Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Non-Coding RNAs (original) (raw)
. Author manuscript; available in PMC: 2007 Nov 21.
SUMMARY
Noncoding RNAs (ncRNA) participate in epigenetic regulation but are poorly understood. Here we characterize the transcriptional landscape of the four human HOX loci at five base pair resolution in eleven anatomic sites, and identify 231 HOX ncRNAs that extend known transcribed regions by more than 30 kilobases. HOX ncRNAs are spatially expressed along developmental axes, possess unique sequence motifs, and their expression demarcate broad chromosomal domains of differential histone methylation and RNA polymerase accessibility. We identified a 2.2 kilobase ncRNA residing in the HOXC locus, termed HOTAIR, which represses transcription in trans across 40 kilobases of the HOXD locus. HOTAIR interacts with Polycomb Repressive Complex 2 (PRC2) and is required for PRC2 occupancy and histone H3 lysine-27 trimethylation of HOXD locus. Thus, transcription of ncRNA may demarcate chromosomal domains of gene silencing at a distance; these results have broad implications for gene regulation in development and disease states.
INTRODUCTION
A distinguishing feature of metazoan genomes is the abundance of noncoding RNA (ncRNAs), which function by means other than directing the production of proteins. In addition to small regulatory RNAs such as miRNAs, recent studies have predicted the existence of long ncRNAs-- ranging from 300 nucleotides (nt) to over 10 kb-- that are spliced, polyadenylated, and are roughly as diverse in a given cell type as protein-coding mRNAs (Bertone et al., 2004; Carninci et al., 2005; Kapranov et al., 2005; Rinn et al., 2003). Long ncRNAs may have diverse roles in gene regulation, especially in epigenetic control of chromatin (Bernstein and Allis, 2005). Perhaps the most prominent example is silencing of the inactive X chromosome by the ncRNA XIST. To normalize the copy number of X chromosomes between male and female cells, transcription of XIST RNA from one of the two female X chromosome is involved in recruiting Polycomb group proteins (PcG) to trimethylate histone H3 on lysine 27 (H3K27me3), rendering the chromosome transcriptionally silent (Plath et al., 2003). It is believed that Polycomb Repressive Complex 2 (PRC2), comprised of H3K27 histone methyl transferase (HMTase) EZH2 and core components Suz12 and EED, initiates this histone modification and subsequently Polycomb Repressive Complex 1 (PRC1) maintains this modification and promotes chromatin compaction (reviewed by (Sparmann and van Lohuizen, 2006). Presently, the mechanism by which XIST ncRNA guides Polycomb activity is unclear. Several PcG proteins possess RNA binding activity, and RNA is required for PcG binding to DNA, suggesting that specific ncRNAs may be critical interfaces between chromatin remodeling complexes and the genome (Bernstein et al., 2006; Zhang et al., 2004).
In addition to dosage compensation, long ncRNAs may also play critical roles in pattern formation and differentiation. In mammals, thirty-nine HOX transcription factors clustered on four chromosomal loci, termed HOXA through HOXD, are essential for specifying the positional identities of cells. The temporal and spatial pattern of HOX gene expression is often correlated to their genomic location within each loci, a property termed colinearity (Kmita and Duboule, 2003; Lemons and McGinnis, 2006). Maintenance of HOX expression patterns is under complex epigenetic regulation. Two opposing groups of histone modifying complexes, the trithorax group (TrxG) of H3K4 HMTase and the PcG H3K27 HMTase, maintain open and closed chromatin domains in the HOX loci, respectively, over successive cell division (Ringrose and Paro, 2007). Transcription of many ncRNAs has been observed in fly, mouse, and human HOX loci (Bae et al., 2002; Bernstein et al., 2005; Carninci et al., 2005; Drewell et al., 2002; Sessa et al., 2006), and three models have been proposed to account for their action based on experiments in Drosophila. First, elegant genetic studies suggested that transcription of ncRNAs altered the accessibility of DNA sequences important for TrxG and PcG binding; the act of intergenic transcription enabled TrxG activation of downstream HOX genes and prevented PcG-mediated silencing (Ringrose and Paro, 2007; Schmitt et al., 2005). Second, the above model has been extended by a recent report that several ncRNAs transcribed 5′ of the Drosophila Hox gene Ubx bind to and recruit the TrxG protein Ash1 to the Ubx promoter, thereby inducing Ubx transcription (Sanchez-Elsner et al., 2006). However, these results have been challenged by the third model of “transcriptional interference”, where transcription of 5′ ncRNAs into the promoters of downstream Hox genes prevents Hox gene expression, leading to transcriptional silencing in cis (Petruk et al., 2006). The extent to which any of these models and alternative mechanisms explain the copious amount of transcription in mammalian HOX loci remain to be discovered. Nonetheless, the large number of HOX ncRNAs, their complex clustering on the chromosomes, and potentially diverse modes of action suggest ncRNAs play a significant role in HOX regulation. By profiling the entire transcriptional and epigenetic landscapes of ~500 kilobase HOX loci, at near nucleotide resolution, we will begin to discern competing models of ncRNA action in humans and reveal potentially new mechanisms of ncRNA function.
Transcriptomic and proteomic analysis of the HOX loci requires pure cell populations with distinct positional identities. Rather than study whole animals where cells of many histologic types and positional identities are intermixed, we and others have observed that primary adult human fibroblasts retain many features of the embryonic pattern of HOX gene expression both in vitro and in vivo (Bernstein et al., 2005; Chang et al., 2002; Rinn et al., 2006). Differential and colinear expression of HOX genes in adult fibroblasts faithfully reflects their position along the anterior-posterior and proximal-distal axes of the developing body (Rinn et al, 2006), and is believed to be important for maintenance of regional identities of skin throughout the lifetime of the animal (Chuong, 2003). The remarkable persistence --over decades-- of the embryonic patterns of HOX gene expression in these human cells suggest the action of a powerful epigenetic machinery operative over the HOX loci. In this study, we create an ultra-high resolution tiling microarray to interrogate the transcriptional and epigenetic landscape of the HOX loci in a unique collection of primary human fibroblasts with eleven distinct positional identities. Our results identify numerous novel human HOX ncRNAs, clarify potential mechanisms of their regulation, and reveal a novel mechanism of ncRNA-assisted transcriptional silencing via the PcG proteins in trans.
RESULTS
Noncoding RNAs of the human HOX loci: Identity, conservation, expression pattern, and sequence motifs
To systematically investigate the transcriptional activity of the human HOX loci, we designed a DNA microarray for all 4 human HOX loci at five base pair (bp) resolution along with 2 megabases of control regions (Supplementary Tables 1). Computational and experimental analysis confirmed the specificity of the tiling array to distinguish highly related HOX sequences (Supplementary Fig. 1, 2).
Because adult primary fibroblasts are differentiated based on their anatomic site of origin and retain canonical features of the embryonic HOX code (Chang et al., 2002; Rinn et al., 2006), we used HOX tiling arrays to profile polyadenylated transcripts from fibroblasts representing 11 distinct positional identities (Fig. 1a). Previously, analytic methods for tiling arrays have allowed present/absent calls of transcripts and binding events, but were less successful in quantification of signal intensity (Bernstein et al., 2005; Bertone et al., 2004). We addressed this challenge by adapting a signal processing algorithm used in computer vision termed Otsu’s method (Otsu, 1979). The method dynamically searches for statistically significant cutoffs between signal and background, and detects contiguous regions of at least 100 bp (20 probes) with signal intensity significantly above background. Averaging the signal intensity over all probes in the called region thus produces a quantitative measure of transcript abundance. Using this algorithm, we identified a total of 407 discrete transcribed regions in the four HOX loci (Supplementary Table 2). We used current genome annotations to partition them into known HOX gene exons, introns, and intergenic transcripts (Fig. 1b). As expected, we detected many transcribed regions that corresponded to known HOX exons and introns (101 and 75, respectively), including exonic transcription for 34 of the 39 HOX genes, thus indicating that these 11 samples encompass the majority of HOX transcriptional activity. In all cases examined, the expression of HOX genes as determined by the tiling array matched that previously determined by cDNA microarray and RT-PCR for these same samples (Chang et al., 2002; Rinn et al., 2006).
Figure 1. The human HOX transcriptome.
