Pim et al., 1997). Studies have shown that cells with extrachromosomal HPV genomes contain more spliced E6 than unspliced (Doorbar et al., 1990; Hummel et al., 1992). This could account for the lower levels of full-length E6 detected in the W12 and HPVEP cells in our assays. The overabundance of full-length E6 in the E6/E7LX cells suggests that the LXSN-16E6/E7 construct is partially dysfunctional in its ability to make the spliced forms. Enhancement of AI growth by Ras in E6/E7LX and not HPVEP cells could also be due to genetic variations in the oncogenes. HPVEP cells contain HPV-16 genomes derived from W12-E (GenBank accession no. AF125673) whereas LXSN-16E6/E7 contains sequences from the prototype genome (GenBank accession no. K02718). Sequence analysis reveals a deviation in W12-E from the prototype—a codon change at amino acid 83 in E6. E6 variants occur in nature and are epidemiologically linked to progression of high-grade lesions to invasive cancers ( Zehbe et al., 1998). The E6 amino acid 83 variant (or E6 L83V) does not differ from prototype in ability to degrade p53 but does enhance MAPK signaling (Lichtig et al., 2006; Zehbe et al., 1998), and this may affect the ability of cells to be transformed by Ras.
Certainly, other HPV proteins besides E6 and E7 could play a role in the response of HPVEP cells to Ras. It is possible, for example, that loss of E2 by integration of the HPV genome constructs in previous studies with Ras ( DiPaolo et al., 1989; Durst et al., 1989,1991;Rhim et al., 1994) or lack of E2 in our E6/E7LX cells played a role in Ras-transformation. Gammoh et al. showed that E2 inhibits transformation by HPV-16 E7 and Ras in rodent cells ( Gammoh et al., 2006). E2 may similarly effect the ability of E6 to cooperate with Ras via transcriptional regulation as well as protein interaction and mislocalization (Grm et al., 2005). In our HPVEP cells, E2 may be limiting the transformation potential of E6 and E7 through protein–protein interactions, mislocalization, and/or repression of transcription, thereby rendering cells more susceptible to Ras-induced growth inhibition than cells without E2.
Several epidemiological studies have reported higher levels of Ras in advanced cervical lesions (reviewed in Mammas et al., 2005) and experimental data show that Ras stimulates malignant transformation of human cells containing integrated high-risk HPV (DiPaolo et al., 1989; Durst et al., 1989,1991;Rhim et al., 1994). Since high-grade lesions are often associated with integration and upregulated oncogene transcription while low-grade lesions more commonly contain extrachromosomal HPV, mutations in Ras may only be tolerated by cells expressing high levels of E6 and E7. Since advanced lesions harboring integrated DNA likely also contain episomal DNA (Arias-Pulido et al., 2006) and also since episomal E2 expression can affect regulation of integrated viral sequences (Pett et al., 2006), episomal HPV may still prevent Rasmediated transformation even when integrated HPV is present. These hypotheses would require further testing using similarly derived episomal and integrated cell lines. Since our experiments were performed using a retroviral construct that expresses high levels of H-RasG12V, we also cannot rule out the possibility that lower levels of mutated H-Ras from the endogenous promoter could lead to enhanced transformation in HPV-infected cells (Hua et al., 1997).
Not surprisingly, cervical cells expressing Ras with episomal HPV-16 or overexpressed 16E6/E7 do not form tumors in mice. In HPV tumor models, Ras has only been shown to induce tumor growth of cells containing integrated genomes (DiPaolo et al., 1989;Durst et al., 1989,1991;Rhim et al., 1994) or by crossing Ras transgenic mice with E6/E7 transgenic mice (Schreiber et al., 2004). Our analysis of cervical keratinocytes agrees with Morales et al. and Hahn et al. who demonstrated that E6/E7 overexpression, hTERT, and Ras were insufficient for tumor growth of human cells in mice (Hahn et al., 2002;Morales et al., 1999). The addition of SV40-small t antigen was required for tumorigenesis (Hahn et al., 2002;Uren et al., 2005). SV40-small t antigen is known to affect the MAP kinase pathway and also inhibit protein phosphatase 2A. Since our cell lines already have active telomerase (Sprague et al., 2002), it would be of interest to determine whether expression of small t might be sufficient for tumor growth in conjunction with Ras expression. Furthermore, in some cervical cancer specimens, Myc, ErbB-2, PTEN, and EGF-R proteins are found to be altered (Pinion et al., 1991;Spandidos et al., 2000) and promote malignant transformation in experimental tumor models (Boehm et al., 2005; Goessel et al., 2005). These are other cellular changes that may be required for full transformation of the HPVEP and E6/E7LX keratinocytes.
