22q11.2 Distal Deletion: A Recurrent Genomic Disorder Distinct from DiGeorge Syndrome and Velocardiofacial Syndrome (original) (raw)
Abstract
Microdeletions within chromosome 22q11.2 cause a variable phenotype, including DiGeorge syndrome (DGS) and velocardiofacial syndrome (VCFS). About 97% of patients with DGS/VCFS have either a common recurrent ∼3 Mb deletion or a smaller, less common, ∼1.5 Mb nested deletion. Both deletions apparently occur as a result of homologous recombination between nonallelic flanking low-copy repeat (LCR) sequences located in 22q11.2. Interestingly, although eight different LCRs are located in proximal 22q, only a few cases of atypical deletions utilizing alternative LCRs have been described. Using array-based comparative genomic hybridization (CGH) analysis, we have detected six unrelated cases of deletions that are within 22q11.2 and are located distal to the ∼3 Mb common deletion region. Further analyses revealed that the rearrangements had clustered breakpoints and either a ∼1.4 Mb or ∼2.1 Mb recurrent deletion flanked proximally by LCR22-4 and distally by either LCR22-5 or LCR22-6, respectively. Parental fluorescence in situ hybridization (FISH) analyses revealed that none of the available parents (11 out of 12 were available) had the deletion, indicating de novo events. All patients presented with characteristic facial dysmorphic features. A history of prematurity, prenatal and postnatal growth delay, developmental delay, and mild skeletal abnormalities was prevalent among the patients. Two patients were found to have a cardiovascular malformation, one had truncus arteriosus, and another had a bicuspid aortic valve. A single patient had a cleft palate. We conclude that distal deletions of chromosome 22q11.2 between LCR22-4 and LCR22-6, although they share some characteristic features with DGS/VCFS, represent a novel genomic disorder distinct genomically and clinically from the well-known DGS/VCF deletion syndromes.
Main Text
Interstitial deletions of chromosome 22q11.2 comprise the most common microdeletion in humans and occur at a frequency of approximately 1:4000–1:8000 live births.1 The well-characterized 22q11.2 deletion syndrome is associated with a wide array of clinical phenotypes including velocardiofacial syndrome (VCFS [MIM 192430]) and DiGeorge syndrome (DGS [MIM 188400]).2 Individuals with the 22q11.2 deletion can have a range of findings, including cardiovascular malformations consisting predominantly of conotruncal heart anomalies, palatal abnormalities, characteristic facial dysmorphic features, learning difficulties, immune deficiency, congenital hypocalcemia, and urogenital abnormalities.2–4
About 90% of patients with the 22q11.2 microdeletion have a common ∼3 Mb deletion, whereas 7% of the patients have a smaller, nested ∼1.5 Mb recurrent deletion.5 Both the ∼3 Mb and the ∼1.5 Mb deletions were found to occur as a result of nonallelic homologous recombination (NAHR),6 utilizing low-copy repeat (LCR) sequences located in the 22q11.2 region as recombination substrates.7,8 This mechanism of rearrangement explains not only the clustered breakpoints and existence of a common recurrent rearrangement among patients with chromosome 22q11.2 deletion, but also the high prevalence of de novo deletions.9 Proximal 22q contains eight LCRs known as LCR22s (Figure 1).8,10 On the basis of the architecture of the 22q11.2 region, it was anticipated that other rearrangements—utilizing distinct LCRs as recombination substrates and leading to different recurrent deletions—might occur in the genomic region.
Figure 1.
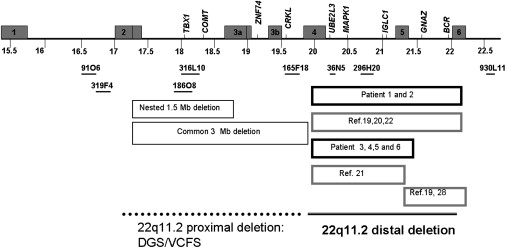
Schematic Overview of the 22q11.2 Region
Numbers below the schematic represent distance (million base units) from the 22p telomere according to NCBI human genome build 36 (2006). LCRs are depicted as gray boxes and labeled according to their number. LCR22-7 and LCR22-8 are located telomeric to the scheme and are not shown. The BAC clones that are included in the array-based CGH are depicted as horizontal black lines. Boxes below the map depict the typical deletions in DGS/VCFS and the different distal deletions reported in patients by this study and in previous reports (ref.). Note that exact deletion borders are sometimes only estimated in previously reported cases as well as in patient 3.