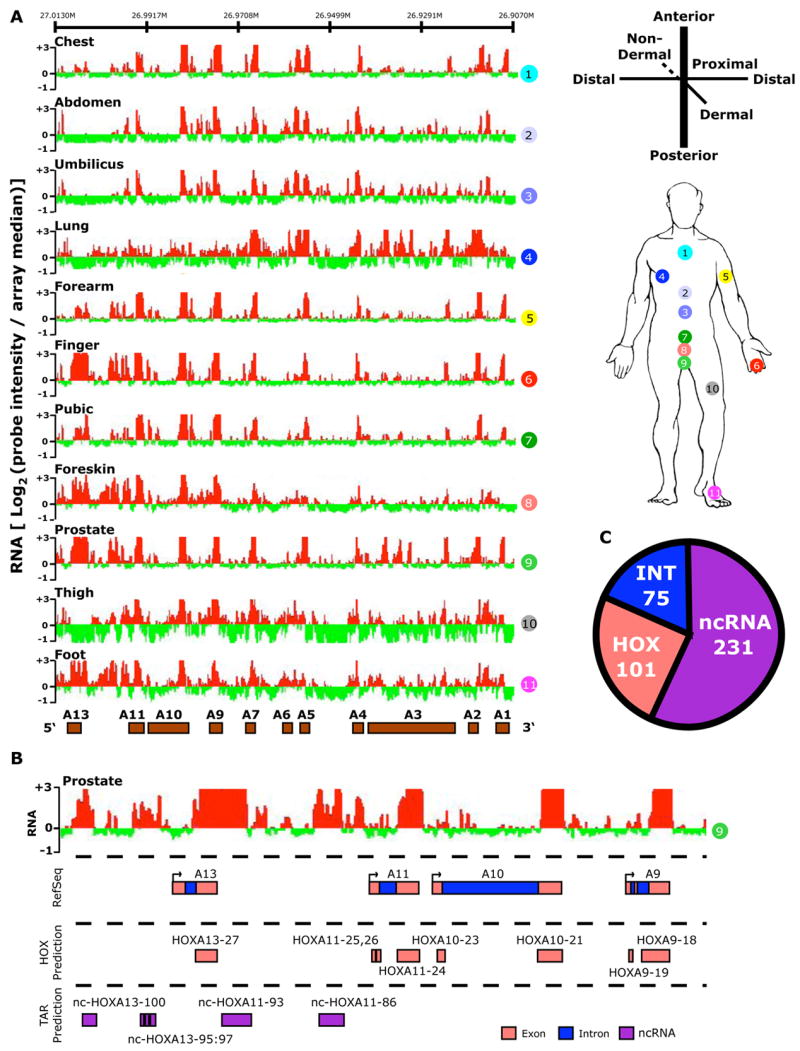
(A) Site-specific transcription of the HOXA locus. Left: The hybridization intensity of 50,532 probes that tile the human HOXA locus for each of the 11 samples (numbered in circles). The intensity of each probe is displayed as the log2 of the ratio of the individual probe intensity divided by the average intensity of all 301,027 probes on the array. The log2 ratio of each probe was averaged over a 100 bp window; red and green bars indicate expression above or below the array mean, respectively. Genomic locations of protein-coding HOX genes are displayed as brown boxes. Right: Anatomic origins of the 11 fibroblast samples with respect to the developmental axes. (B) Transcribed regions were identified by contiguous signals on tiling array, then compared with Refseq sequence to define genic [exonic (pink color) and intronic (blue)] and intergenic transcribed regions (purple). Each predicted HOX exon or intron was named HOXn or int-HOXn, respectively. Intergenic transcribed regions were named as nc-HOXn where n is the HOX paralog located 3′ to the ncRNA on the HOX coding strand. (C) Summary of transcribed regions in all four HOX loci defining the number of HOX genic, intronic, and ncRNA transcribed regions.
Interestingly, the majority of the transcribed regions (231 of 407) arise from intergenic regions (Fig. 1c). By comparison to databases of all known amino-acid sequences, we found that only 13% (29 of 231) of these intergenic transcripts showed any coding potential in all six possible translational frames (Experimental Methods, Supplementary Table 3). In contrast, 88% (84 of 96) of the HOX exon transcripts had coding potential, where the 12% non-coding exonic transcripts corresponded to untranslated exonic regions. While these results do not completely rule out the possibility of new protein coding genes interspersed throughout the HOX loci, these intergenic transcripts are more likely candidate noncoding RNAs. We therefore refer to these intergenic transcripts as HOX ncRNAs. We named each ncRNA by its genomic location, affixing the name of the HOX gene located 3′ to the ncRNA on the HOX coding strand (Fig. 1c). As previously suggested (Sessa et al., 2006), the majority of ncRNAs (74%) demonstrate evidence for opposite-strand transcription from the HOX genes (Supplementary Table 4). Fifteen percent of the ncRNAs we identified are novel while the majority of ncRNAs (85%) have been independently observed by EST sequencing or other means (Methods). Even for the known ncRNAs, our data suggest that almost all ncRNAs are longer than previously believed (Supplementary Table 4). The average extension for previously observed ncRNAs is 202 bases; in total we discovered over 30 kilobases of new transcribed bases in the human HOX loci. Thus, in just 11 hybridizations, we have substantially expanded the number and length of known transcribed regions in the human HOX loci as well as define their expression patterns throughout the human body.
We found several lines of evidence that confirm the biological importance of the HOX ncRNAs. First, comparative analysis with seven vertebrate genomes revealed that some ncRNAs are preferentially conserved in evolution over non-transcribed or intronic HOX sequences. For instance, more than one third of the top 100 conserved transcribed regions in the HOX loci are ncRNAs (Supplementary Fig. 3a). Second, RT-PCR analysis of forty predictions of ncRNA expression levels from the tiling array confirmed a high level of agreement (85%) between array signal intensity and transcript abundance as measured by RT-PCR (Supplementary Fig. 3b).
Third, we found that, like canonical HOX genes, ncRNAs also systematically vary their expression along developmental axes of the body in a manner coordinated with their physical location on the chromosome (Fig. 2 and Supplementary Table 5). 147 of 231 HOX ncRNAs (64%) are differentially expressed along a developmental axis of the body (p<0.05). For instance, 48 HOX ncRNAs are differentially expressed with their neighboring HOX genes along the proximal-distal axis (close or far from the trunk of the body) (p<0.05, Fig. 2a). Strikingly, all 41 transcribed regions (both HOX genes and ncRNAs) that are induced in distal sites belonged to HOX paralogous groups 9–13, and all 30 transcribed regions that are repressed in distal sites belonged to paralogous groups 1–6, precisely recapitulating the evolutionary origin of the two domains from Drosophila Ultrabithorax and Antennepedia complexes, respectively (P<10−19, 2-way chi-square test) (Carroll, 1995). Similarly, 87 HOX ncRNAs are differentially expressed along the anterior-posterior axis (top to bottom of the body), this time with ncRNAs from HOXC9-13 preferentially induced in posterior sites (p<0.05, Fig. 2b). Additionally, we observed 7 HOX genes and 12 ncRNAs that are either expressed in dermal (outside the body) or nondermal (inside the body) fibroblasts (Supplementary Figure 4, Table 5). Systematic comparison of the expression pattern of every ncRNA with its immediate 5′ and 3′ HOX gene neighbor showed that the vast majority of ncRNAs (90%) are coordinately induced with their 3′ HOX genes while only 10% of instances are ncRNA expression anti-correlated with 3′ HOX gene expression (Supplementary Fig. 5).
Figure 2. Site-specific expression and primary sequence motifs of HOX ncRNAs.