In conclusion, we have demonstrated that human cervical keratinocytes containing stable HPV episomes resist transformation by H-RasG12V. We are currently developing a model of how early cervical lesions progress to malignancy using the HPVEP cells in conjunction with other cervical cancer markers. These studies will be important for determining the types of events necessary for HPV-associated transformation and may point to strategies for inhibiting HPV-related cancer.
Materials and methods
Cell culture
Cell cultures were maintained with post-mitotic, γ-irradiated J2 3T3 feeder fibroblasts in E-media: 3 parts Dulbecco’s modified Eagle’s media (Invitrogen, Carlsbad, CA) and 1 part HAMs F-12 nutrient mixture (Invitrogen) supplemented with 10% FBS (Invitrogen), 1% penicillin–streptomycin (Invitrogen), 1.36 ng/ml tri-iodo-thyronine (Sigma, St. Louis, MO), 0.5 μg/ml hydrocortisone (Sigma), 8.4 ng/ml cholera toxin (Sigma), 5.0 μg/ml transferrin (Sigma), 5.0 μg/ml insulin (Sigma), and 4.5 ng/ml human-EGF (Invitrogen). Primary human cervical keratinocytes were isolated as previously described (Blanton et al., 1991). Prior to splitting and sample collection, irradiated fibroblasts were removed using a mixture of 0.05% trypsin, 50 mM HEPES buffer in Hanks balanced salt solution (Invitrogen). Keratinocytes were removed by incubation in 0.05% trypsin/EDTA (Invitrogen) and passed at a 1:4 ratio.
Cell lines
We previously generated human cervical keratinocyte cell lines containing stable HPV-16 episomes (Sprague et al., 2002), named HPVEP cells in this paper. Briefly, primary keratinocytes were co-transfected with circular HPV-16 genomes derived from W12 subclone E cells (W12-E) and a selectable marker (W12-E DNA provided by Paul Lambert, University of Wisconsin). After selection and dilution plating, colonies were isolated by ring cloning to establish isogenic cell lines (labeled B, F, and G). The same primary cells were used for transductions with the retroviral construct LXSN-16E6/E7 (obtained from Denise Galloway, Fred Hutchinson Cancer Research Center ( Halbert et al., 1991)) by a method previously described (Farwell et al., 2000). Isogenic cell lines were generated from ring-cloned colonies (labeled #2, #5, and #6) and named E6/E7LX cells. Both HPVEP and E6/E7LX cells have been passaged over 40 times, equivalent to greater than 100 PD at which point we consider them to be immortalized. Due to the premise of the paper, we used cells at an earlier passage. Cells were used at passage 15 (˜30 PD) for retroviral transductions. The W12 parental cell line (positive for HPV-16 episomes), 1811-NMU-T cells ( Klingelhutz et al., 1993), and C33A cells were used for various controls.
Constitutively active Ras expression was achieved using the pBabe-puro H-RasG12V retroviral construct (obtained from Scott Lowe, Cold Spring Harbor Laboratory). The vector was sequenced to ensure functionality. The concentration for neomycin selection for LXSN transductions was 100 μg/ml G418 (Invitrogen). Puromycin (Sigma) was used for three days at 1 μg/ml after infection of pBabe-puro retroviruses. All collections and experiments with Ras were performed within the first 3 passages after final puromycin selection.
Senescent cell count
HPVEP and E6/E7LX cells were plated in triplicate 60-mm dishes in E-media without feeders and grown to subconfluency. Cell culture microphotographs were taken using a Nikon Eclipse TE2000 inverted microscope. Enlarged and flattened cells, indicative of senescence, were tabulated from a blinded count of triplicate microphotographs. The number of senescentlike cells was calculated as a percentage of the total number of cells counted in each field.