The structure of LCR22-4, LCR22-5, and LCR22-6 have been extensively studied recently11 and revealed the existence of a BCRL module in each LCR, suggesting a potential substrate for NAHR-mediated rearrangement in the distal 22q11.2. However, to date, only a limited number of “atypical” deletions in 22q11.2 have been reported. Most of these deletions were found to be located within the common ∼3 Mb deletion region or to overlap with it.12–19 Only a few “atypical” deletions were found to be located distally to the common deletion region.11,19–22 These distal deletions with the exception of one were detected among patients with clinical presentations related to DGS/VCFS and therefore are subject to ascertainment bias. Not surprising, therefore, is the fact that three out of the four previously described de novo cases of 22q11.2 distal deletions and the two index cases of the affected families presented with cardiovascular defect. Cardiac phenotype has not been detected among the three affected relatives of the familial index cases, implying that cardiac phenotype might actually be less frequent in patients with 22q11.2 distal deletion. However, given that there is no overlap of the genomic intervals between the common DGS/VCFS deletion and the 22q11.2 distal deletions, a similar phenotype shared by these two groups is not obvious. Indeed, searching for “atypical” deletions among patients with “atypical” VCFS (patients that did not have the facial phenotype typical for DGS/VCFS but manifested with malformations and/or developmental delay) identified distal deletions (both within and distal to the DGS/VCFS) in about 5%. Distal deletions have not been detected among 273 patients with conotruncal heart defects.19
We implemented array-based comparative genomic hybridization (CGH) analyses in > 8000 cases of individuals with suspected chromosomal aberrations23 (and unpublished data). The targeted microarrays were designed to cover all the telomeres at 1 Mb interval up to 10 Mb, one bacterial artificial chromosome (BAC) at every chromosome band, over 150 potential microdeletion or microduplication syndromes, and their flanking regions. For detailed information regarding loci covered, see the BCM website listed in the Web Resources. The arrays were manufactured and analyzed with a protocol described previously.24 Sixty patients were identified with rearrangement in the 22q11.2 region. Thirty-three patients (55%) had a ∼3 Mb deletion, one had a ∼1.5 Mb deletion, one had a ∼4 Mb deletion, and six (10%) had deletions distal to the DGS/VCFS common region. Ten (17%) were identified to have a ∼3 Mb duplication, and 9 (15%) had atypical duplications within and distal to the common DGC/VCFS critical region.25 As stated above, in six unrelated cases, the analyses revealed a loss in the 22q11.2 region detected with the same two BAC clones, RP11-36N5 and RP11-296H20 (Figure 1). These clones are located in the proximal long arm of chromosome 22 (22q11.2) just distal to the common ∼3 Mb DGS/VCFS interval. No copy-number variation of any of the five BAC clones located within the recurrent DGS/VCFS ∼3 Mb region was detected by the array-based CGH among these six patients. The array-based CGH confirmed the previously known diagnosis of 47,XYY karyotype in patient 1. No other abnormalities have been found by targeted array-based CGH. Fluorescence in situ hybridization (FISH) analyses using BAC clone RP11-36N5 or RP11-296H20 confirmed a loss at 22q11.2 in all six patients (Figure 2). Furthermore, FISH analysis using the same probes on the parental samples from each of the eleven available parents showed no evidence of a deletion (Figure 2), indicating a de novo deletion in at least five cases and potentially in the sixth.
Figure 2.
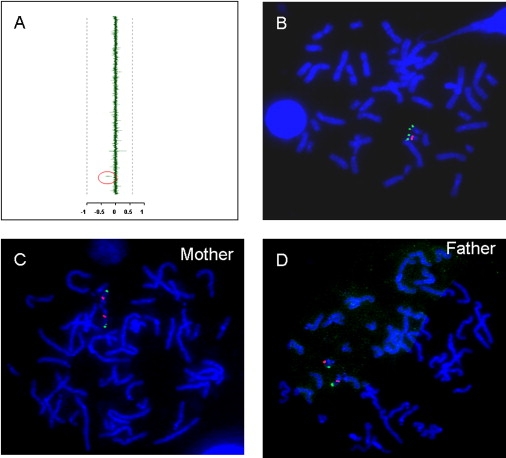
Array-CGH and FISH Findings among Patients and Their Parents
(A) Representative array-CGH output. The mean normalized log2(Cy3/Cy5) ratio of each BAC is plotted on the x axis as dots with error bars and arranged along the vertical axis from chromosome 1 at the top to chromosomes X and Y at the bottom. Decreased copy number was identified for two BAC clones, RP11-36N5 and 296H20 (marked by a red ellipse to the left of the vertical axis), which map between LCR22-4 and LCR22-5 on chromosome 22q11.2, just distal to the telomeric end of the common DGS/VCFS ∼3 Mb deletion.
(B) Representative FISH analysis of deletion case. The two red fluorescence signals represent hybridization using a subtelomeric chromosome 22q FISH probe, and the single green signal represents the hybridization of the RP11-36N5 clone to the normal (nondeleted) chromosome 22. Deletions were confirmed by FISH analyses in all the cases.
(C and D) Representative FISH analyses of normal parents. Metaphase FISH analysis using the same probes detected two normal copies of the distal 22q11.2 probe among the parents.