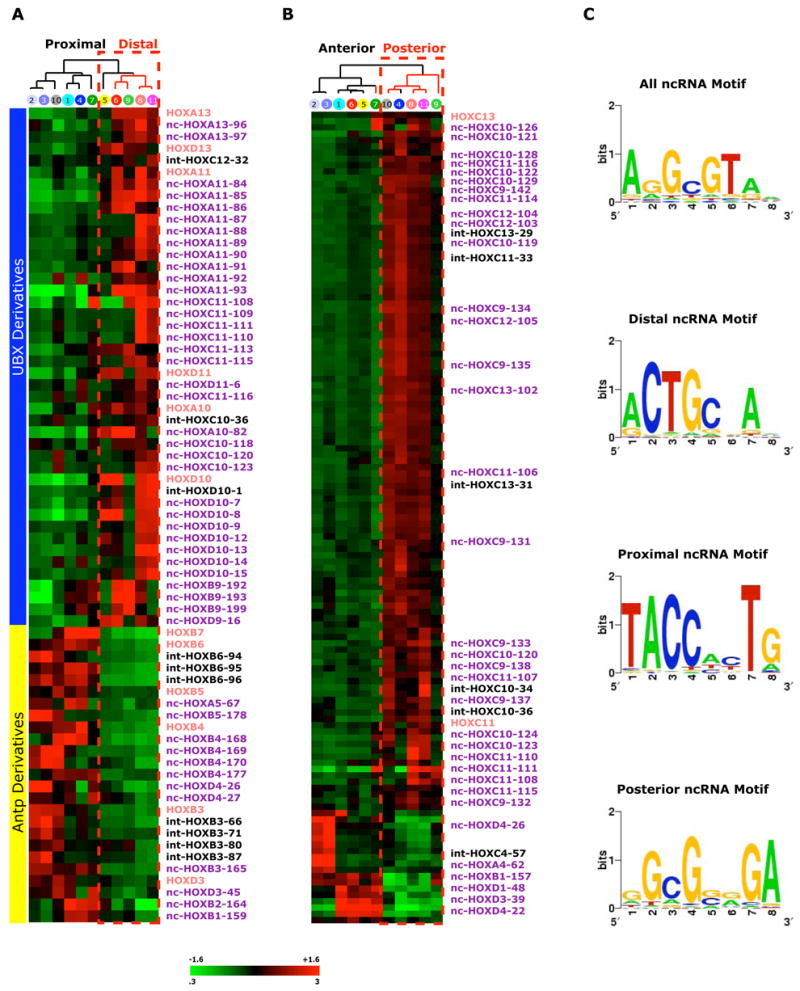
(A) HOX-encoded transcripts differentially expressed along the proximal-distal axis. Sixty transcribed regions (12 HOX genes and 48 ncRNAs) were differentially expressed (P<0.05, Student’s t-test) between distal fibroblast samples (foot, finger, foreskin, and prostate) and all other cells. Expression level of each transcribed region above or below the global median is denoted by the color scale (3 fold to 0.3 fold on linear scale or +1.6 to −1.6 on log2 scale). Transcribed regions were ordered by their position along the chromosome, and samples were hierarchically clustered by similarity of expression of these 60 transcripts. The evolutionary origin of HOX paralogs to fly ultrabithorax (UBX) or antennapedia (Antp) are indicated by blue and yellow boxes, respectively.
(B) HOX encoded transcripts differentially expressed along the anterior-posterior anatomic division. A total of 92 transcripts (6 HOX genes, 86 ncRNAs) were differentially expressed (P<0.05, Student’s t-test) in anterior or posterior primary fibroblast cultures (above or below the umbilicus). Expression of each ncRNA is represented as in (A).
(C) Enriched sequence motifs in HOX ncRNA based on their pattern of expression (p<10−9). Logograms of sequence motifs enriched in the primary sequences of ncRNAs over non-transcribed HOX sequences, or in ncRNAs with distal, proximial, or posterior patterns of expression are shown. ncRNAs expressed in anterior anatomic sites did not share a primary sequence motif more than expected by chance.
Fourth, in addition to their distinctive expression patterns, we found that the ncRNAs also possess specific sequence motifs. Using a discriminative motif finder that we previously developed (Segal et al., 2003), we found that ncRNAs are enriched for specific DNA sequence motifs based on their site-specific expression patterns (p<10−9, Fig. 3c). We identified a sequence motif enriched in ncRNAs over exonic, intronic, or nontranscribed sequences, and further identified sequence motifs for ncRNAs that are expressed in distal, proximal, or posterior sites. These sequence motifs may represent DNA or RNA binding sites for regulatory factors to regulate gene expression in cis. Together, these results establish that the majority of site-specific transcriptional output of the HOX loci consists of ncRNAs. Their evolutionary conservation, differential expression along developmental axes, and distinct primary sequence motifs suggest important and possibly widespread roles for these ncRNA transcripts in HOX gene regulation.
Figure 3. Diametrically opposed chromatin modifications and transcriptional accessibility in the HOXA locus.
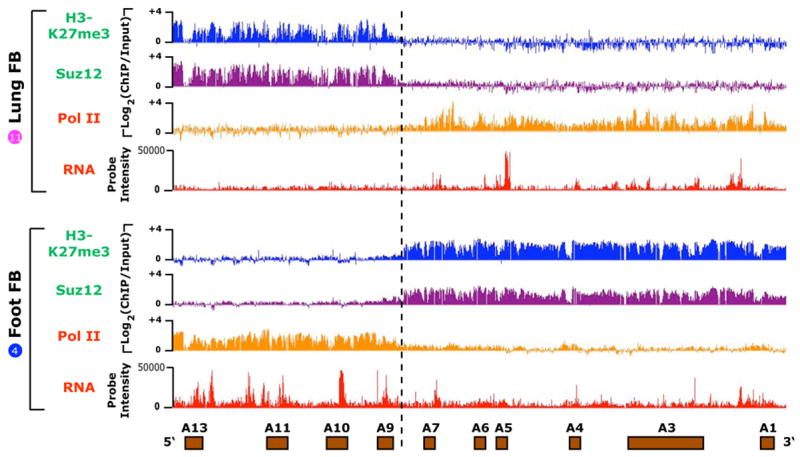
Occupancy of Suz12, H3K27me3, and pol II versus transcriptional activity over ~100 kb of the HOXA locus for primary lung (top) or foot (bottom) fibroblasts (Fb). For chIP data, the log2 ratio of ChIP/Input is plotted on the Y-axis. For RNA data, the hybridization intensity on a linear scale is shown. Dashed line highlights the boundary of opposite configurations of chromatin modifications and intergenic transcription.
Diametrical domains of chromatin modifications demarcated by HOX ncRNAs
The coordinate expression of HOX genes and neighboring ncRNAs raised the possibility that their expression may be regulated by chromatin domains, large contiguous regions of differential chromatin modifications that enable transcriptional accessibility or cause silencing. Such domains, first observed by Bernstein and colleagues for histone H3 lysine 4 dimethylation (H3K4me2) (Bernstein et al., 2005), are a notable and unique feature of HOX loci chromatin (Bracken et al., 2006; Lee et al., 2006; Papp and Muller, 2006; Squazzo et al., 2006). We tested this idea by loci-wide chromatin immunoprecipitation followed by tiling array analysis (ChIP-chip). We found that both HOX and ncRNA transcription fell within broad domains occupied by RNA polymerase II, whereas the transcriptionally silent regions were broadly occupied by the PRC2 component Suz12 and its cognate histone mark, histone H3 trimethylated at lysine 27 (H3K27me3) (P < 10−15, chi square test, Methods) (Fig. 3). Comparison of cells from different anatomic origins showed that the primary DNA sequence can be programmed with precisely the same boundary but in the opposite configuration. For example, in lung fibroblasts the 5′ HOXA locus is occupied by Suz12 but not Pol II, whereas in foreskin fibroblasts this exact same chromatin domain is occupied by Pol II but not Suz12. Thus, positional identity in differentiated cells may be marked by diametric or mutually exclusive domains of chromatin modifications, which switch their configurations around a center of inversion in a site-specific manner.
Interestingly, the boundary of the diametric chromatin domains defined by ChIP-chip is precisely the same as that suggested by our transcriptional analysis. In the HOXA locus, the chromatin boundary and switch between proximal vs. distal expression patterns occurs between HOXA7 and HOXA9. Additional ChIP-chip analysis showed the domain of PolII occupancy precisely overlaps the domain of H3K4 dimethylation, but H3K9 trimehtylation, a histone modification characteristic of constitutive heterochromatin, is not present on any HOX loci in these cells (Supplementary Fig. 6). These results suggest that HOX loci transcription in adult fibroblasts is governed by opposing epigenetic modifications over large chromosomal regions, and further define the locations of specific boundary elements that delimit chromatin domains.
HOTAIR: A noncoding RNA that regulates chromatin silencing in trans
We next asked whether the coordinate transcription of HOX ncRNAs is merely a consequence of the broad chromatin domains, or whether the ncRNAs are actively involved in establishing such domains. To address this question, we analyzed in depth the function of a long ncRNA situated at the boundary of two diametrical chromatin domains in the HOXC locus (Fig. 4a). This ncRNA is transcribed in an antisense manner with respect to the canonical HOXC genes; we therefore named it HOTAIR for HOX Antisense Intergenic RNA. Molecular cloning and Northern blot analysis confirmed that HOTAIR is a 2158 nucleotide, spliced, and polyadenylated transcript; strand specific RT-PCR analysis confirmed that only one strand of HOTAIR which is antisense to HOXC genes is transcribed (Fig. 4b, 4c). Computational analysis of HOTAIR secondary structure did not reveal obvious stem loops suggestive of pre-miRNAs. Northern blot analysis of size-fractionated RNA showed no evidence of small RNA products suggestive of micro- or siRNA production while we readily detected the ubiquitous miRNA let7 in parallel experiments (Fig. 4d).