Crystal violet assay
Cells were plated in triplicate in a 96-well format at a concentration of 2000 cells per 150 μl media and media changed daily. At specific time points, wells were washed with 1× PBS and stained with a solution of 0.5% crystal violet (w/v) in 20% methanol for 15 min. Wells were washed five times with distilled water and allowed to air-dry for 45 min or overnight. The cellassociated dye was extracted with 100 μl of 10% acetic acid and absorbance was read at λ =595 nm. Population doubling time was calculated as [time in hours]/[(log _N_−log _N_O)/log 2]; _N_=absorbance value at day 5 and _N_O =value at day 2.
Southern analysis
Southern analysis was performed as previously described (Sprague et al., 2002). Briefly, whole cell DNA was isolated from later passage clonal cultures using the QIAamp DNA BloodMini Kit (Qiagen, Valencia, CA) and digested either with _Bam_HI or _Bg_lII. DNA was resolved on 1% agarose gels, depurinated in 0.25 M HCl, and blotted onto positively charged Hybond-XL nylon membranes (Amersham Biosciences Corp., Piscataway, NJ) by alkaline transfer with 0.4N NaOH. The different forms of HPV DNA were detected using radioactive probes of PCR-amplified segments of HPV-16 (nt 6226–3873/4471–6000). Known quantities of HPV DNA linearized by _Bam_HI were included to estimate the viral load.
Western analysis
Protein from subconfluent cultures was collected in lysis buffer as previously described (Foster and Galloway, 1996) and quantified using the Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA). Western blots were prepared using 30 μg of protein transferred to polyvinyl difluoride membranes. Blots were probed with primary antibodies for H-Ras (sc-29, Santa Cruz), p-MEK1/2 (Ser217/221; #9121, Cell Signaling Technology, Danvers, MA), p-44/42 MAPK (Thr202/Tyr204; #9101S, Cell Signaling Technology), p53 (Ab-6, Oncogene Research), HPV-16 E7 (#28-0006, Zymed Labs, South San Francisco, CA), p16 (#554070, Pharmingen) or beta-actin (I-19, Santa Cruz) and with the appropriate HRP-conjugated secondary antibodies, either goat anti-mouse IgG (Jackson Immuno-Research Laboratories), goat anti-rabbit IgG (Santa Cruz), or donkey anti-goat IgG (Santa Cruz). Chemiluminescence was detected using the Western Lightning kit (Perkin-Elmer Life Sciences). All films were scanned with a Hewett-Packard PrecisionScan 5300 and Adobe Photoshop software. Membranes were stripped by a 30 min, 50 °C incubation in a solution of 100 mM 2-mercaptoethanol, 2% SDS, and 62.5 mM Tris–HCl at pH 6.7.
Soft agar assay
Polyester membranes of transwell inserts (12-mm diameter, 0.4-μm pore; Costar, Corning, NY) were coated with 100 μl 1% noble agar. After counting cells using a hemacytometer, 1 part of 3.3% noble agar at 45 °C was diluted with 10 parts cell suspension for a final concentration of 700 cells per 400 μl. Triplicate wells for each cell line expressing Ras or vector were seeded with 700 cells. After agar solidification, media were added to the outer wells. After 3 weeks, transwell inserts were photographed using an Olympus stereoscope. Colonies approximately 0.19 mm in diameter or greater were tabulated. For statistical analysis, we compared the overall proportion (_Z_-statistic) of soft agar colonies between the Ras-expressing and vector conditions.
Tumorigenicity
Cells (1×106) in 0.2 ml KSFM were injected subcutaneously into 3-week-old female athymic (nu/nu) mice (Harlan-Sprague-Dawley, Madison, WI). Three mice were used for each Ras-expressing and each vector control cell line. Mice were housed in a pathogen-free barrier facility at The University of Iowa and handled in compliance with federal and institutional regulations.
