Recombination and the nature of bacterial speciation (original) (raw)
. Author manuscript; available in PMC: 2008 Jan 31.
Published in final edited form as: Science. 2007 Jan 26;315(5811):476–480. doi: 10.1126/science.1127573
Abstract
Genetic surveys are uncovering the diversity of bacteria, and are causing the species concepts used to categorize these to be questioned. One difficulty in defining bacterial species arises from the high rates of recombination that results in the transfer of DNA between relatively distantly related bacteria. Barriers to this process, which could be used to define species naturally, are not apparent. Here, we have reviewed conceptual models of bacterial speciation and simulate speciation in silico. Our findings suggest that the rate of recombination and its relation to genetic divergence, have a strong influence on outcomes: we propose that a distinction be made between clonal divergence and sexual speciation. Hence, to make sense of bacterial diversity we need data not only from genetic surveys, but also from experimental determination of selection pressures and recombination rates, and from theoretical models.
Introduction
Bacteria are promiscuous. They often live in environments with an abundant diversity of donor DNA, and studies of the genomes of members of the same, or similar species, indicate the dynamic nature of gene acquisition, loss and transfer (1). It is probably possible, via a series of intermediates and vectors, to transfer genes between any two bacteria. Besides the illegitimate recombinational process that leads to gene acquisition from distantly related sources there is convincing evidence that homologous recombination may frequently replace small regions of the genome of a bacterium with those from other members of the same species or from closely related species (2). The rate of homologous recombination varies greatly: in some species it appears to be rare and leads to the evolution of distinct clonal lineages, whereas in others these localised recombinational imports arise much more frequently than mutations (3). In recent years, extensive homologous recombination has been shown to be so widespread that it may be regarded as the norm rather than the exception.
Nonetheless, surveys of genetic diversity in the bacterial kingdom are revealing that, far from a continuum mediated by promiscuous gene exchange, bacteria seem to form clusters of genetically related strains (species), at least for those genera studied so far (4-6). There is thus uncertainty regarding the nature of bacterial speciation and the influence that homologous recombination exerts upon it (7).
One proposition is that speciation (by which we mean the generation of permanently distinct clusters of closely related bacteria) could arise, not because of fundamental ecological constraints or geographic separation, but rather as a consequence of recombination failing more frequently between DNA sequences that are different than between those that are similar (8-10). In experimental studies of recombination in bacteria from widely differing genera, a consistent pattern of decline in the recombination rate as a function of genetic distance has been observed (Fig. 1A) (11). This effect has been shown to be associated with the various mechanisms that detect the sequence similarities between donor and recipient DNA, principally MutS mediated mismatch repair and RecA mediated recombination (12-14). RecA is involved in initiating recombination between donor and recipient DNA and is thus essential for recombination, while MutS inhibits recombination between mismatched sequences. One mechanism that has received particular attention is the requirement of RecA for minimally efficient processing segments (MEPS), which are short regions of sequence identity located at either end of the donor DNA strand, hypothesized to be required for recombination to occur (15). This mechanism can generate the general relationship seen in fig 1A, and provides a corroborative estimate for the length of MEPS as being between twenty and thirty base pairs (16). Whatever mechanism underlies the decline in recombination with increasing sequence divergence, this relationship results in constraints on recombination operational at the genomic level, potentially allowing species distinctness to emerge as a dynamic corollary to diversification and adaptation (8, 9).
Figure 1.

A, Recombination rate for a range of related donors, as a function of the proportion of sequence which is different (sequence divergence), for a variety of bacterial recipients: ●, Bacillus subtilis, □, Bacillus mojavensis, ◆, Streptococcus pneumoniae and Δ, Escherichia coli. The best fit log-linear curve is shown, with intercept 0.8% and slope 19.8. Data are from (12-14). Slopes for individual named species range from 17.9 for S. pneumoniae to 25.7 for E. coli. B, genome of S. pneumoniae (from (35)) and location of the MLST genes. C, schematic representation of the simulated genomes, in a stochastic neutral model: MLST genes are highlighted.
