Cytotoxic T Lymphocyte–Associated Antigen 4 Plays an Essential Role in the Function of Cd25+Cd4+ Regulatory Cells That Control Intestinal Inflammation (original) (raw)
Abstract
It is now clear that functionally specialized regulatory T (Treg) cells exist as part of the normal immune repertoire, preventing the development of pathogenic responses to both self- and intestinal antigens. Here, we report that the Treg cells that control intestinal inflammation express the same phenotype (CD25+CD45RBlowCD4+) as those that control autoimmunity. Previous studies have failed to identify how CD25+ Treg cells function in vivo. Our studies reveal that the immune-suppressive function of these cells in vivo is dependent on signaling via the negative regulator of T cell activation cytotoxic T lymphocyte–associated antigen 4 (CTLA-4), as well as secretion of the immune-suppressive cytokine transforming growth factor β. Strikingly, constitutive expression of CTLA-4 among CD4+ cells was restricted primarily to Treg cells, suggesting that CTLA-4 expression by these cells is involved in their immune-suppressive function. These findings raise the possibility that Treg cell function contributes to the immune suppression characteristic of CTLA-4 signaling. Identification of costimulatory molecules involved in the function of Treg cells may facilitate further characterization of these cells and development of new therapeutic strategies for the treatment of inflammatory diseases.
Keywords: inflammatory bowel disease, CD4+ T lymphocyte, T lymphocyte suppressor, interleukin 2 receptor, autoimmunity
Introduction
The intestine is home to a huge number of resident bacteria, estimated to be in the order 1014/g of tissue. These are a significant source of both antigens and proinflammatory molecules present in bacterial cell walls. Despite this potential immune stimulus, it is well recognized that immune responses in the intestine remain in a state of controlled inflammation 1. Recently, we have identified a phenotypically and functionally distinct population of regulatory T (Treg) cells capable of controlling intestinal inflammation, and have suggested that these cells play a pivotal role in intestinal homeostasis 2. Transfer of CD45RBhighCD4+ T cells from normal donors to SCID mice led to the development of an inflammatory bowel disease (IBD)-like syndrome characterized by extensive mononuclear cell infiltrates, epithelial cell hyperplasia, and ulceration 3 4 5. Colitis was the result of the differential expansion of Th1 cells driven by enteric bacteria, as disease could be prevented by strategies that inhibited the development of Th1 responses 6 and did not occur in T cell reconstituted mice raised under germ-free conditions 7 8. Importantly, cotransfer of the reciprocal CD45RBlowCD4+ subset with an inoculum of potentially pathogenic CD45RBhighCD4+ cells inhibited disease. Immune suppression was dependent on TGF-β 9 and production of IL-10 10 by T cells contained within the CD45RBlow population. These data indicate that normal mice contain a population of IL-10– and TGF-β–dependent Treg cells that control inflammatory responses in the intestine.
Subpopulations of peripheral CD4+ T cells have also been shown in several different model systems to be essential for the maintenance of tolerance to tissue-specific self-antigens 11 12 13. Further phenotypic characterization of these Treg cells has shown that they express CD25, as transfer of CD4+ T cells depleted of the CD25+ fraction to nude mice led to the development of autoimmune gastritis 14 15. Furthermore, organ-specific autoimmune disease induced by neonatal thymectomy was prevented by transfer of CD25+CD4+ T cells 16. Functional analysis of CD25+CD4+ T cells in vitro showed that this population failed to proliferate or secrete cytokines in response to polyclonal or antigen-specific stimulation, but rather inhibited the activation of normally responsive T cells 17 18. Precisely how CD25+CD4+ T cells mediate immune suppression in vitro has not been elucidated, but the mechanism has been shown to be independent of IL-10 and TGF-β and to require cell to cell contact between responding and regulatory T cell populations.
Several factors are thought to influence the fate of T cells after encounter with antigen, including the strength of the signal delivered after TCR triggering, the balance of proinflammatory versus antiinflammatory cytokines present at the time of T cell encounter with antigen, and the integrated signals delivered by costimulatory molecules present on the T cell surface after interaction with their ligands on APCs (for reviews, see references 19, 20). Two well-characterized costimulatory molecules are Ig superfamily members CD28 and cytotoxic T lymphocyte–associated antigen 4 (CTLA-4), both of which interact with CD80 and CD86 present on APCs. CD28 is present on naive T cells and has been shown to be important for the development of optimal primary responses 21 22 23. In contrast, detectable levels of CTLA-4 are induced only after T cell activation, and its ligation results in inhibition of T cell activation. Cross-linking of CTLA-4 concomitant with TCR signaling inhibits IL-2 gene expression and cell cycle progression 24 25. The significance of signaling via CTLA-4 in vivo is revealed by the fact that administration of anti–CTLA-4 mAb was able to abrogate the induction of tolerance to specific antigens 26, enhance antitumor responses 27, and exacerbate autoimmune reactivity 28 29 30. Most strikingly, CTLA-4–deficient mice developed a fatal lymphoproliferative disease and multiple organ immune pathology, highlighting the essential role that CTLA-4 plays in the normal homeostasis of the immune system 31 32. Recently, cross-linking of CTLA-4 in the presence of TCR ligation has been shown to lead to TGF-β secretion by purified naive CD4+ T cells or CD4+ T cell clones 33, providing an additional mechanism through which CTLA-4 may induce immune suppression.
