Blockade of the Bcr-Abl Kinase Activity Induces Apoptosis of Chronic Myelogenous Leukemia Cells by Suppressing Signal Transducer and Activator of Transcription 5–Dependent Expression of Bcl-XL (original) (raw)
Abstract
Bcr-Abl–expressing leukemic cells are highly resistant to apoptosis induced by chemotherapeutic drugs. Although a number of signaling molecules have been shown to be activated by the Bcr-Abl kinase, the antiapoptotic pathway triggered by this oncogene has not been elucidated. Here, we show that the interleukin 3-independent expression of the antiapoptotic protein, Bcl-xL, is induced by Bcr-Abl through activation of signal transducer and activator of transcription (Stat)5. Inhibition of the Bcr-Abl kinase activity in Bcr-Abl–expressing cell lines and CD34+ cells from chronic myelogenous leukemia (CML) patients induces apoptosis by suppressing the capacity of Stat5 to interact with the bcl-x promoter. Interestingly, after inhibition of the Bcr-Abl kinase, the expression of Bcl-xL is downregulated more rapidly in chronic phase than in blast crisis CML cells, suggesting an involvement of this protein in disease progression. Overall, we describe a novel antiapoptotic pathway triggered by Bcr-Abl that may contribute to the resistance of CML cells to undergo apoptosis.
Keywords: CGP 57148, cell death, blast crisis, promoter, interleukin 3
Introduction
Chronic myelogenous leukemia (CML) is characterized by the reciprocal chromosomal translocation 9:22, which generates the Philadelphia chromosome 1. This event occurs in a hematopoietic stem cell and fuses a truncated bcr gene on chromosome 22 to sequences upstream of the second exon of c-abl on chromosome 9. This translocation generates a novel fusion gene, bcr-abl, that encodes a chimeric protein, p210, with a hyperactive tyrosine kinase.
The observation that production of Bcr-Abl is the initiating event in CML has focused attention on the tyrosine phosphorylation-dependent signaling events triggered by this oncoprotein. Expression of Bcr-Abl in hematopoietic cells induces resistance to apoptosis, growth factor independence, alterations in cell–cell and cell–matrix interactions, and leukemogenesis 2 3 4 5. This phenotype is associated with enhanced expression/activation of several effectors, such as Ras, Rac, Raf-1, phosphatidylinositol-3 kinase (PI-3k), Akt, nuclear factor κB, and signal transducers and activators of transcription (STATs) 6.
Bcr-Abl–expressing leukemic cells are highly resistant to apoptotic cell death induced by chemotherapeutic drugs 7 8. Previously, we have shown that a CML-derived cell line, K562, overexpresses the antiapoptotic protein Bcl-xL, but not Bcl-2, a closely related member of the same family of apoptosis regulators 9. Furthermore, downregulation of Bcl-xL by treatment of K562 cells with differentiation inducers is accompanied by activation of an apoptotic pathway, which is abrogated by the ectopic expression of Bcl-xL. In addition, Bcr-Abl–transfected HL-60 cells express high levels of Bcl-xL and undetectable levels of Bcl-2 10, an expression pattern similar to that shown in K562 cells. These findings suggest that Bcl-xL may contribute to the resistance of Bcr-Abl–expressing cells to apoptosis. Recently, we and others have shown that Bcl-xL is induced by activation of a Stat protein, mainly Stat1, Stat3, and Stat5 11 12 13 14 15 by direct binding to a consensus sequence in the bcl-x gene promoter. Since these Stat proteins appear to be constitutively activated in Bcr-Abl–expressing cells 16 17, we raised the question of whether the antiapoptotic effect of Bcr-Abl was due to the transcriptional upregulation of Bcl-xL by activation of a Stat protein. Here, we show that Bcr-Abl and interleukin (IL)-3 transduce antiapoptotic signals through the Stat5/Bcl-xL pathway in CML cells. Moreover, inhibition of the Bcr-Abl kinase activity blocks this antiapoptotic pathway more rapidly in chronic phase than in blast crisis CML cells. Thus, we describe a novel antiapoptotic transcriptional pathway triggered by the Bcr-Abl kinase activity that may be useful not only to understand the mechanisms of resistance to chemotherapy-induced apoptosis and disease progression in CML, but also to have new molecular targets for antileukemic drug discovery.
Materials and Methods
Samples from CML Patients and Normal Donors.
A total of 100–250 mL of heparinized steady state or mobilized peripheral blood was obtained from patients with CML in blast crisis (n = 5) or chronic phase (n = 5). Mobilized peripheral blood progenitors were obtained from normal donors (n = 5) undergoing mobilization for allogeneic peripheral blood progenitor cell transplantation with G-CSF at doses of 5 mg/kg/12 h subcutaneously. All patients and normal donors signed informed consent according to Guidelines from the Committee for the Protection of Human Subjects at the University of Valencia. All patients were 100% Philadelphia chromosome positive at direct cytogenetic analysis.
Cell Culture.
The CML-derived K562 and K562–Bcl-xL 9 cell lines were maintained in RPMI 1640 medium (Seromed Biochrom KG) supplemented with 10% FCS (Flow Laboratories). Parental Mo7e, Mo7e-Neo, and Mo7e-p210 cell lines 3, were grown in IMDM (GibcoBRL) supplemented with 20% FCS and with (Mo7e and Mo7e-Neo) or without (Mo7e-p210) 5 ng/mL of recombinant human IL-3 (Immunex).
CD34+ cells were selected from the PBMC population by either two passages over the MACS CD34 Isolation Kit (Miltenyi Biotec) as previously described 18 or by a single passage using the CliniMACS separation device (Miltenyi Biotec) according to the manufacturer. After positive selection, the CD34+ populations (>95%) were cultured in IMDM containing 20% FCS. Normal CD34+ cells, and in some experiments, CML cells, were maintained in culture supplemented with recombinant human IL-3 at a final concentration of 100 ng/mL.
When indicated, cells were treated with 2 μM CGP 57148 19, developed and provided by Novartis Inc., or 40 μM tyrphostin AG 555 (CALBIOCHEM) for different time intervals, and then analyzed. Viability and total cell counts were determined at various times by trypan blue exclusion and counting of at least 200 cells from each individual culture.
Cell Transfection.