While this picture of speciation driven by recombinational (i.e., sexual) incompatibility is appealing, especially for the parallels it offers with the biological species concept of Mayr (17), the elucidation of the quantitative detail of recombinational incompatibility is only one aspect of the story (or stories) of bacterial speciation. What drives new strains to cross these ‘soft’ genetic barriers and form new species? How distinct must clusters be for this soft barrier to be effective enough to maintain separation, and for the evolutionary fate of each cluster to be distinct? Is there a consistent mechanism of speciation that applies to all bacteria, irrespective of the rates and mechanisms of recombination, which are known empirically to be extremely variable?
Modeling bacterial diversity
Genetic surveys of bacterial populations usually provide a static picture of the patterns of genotypic clustering, consequently exploring the dynamics of populations requires theoretical models using computer simulations and analytical approximations. Clustering in natural populations can then be compared with those from simulated populations if the genotypes of strains are defined in the same way. Isolates within bacterial populations are commonly characterized by the alleles at seven house-keeping loci (multilocus sequence typing; MLST), where each allele corresponds to a different sequence (18).We have developed a model in which strains are defined in the same way and in which alleles change at defined rates by mutation or recombination. We also showed that genetic diversity in several bacterial pathogens could be explained by this simple model of neutral drift (19).
The use of neutral models of mutation and drift is not a denial of selection, but a recognition that much observed population genetic structure can be explained in simple terms. It makes sense, as a null model, to explore the dynamics of neutral diversification and the conditions under which populations do, or do not, separate into distinct genotypic clusters that mimic the emergence of species. Estimates for the rates of mutation and recombination are available from empirical studies of a variety of bacteria (e.g., (2, 20)), as is the relationship between sequence divergence and recombination rate shown in fig 1A.
We estimated population mutation rates (denoted θ) in the range 1 to 10, while recombination rates (denoted ρ) are more variable, ranging from 0.1 to 100 (19, 20). These values are expressed per gene segment per generation, and are related to the underlying biological mutation and recombination rates (denoted m and r respectively) via a constant known as the effective population size Ne, such that θ=2_mLNe_ and ρ=2_rNe_. Our estimates of θ are based on genes approximately _L_≈500 base pair long, and if we take a plausible estimate of the DNA mutation rate (m) at 5×10-10 per base pair per replication (21), this gives a ballpark estimate for the effective population size N e of 107.
In interpreting the effective population size, note that this is not directly related to the census population size, but is rather a measure of how much neutral diversity the environment can carry. It may be considerably smaller than the census population size, as a result of many factors, such as regular bottlenecks, genome-wide selective sweeps or hierarchical structure (22). Consider for example an infectious agent, such as Streptococcus pneumoniae. Three factors at least result in the effective population size being many orders of magnitude smaller than the actual number of bacteria. First, the bacterial population is divided into distinct populations within individual humans, and is transmitted via small inocula, so that the number of infected people may be a better measure of population size than the number of bacteria. Second, transmission is seasonal, with peaks occurring during the winter months, creating bottlenecks during the low season, so that the effective population size may reflect the number of people infected at the trough. Third, the human contact network is hierarchically structured into communities, communities of communities and so on, so that the effective number of people infected is lower than the actual number of people infected (23). Thus, a population of trillions of bacteria can have a low effective population size. Similar considerations may affect effective population sizes in many environments, such as the partitioning of marine bacteria around nutrient rich coastal regions, seasonal regulation caused by the “bloom-bust” cycle of algal nutrient availability, and local clustering of populations around small particles of nutrients (24). In general, most natural populations of bacteria live in structured environments with well-defined patches of growth, where serious limits exist on the dispersion of novel types between patches. Establishing plausible estimates for Ne for a diverse range of bacteria, as well as identifying the factors which affect it, should be a research priority.