In this study, we have further dissected the phenotype and mechanism of action of Treg cells that control intestinal inflammation. Here, we show a primary role for CD25+CD45RBlowCD4+ cells in inhibition of colitis, and present evidence that ligation of CTLA-4 and production of TGF-β are required for their function in vivo.
Materials and Methods
Mice.
Specific pathogen-free BALB/c, C57BL/6, C57BL/6.SJL.CD45 congenic, C57BL/6.RAG2−/−, and C.B-17 SCID mice were maintained in the Biomedical Services Unit at the John Radcliffe Hospital in microisolator cages with filtered air. Mice were used at 8–12 wk of age.
Antibodies.
The following antibodies were used for cell purification: YTS169, anti–mouse CD8; TIB120, anti–mouse MHC class II (American Type Culture Collection); M1/70, anti–mouse Mac-1 (TIB128; American Type Culture Collection); RA3-6B2, anti–mouse B220 34; FITC-conjugated anti–mouse CD45RB (clone 16A; BD PharMingen); biotinylated anti–mouse CD25 (clone 7D4; BD PharMingen); CyChrome-conjugated anti–mouse CD4 (clone RM4-5; BD PharMingen); and PE-conjugated streptavidin (BD PharMingen). Peridinine chlorophyll protein (PerCP)-conjugated anti–mouse CD4 (clone RM4-5; BD PharMingen), FITC-conjugated anti–mouse CD45.2 (clone 104; BD PharMingen), and PE-conjugated anti–mouse CTLA-4 (CD152) (clone UC10-4F10-11; BD PharMingen) were used for analysis of CTLA-4 expression. For in vivo use, anti–mouse CTLA-4 (clone UC10-4F10-11) and anti–mouse TGF-β mAb (clone 1D11.16.8) were purified from hybridoma supernatant by affinity chromatography, and were shown to contain <1.0 endotoxin unit/mg of protein. Purified hamster IgG was purchased from Jackson Immunoresearch Laboratories.
Purification of T Cell Subsets.
CD4+ T cell subsets were isolated from spleens as described previously 3. In brief, single cell suspensions were depleted of CD8+, MHC class II+, Mac-1+, and B220+ cells by negative selection using sheep anti–rat coated Dynabeads (Dynal). The resulting CD4+-enriched cells were stained with CyChrome-conjugated anti-CD4, FITC-conjugated anti-CD45RB, biotinylated anti-CD25 mAb, and streptavidin-PE. Subpopulations of CD4+ cells were generated by three-color sorting on a FACS Vantage™ (Becton Dickinson). All populations were >98.0% pure on reanalysis.
T Cell Reconstitution and Antibody Treatment.
C.B-17 SCID mice were injected intraperitoneally with sorted CD4+ T cell subpopulations in PBS. Mice received 4 × 105 CD45RBhigh CD4+ cells alone or in combination with other subpopulations. Anti–CTLA-4 mAb (200 μg) was injected intraperitoneally in PBS the day after T cell reconstitution and then on alternate days for 6 wk; mice treated with anti–TGF-β mAb received 2 mg weekly for up to 6 wk.
Histological Examination.
Colons were removed from mice 6–8 wk after T cell reconstitution and fixed in buffered 10% formalin. 6-μm paraffin-embedded sections were cut and stained with hematoxylin and eosin. Inflammation was scored in a blinded fashion on a scale of 0–4, representing no change to severe changes, as described 10.
Analysis of CTLA-4 Expression.
Lymphocyte populations were prepared from spleen and mesenteric lymph nodes of normal and T cell–restored mice. Cells were stained with PerCP-conjugated anti-CD4, FITC-conjugated anti-CD45.2, and PE-conjugated anti–CTLA-4. Before staining with anti–CTLA-4 mAb, the cells were fixed with 2.0% paraformaldehyde and permeabilized using 0.5% saponin (Sigma-Aldrich). Samples were analyzed on a FACSort™ using CELLQuest™ software (Becton Dickinson).
Statistical Analysis.
Colitis scores were compared using Mann-Whitney or Student's t test, and differences were considered statistically significant with P < 0.05.
Results and Discussion
Inhibition of Colitis Is Mediated by CD25_+CD4+_ T Cells.
To further analyze the relationship between Treg cells that control responses to self-antigens and those that regulate responses driven by bacteria, CD45RBlowCD4+ T cells were sorted into CD25+ and CD25− fractions and tested for their ability to inhibit colitis induced by transfer of CD45RBhigh cells. The results clearly demonstrate that control of intestinal inflammation resided predominantly within the CD25+ fraction, as these cells significantly inhibited development of colitis and wasting disease even when transferred at a ratio of 1:8 (CD25+CD45RBlowCD4+/CD45RBhighCD4+ T cells; Table ). Indeed, colons from mice restored with a mixture of CD45RBhighCD4+ and CD25+CD45RBlowCD4+ cells exhibited no detectable pathological changes and were indistinguishable from colons from mice restored with CD45RBhighCD4+ cells and unseparated CD45RBlowCD4+ cells (Fig. 1, a–d). In contrast, CD25−CD45RBlow cells transferred incomplete protection at the highest dose and mediated no significant regulatory function at the lower dose. Despite the fact that CD25−CD45RBlow cells failed to efficiently prevent colitis, they were also not capable of transferring the disease (Table ), indicating that pathogenic effector cells were not present in this population and did not account for its lack of Treg cell function.