The six patients were found to share similar clinical features as well as physical findings (Table 1, Figure 3). Five of the patients were born prematurely and the sixth was born during the 38th week of gestation. Birth weight of four patients was below the 5th percentile for gestational age, and the birth weight of the other two was in the 5th–10th percentile range.26 Five of the patients had postnatal growth restriction as well. One of those (patient 3) was diagnosed has having gluten enteropathy (celiac disease) on the basis of immunological studies. However, this child had growth delay prior to his first gluten exposure (both prenatal and postnatal growth delay). In addition, growth acceleration was not observed after a gluten-free diet had been administered. Interestingly, even the oldest patient (patient 1), who presented with obesity at the time of diagnosis, had a significant postnatal growth delay up to the age of 6 yr. The growth acceleration of this patient may be attributed to the mid-childhood growth acceleration frequently seen in individuals with 47,XYY karyotype.27 A global developmental delay and/or mild mental retardation, more prominent in language, was found in four of the six. Another patient has borderline normal development, and one had normal development. This latter child, however, presented with severe behavioral problems including uncontrolled aggression. Minor skeletal abnormalities were found in four of the patients. These abnormalities included coxa valga and bowed ulnae in one case; small distal phalanx, short and broad fingernails, and bilateral fifth finger clinodactyly in another case; bilateral hip dysplasia in the third case; and contractures and brachydactyly in the fourth. One patient had a cleft palate, and another one was found to have a high arched palate without cleft. Two of the patients had a cardiac defect. One of them (patient 2) had truncus arteriosus and was tested for DGS/VCFS due to the combination of conotruncal heart defect and dysmorphic features. However, FISH analysis did not reveal a deletion in the DGS/VCFS critical region. Another patient had a bicuspid aortic valve, without other structural heart defects (patient 3). This type of heart defect has not been reported to be associated with DGS/VCFS. Echocardiogram was performed in three of the four other cases and did not detect any structural abnormalities. Neonatal hypocalcemia, neonatal seizures, or immunodeficiency was not described among any patient with 22q11.2 distal deletion (in contrast to 77% of the patients with DGS). One of our patients, but none of the previously described cases with 22q11.2 distal deletion, had a history of cleft palate, a common finding among patients with DGS/VCFS.
Table 1.
Clinical and Cytogenetic Finding among Six Patients with 22q11.2 Distal Deletion
| Manifestation | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Total |
|---|---|---|---|---|---|---|---|
| 22q11.2 deletion size (Mb) | ∼2.1 | ∼2.1 | ∼1.4 | ∼1.4 | ∼1.4∗ | ∼1.4 | |
| LCR involved | 4–6 | 4–6 | 4–5 | 4–5 | 4–5∗ | 4–5 | |
| Karyotype | 47,XYY | 46,XY | 46,XY | 46,XY | 46,XX | 46,XY | |
| Parental FISH | P-Nl, M-Nl | P-Nl, M-Nl | P-Nl, M-Nl | P-?, M-Nl | P-Nl, M-Nl | P-Nl, M-Nl | 11-Nl, 1-? |
| Gender | M | M | M | M | F | M | 4M/1F |
| Age (years) | 11 | 3 | 6 | 5 | 3 | 4 | |
| Gestational age (weeks) | 32 | 33 | 38 | 36 | 35 | 37 | |
| Birth weight(centile) | 1050 (3%) | 1360(3%–10%) | 2210(3%–10%) | 1250 (< < 3%) | 1700 (< < 3%) | ||
| Postnatal growth retardation | + | + | + | − | + | + | 5/6 |
| Weight at presentation | 97% | <5% | < < 5% | 60% | < < 5% | < < 5% | |
| Height at presentation (centile) | 20% | <5% | ? | 60% | < < 5% | < < 5% | |
| Developmental delay | + | + | + | − | +/− | + | 4/6 |
| Behavioral problems | + | − | − | + | − | − | 2/6 |
| Cardiovascular defect (type) | + (truncus arteriosus) | + (bicuspid aortic valve) | − | − | − | 2/6 | |
| Smooth philtrum | + | + | + | + | + | + | 6/6 |
| Cleft palate | + | − | − | − | − | − | 1/6 |
| Arched eye brows | + | + | − | + | + | 4/5 | |
| Pointed chin | − | + | + | − | + | 3/5 | |
| Skeletal abnormalities | + | − | − | + | − | + | 3/6 |
| High arched palate | − | + | − | − | − | − | 1/6 |
| Deep-set eyes | − | − | + | − | + | + | 3/6 |
| Hypoplastic alae nasi | − | + | + | − | + | + | 4/6 |
| Recurrent infections | − | − | − | − | − | − | 0/6 |
Figure 3.