K562 cells (3 × 106) were transfected with the pSFFV-Neo expression vector containing a truncated form of Stat5 that lacks the COOH-terminal transactivation domain (Stat5Δ750) and exerts a dominant negative effect 11. pSFFV-Stat5Δ750 (3 μg) or the control pSFFV-Neo vector (3 μg) was mixed with 12 μl of lipofectamine (GibcoBRL) and incubated with the cells for 5 h in the absence of FCS. Then, fresh complete medium was added to the culture and after 24 h of incubation, cells were harvested and analyzed for expression of Stat5Δ750 and Bcl-xL proteins.
Analysis of Apoptotic Cells.
Apoptosis was assessed by several criteria. DNA content was quantified by cell cycle analysis as described elsewhere 20, with slight modifications. Cells (106) were resuspended in the fluorochrome solution (0.1% sodium citrate, 0.01% Triton X-100, and 0.1 mg/mL propidium iodide). After 4 h at 4°C in the dark, fluorescence was measured using a FACScan flow cytometer (Becton Dickinson). The percentage of hypodiploid cells correlates with the extent of apoptosis in the sample. For DNA fragmentation analysis, cells (106) were washed with PBS and pelleted by centrifugation. Genomic DNA was isolated from cell pellets as described previously 9. DNA samples were electrophoresed on a 2% agarose gel and stained with 0.1% ethidium bromide. The early apoptotic cells were detected with annexin V labeled with fluorescein isothiocyanate (PharMingen) by flow cytometry.
Western Blot Analysis.
The expression of Bcl-xL protein was determined by Western blotting as previously described 9. Proteins (30–60 μg) were separated on a 12% polyacrylamide gel, and transferred to nitrocellulose. Blots were blocked with 3% BSA and incubated with rabbit antibodies against Bcl-x (Transduction Laboratories), and mouse anti–β-tubulin (Sigma Chemical Co.), and then incubated with goat anti–rabbit or anti–mouse antibodies conjugated to alkaline phosphatase (Tropix). Bound antibody was detected by a chemiluminescence system (Tropix).
In some experiments, 3–8% NuPAGE Tris-Acetate gels (Novex) were used to separate endogenous Stat5 from transfected Stat5Δ750 proteins.
Immunoprecipitation.
K562 and Mo7e-p210 cells were cultured with or without tyrosine kinase inhibitors (CGP 57148 and tyrphostin AG 555) for 3 h, and were then lysed in 0.5% NP-40–containing solution as previously described 11. Cleared lysates were incubated with mouse monoclonal anti–c-Abl, rabbit anti-Stat5, anti-Stat1, or anti-Stat3 antibodies (Santa Cruz), and protein A/G conjugated to agarose beads (Santa Cruz). Proteins eluted from the agarose beads were electrophoresed and transferred to nitrocellulose. Membranes were incubated with the same primary antibodies used for immunoprecipitation or mouse antiphosphotyrosine (Upstate Biotechnology), and bound antibodies were detected as described above.
Electrophoretic Mobility Shift Assays (EMSAs).
Cells were cultured for 3 h in the presence or absence of tyrosine kinase inhibitors, and were then lysed as previously described 11. In brief, nuclear fractions were resuspended in 20 mM Hepes, pH 7.6, 0.4 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 10 mM sodium molybdate, and protease inhibitors. Nuclear extracts (5 μg of total protein) were incubated with a 32P-labeled double-stranded DNA probe from the promoter region of the bcl-x gene 11 or from the SV40 enhancer, containing a single consensus sequence for Spi-1/PU.1 21. Samples were run on a 5% nondenaturing polyacrylamide gel in 200 mM Tris-borate and 2 mM EDTA. Gels were dried and visualized by autoradiography. Supershifts were performed using rabbit polyclonal antibodies specific for Stat5 (Santa Cruz) or Spi-1 (generously provided by Dr. J. Leon, University of Cantabria, Spain). For competition assays, nuclear extracts containing equal amounts of total protein were preincubated with 100-fold molar excess of either unlabeled bcl-x probe or unlabeled irrelevant DNA fragment.
Results
Bcr-Abl Upregulates Bcl-xL through Activation of Stat5 in K562 Cells.
The activation of Stat5 may play an important role in Bcr-Abl–mediated transformation and leukemogenesis 16; however, the biological consequences of this activation in hematopoietic cells are not elucidated. Recently, we have shown that erythropoietin can induce the expression of the antiapoptotic protein, Bcl-xL, through the binding of Stat5 to the bcl-x promoter, which may contribute to maintain the survival of erythoid progenitors 11. To study whether this antiapoptotic pathway is activated by Bcr-Abl, we treated K562 cells, which have endogenous expression of p210 Bcr-Abl, with CGP 57148, an ATP-competitive inhibitor of the Abl protein kinase that has been shown to efficiently eradicate human CML-derived cells in a mouse model 22. It has been described that a complete inhibition of autophosphorylation of the Bcr-Abl kinase is achieved with CGP 57148 at concentrations between 1 and 10 μM 23. Based on this result, we treated K562 cells with different concentrations of CGP 57148 (from 0.5–4 μM) for 3 h, and found that 2 μM completely abrogated phosphorylation of Bcr-Abl, as assessed by antiphosphotyrosine immunoblots of anti–c-Abl-immunoprecipitated cell lysates (Fig. 1 and data not shown). In addition, at this concentration, CGP 57148 inhibited phosphorylation of Stat5, whereas Stat3 and Stat1 remained phosphorylated. By contrast, in untreated cells or in cells treated for 3 h with 40 μM tyrphostin 555, an inhibitor selective for epidermal growth factor receptor kinase 24, immunoprecipitated Bcr-Abl, Stat5, Stat1, and Stat3 were all phosphorylated (Fig. 1). Since the expression of Stat proteins was the same at all culture conditions, this finding suggests that the loss of Stat5 phosphorylation correlated with inhibition of the Bcr-Abl kinase activity. Furthermore, the expression of Bcl-xL in K562 cells gradually decreased within 48 h of treatment with CGP 57148 (Fig. 2 A), and this correlated with the activation of an apoptotic process, as assessed by cell cycle analysis (Fig. 2 B) and genomic DNA fragmentation analysis (data not shown). After 24 h of treatment, the percentage of cells in the sub-G1 peak of apoptotic cells increased from 14.8% (9.2 ± 3.3, mean ± SD, n = 3) to 38.9% (43.7 ± 5.2) and the G2/M fraction decreased from 27.2 ± 3.3% to 11.6 ± 2.1%. In contrast, the fraction of K562 cells stably transfected with Bcl-xL (constitutive expression driven by the SFFV promoter; 9) that appeared in the sub-G1 cycle phase after 24 h of treatment with CGP 57148 was 11.9% (10.1 ± 2.6), compared with 5.7% (6.6 ± 1.8) in untreated cells (Fig. 2A and Fig. B). However, the G2/M fraction was reduced in these transfected cells (from 28.7 ± 3.8% to 13.1 ± 2.6%), indicating that a constitutive, Stat5 independent expression of Bcl-xL blocked apoptosis induced by inhibition of the Bcr-Abl kinase activity, but not proliferation. Because multiple signal transduction pathways have been shown to be activated by Bcr-Abl 25, we next studied the effects of a dominant negative Stat5 protein on Bcl-xL expression and cell survival (a representative experiment is shown in Fig. 3). A Stat5 protein that lacks the transactivation domain and exerts a dominant negative effect (Stat5Δ750) was transiently transfected in K562 cells. After 24 h of transfection, the expression of Stat5 dominant negative correlated with downregulation of Bcl-xL (Fig. 3 A). Significantly, when we examined transiently transfected Stat5Δ750-expressing cells by annexin V staining and FACS analysis, an increase in the percentage of apoptotic cells (23.8%) was observed relative to empty vector transfectants (5.2%; Fig. 3 B). These results demonstrate that Stat5-dependent signaling is required for preventing loss of Bcl-xL expression and apoptosis in K562 cells.