Table 1.
CD25+CD45RBlowCD4+ but Not CD25−CD45RBlowCD4+ Cells Inhibit Colitis
| Cells injected | |||
|---|---|---|---|
| Phenotype | No. | Percentage of colitis (n) | Percentage of weight change |
| CD45RBhighCD4+ alone | 89 (9) | 78.6 ± 4.9 | |
| CD45RBhighCD4+ | |||
| Plus CD45RBlowCD4+ | 2 × 105 | 0 (5) | 103.3 ± 1.6 |
| Plus CD25+CD45RBlowCD4+ | 2 × 105 | 0 (4) | 105.3 ± 0.8 |
| 1 × 105 | 0 (4) | 107.0 ± 2.5 | |
| 5 × 104 | 0 (5) | 110.3 ± 2.5 | |
| Plus CD25−CD45RBlowCD4+ | 2 × 105 | 38 (8) | 98.5 ± 5.0 |
| 5 × 105 | 60 (10) | 8 6.7 ± 3.5 | |
| CD25+CD45RBlowCD4+ alone | 2 × 105 | 0 (3) | 114.5 ± 6.9 |
| CD25−CD45RBlowCD4+ alone | 2 × 105 | 0 (3) | 107.0 ± 0.6 |
Figure 1.
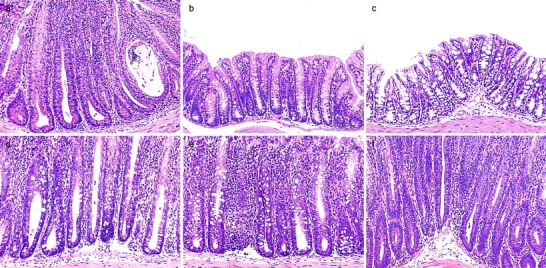
CD4+ regulatory T cells that control intestinal inflammation are CD25+CD45RBlow. (a) Representative photomicrographs show severe colitis in mice that received CD45RBhighCD4+ cells alone. (b) Inhibition of colitis in mice given CD45RBhigh plus 2 × 105 unfractionated CD45RBlowCD4+ cells. (c) Regulatory T cell function is enriched within the CD25+CD45RBlow subset, as 105 CD25+CD45RBlow cells inhibited colitis when transferred with CD45RBhighCD4+ cells, whereas (d) mice restored with a mixture of 105 CD25−CD45RBlow and CD45RBhighCD4+ cells developed severe colitis. Severe colitis in mice restored with (e) CD45RBhigh and 2 × 105 CD45RBlowCD4+ cells or (f) CD45RBhigh and 105 CD25+CD45RBlowCD4+ cells and treated with anti-CTLA-4 mAb. Mice receiving CD45RBhigh and CD45RBlowCD4+ cells or CD45RBhigh and CD25+CD45RBlowCD4+ cells and treated with control hamster IgG were similar to those shown in b. Original magnification: ×80.
Signals through CTLA-4 Are Required for the Function of Treg Cells In Vivo.
These findings indicate that Treg cells that control inflammatory responses in the intestine are enriched within the same phenotypic subset (CD25+CD4+) as those that inhibit T cell activation in vitro and prevent organ-specific autoimmune disease 14 15 16. The fact that the function of the former, at least in vitro, requires a cognate interaction between responding and regulatory populations prompted us to search for cell surface molecules that may be involved in the function of Treg cells in vivo. A potential candidate was CTLA-4, which has been clearly identified as a negative regulator of T cell activation 24 25. To analyze the role of CTLA-4 in the regulation of intestinal inflammation, SCID mice transferred with a mixture of CD45RBhigh and CD45RBlow cells were treated with an anti–CTLA-4 mAb or control hamster IgG. As expected, mice restored with a mixture of CD45RBhigh and CD45RBlow cells and treated with control IgG were significantly protected from both wasting disease and colitis (Fig. 1 b and Fig. 2, a and c). In contrast, mice in the anti–CTLA-4–treated group developed wasting disease with identical kinetics to mice restored with CD45RBhigh cells alone (Fig. 2, a and c). Furthermore, intestinal inflammation occurred with a similar incidence and severity in these mice and had identical characteristics to those observed in mice restored with CD45RBhigh cells alone, including epithelial cell hyperplasia, mucin cell depletion, and pronounced mononuclear cell infiltrates (Fig. 1 e and Fig. 2, a and c). Mice lacking CTLA-4 have been shown to preferentially activate Th2 responses 35; however, there was no evidence that anti–CTLA-4 therapy induced a Th2 response in the colon, as IFN-γ and TNF-α but not IL-4 or IL-10 mRNA were elevated in the colon of anti–CTLA-4–treated mice with colitis (data not shown). Importantly, anti–CTLA-4 treatment also abrogated inhibition of colitis by CD25+CD45RBlow cells, providing direct evidence that the immune-suppressive function of CD25+ Treg cells is dependent on CTLA-4 (Fig. 1 f and Fig. 2 c).