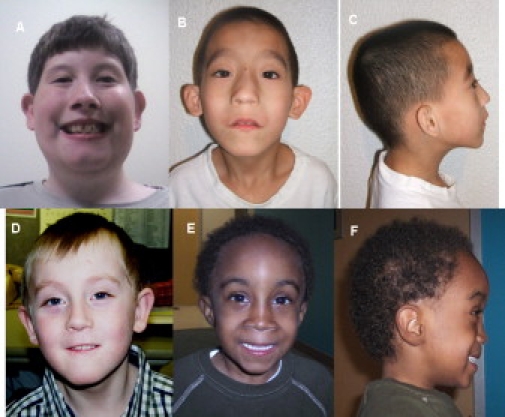
Facial Appearance of Patients with 22q11.2 Distal Deletions
Shared facial dysmorphic features include arched eyebrows, flattened midface, smooth philtrum, and thin upper lip. (A) Patient 1, whose ∼2.1 Mb deletion extends from LCR22-4 through LCR22-6. Note the arched eyebrows and the smooth philtrum. This patient also has sex-chromosome aneuploidy with 47,XYY karyotype.
(B and C) Patient 2, with ∼2.1 Mb deletion. Note the low-set, cuboidal ears, hypoplastic browridges, broad nasal bridge, smooth philtrum, and pointed chin.
(D) Patient 5, whose ∼1.4 Mb deletion extends from LCR 22-4 through LCR22-5. Note the pointed chin in addition to partial ptosis.
(E and F) Patient 6, with ∼1.4 Mb deletion. Note the arched eyebrows, hypoplastic browridges, smooth philtrum, and flat retracted midface.
Physical examination of all of the six patients revealed characteristic multiple dysmorphic facial features (Table 1, Figure 3). The common facial dysmorphic features included arched eyebrows, deep-set eyes, a smooth philtrum, a thin upper lip, hypoplastic alae nasi, and a small, pointed chin.
In order to more precisely map breakpoints of the 22q11.2 deletion and to further explore the molecular mechanism for the deletion, we performed higher-resolution genomic analysis in five of the cases by using high-density array-based oligonucleotide CGH (Agilent 244K Whole Human Genome Oligo Microarray Kit, Agilent Technologies, CA, USA) according to the manufacturer's recommendations. The array-based oligonucleotide CGH revealed that patients 1 and 2 had a ∼2.1 Mb deletion located between LCR22- 4 and LCR22-6. The deletion size in cases 3, 4, and 6 was found to be ∼1.4 Mb and spans the genomic region between LCR22-4 and LCR22-5 (Figure 4). The high-resolution array analyses revealed that all five patients shared a proximal breakpoint. The two patients with the ∼2.1 Mb deletion and the three patients with the nested ∼1.4 Mb deletion shared the same distal breakpoint as well. The three breakpoints coincided with LCR22-4, LCR22-5, and LCR22-6 (based on the sequence data from the genome browser). The parents of patient five did not consent to the Agilent 244K array, and therefore in this case we have used the BCR probe, which maps between LCR22-5 and LCR22-6, for FISH analysis. The patient was found to have two copies of the BCR probe, suggesting that the LCR22-5 to LCR22-6 segment was not deleted in this case.
Figure 4.
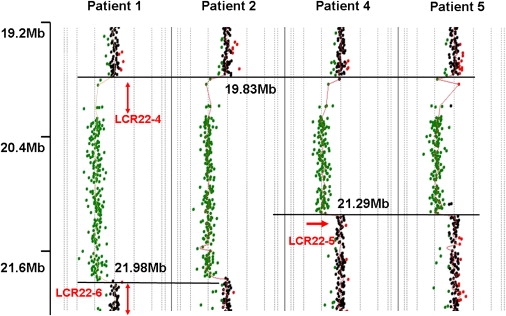
Analysis of 22q11.2-Distal-Deletion Breakpoints by High-Resolution Oligonucleotide Microarray Analysis
Results of Agilent 244K array-based oligonucleotide CGH performed on DNA samples from patients 1, 2, and 5 and 6 revealed similar proximal breakpoints in all of the cases. Red arrows represent the known LCR22s in the region. Black horizontal lines represent the different breakpoints, and the numbers represent genomic distance of the different breakpoints (Megabase units) from 22p telomere according to NCBI human genome build 36 (2006). Note that the proximal breakpoint is common in all of the cases, and the distal breakpoints also coincide for patients 1 and 2 and patients 5 and 6, respectively. The breakpoints are located in the known LCR22s, implying NAHR-based recombination mechanism.
A review of the literature reporting 22q11.2 distal deletions revealed that three of the previously described cases had, similar to our cases, de novo deletions of the LCR22-4 to LCR22-5 segment or the LCR22-4 to LCR22-6 segment,19,21,22 implying a pathogenic role for such deletions. A fourth case20 with the LCR22-4 to LCR22-6 segment deletion was found to be part of an affected family with multiple affected individuals and a variable phenotype. The deletion found in the fifth previously described case19 was smaller (∼0.7 Mb), located between the LCR22-5 and LCR22-6 segment, and did not include the ∼1.4 Mb LCR22-4 to LCR22-5 segment, a genomic segment that was deleted in all of the other previously reported cases and in all of our cases. This patient had a congenital heart defect and normal appearance and psychomotor development. Only very careful investigation revealed minimal dysmorphic features. This deletion was also detected in the patent's healthy father, who had narrow palpebral fissures and mildly low-set ears. Interestingly, recently eight individuals with the BCR deletion (located within LCR22-5 and LCR22-6 interval used as a control probe for chromosome 22) have been detected from among 11,688 individuals that were referred for evaluation of subtelomeric rearrangements.28 All of these individuals had an intact DGS/VCFS region, but the size of the deletions has not been evaluated and the phenotype was not described in detail.