Figure 1.
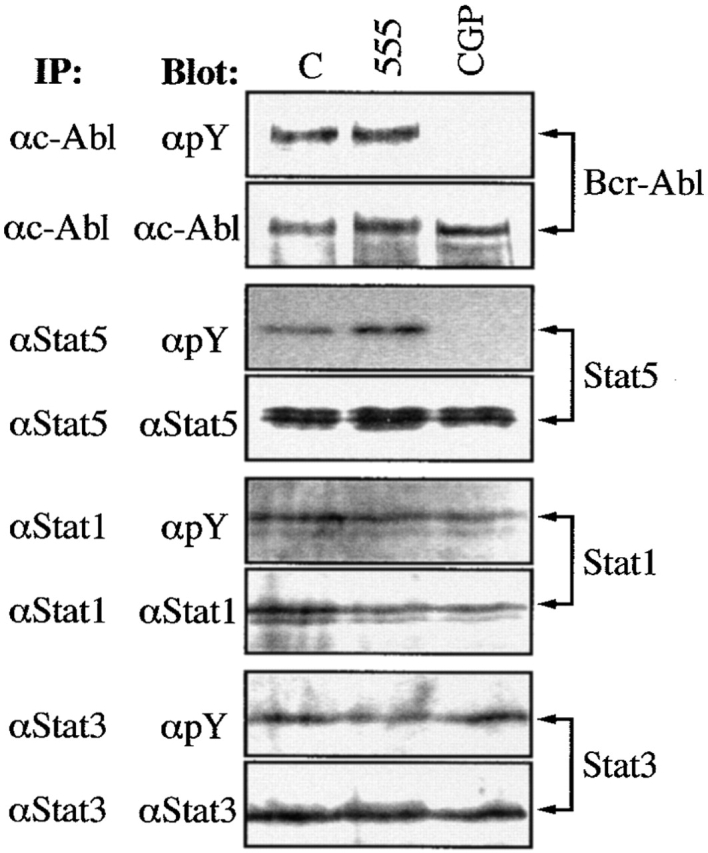
Tyrosine phosphorylation of Bcr-Abl and Stat proteins in K562 cells. Cells were cultured with CGP 57148 or tyrphostin AG 555 for 3 h, and then cell extracts were analyzed by immunoprecipitation (IP) with antibodies specific for c-Abl, Stat5, Stat1, and Stat3, and by subsequent blotting with an antiphosphotyrosine antibody (αpY). The same filters were stripped and reblotted with the same antibodies used for IP. C, Untreated control cells.
Figure 2.
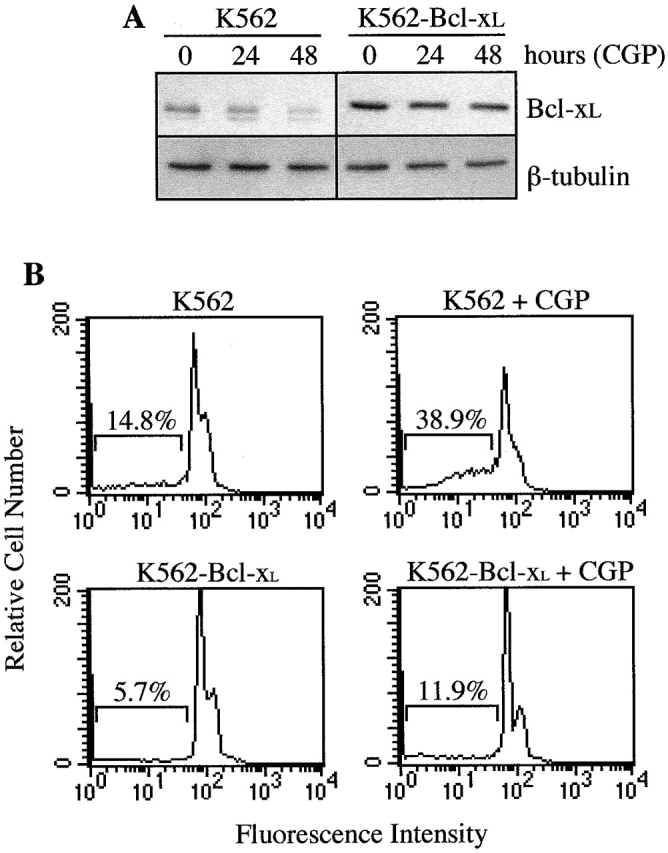
Analysis of Bcl-xL and apoptosis in K562 and K562–Bcl-xL cell lines. (A) Cells were treated for different times with CGP 57148 and analyzed for the expression of Bcl-xL by Western blot. The levels of β-tubulin were also analyzed to assure equal loading. (B) Apoptosis of cells treated with the kinase inhibitor was quantified by cell cycle analysis. Numbers represent the percentage of cells with subdiploid DNA content. Histograms are from a representative experiment (n = 3).