Figure 2.



Anti–CTLA-4 treatment abrogates the function of regulatory T cells. (a) SCID mice were reconstituted with CD45RBhighCD4+ cells alone (□) or in combination with 2 × 105 CD45RBlowCD4+ cells and treated with anti–CTLA-4 mAb (•) or purified hamster IgG (○). Asterisk indicates one of five mice killed on D32. Data represent the mean ± SEM for five mice per group. For CD45RBhigh versus CD45RBhigh plus CD45RBlow cells plus control IgG, P < 0.05. For CD45RBhigh versus CD45RBhigh plus CD45RBlow cells plus anti–CTLA-4, results were not significant (Student's t test). (b) SCID mice were reconstituted with CD45RBhigh cells alone and received anti–CTLA-4 mAb (▪) or control hamster IgG (□). Data represent the mean ± SEM for six mice per group. (c) SCID mice received CD45RBhighCD4+ cells (□), 2 × 105 CD45RBlow cells (⋄), both CD45RBhighCD4+ cells and 2 × 105 CD45RBlow cells (○), or CD45RBhighCD4+ cells in combination with 105 CD25+CD45RBlow CD4+ cells (▵). Mice also received either anti–CTLA-4 (filled symbols) or control hamster IgG (open symbols). Data are pooled from three independent experiments. Significant protection was mediated by CD45RBlow cells (P < 0.01) and CD25+ cells (P < 0.01). Adminstration of anti–CTLA-4 mAb abrogated protection mediated by CD45RBlow cells (P < 0.01) and CD25+ cells (P < 0.05).
Anti–CTLA-4 treatment may act to enhance the pathogenicity of cells within the CD45RBhigh population. However, the kinetics of wasting disease (Fig. 2 b) as well as the incidence and severity of colitis induced by transfer of CD45RBhighCD4+ cells was indistinguishable in anti–CTLA-4–treated or untreated groups (Fig. 2 c). Similarly, anti–CTLA-4 treatment did not lead to the development of a pathogenic T cell population after transfer of CD45RBlowCD4+ T cells, as the majority of mice in this group failed to develop wasting disease or colitis and were not significantly different from untreated mice (Fig. 2 c). The finding that anti–CTLA-4, which inhibits the function of Treg cells, failed to reveal pathogenic cells among the CD45RBlow population further supports the idea that pathogenic effector cells are not present (at least not in a physiologically relevant number) in the antigen-experienced pool. Taken together, these data suggest that anti–CTLA-4 acts to inhibit the function of Treg cells as opposed to enhancing the pathogenicity of CD45RBhigh cells or revealing pathogenic T cells among the CD45RBlow population.
CTLA-4 Is Expressed Predominantly on CD25+CD4+ Cells.
The consensus of opinion on the function of CTLA-4 is that ligation of CTLA-4 uncouples TCR ligation from the downstream signaling events that lead to full T cell activation (for a review, see reference 36). However, more recently, ligation of CTLA-4 has also been shown to induce TGF-β secretion by both naive and effector CD4+ T cells 33, suggesting that CTLA-4 may mediate positive signaling for secretion of immune-suppressive cytokines. Precisely how CTLA-4 is involved in the function of Treg cells that inhibit intestinal inflammation is not known. It may be induced on the progeny of CD45RBhighCD4+ T cells in the presence of Treg cells and serve to transmit a negative signal to these potentially pathogenic cells. Alternatively, it may be present on Treg cells themselves, providing a costimulatory signal required for these cells to mediate their effector function. To address this issue, we have analyzed the constitutive expression of CTLA-4 among splenic CD4+ T cell subsets. Previous studies have shown that although CTLA-4 functions at the cell surface, it is located primarily in endosomal vesicles, from where it has been shown to cycle continuously to and from the cell surface 37 38. To measure both membrane and cytoplasmic CTLA-4 expression, antibody staining was performed on permeabilized cells. Although it has been reported that CTLA-4 is not expressed by CD4+ T cells examined directly ex vivo 39 40, our studies revealed significant CTLA-4 expression among CD25+CD45RBlowCD4+ cells (46.4% ± 1.5; representative staining shown in Fig. 3 b) and lower levels on the CD25− subset (12.5% ± 0.2; Fig. 3 c). Low to undetectable levels were present among CD45RBhighCD4+ cells (Fig. 3 a). These results suggest that CTLA-4 is expressed constitutively among CD4+ T cells analyzed directly ex vivo and that this expression is restricted primarily to Treg cells. Recently, expression of CTLA-4 among CD25+CD4+ cells has been noted 41. However, the significance of this was not examined.
Figure 3.