Clinically, many microdeletion syndromes not only show a high degree of variability among patients with identical deletions29 but also share many common features. Features such as developmental and growth delay, multiple congenital anomalies, and a wide range of dysmorphic features are common in many chromosomal aberrations. Nevertheless, several features of 22q11.2 distal deletion between LCR22-4 and LCR22-6 were shared by the majority of our patients (Table 1), most are distinctive from those observed in DGS/VCFS, and some are characteristic features associated with deletion of a common genomic interval. Prenatal and postnatal growth restriction, for example, was observed in 83% of our patients. Whereas growth restriction is a common feature of chromosomal aberration, many syndromes show no growth restriction or only postnatal growth restriction. Prenatal growth restriction has not been widely described in most patients with DGS/VCFS, and postnatal growth restriction has been described in only about 36% of the patients. Developmental delay and/or mental retardation appear more common among patients with distal 22q11.2 deletion compared with patients having DGS/VCFS. Similarly, skeletal abnormalities among patients with distal 22q11.2 deletion are not only more common than those described among patients with DGS/VCFS,30 but are also different in nature. The typical facial dysmorphic features of patients with 22q11.2 distal deletion are characteristic and do not resemble those of patients with DGS/VCFS.27
Although prematurity is not a common feature of chromosomal aberration, and has not been described as part of DGS/VCFS, at least four of our six patients were born prematurely (between 32 and 36 weeks of gestation), the fifth was born during or after the 37th week of gestation, and the sixth was born during the 38th week of gestation. Given that our cohort is small, there is still a finite probability that the prematurity among our patients happened by chance. The probability of such a random event is low because the percentage of preterm births (<37 completed weeks of gestation) in the United States is about 12.7%.31 Trisomy 18 is the only other chromosomal syndrome that has been described to be associated with prematurity to date. However, the average gestational age at birth of such newborns was calculated to be 38 weeks,32 a distinctly longer term than the average of 35.2 weeks in our patients. This observation may imply the presence of a dosage-sensitive gene that is associated with prematurity and/or low birth weight and is located within the distal 22q11.2 region. Interestingly, one of the genes located in the deleted region, mitogen-activated protein kinase 1 (MAPK1) may be associated with placental development and low birth weight. MAPK1 has a role in one of the four mitogen-activated protein kinase cascades. Knockout studies of a few genes in these cascades, including MAPK1, revealed some associations with abnormal placental development, leading to intrauterine growth restriction and intrauterine death of null mice.33
Although congenital heart defects are common in many chromosomal aberrations, the fact that one of our six patients in addition to one of the four previously described de novo cases and one member of the familial case with 22q11.2 distal deletion had a very specific cardiac defect, truncus arteriosus, is intriguing. This high percentage of truncus arteriosus in 22q11.2 distal deletion may be partially due to ascertainment bias of the previously described cases; nevertheless, distal 22q11.2 may be associated with an increased risk of truncus arteriosus. In fact, although truncus arteriosus is one of the most characteristic heart defects in DGS/VCFS, it exists in only 7% of such patients.34 Truncus arteriosus has not been described yet as a common finding in any other chromosomal aberration or single-gene defect. About one-third of patients with such a cardiac defect have one of the two more common ∼1.5 Mb or ∼3 Mb 22q11.2 recurrent deletions,35,36 but a systematic analysis of other genomic disorders leading to truncus arteriosus in a large number of patients has not been performed. The fact that such a rare heart defect is common in two adjacent but nonoverlapping microdeletion syndromes (DGS/VCFS and 22q11.2 distal deletion), and the fact that this heart defect has not been described commonly with other chromosomal aberration, might be a random finding. An alternative explanation is a position effect of the distal deletion on a gene located in the DGS/VCFS or vice versa responsible for this overlapping cardiac phenotype.37,38 Another explanation for this shared phenotype could be clustering of genes involved in the development of heart in this genomic region.