Figure 3.

Analysis of Bcl-xL and apoptosis in K562 cells transfected with a Stat5 dominant negative. Cells were transiently transfected with 3 μg of pSFFV-Neo (control vector) or pSFFV-Stat5Δ750 (dominant negative) and then analyzed. (A) Western blot analysis with a polyclonal antibody that recognizes both Stat5 and Stat5Δ750 proteins, and with a rabbit anti–Bcl-x antibody. The levels of β-tubulin were used as a loading control. (B) Apoptosis of transfected cells, measured by staining with annexin V-FITC and analysis by flow cytometry. Histograms are from a representative experiment (n = 3).
Blockade of the Bcr-Abl Kinase Inhibits the Stat5/Bcl-xL Antiapoptotic Pathway in Bcr-Abl–Transfected Cells.
The effects of CGP 57148 on the inhibition of the Stat5/Bcl-xL antiapoptotic pathway were compared in IL-3–independent Bcr-Abl–transfected Mo7e cells (Mo7e-p210) versus the control IL-3–dependent Mo7e and Mo7e-Neo cells 3. As shown in Fig. 4, CGP 57148 (2 μM) inhibited phosphorylation of Bcr-Abl and Stat5 in Mo7e-p210 cells, whereas Stat3 and Stat1 remained phosphorylated, a result similar to that obtained in K562 cells. In addition, after an exposure to CGP 57148 for 3 h, complete inhibition of the Stat5–DNA binding complex was observed in Mo7e-p210 cells, but not in Mo7e-Neo cells, as measured in EMSA using an oligonucleotide that corresponds to a consensus Stat5 sequence of the bcl-x promoter (Fig. 5). The specificity of the Stat5 binding was confirmed using anti-Stat5 antibodies that supershifted all of the DNA protein complexes in Mo7e-p210 and Mo7e-Neo cells (Fig. 5). Furthermore, treatment of cells with AG 555 or preincubation of the nuclear extracts with a 100-fold molar excess of a nonspecific probe did not affect the Stat5-DNA binding; however, a 100-fold molar excess of the unlabeled Stat5-specific probe inhibited the formation of this complex in both cell populations. To further control the specificity of the inhibitor, nuclear extracts from Mo7e-p210 cells were analyzed by EMSA with a probe containing the binding site for Spi-1/PU.1, a transcription factor of the Ets family, expressed in myeloid cells 21. Fig. 5 shows that the protein–DNA binding complex was detected both in controls (untreated cells or cells treated with AG 555) and in cells treated with CGP 57148.
Figure 4.

Tyrosine phosphorylation of Bcr-Abl and Stat proteins in Mo7e-p210 cells. Cells were cultured with CGP 57148 or tyrphostin AG 555 for 3 h, and then cell extracts were analyzed by immunoprecipitation (IP) with antibodies specific for c-Abl, Stat5, Stat1, and Stat3, and by subsequent blotting with an antiphosphotyrosine antibody (αpY). The same filters were stripped and reblotted with the same antibodies used for IP. C, Untreated control cells.
Figure 5.
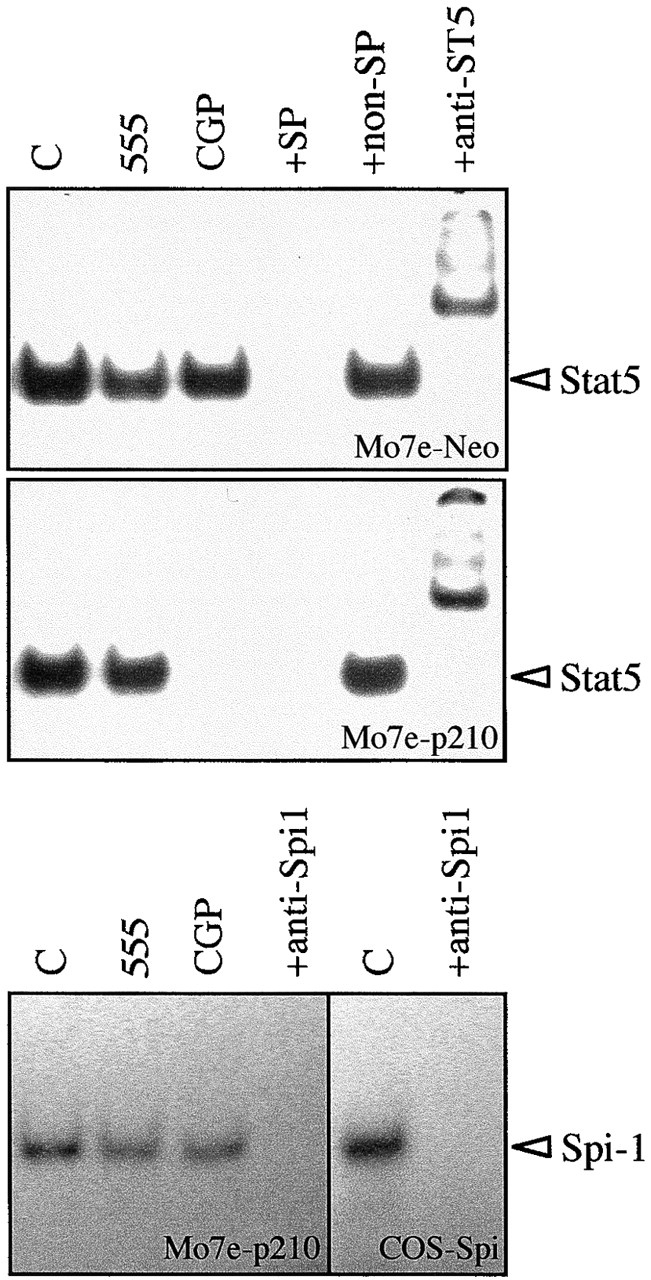
Binding of Stat5 to the bcl-x promoter in Mo7e-Neo and Mo7e-p210 cell lines. Cells were treated for 3 h with CGP 57148 or tyrphostin AG 555, and formation of Stat5-DNA complexes was determined by an EMSA using a radiolabeled bcl-x probe. DNA binding activity of Spi-1/PU.1 in Mo7e-p210 cells was also analyzed as a specificity control. Nuclear extracts from untreated cells (C) were preincubated with antibodies specific for Stat5 (+anti-ST5) or Spi-1 (anti-Spi1), and with an excess of an unlabeled bcl-x probe as a specific competitor (+SP) or with an irrelevant nonspecific probe (+non-SP). Extracts from COS cells transfected with a Spi-1 expression vector (COS-Spi) were used as positive controls.