To explore speciation, we extended our previous model (19) to simulate simplified genomes (Fig. 1C), using an effective population size _N_e = 105, a population mutation rate θ=2 and defining each strain by the alleles at a larger number of loci (=70), to counter the effect that occasional recombination at a single locus has in distorting relationships between otherwise divergent or similar strains (25). This model ignores several heterogeneities that may arise in populations (e.g., fitness, ecology and recombination rate), but may nonetheless provide a first-draft description of the generation of diversity by drift. Our choice of parameters and model structure is an inevitable compromise between plausibility and computational limitations, achieved principally by reducing the effective population size and using an approximation algorithm for modeling mutation of DNA sequences (25).
The clonal-sexual threshold
The most salient feature of this simple model is a sharp transition in population structure with increasing rates of homologous recombination. When recombination rates are low, the population is effectively clonal in structure. In some sense, each clone has a separate evolutionary fate, since novel alleles that arise are unlikely to spread horizontally through the population. A feature of neutral population structure in the clonal region is strong genotypic clustering (Fig. 2A,B). These clusters are unstable, and the long-term dynamics are characterized by a constant process in which major clusters regularly emerge by chance success, split, drift apart and eventually become extinct (Fig. 2C).
Figure 2.

Simulated genetic structure of a clonal population (A-C) and sexual population (D-F). All populations are evolving under neutral drift and are homogeneously mixing. Genetic maps (A,D,G), which are determined by principal co-ordinate analysis (36), represent the genetic distances between 1,000 randomly chosen isolates from the simulated population after 106 generations have elapsed. Co-ordinates are expressed in units of sequence divergence. An alternative way to represent clustering is the distribution of sequence divergence between pairs of isolates in the population (B, E, H). The thin lines show the distance between five random strains and all the other strains in the sample, while the thick red line shows the distribution of all the pairwise distances (thick red line). Where there is little clustering (E), all pairwise distances are similar and the distribution has a single peak, while where there is strong clustering (B, H), the distribution has multiple peaks corresponding to pairwise comparisons within and between clusters. (C, F, I) show this distribution of pairwise comparisons evolving over 106 generations. To normalise the distribution, pairs of isolates are compared for the number of alleles that are different, between 0 and 70, rather than for the proportion of base pairs, as in (B, E, H). The height of the distribution is represented by color shade, ranging from black (0.0) to red (>0.1), so that peaks in the (B, E, H) correspond to red shaded areas in (C, F, I). C and I show clusters moving apart, visible as red peaks moving up through time. When clusters split, a new peak appears at the bottom, while extinctions are apparent from peaks disappearing. F shows instead more stable population structure with a stable diffuse cluster being maintained throughout the simulation. Parameter values for θ and ρ, the population mutation and recombination rates, are θ=2, ρ=0.01 (A-C), ρ=20*10-18_x_, where x is the sequence divergence (D-F). We also explored under which conditions clustering could occur in the presence of high recombination rates (G-I). Clusters with high within cluster recombination can be generated, mimicking spontaneous speciation (G-I), but require that recombination rate declines as a function of sequence divergence at a very rapid rate uncharacteristic of most bacteria studied to date, such that ρ=20*10-300_x_.
When recombination rates are increased to values between 0.25- and 2-fold the mutation rate (per locus), a threshold is passed where clusters no longer diverge, but are constantly reabsorbed into the parent population by the cohesive force of recombination. Alleles can succeed through horizontal spread even when the parental lineage does not. The degree of clustering is much reduced compared with the clonal situation (Fig 2D,E) and dynamic analysis (Fig 2F) reveals that what clustering there is (Fig 2E) is transient.
It is worth noting that in both situations, the degree of diversity at each locus is the same, and is governed by the balance between extinction and mutation. The sexual population contains more distinct genotypes, based on different combinations of a similar number of alleles. Recombination is sufficiently frequent that the fate of alleles at one locus is not tied to their association with alleles at other loci. In the clonal situation, in contrast, clusters regularly become extinct (Fig 2C), and extinction of clones is the principal regulator of diversity as a whole. Clustering can be defined as over-dispersion of the genetic distances between isolates (Fig 2B,E,H), and a measure of this is the index of association (26). In earlier work, we showed how to calculate this for neutral models (without the dependence on sequence divergence of Fig. 1) (19), and have shown that the threshold between clonal and sexual regimes holds for a wide range of parameters (20). The transition between clonal and sexual population structure is studied in more detail in the accompanying Supplementary Online Material (27).