Given that three de novo cases and one familial case (with three affected individuals) have been reported with LCR22-4 to LCR22-6 deletions, we have examined the characteristic phenotype of 22q11.2 distal deletion among these patients. Unfortunately, birth weight, indicating prenatal growth restriction, has been reported in only one-sixth of them (and was normal for gestational age).22 However, height at presentation was reported among one-third of the de novo cases and two-thirds of individuals from the affected family.20,22 In all of them, the height was reported to be at the 10th centile. Another previously described case reported short stature.21 These data imply that postnatal growth restriction and short stature are common features of this syndrome. Information regarding the gestational age at birth was provided for only two of the previously described cases. Both children were born prematurely after 35 weeks of gestation.21,22 Congenital heart defects and palate abnormalities were prevalent among previously described cases with higher prevalence than among our patients, in part because of to ascertainment bias. Similar to our patients, learning difficulties and/or psychomotor developmental delay was prevalent among the majority of the previous described cases. The overall data suggest, however, that severe mental retardation is not common among patients with 22q11.2 distal deletion. On the basis of on available photographs and clinical description of published cases, it seems that most of them share the characteristic facial features of 22q11.2 distal deletion. Two previously described patients19,20 had choanal atresia and/or stenosis, and one had immunodeficiency.20 Such phenotypes have not been detected among our patients and may be subject to ascertainment bias because these features are prevalent in DGS/VCFS27 and were even part of the inclusion criteria in one of the previous study.19
On the basis of oligonucleotide array-CGH analysis (Figure 4) and information obtained from the NCBI genome (build 36, 2006), it is apparent that 22q11.2 distal deletions involve segments that extend between known LCRs: LCR22-4 to LCR22-5 or LCR22-6. Recent investigation of LCR22-5 and LCR22-6 structure and organization11 revealed that both include _BCRL_-containing modules and that LCR22-5 includes a _GGTL_-containing module as well. Both modules are part of LCR22-4 structure.7 In all of the cases that were analyzed with a high-resolution oligonucleotide microarray, breakpoint analyses revealed similar junctions. This breakpoint is located in the proximal part of the three relevant LCR22s: LCR22-4, LCR22-5, and LCR22-6 (Figure 4). This proximal portion of these LCRs contains the BCRL module in all of them.7,11 This finding implies not only that these LCRs have a role in the mechanism of the rearrangement by NAHR but also that the BCRL module specifically may be utilized as a substrate in NAHR causing 22q11.2 distal deletions. The fact that de novo rearrangements were found in the great majority of 22q11.2 distal deletions supports the role of NAHR in these deletions, because one would expect to find a high rate of de novo arrangement in regions that have high genome complexity and are subjected to NAHR.39
We could not detect a clear difference in the severity or the phenotype between the larger ∼2.1 Mb deletion and the smaller nested ∼1.4 Mb deletion. However, more patients must be characterized in order to determine whether a phenotypic difference exists between these two sizes of deletion. Molecularly, the ∼1.4 Mb LCR22-4 to LCR22-5 segment contains at least 17 annotated genes, and the ∼0.7 Mb LCR22-5 to LCR22-6 contains at least four additional genes. Many of the genes in this genomic region have not been extensively studied. We do not know yet which of the genes in this region are sensitive to the dosage effect generated by the copy-number variation due to the deletion or the potential influence of such a dosage effect.
In conclusion, 22q11.2 distal deletion is a recurrent microdeletion syndrome likely to result from NAHR. This newly recognized genomic disorder has specific characteristics including prematurity, prenatal and postnatal growth restriction, learning difficulties and/or developmental delay, characteristic facial features, skeletal abnormalities, and increased incidence of a specific type of heart defect, truncus arteriosus. The phenotype is, in many ways, distinct from the phenotype of the adjacently located microdeletion syndrome DGS/VCFS. The two cytogenetically and clinically distinctive syndromes share an overlapping cardiac phenotype. We suggest, therefore, that individuals with the characteristic features of 22q11.2 LCR22-4 to LCR22-6 deletion and patients with a less suggestive DGS/VCFS phenotype and negative DGS/VCFS FISH results, as well as every individual with truncus arteriosus, should be tested for the 22q11.2 distal deletion. However, given that dysmorphic features, short stature, heart defects, and developmental delay are common findings in many chromosomal aberrations, array CGH23,39 is currently the most efficient and cost-effective means of testing for submicroscopic chromosomal aberrations associated with genomic disorders.