Figure 4. HOTAIR, an antisense intergenic long ncRNA of the HOXC locus.
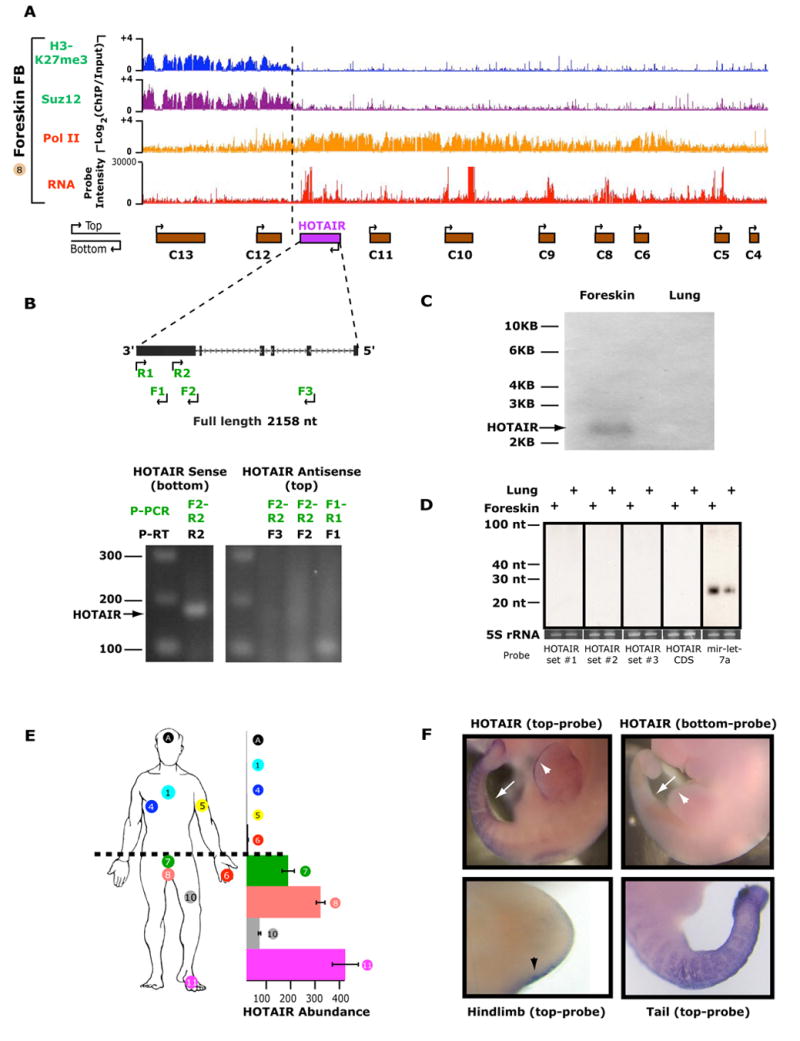
(A) Genomic location of HOTAIR at the boundary of two chromatin domains. ChIP-chip and RNA expression on tiling array are as shown in Fig. 3.
(B) Strand specific RT-PCR shows exclusive expression of HOTAIR from the strand opposite to HOXC genes (bottom). Primers for reverse transcription (P-RT) and PCR (P-PCR) were designed to specifically target either the top (primers F1–F3) or bottom strand (primer R1) of HOTAIR.
(C) Northern blot analysis of HOTAIR in lung and foreskin fibroblast RNA.
(D) Size-fractionated small RNA was probed with pools of oligonucleotides spanning HOTAIR (sets #1–3), full length antisense HOTAIR (CDS), or a probe against miRNA let7a.
(E) Posterior and distal expression of HOTAIR in human fibroblasts as measured by qRT-PCR. The site of origin of each fibroblast sample is indicated by the sample number on the anatomic cartoon. “A” is derived from the scalp. The relative abundance of HOTAIR in each position, relative to scalp (most anterior) is shown on the X-axis.
(F) Whole mount in situ hybridization using HOTAIR sense (bottom strand) or antisense (top strand) probes in embryonic day 10.5 whole mount embryos. (top panels) and the hind limb and tail (bottom left and right panels, respectively). Expression of HOTAIR in posterior hindlimb (arrowhead) and tail (arrow) are highlighted.
Our tiling array data suggested that HOTAIR is preferentially expressed in posterior and distal sites, and indeed this expression pattern is confirmed by additional RT-PCR experiments (Fig. 4e). In situ hybridization of developing mouse embryos confirmed that HOTAIR is expressed in posterior and distal sites, indicating the conservation of anatomic expression pattern from development to adulthood (Fig. 4f). Interestingly, this transcript has very high nucleotide conservation in vertebrates (99.5%, 95%, 90%, and 85% sequence identity in chimp, macaque, mouse, and dog genomes, respectively), yet is riddled with stop codons with little amino-acid sequence conservation amongst vertebrates (Supplementary Experimental Methods). These results suggest that HOTAIR may function as a long ncRNA.
HOTAIR ncRNA may regulate gene expression in HOX loci in cis or trans; alternatively, it may be the act of antisense transcription in the HOXC locus rather than the ncRNA itself that has a functional role in gene regulation. To distinguish between these possibilities, we depleted HOTAIR ncRNA by RNA interference in primary human fibroblasts, and determined the consequences on the transcriptional landscape of the HOX loci. Strikingly, while siRNA-mediated depletion of HOTAIR had little effect on transcription of the HOXC locus on chromosome 12 compared to wild-type and control siRNA targeting GFP, depletion of HOTAIR lead to dramatic transcriptional activation of the HOXD locus on chromosome 2 spanning over 40 Kb, including HOXD8, HOXD9, HOXD10, HOXD11, and multiple ncRNAs (Fig. 5a, b, Supplementary Fig. 7). To ensure that this was not an off-target effect of RNA interference, we employed four independent siRNA sequences targeting HOTAIR. Each siRNA depleted HOTAIR ncRNA and led to concomitant HOXD10 activation as determined by quantitative RT-PCR (Fig. 5c, d). These observations indicate that HOTAIR ncRNA is required to maintain a transcriptionally silent chromosomal domain in trans on the HOXD locus.
Figure 5. Loss of HOTAIR results in transcriptional induction of HOXD locus.
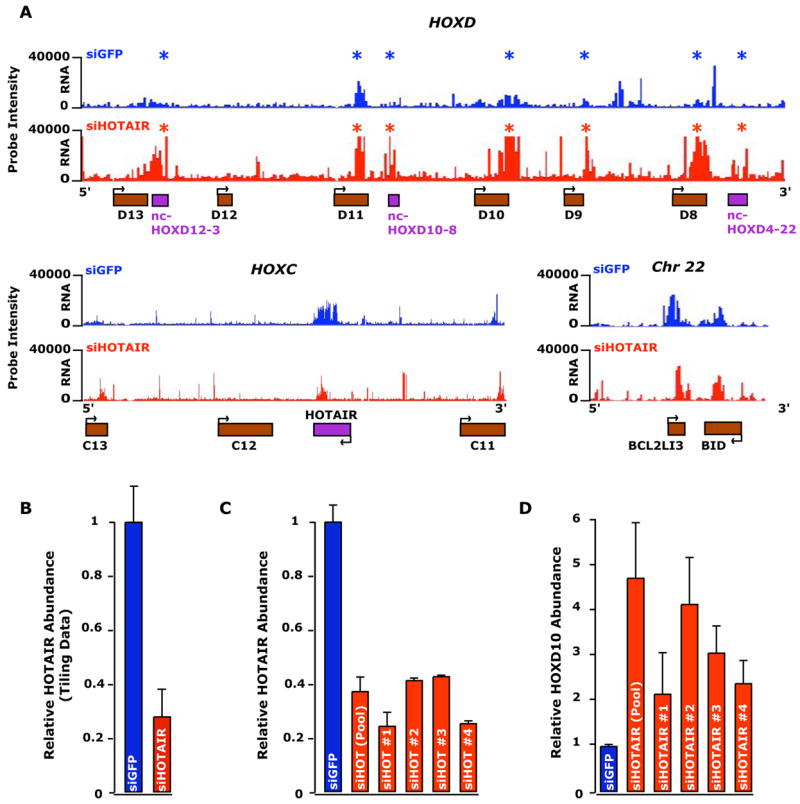
(A) RNA expression profiles of HOXD locus (top), HOXC locus surrounding HOTAIR (bottom left), and a control region on chromosome 22 (bottom right) following transfection of siRNA targeting GFP (siGFP) or a pool of four siRNAs targeting HOTAIR (siHOTAIR). Intensities of RNA hybridized to the tiling array from the siGFP and the siHOTAIR transfections are plotted on a linear scale in blue and red respectively. *, genes with significant increased transcription.