Real-time RT-PCR
Total RNA was collected using TRIzol reagent (Invitrogen) and treated with 1U per μg RNA of DNase I AMP-grade (Invitrogen) per manufacturer’s instructions. The RT reaction consisted of 1.7 μg RNA, 2 μl random hexamers, and RetroScript kit reagents (Ambion, Austin, TX) according to instructions. RNA was reverse transcribed for 1 h at 42 °C with inactivation at 92 °C for 10 min. Real-time PCR was performed on cDNA equivalent to 150 ng RNA in triplicate 25 μl reactions using SYBR Green PCR master mix (#4309155, Applied Biosystems, Foster City, CA) containing 250 nM forward and reverse primers for E6 or E7. Unspliced HPV16-E6 was detected as described (Lanham et al., 2001). _E_6 forward: 5′-CAA ACC GTT GTG TGA TTT GTT AAT TA-3′; E6 reverse: 5′-GCT TTT TGT CCA GAT GTC TTT GC-3′ (Integrated DNA Technologies, Coralville, IA). Sequences for HPV16-E7 primers were provided by Sara Isabel Pai (Johns Hopkins Medical Institution, MA) (Walboomers et al., 1999). E7 forward: 5′-CCG GAC AGA GCC CAT TAC AA-3′; E7 reverse: 5′-CGA ATG TCT ACG TGT GTG CTT TG-3′ (Integrated DNA Technologies). Standardization was based on 18S levels from reactions using primers at 75 nM final concentration. 18S forward: 5′-CCT TGG ATG TGG TAG CCC GTT T-3′; 18S reverse: 5′-AAC TTT CGA TGG TAG TCG CCG-3′ (Sigma-Genosys, The Woodlands, TX). PCR amplification began at 94 °C for 10 min prior to 30 cycles of 94 °C for 40 s, 60 °C for 40 s, and 72 °C for 1 min. Real-time PCR data were analyzed using the ABI 7000 Sequence Detection System and ABI Prism 7000 SDS software (Applied Biosystems). Fold differences in oncogene mRNA levels were calculated relative to the W12 cell line by using the equation:
2∧(ΔΔCT) where ΔΔCT = ΔCTW12 − ΔCTcell line.
Acknowledgments
This work was supported by NIH grant #AG18265 awarded to A.J.K., a VA Merit Award to J.H.L., and NIH grant #CA66081 to F.E.D. We thank the members of the Klingelhutz laboratory for useful discussion and technical assistance, the Turek laboratory for cell culture advice, Mary Anderson and the Domann laboratory for qRT-PCR expertise, and Martine Dunnwald, the University of Iowa Central Microscopy Research Facility and the Flow Cytometry Facility for equipment usage and technical assistance.
References
- Alonio LV, Picconi MA, Dalbert D, Mural J, Bartt O, Bazan G, Dominguez M, Teyssie AR. Ha-ras oncogene mutation associated to progression of papillomavirus induced lesions of uterine cervix. J Clin Virol. 2003;27:263–269. doi: 10.1016/s1386-6532(02)00181-6. [DOI] [PubMed] [Google Scholar]
- Andersson S, Safari H, Mints M, Lewensohn-Fuchs I, Gyllensten U, Johansson B. Type distribution, viral load and integration status of high-risk human papillomaviruses in pre-stages of cervical cancer (CIN) Br J Cancer. 2005;92:2195–2200. doi: 10.1038/sj.bjc.6602648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Pulido H, Peyton CL, Joste NE, Vargas H, Wheeler CM. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J Clin Microbiol. 2006;44:1755–1762. doi: 10.1128/JCM.44.5.1755-1762.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Mougin C, Laurent R, Lab M. Oncogene activation: an informative marker for the human papillomavirus-lesions severity. Cancer Detect Prev. 1994;18:273–282. [PubMed] [Google Scholar]
- Bihani T, Mason DX, Jackson TJ, Chen SC, Boettner B, Lin AW. Differential oncogenic Ras signaling and senescence in tumor cells. Cell Cycle. 2004;3:1201–1207. [PubMed] [Google Scholar]
- Blanton RA, Perez-Reyes N, Merrick DT, McDougall JK. Epithelial cells immortalized by human papillomaviruses have premalignant characteristics in organotypic culture. Am J Pathol. 1991;138:673–685. [PMC free article] [PubMed] [Google Scholar]