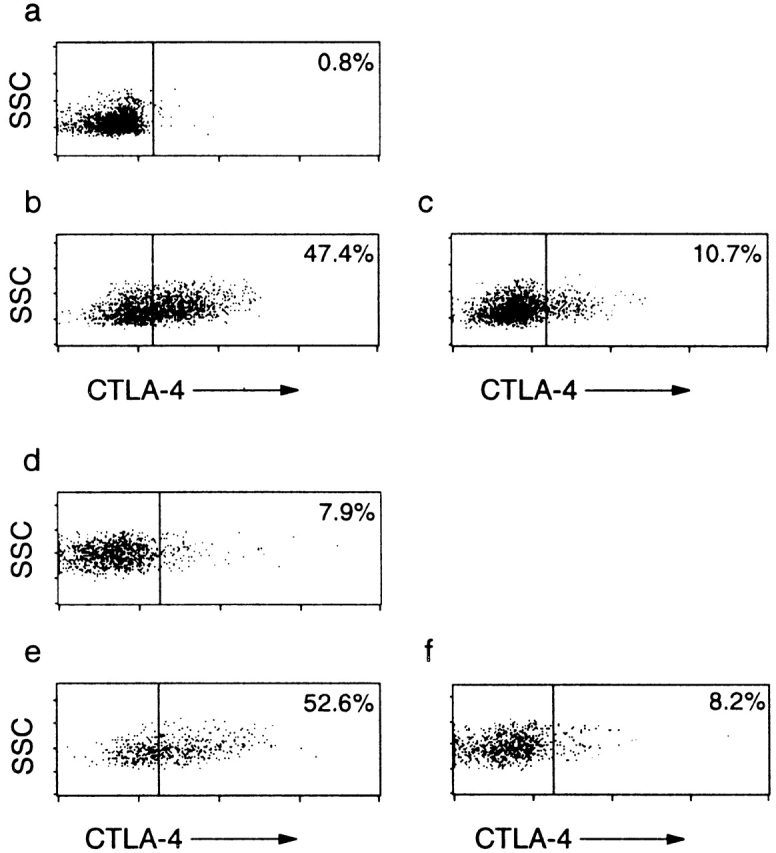
CTLA-4 is expressed constitutively on CD25+CD45RBlow CD4+ cells. (a–c) CTLA-4 staining on permeabilized (a) CD45RBhighCD4+, (b) CD25+CD45RBlowCD4+, and (c) CD25−CD45RBlowCD4+ splenocytes. SSC, side scatter. (d–f) CTLA-4 expression on the progeny of (e) CD25+CD45RBlowCD4+ and (d and f) CD45RBhighCD4+ cells after transfer into immunodeficient mice. C57BL/6 recombination activating gene (RAG)2−/− mice received CD45RBhighCD4+ cells from C57BL/6 mice (CD45.2) (d) alone or (e and f) in combination with 105 CD25+CD45RBlowCD4+ cells from C57BL/6.SJL.CD45 congenic mice (CD45.1). 4 wk after transfer, cells from the mesenteric lymph nodes were stained for CTLA-4 and congenic marker expression. Results are representative of four mice (CD45RBhigh alone) and seven mice (CD45RBhigh plus CD25+) from two independent experiments.
To investigate whether inhibition of colitis by CD25+ CD45RBlow cells led to induction of expression of CTLA-4 among the progeny of CD45RBhigh cells that expand in the immunodeficient recipients, pathogenic and regulatory T cell populations were isolated from donors congenic for CD45 allotypes and transferred to immune-deficient recipients. 4 wk after T cell reconstitution, CTLA-4 expression on the progeny of CD45RBhighCD4+ or CD25+CD45RBlow CD4+ cells present in the mesenteric lymph nodes was analyzed. In mice restored with CD45RBhigh cells alone, which would normally develop colitis, CTLA-4 was expressed on a minority of cells (6.4 ± 2.4%; representative staining shown in Fig. 3 d). The picture was similar in mice given a mixture of pathogenic and Treg cells, as under these circumstances only 7.6 ± 1.8% (representative staining shown in Fig. 3 f) of the progeny of CD45RBhighCD4+ T cells expressed CTLA-4. The fact that the progeny of CD45RBhigh cells expressed similar levels of CTLA-4 whether they were able to induce colitis (transfer of CD45RBhigh cells alone) or when they were suppressed (transfer of CD45RBhigh cells plus CD25+ cells) argues against the hypothesis that it is induced CTLA-4 expression on potentially pathogenic cells in the presence of Treg cells that accounts for the requirement for CTLA-4 in the mechanism of immune suppression. Consistent with this, CTLA-4 expression remained high (43.7% ± 7.8, Fig. 3 e) on the progeny of CD25+ cells as they exerted immune suppression in vivo, providing support for the idea that it is CTLA-4 signaling on Treg cells themselves that is crucial for their function. As the highest levels of CTLA-4 expression were restricted to Treg cells, it was possible that anti–CTLA-4 treatment selectively depleted these cells. However, using the congenic transfer system, it was possible to monitor the progeny of Treg cells in mice treated with anti–CTLA-4. Similar numbers of CD25+CD4+ cells were present in anti–CTLA-4–treated mice, indicating that the antibody did not lead to the wholesale depletion of Treg cells (data not shown).
The Function of CD25+ Treg Cells In Vivo Is Dependent on TGF-β Production.