Next, we analyzed the levels of Bcl-xL protein in the different cell populations. As shown in Fig. 6, CGP 57148 inhibited the expression of Bcl-xL and induced loss of cell viability in Mo7e-p210, but not in parental Mo7e or Mo7e-Neo cells. Treatment of Mo7e-p210 cells with CGP 57148 for 24 h drastically reduced the expression of Bcl-xL, which continued to decrease by 48 h of treatment (Fig. 6 A). This pattern of expression correlated with the progressive loss of Mo7e-p210 cell viability within five days of treatment with the inhibitor (Fig. 6 B), due to the activation of an apoptotic process as assessed by genomic DNA fragmentation analysis (data not shown). Because Mo7e cells become IL-3 independent upon transfection with Bcr-Abl cDNA, and do not undergo apoptosis in the absence of IL-3, we examined whether the antiapoptotic pathway inhibited by CGP 57148 could be restored by culturing Mo7e-p210 in the presence of IL-3. As shown in Fig. 6, in the presence of IL-3, the Bcr-Abl kinase inhibitor did not affect the levels of Bcl-xL within 48 h of treatment, and this correlated with activation of Stat5 as assessed by EMSA (data not shown). Furthermore, under these culture conditions, cell viability was maintained by the first three to four days, and it was slightly reduced by the fifth day (64%), a time point to which most of the Mo7e-p210 cells (90%) treated with CGP 57148 in the absence of IL-3 were dead, suggesting that IL-3 and Bcr-Abl may activate the same antiapoptotic pathway.
Figure 6.

Analysis of Bcl-xL and cell viability in Mo7e, Mo7e-Neo, and Mo7e-p210 cell lines. Mo7e-p210 cells were treated for different times with CGP 57148 or tyrphostin AG 555 in the presence or absence of IL-3. Mo7e and Mo7e-Neo (Bcr-Abl−) cells were also incubated with the inhibitors in the presence of IL-3. (A) At the indicated times, cells were harvested and analyzed for the expression of Bcl-xL by Western blot. The levels of β-tubulin were also analyzed to assure equal loading. C, Untreated control cells. (B) Viability of the Mo7e cell lines was measured by trypan blue dye exclusion at different times of incubation with CGP 57148. All data points represent the mean of triplicate cultures ± SD.
Inhibition of the Bcr-Abl Kinase Activity Downregulates the Expression of Bcl-xL and Induces Apoptosis in CML Cells from Patients in Chronic Phase and Blast Crisis.
To extend these findings to a more clinically relevant cellular model, we treated CD34+ progenitor cells from normal donors and patients with chronic phase CML and blast crisis CML with CGP 57148. Treatment of blast crisis and chronic phase CML cells with CGP 57148 for 3 h in the absence of growth factors completely abrogated the binding of Stat5 to the Stat5-specific sequence of the bcl-x promoter; however, treatment with the control tyrosine kinase inhibitor, AG 555, did not affect the formation of the Stat5-DNA binding complex (Fig. 7 shows a representative experiment). The specificity of the Stat5 binding was confirmed using antibodies against Stat5. As shown in Fig. 7, all of the DNA–protein complexes were supershifted by pretreatment of nuclear extracts with the antibodies. This pattern of Stat5 activation in CML primary cells thus, is similar to that of Mo7e-p210 cells described above.
Figure 7.
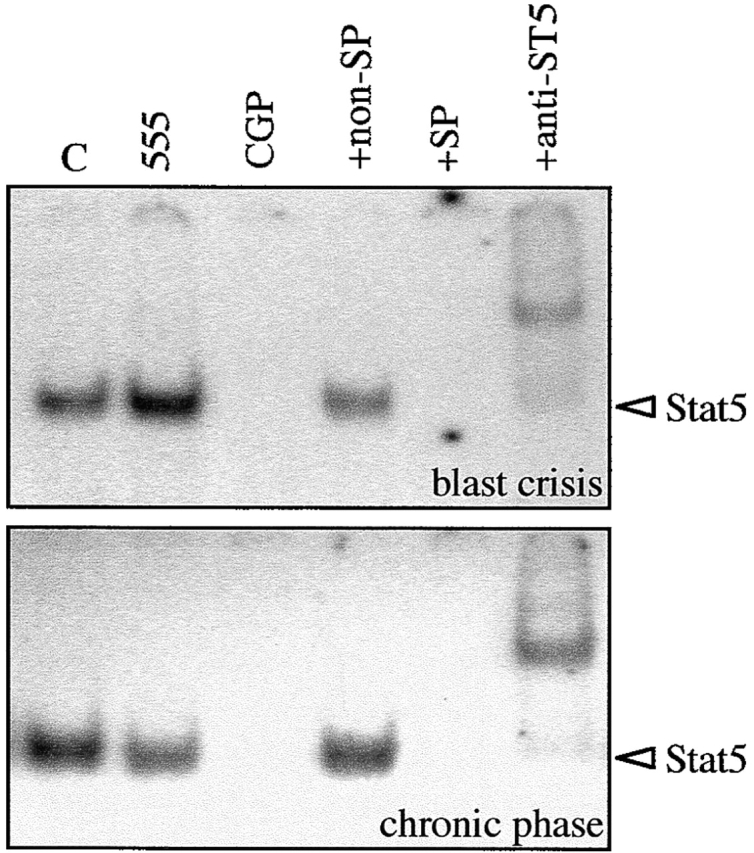
Binding of Stat5 to the bcl-x promoter in blast crisis and chronic phase CML cells. CD34+ cells were isolated from the peripheral blood of patients with CML in blast crisis and chronic phase, and treated for 3 h with CGP 57148 or tyrphostin AG 555. An EMSA was performed using a radiolabeled bcl-x probe. Nuclear extracts from untreated cells (C) were preincubated with antibodies specific for Stat5 (+anti-ST5), and with an excess of an unlabeled bcl-x probe as a specific competitor (+SP) or with an irrelevant nonspecific probe (+non-SP).