(B) qRT-PCR measuring the relative abundance of the HOTAIR transcript in the primary foreskin samples show in (A).
(C, D) qRT-PCR measuring the relative abundance of the HOTAIR (C) and HOXD10 (D) transcripts after depletion of four individual siRNAs to HOTAIR and the pool.
HOTAIR ncRNA enhances PRC2 activity at the HOXD locus
To investigate the molecular mechanisms involved in the HOTAIR dependant silencing of the HOXD locus, we used chromatin immunoprecipitation to interrogate changes to the HOXD chromatin structure upon depletion of HOTAIR. Our previous ChIP-chip experiments indicated that in primary foreskin fibroblasts, the entire HOXD locus was occupied by both Suz12 and H3K27me3. Depletion of HOTAIR followed by ChIP-chip revealed substantial and global loss of H3K27Me3 occupancy over the HOXD locus, with the greatest loss residing in the intergenic region between HOXD4 and HOXD8 (Fig. 6a). HOTAIR depletion also led to a modest but consistent loss of Suz12 occupancy of the HOXD locus (Fig. 6B, Supplementary Fig. 8). Importantly, occupancy of H3K27me3 and Suz12 across the silent HOXB locus was not affected by HOTAIR depletion in these cells. These results suggest that HOTAIR is selectively required to target PRC2 occupancy and activity to silence transcription of the HOXD locus.
Figure 6. HOTAIR is required for H3K27 trimethylation and Suz12 occupancy of the HOXD locus.
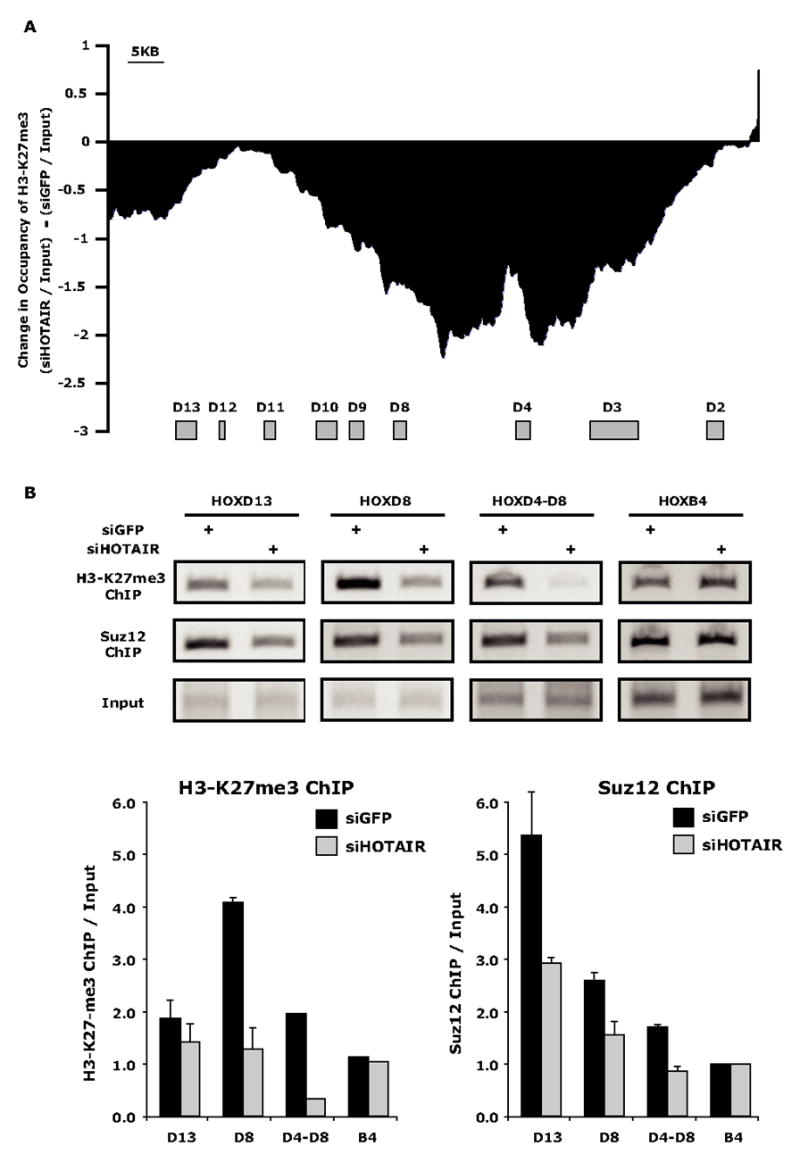
(A) Change in H3K27me3 ChIP-chip signal over the HOXD locus caused by depletion of HOTAIR compared to control siRNA against GFP. The location of HOXD genes are indicated by brown boxes.
(B) ChIP of H3K27me3 and Suz12 of select promoters across the HOXD locus after siRNA treatment targeting GFP or HOTAIR. Bottom: quantitation of ChIP assays (mean ± standard error).
Because PcG protein binding to chromatin can involve RNA and HOTAIR ncRNA is required for PRC2 function (i.e. H3K27 trimethylation), we reasoned that HOTAIR may bind to PRC2 and directly regulate Polycomb function. Indeed, native immunoprecipitation of Suz12 from nuclear extracts of two types of primary fibroblasts retrieved associated endogenous HOTAIR ncRNA as detected by RT-PCR, but not non-specific U1 RNA or DNA (Fig. 7a). HOTAIR ncRNA was not retrieved by immunoprecipitation of YY1, which has been suggested to be a component of PRC1 ((Sparmann and van Lohuizen, 2006). Suz12 also did not associate with the neighboring HOXC10 mRNA, indicating that PRC2 binds selectively to HOXC-derived transcripts (Supplementary Fig. 9). In the reciprocal experiment, we prepared purified biotinylated sense or antisense HOTAIR RNA by in vitro transcription, and probed nuclear extracts of HeLa cells to identify HOTAIR binding proteins. HOTAIR ncRNA retrieved PRC2 components Suz12 and EZH2 but not YY1 (Fig. 7b). Antisense HOTAIR RNA did not retrieve any of the above proteins, indicating that the binding conditions are highly specific. Collectively, these experiments indicate that HOTAIR is physically associated with PRC2 either directly or indirectly; loss of this interaction may reduce the ability of PRC2 to methylate histone tails and silence transcription at the HOXD locus.
Figure 7. HOTAIR ncRNA binds Polycomb Repressive Complex 2.