- Boehm JS, Hession MT, Bulmer SE, Hahn WC. Transformation of human and murine fibroblasts without viral oncoproteins. Mol Cell Biol. 2005;25:6464–6474. doi: 10.1128/MCB.25.15.6464-6474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti O, Veeraraghavalu K, Tergaonkar V, Liu Y, Androphy EJ, Stanley MA, Krishna S. Human papillomavirus type 16 E6 amino acid 83 variants enhance E6-mediated MAPK signaling and differentially regulate tumorigenesis by notch signaling and oncogenic. Ras J Virol. 2004;78:5934–5945. doi: 10.1128/JVI.78.11.5934-5945.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordova-Alarcon E, Centeno F, Reyes-Esparza J, Garcia-Carranca A, Garrido E. Effects of HRAS oncogene on cell cycle progression in a cervical cancer-derived cell line. Arch Med Res. 2005;36:311–316. doi: 10.1016/j.arcmed.2005.04.001. [DOI] [PubMed] [Google Scholar]
- DiPaolo JA, Woodworth CD, Popescu NC, Notario V, Doniger J. Induction of human cervical squamous cell carcinoma by sequential transfection with human papillomavirus 16 DNA and viral Harvey ras. Oncogene. 1989;4:395–399. [PubMed] [Google Scholar]
- Doorbar J, Parton A, Hartley K, Banks L, Crook T, Stanley M, Crawford L. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology. 1990;178:254–262. doi: 10.1016/0042-6822(90)90401-c. [DOI] [PubMed] [Google Scholar]
- Durst M, Gallahan D, Jay G, Rhim JS. Glucocorticoid-enhanced neoplastic transformation of human keratinocytes by human papillomavirus type 16 and an activated ras oncogene. Virology. 1989;173:767–771. doi: 10.1016/0042-6822(89)90595-3. [DOI] [PubMed] [Google Scholar]
- Durst M, Bosch FX, Glitz D, Schneider A, zur Hausen H. Inverse relationship between human papillomavirus (HPV) type 16 early gene expression and cell differentiation in nude mouse epithelial cysts and tumors induced by HPV-positive human cell lines. J Virol. 1991;65:796–804. doi: 10.1128/jvi.65.2.796-804.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell DG, Shera KA, Koop JI, Bonnet GA, Matthews CP, Reuther GW, Coltrera MD, McDougall JK, Klingelhutz AJ. Genetic and epigenetic changes in human epithelial cells immortalized by telomerase. Am J Pathol. 2000;156:1537–1547. doi: 10.1016/S0002-9440(10)65025-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, Lowe SW. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol. 2002;22:3497–3508. doi: 10.1128/MCB.22.10.3497-3508.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores ER, Allen-Hoffmann BL, Lee D, Sattler CA, Lambert PF. Establishment of the human papillomavirus type 16 (HPV-16) life cycle in an immortalized human foreskin keratinocyte cell line. Virology. 1999;262:344–354. doi: 10.1006/viro.1999.9868. [DOI] [PubMed] [Google Scholar]
- Foster SA, Galloway DA. Human papillomavirus type 16 E7 alleviates a proliferation block in early passage human mammary epithelial cells. Oncogene. 1996;12:1773–1779. [PubMed] [Google Scholar]
- Frattini MG, Lim HB, Laimins LA. In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differentiation-dependent late expression. Proc Natl Acad Sci USA. 1996;93:3062–3067. doi: 10.1073/pnas.93.7.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini MG, Lim HB, Doorbar J, Laimins LA. Induction of human papillomavirus type 18 late gene expression and genomic amplification in organotypic cultures from transfected DNA templates. J Virol. 1997;71:7068–7072. doi: 10.1128/jvi.71.9.7068-7072.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiotti D, Chung J, Iglesias M, Nees M, Baker PD, Evans CH, Woodworth CD. Tumor necrosis factor-alpha promotes human papillomavirus (HPV) E6/E7 RNA expression and cyclin-dependent kinase activity in HPV-immortalized keratinocytes by a ras-dependent pathway. Mol Carcinog. 2000;27:97–109. doi: 10.1002/(sici)1098-2744(200002)27:2<97::aid-mc5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Gammoh N, Grm HS, Massimi P, Banks L. Regulation of human papillomavirus type 16 E7 activity through direct protein interaction with the E2 transcriptional activator. J Virol. 