To define the antiapoptotic pathway in CML CD34+ cells, the expression of Bcl-xL and the percentage of viable cells were compared in blast crisis and chronic phase CML cells versus normal CD34+ cells after treatment with CGP 57148 (Fig. 8). Normal cells, cultured in the presence of IL-3, did not show any variation in the levels of Bcl-xL at any time of treatment. In contrast, both chronic phase and blast crisis CML cells showed a downregulated expression of Bcl-xL (Fig. 8 A shows a representative experiment). However, it is noteworthy that, whereas Bcl-xL protein virtually disappeared in chronic phase within 24 h of treatment, it was readily detectable, although at lower levels, in blast crisis after 48 h of treatment with the kinase inhibitor. This loss of Bcl-xL expression was restored in the presence of IL-3 in both cell populations. Furthermore, this pattern of expression correlated with cell viability. Viability of normal CD34+ cells was maintained during the treatment with CGP 57148. In contrast, CML CD34+ cells progressively died within four days of treatment; however, as shown previously for Mo7e-p210, cell viability was restored by addition of IL-3 to the culture. Consistent with the protein analysis data, blast crisis CML cells died at a slower rate. By 24 h of treatment with the inhibitor, viability of blast crisis cells decreased from 76% (day 0) to 60%, whereas viability of chronic phase cells was reduced from 65% (day 0) to 23% (Fig. 8 B).
Figure 8.

Analysis of Bcl-xL and cell viability in chronic phase and blast crisis CML cells. CD34+ cells were isolated from the peripheral blood of patients with CML in chronic phase or in blast crisis and treated with tyrphostin AG 555 or CGP 57148 in the presence or absence of IL-3. Normal CD34+ cells were treated with the same inhibitors in the presence of IL-3. (A) At the indicated times, cells were analyzed for the expression of Bcl-xL by Western blot. The levels of β-tubulin were also analyzed to assure equal loading. C, Untreated control cells. (B) Viability of normal and CML cells was measured by trypan blue dye exclusion at different times of incubation with CGP 57148. All data points represent the mean of triplicate cultures ± SD.
Discussion
Evidence is emerging to support the idea that Bcr-Abl exerts an antiapoptotic effect 8 10 20 that may contribute to the resistance of Bcr-Abl expressing cells to high doses of antileukemic drugs such as Ara-C or etoposide 26. However, the signaling pathway triggered by Bcr-Abl to inhibit apoptosis has not been elucidated. Here, we have shown that treatment of both Bcr-Abl–expressing cell lines and CD34+ CML cells with CGP 57148, a selective inhibitor of the Bcr-Abl kinase activity, blocks activation of Stat5, a transcription factor recently shown to induce the expression of Bcl-xL in response to erythropoietin and IL-3 in hematopoietic progenitors 11 12 13. Consistent with this, we found that inactivation of Stat5 in CML-derived cells by treatment with the kinase inhibitor or transfection with a Stat5 dominant negative, downregulates the expression of Bcl-xL and induces an apoptotic process. Stat5 activation has been involved in antiapoptotic activity and cell cycle progression induced by Bcr-Abl, and it has been suggested that the constitutive activation of Stat5 is important for Bcr-Abl–mediated leukemogenesis in vitro and in vivo 16. Stat5 is phosphorylated on tyrosine residues in hematopoietic cells in response to growth factors such as IL-3, IL-5, GM-CSF, and Epo 27 28, and this may represent an important point of convergence between the Bcr-Abl and IL-3 signaling pathways that could contribute to the known overlap in the biological effects of IL-3 and Bcr-Abl 17. Consistent with this, we found that inhibition of the Stat5/Bcl-xL antiapoptotic pathway by treatment of Bcr-Abl–transfected Mo7e cells and CD34+ CML cells with CGP 57148 could be overcome in the presence of IL-3, which induced activation of Stat5 and upregulated the expression of Bcl-xL. In addition, recently it has been shown that IL-3 regulates the expression of Bcl-xL by Stat5 in a bone marrow-derived cell line 13. These findings indicate that IL-3 and Bcr-Abl are transducing survival signals through the same pathway that involves activation of Stat5 and expression of Bcl-xL. Nevertheless, since it has been shown that Bcl-xL can be phosphorylated in some cell systems 29 we cannot rule out the possibility that Bcr-Abl may also mediate phosphorylation of Bcl-xL, which could play a role in modulating Bcl-xL function.
CML is characterized by a stable phase in which the expanded myeloid population undergo complete differentiation and a blast crisis or accelerated phase that follows the chronic phase, in which the myeloid cells are unable to differentiate, resulting in an acute leukemia. Although chromosomal alterations other than the t(9;22) translocation are detected in blast crisis CML cells, a specific molecular marker of this phase of the disease has not been characterized yet. In this study, we have shown that the expression of Bcl-xL is inhibited at earlier times in chronic phase than in blast crisis CML cells after blockade the Bcr-Abl kinase activity, and this correlates with a more rapid loss of cell viability. This finding suggests that additional genetic abnormalities may contribute to the resistance of blast crisis CML cells to undergo apoptosis. Recently, the biological effects of high and low levels of Bcr-Abl expression have been compared in growth factor-dependent myeloid cells, demonstrating that, although low levels of Bcr-Abl were sufficient to render these cell lines growth factor independent and tumorigenic, higher levels were mandatory for additional protection against apoptosis 30. According to this, a prolonged expression of Bcl-xL could be due to increased levels of Bcr-Abl, which is consistent with the evidence that the expression of bcr-abl mRNA frequently increases with disease progression and that the duplication of the Philadelphia chromosome represents the most frequent karyotypic abnormality in blast crisis CML 31 32. Alternatively, activation of Stat proteins other than Stat5 could contribute to prolong the expression of Bcl-xL in blast crisis, although it would not be sufficient to maintain the levels of this antiapoptotic protein. It has been shown that most of the Bcr-Abl–induced Stat activity is due to Stat5 (16, this paper); however, Stat1 and Stat3 are also activated in hematopoietic cell lines expressing Bcr-Abl and in CML cells 33. Stat1 and Stat3 are able to bind the bcl-x promoter and to induce transactivation of this gene in cardiac myocytes 14, and myeloma cells 15, respectively. Consequently, an increased phosphorylation of at least one of these Stat family members could extend cell survival in blast crisis CML cells. A third possibility is that prolonged expression of Bcl-xL is due to an increase in the Bcl-xL protein half-life. In line with this, it has been described that the decreased protein levels of Bcl-xL in a T cell clone after growth factor withdrawal is due to the cleavage of Bcl-xL by caspase-3, which results in accelerated apoptotic cell death 34. Thus, inhibition of this enzymatic activity could account for an increased Bcl-xL protein half-life in blast crisis CML cells.