Extensive analysis of the immune-suppressive function of CD25+ Treg cells in vitro has shown no demonstrable role for the immune-suppressive cytokine TGF-β 17 18. This was somewhat at odds with our previous observations that the ability of CD45RBlow cells to inhibit colitis was absolutely dependent on TGF-β 9. It is possible that this difference may reflect a comparison of the function of CD25+ cells with unseparated CD45RBlow cells. Given that we now show that Treg cells that most efficiently inhibit colitis are also CD25+, we directly tested whether TGF-β was required for the function of these cells in vivo. As with unseparated CD45RBlow cells, administration of anti–TGF-β to mice usually protected from colitis by transfer of a mixture of CD45RBhigh and CD25+CD45RBlow cells led to abrogation of suppression and induction of severe colitis in the recipients (Fig. 4). These data indicate that in contrast to immune suppression in vitro, immune suppression in vivo was absolutely dependent on TGF-β.
Figure 4.

Regulation of colitis by CD25+CD4+ cells is TGF-β dependent. SCID mice received CD45RBhighCD4+ cells alone (□) or in combination with 105 CD25+ cells (▵). Mice also received either anti–TGF-β (filled symbols) or PBS (open symbols). Significant protection mediated by CD25+ cells (P < 0.01) abrogated by treatment with anti–TGF-β mAb (P < 0.02).
Data reported herein demonstrate that bacteria-driven inflammatory responses in the intestine are regulated by the same phenotypic subset of regulatory T cells (CD25+) as control autoimmune responses to tissue-specific self-antigens 14 15 16 and mediate inhibition of T cell activation in vitro 17 18. Previous studies have failed to reveal the mechanism of action of CD25+ Treg cells, which at least in vitro are thought to act via a cognate interaction between responding and regulatory T cells. Our data identify both CTLA-4 and TGF-β as essential components of the immune-suppressive functions of CD25+CD4+ Treg cells that control intestinal inflammation. Consistent with these studies, Takahashi et al. 42 have reported that signaling via CTLA-4 is also required for immune suppression in vitro and that treatment of normal mice with anti–CTLA-4 and anti–CD25 mAbs induces autoimmune disease. The finding that CTLA-4 plays an essential role in the function of Treg cells that control immune responses towards intestinal antigens as well as tissue specific self-antigens suggests that similar functional subsets of Treg cells mediate these diverse roles, emphasizing the central role that Treg cells and CTLA-4 signaling play in the control of the immune response.
There is now overwhelming evidence that CTLA-4 can act as a negative regulator of T cell activation. Blockade of CTLA-4 by injection of anti–CTLA-4 mAbs has been shown to lead to increased T cell–mediated immunity in several model systems, including antigen-specific responses 26, tumor immunity 27, parasitic infection 43, and autoimmune disease 28 29 30. The consensus of opinion is that CTLA-4 acts in a cell-autonomous fashion, and that its expression on potentially reactive T cells results in their inhibition. Our observation that CTLA-4 is required for the function of Treg cells in vivo describes a novel property of CTLA-4, which raises the possibility that the immune stimulatory consequences of CTLA-4 blockade may be in part due to the inhibition of the function of Treg cells. The findings that: (a) anti–CTLA-4 treatment abrogates the function of Treg cells in vivo but does not enhance the pathogenicity of CD45RBhigh cells when transferred alone; (b) CTLA-4 is constitutively expressed by a high proportion of Treg cells; and (c) potentially pathogenic T cells whose function has been suppressed in vivo express similar levels of CTLA-4 as uninhibited pathogenic T cells capable of inducing colitis support the hypothesis that it is CTLA-4 expression on Treg cells themselves that is important for their function. However, these data do not rule out the possibility that in mixtures of pathogenic and Treg cells, expression of CTLA-4 by the progeny of pathogenic T cells plays a role in the mechanism of immune suppression. The definitive answer requires transfer of T cell subsets from CTLA-4−/− mice. However, this experimental approach is confounded by the fact that these mice die at 3–4 wk with fatal immune pathology and have highly distorted T cell subsets 31 32. Recently, immune pathology induced in recipients after transfer of CTLA-4−/− bone marrow was shown to be inhibited by transfer of normal bone marrow, indicating that CTLA-4−/− T cells can be inhibited by normal T cells 44. These results, taken together with our own, raise the possibility that costimulation of immune-suppressive Treg cells as opposed to negative signaling of reactive T cells accounts for some of the inhibitory effects of CTLA-4 signaling.
Both cell contact–dependent signals 17 18 45 and secretion of immune-suppressive cytokines 9 10 46 have been shown to be involved in the function of Treg cells. The finding that CD25+ Treg cells that control intestinal inflammation are dependent on both TGF-β and CTLA-4 suggests that a cell surface molecule that negatively regulates T cell activation and secretion of immune-suppressive cytokines may be linked and involved in the function of the same subset of Treg cells. Cross-linking of CTLA-4 in the presence of TCR-mediated signals has been shown to induce TGF-β secretion, providing one possible mechanism for the linkage 33. The explanation of why TGF-β is involved in the in vivo function of Treg cells but not in their in vitro function is not clear. Obviously, immune suppression in vitro represents a very simplified read out of the in vivo phenomenon, and it may be that in the latter, TGF-β plays an important role in limiting the expansion or homing of potentially pathogenic cells.