2006;80:1787–1797. doi: 10.1128/JVI.80.4.1787-1797.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giarre M, Caldeira S, Malanchi I, Ciccolini F, Leao MJ, Tommasino M. Induction of pRb degradation by the human papillomavirus type 16 E7 protein is essential to efficiently overcome p16INK4a-imposed G1 cell cycle Arrest. J Virol. 2001;75:4705–4712. doi: 10.1128/JVI.75.10.4705-4712.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goessel G, Quante M, Hahn WC, Harada H, Heeg S, Suliman Y, Doebele M, von Werder A, Fulda C, Nakagawa H, Rustgi AK, Blum HE, Opitz OG. Creating oral squamous cancer cells: a cellular model of oral-esophageal carcinogenesis. Proc Natl Acad Sci USA. 2005;102:15599–15604. doi: 10.1073/pnas.0409730102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grm HS, Massimi P, Gammoh N, Banks L. Crosstalk between the human papillomavirus E2 transcriptional activator and the E6 oncoprotein. Oncogene. 2005;24:5149–5164. doi: 10.1038/sj.onc.1208701. [DOI] [PubMed] [Google Scholar]
- Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, DeCaprio JA, Weinberg RA. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111–2123. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert CL, Demers GW, Galloway DA. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65:473–478. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hua VY, Wang WK, Duesberg PH. Dominant transformation by mutated human ras genes in vitro requires more than 100 times higher expression than is observed in cancers. Proc Natl Acad Sci USA. 1997;94:9614–9619. doi: 10.1073/pnas.94.18.9614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel M, Hudson JB, Laimins LA. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J Virol. 1992;66:6070–6080. doi: 10.1128/jvi.66.10.6070-6080.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S, Allen-Hoffmann BL, Lambert PF. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol. 1995;69:2989–2997. doi: 10.1128/jvi.69.5.2989-2997.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz ME, McCormick F. Signal transduction from multiple Ras effectors . Curr Opin Genet Dev. 1997;7:75–79. doi: 10.1016/s0959-437x(97)80112-8. [DOI] [PubMed] [Google Scholar]
- Klingelhutz AJ, Smith PP, Garrett LR, McDougall JK. Alteration of the DCC tumor-suppressor gene in tumorigenic HPV-18 immortalized human keratinocytes transformed by nitrosomethylurea. Oncogene. 8:1993. 95–99. [PubMed] [Google Scholar]
- Lanham S, Herbert A, Watt P. HPV detection and measurement of HPV-16, telomerase, and survivin transcripts in colposcopy clinic patients. J Clin Pathol. 2001;54:304–308. doi: 10.1136/jcp.54.4.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov M, Kubo Y, Cai T, Dajee M, Tarutani M, Lin Q, Fang M, Tao S, Green CL, Khavari PA. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat Med. 2002;8:1105–1114. doi: 10.1038/nm779. [DOI] [PubMed] [Google Scholar]
- Lee JH, Yi SM, Anderson ME, Berger KL, Welsh MJ, Klingelhutz AJ, Ozbun MA. Propagation of infectious human papillomavirus type 16 by using an adenovirus and Cre/LoxP mechanism. Proc Natl Acad Sci USA. 2004;101:2094–2099. doi: 10.1073/pnas.0308615100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtig H, Algrisi M, Botzer LE, Abadi T, Verbitzky Y, Jackman A, Tommasino M, Zehbe I, Sherman L. HPV16 E6 natural variants exhibit different activities in functional assays relevant to the carcinogenic potential of E6. Virology. 2006;350:216–227. doi: 10.1016/j.virol.2006.01.038. [DOI] [PubMed] [Google Scholar]
- Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12:3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammas IN, Zafiropoulos A, Spandidos DA. Involvement of the ras genes in female genital tract cancer. Int J Oncol. 2005;26:1241–1255. [PubMed] [Google Scholar]