In conclusion, we have described a novel Bcr-Abl–induced antiapoptotic pathway in CML cells that may contribute to malignant progression by conferring a survival advantage through suppression of apoptotic cell death. Furthermore, this pathway, which induces expression of Bcl-xL by activation of Stat5, represents an attractive target for drug discovery with the potential of inducing cell death of human leukemic cells.
Acknowledgments
The authors thank Dr. John Dick (University of Toronto, Canada), for providing Mo7e-Neo and Mo7e-p210 cell lines; Novartis Inc. (Basel, Switzerland), for CGP 57148; and Dr. Catherine Verfaillie (University of Minnesota, Minneapolis, MN) for critical reading of the manuscript.
This work was supported by Comision Interministerial de Ciencia y Tecnologia grants SAF96/0174 and SAF99/0139 to J.L. Fernandez-Luna, and by Fondo de Investigaciones Sanitarias grant FIS98/0863 to F. Prosper. E.J. Andreu is a recipient of a fellowship from the Fondo de Investigaciones Sanitarias. C. Sanz is a recipient of a fellowship from the Fundacion Marques de Valdecilla.
Footnotes
M. Horita and E.J. Andreu contributed equally to this work.
A. Benito's present address is Department of Pathology, University of Michigan Medical School, 1500 E. Medical Center Dr., Ann Arbor, MI 48109.
Abbreviations used in this paper: CML, chronic myelogenous leukemia; EMSA, electrophoretic mobility shift assay; IL, interleukin; Stat, signal transducer and activator of transcription.
References
- Shtivelman E., Lifshitz B., Gale R.P., Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukemia. Nature. 1985;315:550–554. doi: 10.1038/315550a0. [DOI] [PubMed] [Google Scholar]
- Bedi A., Zehnbauer B.A., Barberand J.P., Sharkis S.J. Inhibition of apoptosis by BCR/ABL in chronic myelogenous leukemia. Blood. 1994;83:2038–2044. [PubMed] [Google Scholar]
- Sirard C., Laneuville P., Dick J. Expression of BCR/ABL abrogates factor-dependent growth of human hematopoietic M07E cells by an autocrine mechanism. Blood. 1994;83:1575–1585. [PubMed] [Google Scholar]
- Verfaille C.M., McCarthy J.B., McGlave P.B. Mechanisms underlying abnormal trafficking of malignant progenitors in chronic myelogenous leukemia. J. Clin. Invest. 1992;90:1232–1249. doi: 10.1172/JCI115985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorski T., Nieborowska-Skorska M., Wlodarski P., Wasik M., Trotta R., Kanakaraj P., Salomoni P., Antonyak M., Martinez R., Majewski M. The SH3 domain contributes to BCR/ABL-dependent leukemogenesis in vivorole in adhesion, invasion and homing. Blood. 1998;91:406–418. [PubMed] [Google Scholar]
- Sattler M., Salgia R. Activation of hematopoietic growth factor signal transduction pathways by the human oncogene BCR/ABL. Cytokine Growth Factor Rev. 1997;8:63–79. doi: 10.1016/s1359-6101(96)00047-0. [DOI] [PubMed] [Google Scholar]
- Bedi A., Barber J.P., Bedi G.C., el-Deiry W.S., Sidransky D., Vala M.S., Akhtar A.J., Hilton J., Jones R.J. Bcr-Abl–mediated inhibition of apoptosis with delay of G2/M transition after DNA damage. A mechanism of resistance to multiple anticancer agents. Blood. 1995;86:1148–1158. [PubMed] [Google Scholar]
- McGahon A., Bissonnette R., Schmitt M., Cotter K.M., Green D.R., Cotter T.G. Bcr-Abl maintains resistance of chronic myelogenous leukemia cells to apoptotic cell death. Blood. 1994;83:1179–1187. [PubMed] [Google Scholar]
- Benito A., Silva M., Grillot D., Nuñez G., Fernandez-Luna J.L. Apoptosis induced by erythroid differentiation of human leukemia cell lines is inhibited by Bcl-xL . Blood. 1996;87:3837–3843. [PubMed] [Google Scholar]
- Amarante-Mendes G.P., Naekyung Kim C., Liu L., Huang Y., Perkins C.L., Green D.R., Bhalla K. Bcr-Abl exerts its antiapoptotic effect against diverse apoptotic stimuli through blockage of mitochondrial release of cytochrome C and activation of caspase-3. Blood. 1998;91:1700–1705. [PubMed] [Google Scholar]
- Silva M., Benito A., Sanz C., Prosper F., Ekhterae D., Nunez G., Fernandez-Luna J.L. Erythropoietin can induce the expression of Bcl-xL through Stat5 in erythropoietin-dependent progenitor cell lines. J. Biol. Chem. 1999;274:22165–22169. doi: 10.1074/jbc.274.32.22165. [DOI] [PubMed] [Google Scholar]
- Socolovsky M., Fallon A., Wang S., Brugnara C., Lodish H.F. Fetal anemia and apoptosis of red cell progenitors in Stat5a−/−5b−/− micea direct role for Stat5 in Bcl-xL induction. Cell. 1999;98:181–191. doi: 10.1016/s0092-8674(00)81013-2. [DOI] [PubMed] [Google Scholar]
- Dumon S., Santos S.C., Debierre-Grockiego F., Gouilleux-Gruart V., Cocault L., Boucheron C., Mollat P., Gisselbrecht S., Gouilleux F. IL-3 dependent regulation of Bcl-xL gene expression by Stat5 in a bone marrow derived cell line. Oncogene. 1999;18:4191–4199. doi: 10.1038/sj.onc.1202796. [DOI] [PubMed] [Google Scholar]