Diversity-driven speciation in sexual bacteria?
In populations with high rates of recombination, the reduced rate of recombination between two closely-related species, compared to that within each species, provides a mechanism of sexual isolation that can maintain the separation of species, but it is unclear whether the relationship between divergence and recombination rate is sufficient to cause species to arise by drift. In other words, is it conceptually plausible that chance variation would occasionally result in strains arising that are sufficiently different from the founder population that they no longer recombine with the founders frequently enough to maintain genetic proximity, and thus become sufficiently genetically isolated to form a new species? Our simulations suggest that although this type of distance-scaled recombination can lead to the emergence of separate populations, this only occurs under conditions in which the recombination rate declines with divergence more rapidly than is suggested by experimentation (Fig 2G-I). For values of this decline consistent with Fig 1A, we did not observe distinct populations emerging in our simulations for the reason that the amount of variability within simulated populations is too low for the recombination rate to vary appreciably (Fig 2D-F). Thus, while this conceptual model is appealing, it is not supported by the quantitative detail of the interplay between genetic diversification and sexual isolation.
Experimental studies of the relationship between sequence divergence and recombination have focused on interspecific transfer of DNA, i.e., between organisms that are up to ∼20% divergent, and are already presumed to be at least somewhat sexually isolated. For the process of speciation modeled here, we are initially interested in the process of intraspecific transfer, and so the most important question is how very small amounts of sequence divergence, up to 5%, affects recombination. We know that bacteria may vary in their mechanisms of recombination, and hence the pattern shown in Fig 1A may not be universal. In a yeast, for example, a different relation between genetic distance and recombination rates has been observed (28), where the recombination rate declines very rapidly for the first few mismatches (85% reduction for 5 base pairs), by a mechanism linked to the MutS mismatch repair system, and that this mechanism then saturates so that the decline thereafter follows a very similar log-linear relationship to that seen in those bacteria studied to date (Fig 1A). Similarly, there are anomalies, such as the reported 106 reduction in recombination rate (using phage-mediated transduction) between Salmonella enterica serovar Typhimurium and S. enterica serovar Typhi, which are only about 2% divergent (8, 29). Thus, before conclusions can be reached about the feasibility of speciation occurring by distance-scaled recombination, details of the dependence of the recombination rate on sequence divergence must be known. (11).
Using methods based on MLST (2) we can identify strains from natural populations of bacteria separated by single recombination events and calculate the divergence between the ancestral and inserted allele (30). In those species supporting sufficient levels of sequence diversity, such as Neisseria meningitidis and S. pneumoniae, these may frequently be highly divergent: over 5%. This demonstrates that, at least within some species, extensive sequence divergence is no bar to recombination. Mechanisms of reproductive isolation other than sequence divergence certainly exist, such as niche differentiation, differences in DNA exchange by phage-mediated transduction owing to incompatibility in susceptibility to phage infection or restriction-modification systems, or differences in transformability in response to hormones (11, 31). These mechanisms have not yet been implicated in the process of bacterial speciation, but their impact could be profound.
Slow allopatric speciation in sexual bacteria
So far we have considered only the case of a single population. Prolonged physical separation (allopatry) will reduce mixing and recombination between bacteria, and by random accumulation of mutations, two separated populations will genetically diverge at twice the mutation rate (2_m_). As this happens, the intrinsic capacity for recombination between the populations is reduced. The question then arises at what point should they be termed species?