The results of this study identify signaling via CTLA-4 as essential for the function of Treg cells, providing a new mechanism by which ligation of CTLA-4 may induce immune suppression. Induction of Treg cells is one of the host's natural mechanisms for controlling immune responses. The finding that CTLA-4 plays a nonredundant role in the functioning of these cells will open up new avenues of investigation for the development of therapeutic strategies that seek to manipulate Treg cells.
Acknowledgments
The authors wish to thank Professor D. Mason and Professor K.J. Wood for their critical review of this manuscript. We are indebted to N. Rust for excellent technical assistance with cell sorting, S. Biddolph for processing of histological samples, and C. Hetherington and staff for care of experimental animals. We gratefully acknowledge Dr. J. Bluestone for the UC10-4F10-11 hybridoma.
S. Read and F. Powrie are funded by the Wellcome Trust. V. Malmström is funded by the Wenner Green Foundation.
References
- Tlaskalova H., Kamarytova V., Mandel L., Prokesova L., Kruml J., Lanc A., Miler I. The immune response of germ-free piglets after peroral monocontamination with living Escherichia coli strain 086. I. The fate of antigen, dynamics and site of antibody formation, nature of antibodies and formation of heterohaemagglutinins. Folia Biol. (Praha). 1970;16:177–187. [PubMed] [Google Scholar]
- Powrie F. T cells in inflammatory bowel diseaseprotective and pathogenic roles. Immunity. 1995;3:171–174. doi: 10.1016/1074-7613(95)90086-1. [DOI] [PubMed] [Google Scholar]
- Powrie F., Leach M.W., Mauze S., Caddle L.B., Coffman R.L. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 1993;5:1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- Morrissey P.J., Charrier K., Braddy S., Liggitt D., Watson J.D. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J. Exp. Med. 1993;178:237–244. doi: 10.1084/jem.178.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach M.W., Bean A.G., Mauze S., Coffman R.L., Powrie F. Inflammatory bowel disease in C.B-17 scid mice reconstituted with the CD45RBhigh subset of CD4+ T cells. Am. J. Pathol. 1996;148:1503–1515. [PMC free article] [PubMed] [Google Scholar]
- Powrie F., Leach M.W., Mauze S., Menon S., Caddle L.B., Coffman R.L. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–562. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- Powrie F., Mauze S., Coffman R.L. CD4+ T-cells in the regulation of inflammatory responses in the intestine. Res. Immunol. 1997;148:576–581. doi: 10.1016/s0923-2494(98)80152-1. [DOI] [PubMed] [Google Scholar]
- Aranda R., Sydora B.C., McAllister P.L., Binder S.W., Yang H.Y., Targan S.R., Kronenberg M. Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J. Immunol. 1997;158:3464–3473. [PubMed] [Google Scholar]
- Powrie F., Carlino J., Leach M.W., Mauze S., Coffman R.L. A critical role for transforming growth factor β but not interleukin 4 in the suppression of T helper type 1–mediated colitis by CD45RBlow CD4+ T cells. J. Exp. Med. 1996;183:2669–2674. doi: 10.1084/jem.183.6.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asseman C., Mauze S., Leach M.W., Coffman R.L., Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowell D., Mason D. Evidence that the T cell repertoire of normal rats contains cells with the potential to cause diabetes. Characterization of the CD4+ T cell subset that inhibits this autoimmune potential. J. Exp. Med. 1993;177:627–636. doi: 10.1084/jem.177.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saoudi A., Seddon B., Heath V., Fowell D., Mason D. The physiological role of regulatory T cells in the prevention of autoimmunitythe function of the thymus in the generation of the regulatory T cell subset. Immunol. Rev. 1996;149:195–216. doi: 10.1111/j.1600-065x.1996.tb00905.x. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S., Fukuma K., Kuribayashi K., Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J. Exp. Med. 1985;161:72–87. doi: 10.1084/jem.161.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S., Sakaguchi N., Asano M., Itoh M., Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- Suri-Payer E., Amar A.Z., Thornton A.M., Shevach E.M. CD4+CD25+ T cells inhibit both the induction and effector function of autoreactive T cells and represent a unique lineage of immunoregulatory cells. J. Immunol. 1998;160:1212–1218. [PubMed] [Google Scholar]
- Asano M., Toda M., Sakaguchi N., Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 1996;184:387–396. doi: 10.1084/jem.184.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton A.M., Shevach E.M. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Kuniyasu Y., Toda M., Sakaguchi N., Itoh M., Iwata M., Shimizu J., Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cellsinduction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 1998;10:1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- Van Parijs L., Abbas A.K. Homeostasis and self-tolerance in the immune systemturning lymphocytes off. Science. 1998;280:243–248. doi: 10.1126/science.280.5361.243. [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A., Sallusto F. From synapses to immunological memorythe role of sustained T cell stimulation. Curr. Opin. Immunol. 2000;12:92–98. doi: 10.1016/s0952-7915(99)00056-4. [DOI] [PubMed] [Google Scholar]