- Fujio Y., Kunisada K., Hirota H., Yamahuchi-Takihara K., Kishimoto T. Signals through gp130 upregulate bcl-x gene expression via Stat1-binding cis-element in cardiac myocytes. J. Clin. Invest. 1997;99:2898–2905. doi: 10.1172/JCI119484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catlett-Falcone R., Landowski T.H., Oshiro M.M., Turkson J., Levitzki A., Savino R., Ciliberto G., Moscinski L., Fernandez-Luna J.L., Nunez G. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- Nieborowska-Skorska M., Wasik M.A., Slupianek A., Salomoni P., Kitamura T., Calabretta B., Skorski T. Signal transducer and activator of transcription (STAT)5 activation by Bcr-Abl is dependent on intact Src homology (SH)3 and SH2 domains of Bcr-Abl and is required for leukemogenesis. J. Exp. Med. 1999;189:1229–1242. doi: 10.1084/jem.189.8.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlesso N., Frank D.A., Griffin J.D. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr-Abl. J. Exp. Med. 1996;183:811–820. doi: 10.1084/jem.183.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosper F., Stroncek D., McCarthy J.B., Verfaillie C.M. Mobilization and homing of peripheral blood progenitors is related to reversible downregulation of alpha4 beta1 integrin expression and function. J. Clin. Invest. 1998;101:2456–2467. doi: 10.1172/JCI188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker B.J., Tamura S., Buchdunger E., Ohno S., Segal G.M., Fanning S., Zimmermann J., Lydon N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- Amarante-Mendes G.P., McGahon A.J., Nishioka W.K., Afar D.E., Witte O.N., Green D.R. Bcl-2–independent Bcr-Abl–mediated resistance to apoptosisprotection is correlated with upregulation of Bcl-xL . Oncogene. 1998;16:1383–1390. doi: 10.1038/sj.onc.1201664. [DOI] [PubMed] [Google Scholar]
- Gutierrez P., Delgado M.D., Richard C., Moreau-Gachelin F., Leon J. Interferon induces up-regulation of Spi-1/PU.1 in human leukemia K562 cells. Biochem. Biophys. Res. Commun. 1997;240:862–868. doi: 10.1006/bbrc.1997.7760. [DOI] [PubMed] [Google Scholar]
- le Coutre P., Mologni L., Cleris L., Marchesi E., Buchdunger E., Giardini R., Formelli F., Gambacorti-Passerini C. In vivo eradication of human Bcr-Abl–positive leukemia cells with an Abl kinase inhibitor. J. Natl. Cancer Inst. 1999;91:163–168. doi: 10.1093/jnci/91.2.163. [DOI] [PubMed] [Google Scholar]
- Carroll M., Ohno-Jones S., Tamura S., Buchdunger E., Zimmermann J., Lydon N., Gilliland D., Druker B.J. CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins. Blood. 1997;90:4947–4952. [PubMed] [Google Scholar]
- Gazit A., Osherov N., Posner I., Yaish P., Poradosu E., Gilon C., Levitzki A. Tyrphostins. 2. Heterocyclic and alpha-substituted benzylidenemalononitrile tyrphostins as potent inhibitors of EGF receptor and Erb2/neu tyrosine kinases. J. Med. Chem. 1991;34:1896–1907. doi: 10.1021/jm00110a022. [DOI] [PubMed] [Google Scholar]
- Gotoh A., Broxmeyer H.A. The function of Bcr-Abl and related proto-oncogenes. Curr. Opin. Hematol. 1997;4:3–11. doi: 10.1097/00062752-199704010-00002. [DOI] [PubMed] [Google Scholar]
- Ray S., Bullock G., Nunez G., Tang C., Ibrado A.M., Huang Y., Bhalla K. Enforced expression of Bcl-xS induces differentiation and sensitizes CML-blast crisis K562 cells to Ara-C mediated differentiation and apoptosis. Cell Growth Differ. 1996;7:1617–1623. [PubMed] [Google Scholar]
- Mui A.L., Wakao H., O'Farrell A.M., Harada N., Miyajima A. IL-3, GM-CSF, and IL-5 transduce signals through two Stat5 homologs. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:1166–1175. doi: 10.1002/j.1460-2075.1995.tb07100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihle J.N. Cytokine receptor signalling. Nature. 1995;377:591–594. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- Poruchynsky M.S., Wang E., Rudin C.M., Blagosklonny M.V., Fojo T. Bcl-xL is phosphorylated in malignant cells following microtubule disruption. Cancer Res. 1998;58:3331–3338. [PubMed] [Google Scholar]
- Cambier N., Chopra R., Strasser A., Metcalf D., Elefanty A.G. Bcr-Abl activates pathways mediating cytokine independence and protection against apoptosis in murine hematopoietic cells in a dose-dependent manner. Oncogene. 1998;16:335–348. doi: 10.1038/sj.onc.1201490. [DOI] [PubMed] [Google Scholar]
- Beck Z., Bacsi A., Kovacs E., Kiss J., Kiss A., Balogh E., Telek B., Toth F.D., Andirko I., Olah E. Changes in oncogene expression implicated in evolution of chronic granulocytic leukemia from its chronic phase to acceleration. Leuk. Lymphoma. 1998;30:293–306. doi: 10.3109/10428199809057542. [DOI] [PubMed] [Google Scholar]
- Parreira L., Kearney L., Rassool F., Babapulle V.B., Matutes E., Parreira A., Tavares de Castro J., Goldman J.M., Catovsky D. Correlation between chromosomal abnormalities and blast phenotype in the blast crisis of Ph-positive CGL. Cancer Genet. Cytogenet. 1986;22:29–34. doi: 10.1016/0165-4608(86)90134-2. [DOI] [PubMed] [Google Scholar]
- Chai S.K., Nichols G.L., Rothman P. Constitutive activation of Jaks and Stats in Bcr-Abl–expressing cell lines and peripheral blood cells derived from leukemic patients. J. Immunol. 1997;159:4720–4728. [PubMed] [Google Scholar]
- Fujita N., Nagahashi A., Nagashima K., Rokudai S., Tsuruo T. Acceleration of apoptotic cell death after the cleavage of Bcl-xL protein by caspase-3–like proteases. Oncogene. 1998;17:1295–1304. doi: 10.1038/sj.onc.1202065. [DOI] [PubMed] [Google Scholar]