For sexual populations (above the critical recombination threshold) speciation can be said to have occurred when the populations fail to blend even if the barrier isolating them was removed. If the rate at which two populations can exchange genes depends on the genetic distance between the populations, then if this distance is below a threshold recombination can cause distinct populations to converge and blend. If on the other hand this genetic distance is above a threshold, then recombinational incompatibility between the populations is such that the populations can never blend, and could legitimately be considered distinct species (27). Thus, the degree of divergence induced by allopatry or other mechanisms of separation required for speciation to occur is not a constant, but depends on the rate of recombination between similar genotypes. When separation is not sufficient to cause speciation, and sympatry is restored, blending will occur more rapidly than allopatric divergence (Fig 3); however genetic diversity is transiently enhanced owing to the long-term persistence of alleles from both populations. Separation thresholds and the dynamics of blending are explored further in (27). In summary, simple allopatry will only generate distinct clusters of strains over very long periods.
Figure 3.
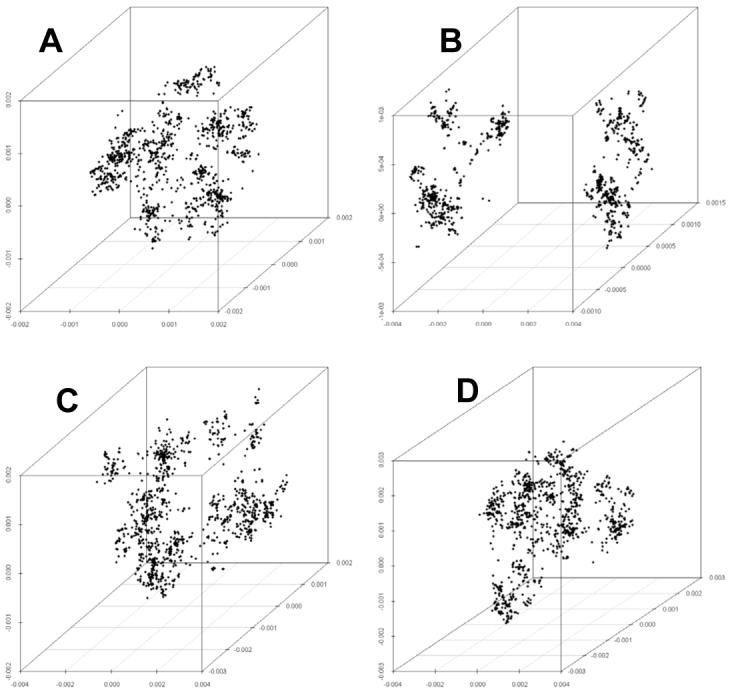
Genetic maps of a population temporarily divided by a strong barrier. With parameters as in Fig 2 for the sexual population, a split is introduced after 300,000 generations (A). After 300,000 generations apart, the populations have drifted and are clearly distinct (B). At this point the populations are re-united; after 10,000 generations, little distinction remains (C), and after a further 10,000 generations no remnants of the separation are evident (D).
Comparison to multi-locus sequence analysis
The inferred genetic map for a sample of bacteria from the mitis group _Streptococc_i (Fig 4) (30) was obtained from the sequences of six of the seven genes that define the Streptococcal MLST scheme and calculating the matrix of sequence divergence between isolates. ddl is excluded because it is linked to genes determining penicillin resistance, which undergo interspecific transfer more frequently than others (this is an interesting example of selection directly affecting the genetic interrelatedness of populations, albeit at one locus). Named species are currently defined by a strict series of phenotypic tests and these indeed correspond to clear clusters of related bacteria. However, these clusters are not uniform, for example S. pneumoniae is less divergent than the other named species.
Figure 4.