Web Resources
The URLs for data presented herein are as follows:
- Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
- OMIM, http://www.ncbi.nlm.nih.gov/Omim/ (for DiGeorge syndrome and velocardiofacial syndrome)
- BCM, http://www.bcm.edu/cma/table.htm
References
- 1.Scambler P.J. The 22q11 deletion syndromes. Hum. Mol. Genet. 2000;9:2421–2426. doi: 10.1093/hmg/9.16.2421. [DOI] [PubMed] [Google Scholar]
- 2.Ryan A.K., Goodship J.A., Wilson D.I., Philip N., Levy A., Seidel H., Schuffenhauer S., Oechsler H., Belohradsky B., Prieur M. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: A European collaborative study. J. Med. Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shprintzen R.J., Goldberg R.B., Lewin M.L., Sidoti E.J., Berkman M.D., Argamaso R.V., Young D. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft Palate J. 1978;15:56–62. [PubMed] [Google Scholar]
- 4.Emanuel B.S., McDonald-McGinn D., Saitta S.C., Zackai E.H. The 22q11.2 deletion syndrome. Adv. Pediatr. 2001;48:39–73. [PubMed] [Google Scholar]
- 5.Carlson C., Sirotkin H., Pandita R., Goldberg R., McKie J., Wadey R., Patanjali S.R., Weissman S.M., Anyane-Yeboa K., Warburton D. Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am. J. Hum. Genet. 1997;61:620–629. doi: 10.1086/515508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stankiewicz P., Lupski J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 7.Shaikh T.H., Kurahashi H., Saitta S.C., O'Hare A.M., Hu P., Roe B.A., Driscoll D.A., McDonald-McGinn D.M., Zackai E.H., Budarf M.L., Emanuel B.S. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: Genomic organization and deletion endpoint analysis. Hum. Mol. Genet. 2000;9:489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- 8.Edelmann L., Pandita R.K., Spiteri E., Funke B., Goldberg R., Palanisamy N., Chaganti R.S., Magenis E., Shprintzen R.J., Morrow B.E. A common molecular basis for rearrangement disorders on chromosome 22q11. Hum. Mol. Genet. 1999;8:1157–1167. doi: 10.1093/hmg/8.7.1157. [DOI] [PubMed] [Google Scholar]
- 9.Wilson D.I., Goodship J.A., Burn J., Cross I.E., Scambler P.J. Deletions within chromosome 22q11 in familial congenital heart disease. Lancet. 1992;340:573–575. doi: 10.1016/0140-6736(92)92107-q. [DOI] [PubMed] [Google Scholar]
- 10.McDermid H.E., Morrow B.E. Genomic disorders on 22q11. Am. J. Hum. Genet. 2002;70:1077–1088. doi: 10.1086/340363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaikh T.H., O'Connor R.J., Pierpont M.E., McGrath J., Hacker A.M., Nimmakayalu M., Geiger E., Emanuel B.S., Saitta S.C. Low copy repeats mediate distal chromosome 22q11.2 deletions: Sequence analysis predicts breakpoint mechanisms. Genome Res. 2007;17:482–491. doi: 10.1101/gr.5986507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amati F., Conti E., Novelli A., Bengala M., Diglio M.C., Marino B., Giannotti A., Gabrielli O., Novelli G., Dallapiccola B. Atypical deletions suggest five 22q11.2 critical regions related to the DiGeorge/velo-cardio-facial syndrome. Eur. J. Hum. Genet. 1999;7:903–909. doi: 10.1038/sj.ejhg.5200399. [DOI] [PubMed] [Google Scholar]
- 13.Kurahashi H., Tsuda E., Kohama R., Nakayama T., Masuno M., Imaizumi K., Kamiya T., Sano T., Okada S., Nishisho I. Another critical region for deletion of 22q11: A study of 100 patients. Am. J. Med. Genet. 1997;72:180–185. doi: 10.1002/(sici)1096-8628(19971017)72:2<180::aid-ajmg10>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 14.O'Donnell H., McKeown C., Gould C., Morrow B., Scambler P. Detection of an atypical 22q11 deletion that has no overlap with the DiGeorge syndrome critical region. Am. J. Hum. Genet. 1997;60:1544–1548. doi: 10.1016/S0002-9297(07)64250-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Minaur S., Fantes J., Murray R.S., Porteous M.E., Strain L., Burns J.E., Stephen J., Warner J.P. A novel atypical 22q11.2 distal deletion in father and son. J. Med. Genet. 2002;39:E62. doi: 10.1136/jmg.39.10.e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McQuade L., Christodoulou J., Budarf M., Sachdev R., Wilson M., Emanuel B., Colley A. Patient with a 22q11.2 deletion with no overlap of the minimal DiGeorge syndrome critical region (MDGCR) Am. J. Med. Genet. 1999;86:27–33. [PubMed] [Google Scholar]
- 17.Kerstjens-Frederikse W.S., Kurahashi H., Driscoll D.A., Budarf M.L., Emanuel B.S., Beatty B., Scheidl T., Siegel-Bartelt J., Henderson K., Cytrynbaum C. Microdeletion 22q11.2: Clinical data and deletion size. J. Med. Genet. 1999;36:721–723. [PMC free article] [PubMed] [Google Scholar]