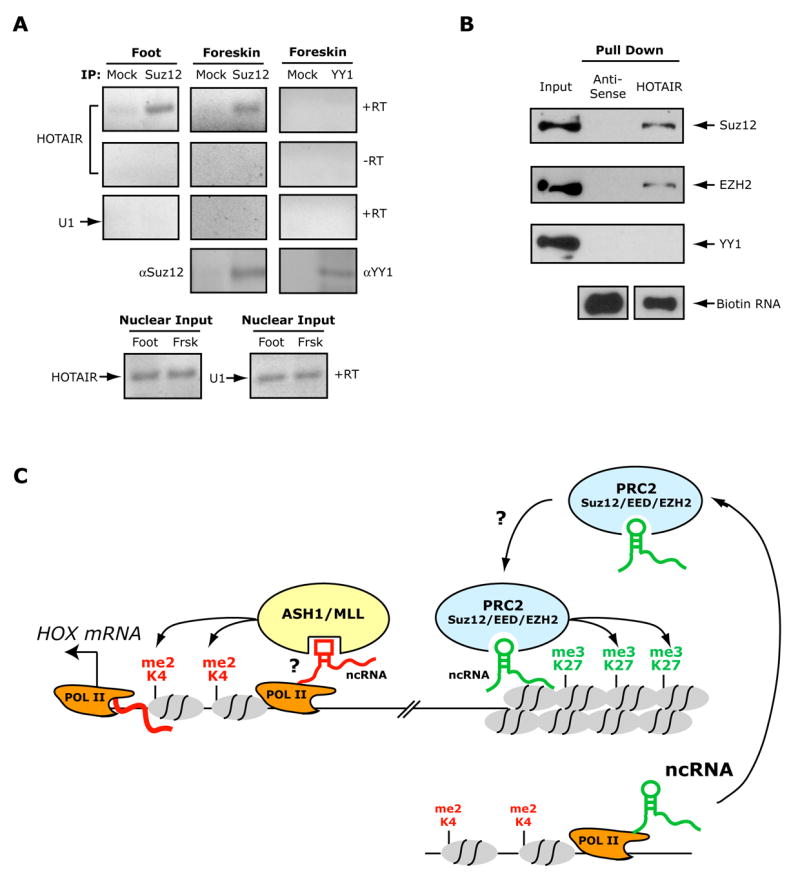
(A) Immunoprecipitation of Suz12 retrieves endogenous HOTAIR. Nuclear extracts of foot or foreskin fibroblasts were immunoprecipiated by IgG (lanes 1, 3, 5), anti-Suz12 (lanes 2, 4), or anti-YY1 (lane 6). Co-precipitated RNAs were detected by RT-PCR using primers for HOTAIR (rows 1 and 2) or U1 small nuclear RNA (row 3). To demonstrate that the HOTAIR band was not due to DNA contamination, each RT-PCR was repeated without reverse transcriptase (-RT, row 2). Immunoprecipitation of Suz12 and YY1 were successful as demonstrated by IP-western using the cognate antibodies (row 4). RT-PCR of nuclear extracts demonstrated equal input RNAs (row 5).
(B) In vitro transcribed HOTAIR retrieves PRC2 subunits. Immunoblot analysis of the indicated proteins is shown; five percent of input extract (5 μg) was loaded as input control.
(C) Model of long ncRNA regulation of chromatin domains via histone modification enzymes. Transcription of ncRNAs in cis may increase the accessibility of TrxG proteins such as ASH1 or MLL or directly recruit them, leading to H3K4 methylation and transcriptional activation of the downstream HOX gene(s). In contrast, recruitment of PRC2 is programmed by ncRNAs produced in trans, which targets PRC2 activity by yet incompletely defined mechanisms to target loci. PRC2 recruitment leads to H3K27 methylation and transcriptional silencing of neighboring HOX genes.
DISCUSSION
Panoramic views of the HOX loci by ultra high resolution tiling arrays
By analyzing the transcriptional and epigenetic landscape of the HOX loci at high resolution in cells with many distinct positional identities, we were afforded a panoramic view of multiple layers of regulation involved in maintenance of site-specific gene expression. The HOX loci are demarcated by broad chromosomal domains of transcriptional accessibility, marked by extensive occupancy of RNA polymerase II and H3K4 dimethylation and, in a mutually exclusive fashion, by occupancy of PRC2 and H3K27me3. The active, PolII-occupied chromosomal domains are further punctuated by discrete regions of transcription of protein-coding HOX genes and a large number of long ncRNAs. Our results confirm the existence of broad chromosomal domains of histone modifications and occupancy of HMTases over the Hox loci observed by previous investigators (Bernstein et al., 2005; Boyer et al., 2006; Guenther et al., 2005; Lee et al., 2006; Squazzo et al., 2006), and extend on these observation in several important ways.
First, by comparing the epigenetic landscape of cells with distinct positional identities, we showed that the broad chromatin domains can be programmed with precisely the same boundary but with diametrically opposite histone modifications and consequences on gene expression. Our data thus functionally pinpoint the locations of chromatin boundary elements in the HOX loci, the existence of some of which have been predicted by genetic experiments (Kmita et al., 2000). One such boundary element appears to reside between HOXA7 and HOXA9. This genomic location is also the switching point in the expression of HOXA genes between anatomically proximal versus distal patterns and is the boundary of different ancestral origins of HOX genes, raising the possibility that boundary elements are features demarcating the ends of ancient transcribed regions. Second, the ability to monitor 11 different HOX transcriptomes in the context of the same cell type conferred the unique ability to characterize changes in ncRNA regulation that reflect their position in the human body. This unbiased analysis identified more than 30 kb of new transcriptional activity, revealed ncRNAs conserved in evolution, mapped their anatomic patterns of expression, and uncovered enriched ncRNA sequence motifs correlated with their expression pattern—insights which could not be gleamed from examination of EST sequences alone (Sessa et al., 2006). Our finding of a long ncRNA that acts in trans to repress HOX genes in a distant locus is mainly due to the ability afforded by the tiling array to comprehensively examine the consequence of any perturbation over all HOX loci. The expansion of a handful of Hox-encoded ncRNAs in Drosophila to hundreds of ncRNAs in human HOX loci suggests increasingly important and diverse roles for these regulatory RNAs.
An important limitation of the tiling array approach is that while we have improved identification of transcribed regions, the data does not address the connectivity of these regions. The precise start, end, patterns of splicing, and regions of double-stranded overlap between ncRNAs will need to be addressed by detailed molecular studies in the future.
ncRNA transcription and HOX gene expression
Noncoding RNAs are emerging as regulatory molecules in specifying specialized chromatin domains (Bernstein and Allis, 2005), but the prevalence of different mechanism by which they act is not known. In Drosophila, transcription of ncRNAs was proposed to induce HOX gene expression by activation of cis regulatory elements (Schmitt et al., 2005) or by ncRNA-mediated recruitment of the TrxG protein Ash1 (Sanchez-Elsner et al., 2006). However, an alternative model, termed “transcriptional interference”, argues that ncRNA transcription prevents the expression of 3′ located Hox genes (Petruk et al., 2006). These two classes of models make opposite predictions on the correlation between expression of 5′ ncRNA and the 3′ HOX gene. Our finding of widespread position-specific ncRNAs that flank and are coordinately induced with neighboring human HOX genes is consistent with models of cis activation by ncRNA transcription. Only 10% of HOX ncRNAs demonstrate anti-correlated expression pattern with their cognate 3′ HOX genes (Supplementary Fig. 5), suggesting that transcriptional interference is not the main mode of ncRNA action, at least in the cell types that we studied. Our results are also consistent with a recent analysis of HOX gene activation during teratocarcinoma cell differentiation, where transcription of certain 5′ ncRNAs immediately preceded HOX gene activation (Sessa et al., 2006). Transcriptional interference may be a more prominent mechanism during embryonic development, where its role in Hox gene expression was documented in Drosophila (Petruk et al., 2006).
Our results uncovered a new mechanism whereby transcription of ncRNA dictates transcriptional silencing of a distant chromosomal domain. The four HOX loci demonstrate complex cross regulation and compensation during development (Kmita and Duboule, 2003; Lemons and McGinnis, 2006). For instance, deletion of the entire HOXC locus exhibits a milder phenotype than deletion of individual HOXC genes, suggesting that there is negative feedback within the locus (Suemori and Noguchi, 2000). Multiple 5′ HOX genes, including HOXC genes, are expressed in developing limbs (Nelson et al., 1996), and deletion of multiple HOXA and HOXD genes are required to unveil limb patterning defects (Zakany et al., 1997). Our results suggest that deletion of the 5′ HOXC locus, which encompass HOTAIR, may lead to transcriptional induction of the homologous 5′ HOXD genes, thereby restoring the total dosage of HOX transcription factors. How HOX ncRNAs may contribute to cross-regulation among HOX genes should be addressed in future studies.
HOTAIR ncRNA is involved in Polycomb Repressive Complex 2-mediated silencing of chromatin
Because many HMTase complexes lack DNA binding domains but possess RNA binding motifs, it has been postulated that ncRNAs may guide specific histone modification activities to discrete chromatin loci (Bernstein and Allis, 2005; Sun and Zhang, 2005). We have shown that HOTAIR ncRNA binds PRC2 and is required for robust H3K27 trimethylation and transcriptional silencing of the HOXD locus. HOTAIR may therefore be one of the long sought after RNAs that interface the Polycomb complex with target chromatin. A potentially attractive model of epigenetic control is the programming of active or silencing histone modifications by specific noncoding RNAs (Fig. 7c). Just as transcription of certain ncRNA can facilitate H3K4 methylation and activate transcription of the downstream Hox genes (Sanchez-Elsner et al., 2006; Schmitt et al., 2005), distant transcription of other ncRNAs may target the H3K27 HMTase PRC2 to specific genomic sites, leading to silencing of transcription and establishment of facultative heterochromatin. In this view, extensive transcription of ncRNAs is both functionally involved in the demarcation of active and silent domains of chromatin as well as being a consequence of such chromatin domains.