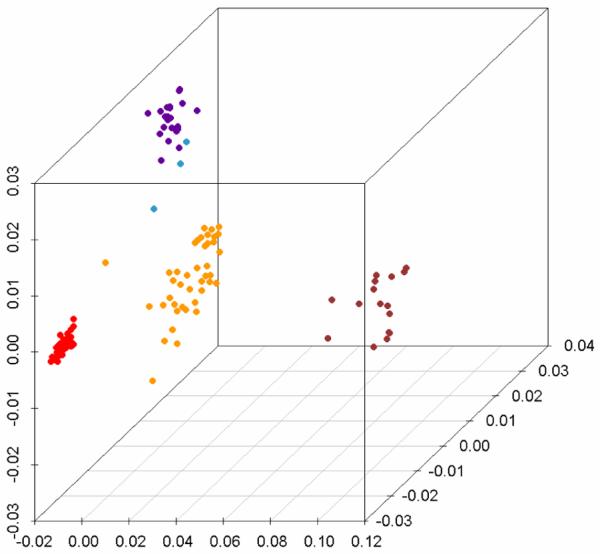
Genetic map of the Streptococcus genus, based on concatenated sequences of MLST genes (excluding ddl). Samples from four named species are highlighted as: red, S. pneumoniae, yellow, S. pseudopneumoniae, purple, S. mitis and brown, S. oralis. The three light blue dots represent strains for which the named species status could not be assessed.
For S. pneumoniae the recombination rate has been estimated to be roughly three times the mutation rate (per locus) (19), i.e., above the clonal/sexual threshold, and thus should behave as a sexual population. The distance between species is quite variable. The divergence between S. pneumoniae and S. oralis is over 10%, and thus, based on Fig 1A, we presume that the recombination rate between them is suppressed approximately 100-fold. Thus, even if opportunities for recombination between these were as frequent as intraspecific recombination, they would not blend owing to genetic divergence. By contrast, the divergence between S. pneumoniae and S. pseudopneumoniae is about 3%, so that interspecific recombination should only be reduced four-fold relative to intraspecific recombination. In sympatry, this is not sufficiently divergent to prevent blending. Interestingly, both types of streptococci appear to share a very similar lifestyle within the human nasopharynx and we thus hypothesise that a mechanism must act to separate the two populations, and that they could thus be considered nascent species. Speciation could be considered complete once these populations have diverged enough for blending by sympatric recombination to be genetically impossible.
Conclusions
Our model is a grossly oversimplified caricature of genetic diversification and speciation, but nonetheless gives some insight into the interplay between mutation, recombination and genetic divergence. For the case of diversity generated by neutral drift, we have derived a simple phenomenology of species. If recombination is less common than mutation, the situation is essentially clonal, and the population is characterized by a high degree of clustering. In this case, we expect that while natural selection and geographic structure will act to influence a process of clustering which may be inherent to clonal populations, they do not actually cause the clustering. If recombination is more frequent than this, then a threshold is crossed and recombination starts to act as a cohesive force on the population, by breaking linkage between alleles and reducing genetic clustering. Such a situation could in principle lead to dynamic speciation by chance drift, but only if the amount of variation within the population is sufficient for recombination rates to vary appreciably between members of the population. Based on current estimates for the species we have studied, this does not occur, but should not be ruled out. Thus, in general, bacteria can and do form sexual species, and mechanisms involving allopatry or niche specialization must be invoked in speciation. In this case, the situation is largely analogous to speciation in higher organisms, without the complications associated with sexual mating choice (32).
In our analysis we have not discussed the role natural selection may play in driving speciation. This is not because we do not believe selection to be important, quite the contrary. Rather it is instructive to understand the dynamics of neutral diversification and speciation to then understand how different types of selection, might influence this process. Also, we might plausibly hypothesize that even in a structured adaptive landscape, adaptation to different niches may involve selection at a small proportion of loci, and thus that the generation of genomic barriers to recombination arises by the accumulation of selectively neutral mutations, a process governed by simple rules not dissimilar to those described here. In this sense, we may expect our results to be applicable to much larger values of the effective population size, where selective forces are amplified relative to drift. Some additional simulations and discussion of the effect of increasing Ne are in (27).The derivation of analytical approximations to the processes of cluster dynamics (i.e., splitting, extinction, blending and relative drift) described here will help in exploring this further.
An alternative perspective on bacterial speciation has been provided by Cohan who identified the clonal-sexual threshold for neutral drift, but has emphasized that the threshold for sharing adaptive polymorphisms is much higher (33), leading to the notion that populations may be adaptively distinct but indistinguishable using neutral markers. These studies have emphasized the role of adaptive mutations in designating “ecotypes” as putative species (34). Our analyses suggest that for populations with recombination rate above the sexual threshold, ecotypes could rapidly blend should the adaptive landscape change and the barriers between niches be removed, and that below the sexual threshold, differentiation into distinct genetic clusters arises even in the absence of selection.