- Shahinian A., Pfeffer K., Lee K.P., Kundig T.M., Kishihara K., Wakeham A., Kawai K., Ohashi P.S., Thompson C.B., Mak T.W. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- Allison J.P. CD28-B7 interactions in T-cell activation. Curr. Opin. Immunol. 1994;6:414–419. doi: 10.1016/0952-7915(94)90120-1. [DOI] [PubMed] [Google Scholar]
- June C.H., Bluestone J.A., Nadler L.M., Thompson C.B. The B7 and CD28 receptor families. Immunol. Today. 1994;15:321–331. doi: 10.1016/0167-5699(94)90080-9. [DOI] [PubMed] [Google Scholar]
- Walunas T.L., Bakker C.Y., Bluestone J.A. CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 1996;183:2541–2550. doi: 10.1084/jem.183.6.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel M.F., Allison J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez V.L., Van Parijs L., Biuckians A., Zheng X.X., Strom T.B., Abbas A.K. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. 1997;6:411–417. doi: 10.1016/s1074-7613(00)80284-8. [DOI] [PubMed] [Google Scholar]
- Leach D.R., Krummel M.F., Allison J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- Karandikar N.J., Vanderlugt C.L., Walunas T.L., Miller S.D., Bluestone J.A. CTLA-4a negative regulator of autoimmune disease. J. Exp. Med. 1996;184:783–788. doi: 10.1084/jem.184.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin P.J., Maldonado J.H., Davis T.A., June C.H., Racke M.K. CTLA-4 blockade enhances clinical disease and cytokine production during experimental allergic encephalomyelitis. J. Immunol. 1996;157:1333–1336. [PubMed] [Google Scholar]
- Luhder F., Hoglund P., Allison J.P., Benoist C., Mathis D. Cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) regulates the unfolding of autoimmune diabetes. J. Exp. Med. 1998;187:427–432. doi: 10.1084/jem.187.3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse P., Penninger J.M., Timms E., Wakeham A., Shahinian A., Lee K.P., Thompson C.B., Griesser H., Mak T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- Tivol E.A., Borriello F., Schweitzer A.N., Lynch W.P., Bluestone J.A., Sharpe A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Chen W., Jin W., Wahl S.M. Engagement of cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) induces transforming growth factor β (TGF-β) production by murine CD4+ T cells. J. Exp. Med. 1998;188:1849–1857. doi: 10.1084/jem.188.10.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman R.L. Surface antigen expression and immunoglobulin gene rearrangement during mouse pre-B cell development. Immunol. Rev. 1982;69:5–23. doi: 10.1111/j.1600-065x.1983.tb00446.x. [DOI] [PubMed] [Google Scholar]
- Oosterwegel M.A., Mandelbrot D.A., Boyd S.D., Lorsbach R.B., Jarrett D.Y., Abbas A.K., Sharpe A.H. The role of CTLA-4 in regulating Th2 differentiation. J. Immunol. 1999;163:2634–2639. [PubMed] [Google Scholar]
- Chambers C.A., Allison J.P. Costimulatory regulation of T cell function. Curr. Opin. Cell Biol. 1999;11:203–210. doi: 10.1016/s0955-0674(99)80027-1. [DOI] [PubMed] [Google Scholar]
- Linsley P.S., Bradshaw J., Greene J., Peach R., Bennett K.L., Mittler R.S. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity. 1996;4:535–543. doi: 10.1016/s1074-7613(00)80480-x. [DOI] [PubMed] [Google Scholar]
- Alegre M.L., Noel P.J., Eisfelder B.J., Chuang E., Clark M.R., Reiner S.L., Thompson C.B. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J. Immunol. 1996;157:4762–4770. [PubMed] [Google Scholar]
- Krummel M.F., Allison J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walunas T.L., Lenschow D.J., Bakker C.Y., Linsley P.S., Freeman G.J., Green J.M., Thompson C.B., Bluestone J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Metzler B., Burkhart C., Wraith D.C. Phenotypic analysis of CTLA-4 and CD28 expression during transient peptide-induced T cell activation in vivo. Int. Immunol. 1999;11:667–675. doi: 10.1093/intimm/11.5.667. [DOI] [PubMed] [Google Scholar]
- Takahashi T., Tagami T., Yamazaki S., Uede T., Shimizu J., Sakaguchi N., Mak T.W., Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte–associated antigen 4. J. Exp. Med. 2000;192:303–309. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy K., Camberis M., Gros G.L. Protective immunity to nematode infection is induced by CTLA-4 blockade. J. Exp. Med. 1997;186:183–187. doi: 10.1084/jem.186.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann M.F., Kohler G., Ecabert B., Mak T.W., Kopf M. Cutting edgelymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J. Immunol. 1999;163:1128–1131. [PubMed] [Google Scholar]
- Read S., Mauze S., Asseman C., Bean A., Coffman R., Powrie F. CD38+ CD45RBlow CD4+ T cellsa population of T cells with immune regulatory activities in vitro. Eur. J. Immunol. 1998;28:3435–3447. doi: 10.1002/(SICI)1521-4141(199811)28:11<3435::AID-IMMU3435>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Seddon B., Mason D. Regulatory T cells in the control of autoimmunitythe essential role of transforming growth factor β and interleukin 4 in the prevention of autoimmune thyroiditis in rats by peripheral CD4+CD45RC− cells and CD4+CD8− thymocytes. J. Exp. Med. 1999;189:279–288. doi: 10.1084/jem.189.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]