- Mantovani F, Banks L. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene. 2001;20:7874–7887. doi: 10.1038/sj.onc.1204869. [DOI] [PubMed] [Google Scholar]
- Mason DX, Jackson TJ, Lin AW. Molecular signature of oncogenic ras-induced senescence. Oncogene. 2004;23:9238–9246. doi: 10.1038/sj.onc.1208172. [DOI] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Wilson S, Mullikin B, Suzich J, Meyers C. Human papillomavirus type 45 propagation, infection, and neutralization. Virology. 2003;312:1–7. doi: 10.1016/s0042-6822(03)00312-x. [DOI] [PubMed] [Google Scholar]
- Medina-Martinez O, Vallejo V, Guido MC, Garcia-Carranca A. Ha-ras oncogene-induced transcription of human papillomavirus type 18 E6 and E7 oncogenes. Mol Carcinog. 1997;19:83–90. [PubMed] [Google Scholar]
- Meyers C, Mayer TJ, Ozbun MA. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA. J Virol. 1997;71:7381–7386. doi: 10.1128/jvi.71.10.7381-7386.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda EI, Santana C, Rojas E, Hernandez S, Ostrosky-Wegman P, Garcia-Carranca A. Induced mitotic death of HeLa cells by abnormal expression of c-H-ras. Mutat Res. 1996;349:173–182. doi: 10.1016/0027-5107(95)00164-6. [DOI] [PubMed] [Google Scholar]
- Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, White MA, Wright WE, Shay JW. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat Genet. 1999;21:115–118. doi: 10.1038/5063. [DOI] [PubMed] [Google Scholar]
- Munger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, Zacny VL. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene. 2001;20:7888–7898. doi: 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- Ngan HY, Liu SS, Yu H, Liu KL, Cheung AN. Proto-oncogenes and p53 protein expression in normal cervical stratified squamous epithelium and cervical intra-epithelial neoplasia. Eur J Cancer. 1999;35:1546–1550. doi: 10.1016/s0959-8049(99)00166-5. [DOI] [PubMed] [Google Scholar]
- Nicke B, Bastien J, Khanna SJ, Warne PH, Cowling V, Cook SJ, Peters G, Delpuech O, Schulze A, Berns K, Mullenders J, Beijersbergen RL, Bernards R, Ganesan TS, Downward J, Hancock DC. Involvement of MINK, a Ste20 family kinase, in Ras oncogene-induced growth arrest in human ovarian surface epithelial cells. Mol Cell. 2005;20:673–685. doi: 10.1016/j.molcel.2005.10.038. [DOI] [PubMed] [Google Scholar]
- O’Leary JJ, Landers RJ, Silva I, Uhlmann V, Crowley M, Healy I, Luttich K. Molecular analysis of ras oncogenes in CIN III and in stage I and II invasive squamous cell carcinoma of the uterine cervix. J Clin Pathol. 1998;51:576–582. doi: 10.1136/jcp.51.8.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pett MR, Herdman MT, Palmer RD, Yeo GS, Shivji MK, Stanley MA, Coleman N. Selection of cervical keratinocytes containing integrated HPV16 associates with episome loss and an endogenous antiviral response. Proc Natl Acad Sci USA. 2006;103:3822–3827. doi: 10.1073/pnas.0600078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pim D, Massimi P, Banks L. Alternatively spliced HPV-18 E6* protein inhibits E6 mediated degradation of p53 and suppresses transformed cell growth. Oncogene. 1997;15:257–264. doi: 10.1038/sj.onc.1201202. [DOI] [PubMed] [Google Scholar]
- Pinion SB, Kennedy JH, Miller RW, MacLean AB. Oncogene expression in cervical intraepithelial neoplasia and invasive cancer of cervix. Lancet. 1991;337:819–820. doi: 10.1016/0140-6736(91)92518-7. [DOI] [PubMed] [Google Scholar]
- Rhim JS, Webber MM, Bello D, Lee MS, Arnstein P, Chen LS, Jay G. Stepwise immortalization and transformation of adult human prostate epithelial cells by a combination of HPV-18 and v-Ki-ras. Proc Natl Acad Sci USA. 1994;91:11874–11878. doi: 10.1073/pnas.91.25.11874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riou G, Barrois M, Sheng ZM, Duvillard P, Lhomme C. Somatic deletions and mutations of c-Ha-ras gene in human cervical cancers. Oncogene. 1988;3:329–333. [PubMed] [Google Scholar]