Our model highlights the importance of a detailed quantitative description of the processes that drive speciation. The simulations used here are based on generic plausible parameters, but further work is required to produce simulations properly calibrated to individual sets of experimental observations. For example, while the log-linear relation observed in Fig 1 seems general, and to be strikingly similar among bacteria as different as Streptococci, Haemophilus and Bacillus species, more effort is needed to measure recombination rates between closely related bacteria, as exceptions and anomalies have been documented in some systems (11, 28, 29), and also to estimate gene flow within and between natural populations. Examination of the Streptococci (Fig 4) reveals a diversity of patterns between relatively closely related species, as well as apparent asymmetries in gene flow which are not easily explained by simple models. More work is also required to explore the interplay between recombination and adaptation in more realistic selective landscapes, including in particular the role of epistatic interactions which can promote diversity and limit the scope for genome-wide selective sweeps.
In our opinion, understanding the nature and organization of genetic diversity can only be achieved by taking a multifaceted approach to the problem. Genetic surveys can reveal the extent and nature of the diversity that surrounds us. Careful experimentation can highlight potential mechanisms for creating the observed patterns. Theoretical models can then be used to explore whether the link between mechanisms and observation is plausible. Since the technological capacity for sequencing and simulating sequences are both growing exponentially, the ability to link those into a consistent picture may soon only be limited by our imagination (37).
Supplementary Material
Supplementary Online Material
References and notes
- 1.Ochman H, Lawrence JG, Groisman EA. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 2.Feil EJ, et al. P Natl Acad Sci USA. 2001;98:182–187. doi: 10.1073/pnas.98.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spratt BG, Hanage WP, Feil EJ. Curr Opin Microbiol. 2001;4:602–606. doi: 10.1016/s1369-5274(00)00257-5. [DOI] [PubMed] [Google Scholar]
- 4.Acinas SG, et al. Nature. 2004;430:551–554. doi: 10.1038/nature02649. [DOI] [PubMed] [Google Scholar]
- 5.Hanage WP, Fraser C, Spratt BG. BMC Biol. 2005;3 doi: 10.1186/1741-7007-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Venter JC, et al. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- 7.Gevers D, et al. Nat Rev Microbiol. 2005;3:733–739. doi: 10.1038/nrmicro1236. [DOI] [PubMed] [Google Scholar]
- 8.Falush D, et al. Phil Trans Roy Soc London B. 2006;361:2045–2053. doi: 10.1098/rstb.2006.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanage W, Spratt BG, Turner KME, Fraser C. Phil Trans Roy Soc London B. 2006;361:2039–2044. doi: 10.1098/rstb.2006.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lawrence JG. Theor Popul Biol. 2002;61:449–460. doi: 10.1006/tpbi.2002.1587. [DOI] [PubMed] [Google Scholar]
- 11.Majewski J. FEMS Microbiol Lett. 2001;199:161–9. doi: 10.1111/j.1574-6968.2001.tb10668.x. [DOI] [PubMed] [Google Scholar]
- 12.Vulic M, Dionisio F, Taddei F, Radman M. P Natl Acad Sci USA. 1997;94:9763–9767. doi: 10.1073/pnas.94.18.9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Majewski J, Zawadzki P, Pickerill P, Cohan FM, Dowson CG. J Bacteriol. 2000;182:1016–1023. doi: 10.1128/jb.182.4.1016-1023.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zawadzki P, Roberts MS, Cohan FM. Genetics. 1995;140:917–932. doi: 10.1093/genetics/140.3.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen P, Huang HV. Genetics. 1986;112:441–457. doi: 10.1093/genetics/112.3.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majewski J, Cohan FM. Genetics. 1999;153:1525–1533. doi: 10.1093/genetics/153.4.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mayr E. Systematics and the origin of species. New York: Columbia University Press; 1942. [Google Scholar]