Several lines of evidence suggest that HOTAIR functions as a bona fide long ncRNA to mediate transcriptional silencing. First, we detected full length HOTAIR in vivo and in primary cells, but not small RNAs derived from HOTAIR indicative of miRNA or siRNA production. Second, depletion of full length HOTAIR led to loss of HOXD silencing and H3K27 trimethyation by PRC2, and third, endogenous or in vitro transcribed full length HOTAIR ncRNA physically associated with PRC2. While these results do not rule out the possibility that RNA interference pathways may be subsequently involved in PcG function (Grimaud et al., 2006; Kim et al., 2006), they support the notion that the long ncRNA form of HOTAIR is functional. The role of HOTAIR is reminiscent of XIST, another long ncRNA shown to be involved in transcriptional silencing of the inactive X chromosome (Plath et al., 2003). An important difference between HOTAIR and XIST is the strictly _cis_-acting nature of XIST. To our knowledge, HOTAIR is the first example of a long ncRNA that can act in trans to regulate a chromatin domain. While we have observed a trans repressive role for HOTAIR, our data do not permit us to rule out a _cis-_repressive role in the HOXC locus. Our siRNA-mediated depletion of HOTAIR was substantial but incomplete; further, the proximity between the site of HOTAIR transcription and the neighboring HOXC locus may ensure significant exposure to HOTAIR even if the total pool of HOTAIR in the cell were depleted. The precise location of HOTAIR at the boundary of a silent chromatin domain in the HOXC locus makes a _cis_-repressive role a tantalizing possibility. Judicious gene targeting of HOTAIR may be required to address its role in _cis_-regulation of chromatin.
The discovery of a long ncRNA that can mediate epigenetic silencing of a chromosomal domain in trans has several important implications. First, ncRNA guidance of PRC2-mediated epigenetic silencing may operate more globally than just in the HOX loci, and it is possible that other ncRNAs may interact with chromatin modification enzymes to regulate gene expression in trans. Second, PcG proteins are important for stem cell pluripotency and cancer development (Sparmann and van Lohuizen, 2006); these PcG activities may also be guided by stem cell or cancer-specific ncRNAs. Third, Suz12 contains a zinc finger domain, a structural motif that can bind RNA (Hall, 2005), and EZH2 and EED both have in vitro RNA binding activity (Denisenko et al., 1998). The interaction between HOTAIR and PRC2 may also be indirect and mediated by additional factors. Detailed studies of HOTAIR and PRC2 subunits are required to elucidate the structural features that establish the PRC2 interaction with HOTAIR. As we illustrated here, high throughput approaches for the discovery and characterization of ncRNAs may aid in dissecting the functional roles of ncRNAs in these diverse and important biological processes.
EXPERIMENTAL PROCEDURES
Tiling array design, hybridization, signal processing, RT-PCR validation of ncRNAs, and motif analysis are described in Supplementary Data.
Chromatin immunoprecipitation
Conventional ChIP and ChIP-chip were performed using anti-H3K27me3 (Upstate Cell Signaling cat# 07–449), anti-Suz12 (Abcam cat# 12,201), anti-PolII (Covance cat# MMS-126R), anti-H3K4me2 (Abcam cat# ab7766), anti-H3K9me3 (Abcam cat# ab1186), and Whole Genome Amplification kit (Sigma) as previously described (Squazzo et al., 2006).
HOTAIR cloning and sequence analysis
5′ and 3′ RACE were performed using the RLM Race kit (Ambion) as recommended by the manufacturer.
HOTAIR expression analysis
In situ hybridizations of C57BL/6 mouse embryo using human HOTAIR sequence 164–666 (clone 7T) (Albrecht, 1997), Northern blot using full length HOTAIR, qRT-PCR with SYBR Green (forward HOTAIR, GGGGCTTCCTTGCTCTTCTTATC; reverse, GGTAGAAAAAGCAACCACGAAGC), and Taqman analysis of HOXD10 expression (Applied Biosystems, cat# Hs00157974_m1) were as described (Rinn et al., 2004). Small RNA Northern blotting was as described (Lau et al., 2001) with the following modifications: 15 μg of small RNA retained total RNA (mirVana miRNA isolation kit, Ambion) was denatured in Novex sample loading buffer and loaded onto 15% TBE-urea gel in Novex running buffer (Invitrogen). RNA was transferred onto Hybond-XL membrane (Amersham) and probed with pools of 32P-gammaATP end-labeled 40mer oligos spanning HOTAIR sequence 1–400 (set 1), 401–800 (set 2), 801–1200 (set 3), full length HOTAIR probe, or a probe for microRNA let-7a (AACTATACAACCTACTACCTCA) as positive control.
RNA interference
Foreskin fibroblasts were transfected with 50nM of siRNAs targeting HOTAIR (#1 GAACGGGAGUACAGAGAGAUU; #2 CCACAUGAACGCCCAGAGAUU; #3 UAACAAGACCAGAGAGCUGUU; #4 GAGGAAAAGGGAAAAUCUAUU) or siGFP (CUACAACAGCCACAACGUCdTdT) using Dharmafect 3 (Dharmacon, Lafayette, CO, USA) per the manufacturer’s direction. Total RNA was harvested for total RNA 72 hours later for microarray analysis as previously described (Rinn et al., 2006).
RNA immunoprecipitation
Foreskin and foot fibroblasts were grown as previously described (Rinn et al., 2006). 10^7 cells were harvested by trypsinization and resuspended in 2ml PBS, 2ml nuclear isolation buffer (1.28 M sucrose; 40 mM Tris-HCl Ph 7.5; 20 mM MgCl2; 4% Triton X-100) and 6ml water on ice for 20 min (with frequent mixing). Nuclei were pelleted by centrifugation at 2,500G for 15 min. Nuclear pellet was resuspended in 1ml RIP buffer [150mM Kcl, 25mM Tris pH 7.4, 5mM EDTA, .5mM DTT, .5% NP40, 9ug/ml leupeptin, 9ug/ml pepstatin, 10ug/ml chymostatin, 3ug/ml aprotinin, 1 mM PMSF, 100 U/ml SUPERASin (Ambion)]. Resuspended nuclei were split into two fractions of 500 μl each (for Mock and IP) and were mechanically sheared using a dounce homogenizer with 15–20 strokes. Nuclear membrane and debris were pelleted by centrifugation at 13,000 RPM for 10 min. Antibody to Suz12 (Abcam cat# 12,201), YY1 (Santa Cruz Biotechnology cat# sc1703) or FLAG epitope (Mock IP, Sigma) was added to supernatant (Suz12 : 6μg, YY1 : 10μg) and incubated for 2hrs at 4C with gentle rotation. 40 μl of protein A/G beads were added and incubated for 1hr at 4C with gentle rotation. Beads were pelleted at 2,500 RPM for 30 sec, the supernatant was removed and beads were resuspended in 500 μl RIP buffer and repeated for a total of 3 RIP washes and followed by 1 wash in PBS. Beads were resuspended in 1ml of Trizol. Co-precipitated RNAs were isolated and RT-PCR for HOTAIR (Forward, GGGGCTTCCTTGCTCTTCTTATC; reverse GGTAGAAAAAGCAACCACGAAGC) or U1 (forward, ATACTTACCTGGCAGGGGAG; reverse, CAGGGGGAAAGCGCGAACGCA) performed as described (Rinn et al., 2006). Protein isolated by the beads was detected by Western blot analysis.
HOTAIR RNA pull down of PcG proteins
Biotin-labeled, full length HOTAIR RNA and antisense HOTAIR fragment (clone 7T) were prepared with the Biotin RNA Labeling Mix (Roche) and T7 RNA polymerase (Stratagene). Biotinylated RNAs were treated with RNase-free DNase I and purified on G-50 Sephadex Quick Spin columns (Roche). 10 pmol biotinylated RNA was heated to 60 C for 10 min and slow cooled to 4 C. RNA was mixed with 100 μg of pre-cleared transcription and splicing-competent HeLa nuclear extract (Gozani et al., 1994) in RIP buffer supplemented with tRNA (0.1 μg/ul) and incubated at 4 C for 1 hour. 60 μl washed Streptavidin agarose beads (Invitrogen) were added to each binding reaction and further incubated at 4 C for 1 hr. Beads were washed briefly 5 times in Handee spin columns (Pierce), boiled in SDS buffer, and the retrieved protein visualized by immunoblotting.