- Romanczuk H, Howley PM. Disruption of either the E1 or the E2 regulatory gene of human papillomavirus type 16 increases viral immortalization capacity. Proc Natl Acad Sci USA. 1992;89:3159–3163. doi: 10.1073/pnas.89.7.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagae S, Kudo R, Kuzumaki N, Hisada T, Mugikura Y, Nihei T, Takeda T, Hashimoto M. Ras oncogene expression and progression in intraepithelial neoplasia of the uterine cervix. Cancer. 1990;66:295–301. doi: 10.1002/1097-0142(19900715)66:2<295::aid-cncr2820660217>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Schreiber K, Cannon RE, Karrison T, Beck-Engeser G, Huo D, Tennant RW, Jensen H, Kast WM, Krausz T, Meredith SC, Chen L, Schreiber H. Strong synergy between mutant ras and HPV16 E6/E7 in the development of primary tumors. Oncogene. 2004;23:3972–3979. doi: 10.1038/sj.onc.1207507. [DOI] [PubMed] [Google Scholar]
- Sedman SA, Barbosa MS, Vass WC, Hubbert NL, Haas JA, Lowy DR, Schiller JT. The full-length E6 protein of human papillomavirus type 16 has transforming and trans-activating activities and cooperates with E7 to immortalize keratinocytes in culture. J Virol. 1991;65:4860–4866. doi: 10.1128/jvi.65.9.4860-4866.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Skinner J, Bounacer A, Bond JA, Haughton MF, deMicco C, Wynford-Thomas D. Opposing effects of mutant ras oncoprotein on human fibroblast and epithelial cell proliferation: implications for models of human tumorigenesis. Oncogene. 2004;23:5994–5999. doi: 10.1038/sj.onc.1207798. [DOI] [PubMed] [Google Scholar]
- Spandidos DA, Dokianakis DN, Kallergi G, Aggelakis E. Molecular basis of gynecological cancer. Ann N Y Acad Sci. 2000;900:56–64. doi: 10.1111/j.1749-6632.2000.tb06216.x. [DOI] [PubMed] [Google Scholar]
- Sprague DL, Phillips SL, Mitchell CJ, Berger KL, Lace M, Turek LP, Klingelhutz AJ. Telomerase activation in cervical keratinocytes containing stably replicating human papillomavirus type 16 episomes. Virology. 2002;301:247–254. doi: 10.1006/viro.2002.1542. [DOI] [PubMed] [Google Scholar]
- Stanley MA, Browne HM, Appleby M, Minson AC. Properties of a non-tumorigenic human cervical keratinocyte cell line. Int J Cancer. 1989;43:672–676. doi: 10.1002/ijc.2910430422. [DOI] [PubMed] [Google Scholar]
- Takaoka M, Harada H, Deramaudt TB, Oyama K, Andl CD, Johnstone CN, Rhoades B, Enders GH, Opitz OG, Nakagawa H. Ha-Ras (G12V) induces senescence in primary and immortalized human esophageal keratinocytes with p53 dysfunction. Oncogene. 2004;23:6760–6768. doi: 10.1038/sj.onc.1207923. [DOI] [PubMed] [Google Scholar]
- Thomas JT, Oh ST, Terhune SS, Laimins LA. Cellular changes induced by low-risk human papillomavirus type 11 in keratinocytes that stably maintain viral episomes. J Virol. 2001;75:7564–7571. doi: 10.1128/JVI.75.16.7564-7571.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren A, Fallen S, Yuan H, Usubutun A, Kucukali T, Schlegel R, Toretsky JA. Activation of the canonical Wnt pathway during genital keratinocyte transformation: a model for cervical cancer progression. Cancer Res. 2005;65:6199–6206. doi: 10.1158/0008-5472.CAN-05-0455. [DOI] [PubMed] [Google Scholar]
- Van Le L, Stoerker J, Rinehart CA, Fowler WC. H-ras codon 12 mutation in cervical dysplasia. Gynecol Oncol. 1993;49:181–184. doi: 10.1006/gyno.1993.1104. [DOI] [PubMed] [Google Scholar]
- Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Zehbe I, Wilander E, Delius H, Tommasino M. Human papillomavirus 16 E6 variants are more prevalent in invasive cervical carcinoma than the prototype. Cancer Res. 1998;58:829–833. [PubMed] [Google Scholar]