- 18.Maiden MCJ, et al. P Natl Acad Sci USA. 1998;95:3140–3145. [Google Scholar]
- 19.Fraser C, Hanage WP, Spratt BG. P Natl Acad Sci USA. 2005;102:1968–1973. doi: 10.1073/pnas.0406993102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanage WP, Fraser C, Spratt BG. J Theor Biol. 2006;239:210–219. doi: 10.1016/j.jtbi.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 21.Drake JW, Charlesworth B, Charlesworth D, Crow JF. Genetics. 1998;148:1667–86. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maynard Smith J. Proc Roy Soc London B. 1991;245:37–41. [Google Scholar]
- 23.Watts DJ, Dodds PS, Newman MEJ. Science. 2002;296:1302–1305. doi: 10.1126/science.1070120. [DOI] [PubMed] [Google Scholar]
- 24.Thompson JR, et al. Science. 2005;307:1311–1313. doi: 10.1126/science.1106028. [DOI] [PubMed] [Google Scholar]
- 25.Briefly, the model is defined as follows. It is a neutral Fisher-Wright model, i.e. in each discrete generation, Ne isolates are sampled with replacement from the previous generation with equal probability. Each isolate has a genotype defined by 70 alleles, each allele is assigned co-ordinates within an abstract 6 dimensional sequence space. Mutation and recombination occurs with probability Lm and r per gene, respectively, where L is the length of the gene (=600 b.p.). If mutation occurs, one of the six co-ordinates in incremented by 1. At the start of the simulation, all co-ordinates are set to 1. Distances in the abstract sequence space are related to real genetic distances by using a probabilistic model relating these. Based on considering the probability of mutations recurring at the same location in real sequences, sequence (Hamming) distance is estimated as h_=L(1-(1-1/L)D) where D is the block distance in the abstract sequence space, a measure of the number of mutations that separates two alleles. Recombination succeeds with probability matched to the relation observed in Fig 1A, namely 10-18 x, where x is the local sequence divergence, equal to the Hamming distance estimated at the altered gene and its two neighbours divided by the total length being compared (=3_L).
- 26.Maynard Smith J, Smith NH, Orourke M, Spratt BG. P Natl Acad Sci USA. 1993;90:4384–4388. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Materials and methods are available as supporting material on Science Online.
- 28.Datta A, Hendrix M, Lipsitch M, JinksRobertson S. P Natl Acad Sci USA. 1997;94:9757–9762. doi: 10.1073/pnas.94.18.9757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zahrt TC, Maloy S. Proc Natl Acad Sci U S A. 1997;94:9786–91. doi: 10.1073/pnas.94.18.9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanage WP, Fraser C, Spratt BG. Phil Trans Royal Soc London B. 2006;361 doi: 10.1098/rstb.2006.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Havarstein LS, Hakenbeck R, Gaustad P. J Bacteriol. 1997;179:6589–6594. doi: 10.1128/jb.179.21.6589-6594.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schluter D. Trends Ecol Evol. 2001;16:372–380. doi: 10.1016/s0169-5347(01)02198-x. [DOI] [PubMed] [Google Scholar]
- 33.Cohan FM. Am Nat. 1994;143:965–986. [Google Scholar]
- 34.Cohan FM. Annu Rev Microbiol. 2002;56:457–487. doi: 10.1146/annurev.micro.56.012302.160634. [DOI] [PubMed] [Google Scholar]
- 35.Tettelin H, et al. Science. 2001;293:498–506. doi: 10.1126/science.1061217. [DOI] [PubMed] [Google Scholar]
- 36.R Development Core Team . R: A language and environment for statistical computing. 2005. [Google Scholar]
- 37.C. F. is funded by a Royal Society University Research Fellowship. W.P.H. and B.G.S. thank the Wellcome Trust for funding. The authors acknowledge useful comments from Daniel Falush and Derek J. Smith. The authors have no conflict of interest.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Online Material