URLs
All primary data are available at the Stanford Microarray Database (http://genome-www5.stanford.edu/) and Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/). Full length sequence of human HOTAIR ncRNA has been deposited in Genebank (Accession # DQ926657 (bankit841140)).
Supplementary Material
01
02
03
04
05
06
Acknowledgments
We thank A.S. Adler, M.L. Cleary, O. Gozani, D. Herschlag, D. Hogan, P.A. Khavari, A.E. Oro, O.J. Rando, and C. Woo for discussion and critical review of the manuscript. Supported by grants from the National Institutes of Health (J.A.H., P.J.F., E.S., H.Y.C.), Israel Science Foundation (M.K, E.S.), and National Science Foundation (J.K.W.). E.S. is the incumbent of the Soretta and Henry Shapiro Career Development Chair; J.L.R. is a Fellow and H.Y.C. is the Kenneth G. and Elaine A. Langone Scholar of the Damon Runyon Cancer Research Foundation. The authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albrecht UHJA, Lin H. Visualization of gene expression patterns by in situ hybridization. Boca Raton, FL: CRC Press; 1997. [Google Scholar]
- Bae E, Calhoun VC, Levine M, Lewis EB, Drewell RA. Characterization of the intergenic RNA profile at abdominal-A and Abdominal-B in the Drosophila bithorax complex. Proc Natl Acad Sci U S A. 2002;99:16847–16852. doi: 10.1073/pnas.222671299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert J, McMahon S, Karlsson EK, Kulbokas EJ, 3rd, Gingeras TR, et al. Cell. Vol. 120. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse; pp. 169–181. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Allis CD. RNA meets chromatin. Genes Dev. 2005;19:1635–1655. doi: 10.1101/gad.1324305. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Duncan EM, Masui O, Gil J, Heard E, Allis CD. Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Molecular and cellular biology. 2006;26:2560–2569. doi: 10.1128/MCB.26.7.2560-2569.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, Weissman S, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Carroll SB. Homeotic genes and the evolution of arthropods and chordates. Nature. 1995;376:479–485. doi: 10.1038/376479a0. [DOI] [PubMed] [Google Scholar]
- Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, Brown PO. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99:12877–12882. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong CM. Homeobox genes, fetal wound healing, and skin regional specificity. The Journal of investigative dermatology. 2003;120:9–11. doi: 10.1046/j.1523-1747.2003.00002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisenko O, Shnyreva M, Suzuki H, Bomsztyk K. Point mutations in the WD40 domain of Eed block its interaction with Ezh2. Molecular and cellular biology. 1998;18:5634–5642. doi: 10.1128/mcb.18.10.5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewell RA, Bae E, Burr J, Lewis EB. Transcription defines the embryonic domains of cis-regulatory activity at the Drosophila bithorax complex. Proc Natl Acad Sci U S A. 2002;99:16853–16858. doi: 10.1073/pnas.222671199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaud C, Bantignies F, Pal-Bhadra M, Ghana P, Bhadra U, Cavalli G. RNAi components are required for nuclear clustering of Polycomb group response elements. Cell. 2006;124:957–971. doi: 10.1016/j.cell.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Jenner RG, Chevalier B, Nakamura T, Croce CM, Canaani E, Young RA. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci U S A. 2005;102:8603–8608. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TM. Multiple modes of RNA recognition by zinc finger proteins. Current opinion in structural biology. 2005;15:367–373. doi: 10.1016/j.sbi.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Kapranov P, Drenkow J, Cheng J, Long J, Helt G, Dike S, Gingeras TR. Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res. 2005;15:987–997. doi: 10.1101/gr.3455305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Villeneuve LM, Morris KV, Rossi JJ. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nature structural & molecular biology. 2006;13:793–797. doi: 10.1038/nsmb1142. [DOI] [PubMed] [Google Scholar]
- Kmita M, Duboule D. Organizing axes in time and space; 25 years of colinear tinkering. Science. 2003;301:331–333. doi: 10.1126/science.1085753. [DOI] [PubMed] [Google Scholar]
- Kmita M, Kondo T, Duboule D. Targeted inversion of a polar silencer within the HoxD complex re-allocates domains of enhancer sharing. Nature genetics. 2000;26:451–454. doi: 10.1038/82593. [DOI] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemons D, McGinnis W. Genomic evolution of Hox gene clusters. Science. 2006;313:1918–1922. doi: 10.1126/science.1132040. [DOI] [PubMed] [Google Scholar]
- Nelson CE, Morgan BA, Burke AC, Laufer E, DiMambro E, Murtaugh LC, Gonzales E, Tessarollo L, Parada LF, Tabin C. Analysis of Hox gene expression in the chick limb bud. Development (Cambridge, England) 1996;122:1449–1466. doi: 10.1242/dev.122.5.1449. [DOI] [PubMed] [Google Scholar]
- Papp B, Muller J. Histone trimethylation and the maintenance of transcriptional ON and OFF states by trxG and PcG proteins. Genes Dev. 2006;20:2041–2054. doi: 10.1101/gad.388706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruk S, Sedkov Y, Riley KM, Hodgson J, Schweisguth F, Hirose S, Jaynes JB, Brock HW, Mazo A. Transcription of bxd noncoding RNAs promoted by trithorax represses Ubx in cis by transcriptional interference. Cell. 2006;127:1209–1221. doi: 10.1016/j.cell.2006.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300:131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- Ringrose L, Paro R. Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development (Cambridge, England) 2007;134:223–232. doi: 10.1242/dev.02723. [DOI] [PubMed] [Google Scholar]
- Rinn JL, Bondre C, Gladstone HB, Brown PO, Chang HY. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet. 2006;2:e119. doi: 10.1371/journal.pgen.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Euskirchen G, Bertone P, Martone R, Luscombe NM, Hartman S, Harrison PM, Nelson FK, Miller P, Gerstein M, et al. The transcriptional activity of human Chromosome 22. Genes Dev. 2003;17:529–540. doi: 10.1101/gad.1055203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Rozowsky JS, Laurenzi IJ, Petersen PH, Zou K, Zhong W, Gerstein M, Snyder M. Major molecular differences between mammalian sexes are involved in drug metabolism and renal function. Developmental cell. 2004;6:791–800. doi: 10.1016/j.devcel.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Sanchez-Elsner T, Gou D, Kremmer E, Sauer F. Noncoding RNAs of trithorax response elements recruit Drosophila Ash1 to Ultrabithorax. Science. 2006;311:1118–1123. doi: 10.1126/science.1117705. [DOI] [PubMed] [Google Scholar]
- Schmitt S, Prestel M, Paro R. Intergenic transcription through a polycomb group response element counteracts silencing. Genes Dev. 2005;19:697–708. doi: 10.1101/gad.326205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Yelensky R, Koller D. Genome-wide discovery of transcriptional modules from DNA sequence and gene expression. Bioinformatics. 2003;19(Suppl 1):i273–282. doi: 10.1093/bioinformatics/btg1038. [DOI] [PubMed] [Google Scholar]
- Sessa L, Breiling A, Lavorgna G, Silvestri L, Casari G, Orlando V. Noncoding RNA synthesis and loss of Polycomb group repression accompanies the colinear activation of the human HOXA cluster. Rna. 2006 doi: 10.1261/rna.266707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- Squazzo SL, O’Geen H, Komashko VM, Krig SR, Jin VX, Jang SW, Margueron R, Reinberg D, Green R, Farnham PJ. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res. 2006 doi: 10.1101/gr.5306606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suemori H, Noguchi S. Hox C cluster genes are dispensable for overall body plan of mouse embryonic development. Dev Biol. 2000;220:333–342. doi: 10.1006/dbio.2000.9651. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zhang H. A unified mode of epigenetic gene silencing: RNA meets polycomb group proteins. RNA biology. 2005;2:8–10. doi: 10.4161/rna.2.1.1465. [DOI] [PubMed] [Google Scholar]
- Zakany J, Fromental-Ramain C, Warot X, Duboule D. Regulation of number and size of digits by posterior Hox genes: a dose-dependent mechanism with potential evolutionary implications. Proc Natl Acad Sci U S A. 1997;94:13695–13700. doi: 10.1073/pnas.94.25.13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Christoforou A, Aravind L, Emmons SW, van den Heuvel S, Haber DA. The C. elegans Polycomb gene SOP-2 encodes an RNA binding protein. Molecular cell. 2004;14:841–847. doi: 10.1016/j.molcel.2004.06.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01
02
03
04
05
06