- 18.Levy A., Demczuk S., Aurias A., Depetris D., Mattei M.G., Philip N. Interstitial 22q11 microdeletion excluding the ADU breakpoint in a patient with DiGeorge syndrome. Hum. Mol. Genet. 1995;4:2417–2419. doi: 10.1093/hmg/4.12.2417. [DOI] [PubMed] [Google Scholar]
- 19.Rauch A., Zink S., Zweier C., Thiel C.T., Koch A., Rauch R., Lascorz J., Huffmeier U., Weyand M., Singer H., Hofbeck M. Systematic assessment of atypical deletions reveals genotype-phenotype correlation in 22q11.2. J. Med. Genet. 2005;42:871–876. doi: 10.1136/jmg.2004.030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rauch A., Pfeiffer R.A., Leipold G., Singer H., Tigges M., Hofbeck M. A novel 22q11.2 microdeletion in DiGeorge syndrome. Am. J. Hum. Genet. 1999;64:659–666. doi: 10.1086/302235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saitta S.C., McGrath J.M., Mensch H., Shaikh T.H., Zackai E.H., Emanuel B.S. A 22q11.2 deletion that excludes UFD1L and CDC45L in a patient with conotruncal and craniofacial defects. Am. J. Hum. Genet. 1999;65:562–566. doi: 10.1086/302514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mikhail F.M., Descartes M., Piotrowski A., Andersson R., Diaz de Stahl T., Komorowski J., Bruder C.E., Dumanski J.P., Carroll A.J. A previously unrecognized microdeletion syndrome on chromosome 22 band q11.2 encompassing the BCR gene. Am. J. Med. Genet. A. 2007;143:2178–2184. doi: 10.1002/ajmg.a.31882. [DOI] [PubMed] [Google Scholar]
- 23.Lu X., Shaw C.A., Patel A., Li J., Cooper M.L., Wells W.R., Sullivan C.M., Sahoo T., Yatsenko S.A., Bacino C.A. Clinical implementation of chromosomal microarray analysis: Summary of 2513 postnatal cases. PLoS ONE. 2007;2:e327. doi: 10.1371/journal.pone.0000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai W.W., Mao J.H., Chow C.W., Damani S., Balmain A., Bradley A. Genome-wide detection of chromosomal imbalances in tumors using BAC microarrays. Nat. Biotechnol. 2002;20:393–396. doi: 10.1038/nbt0402-393. [DOI] [PubMed] [Google Scholar]
- 25.Ou, Z., Berg, J.S., Yonath, H., Enciso, V.B., Miller, D.T., Picker, J., Lenzi, T., Keegan, C.E., Sutton, V.R., Belmont, J., et al. Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet. Med., in press. [DOI] [PubMed]
- 26.Fenton T.R. A new growth chart for preterm babies: Babson and Benda's chart updated with recent data and a new format. BMC Pediatr. 2003;3:13. doi: 10.1186/1471-2431-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones K.L. Elsevier Saunders; Philadelphia, PA: 2006. Smith's Recognizable Patterns of Human Malformation, Volume 6. [Google Scholar]
- 28.Ravnan J.B., Tepperberg J.H., Papenhausen P., Lamb A.N., Hedrick J., Eash D., Ledbetter D.H., Martin C.L. Subtelomere FISH analysis of 11 688 cases: An evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J. Med. Genet. 2006;43:478–489. doi: 10.1136/jmg.2005.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potocki L., Shaw C.J., Stankiewicz P., Lupski J.R. Variability in clinical phenotype despite common chromosomal deletion in Smith-Magenis syndrome. Genet. Med. 2003;5:430–434. doi: 10.1097/01.gim.0000095625.14160.ab. [DOI] [PubMed] [Google Scholar]
- 30.Ming J.E., McDonald-McGinn D.M., Megerian T.E., Driscoll D.A., Elias E.R., Russell B.M., Irons M., Emanuel B.S., Markowitz R.I., Zackai E.H. Skeletal anomalies and deformities in patients with deletions of 22q11. Am. J. Med. Genet. 1997;72:210–215. doi: 10.1002/(sici)1096-8628(19971017)72:2<210::aid-ajmg16>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 31.Hamilton B.E., Minino A.M., Martin J.A., Kochanek K.D., Strobino D.M., Guyer B. Annual summary of vital statistics: 2005. Pediatrics. 2007;119:345–360. doi: 10.1542/peds.2006-3226. [DOI] [PubMed] [Google Scholar]
- 32.Goc B., Walencka Z., Wloch A., Wojciechowska E., Wiecek-Wlodarska D., Krzystolik-Ladzinska J., Bober K., Swietlinski J. Trisomy 18 in neonates: Prenatal diagnosis, clinical features, therapeutic dilemmas and outcome. J. Appl. Genet. 2006;47:165–170. doi: 10.1007/BF03194617. [DOI] [PubMed] [Google Scholar]
- 33.Aouadi M., Binetruy B., Caron L., Le Marchand-Brustel Y., Bost F. Role of MAPKs in development and differentiation: Lessons from knockout mice. Biochimie. 2006;88:1091–1098. doi: 10.1016/j.biochi.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 34.McDonald-McGinn D.M., Kirschner R., Goldmuntz E., Sullivan K., Eicher P., Gerdes M., Moss E., Solot C., Wang P., Jacobs I. The Philadelphia story: The 22q11.2 deletion: Report on 250 patients. Genet. Couns. 1999;10:11–24. [PubMed] [Google Scholar]
- 35.Goldmuntz E., Clark B.J., Mitchell L.E., Jawad A.F., Cuneo B.F., Reed L., McDonald-McGinn D., Chien P., Feuer J., Zackai E.H. Frequency of 22q11 deletions in patients with conotruncal defects. J. Am. Coll. Cardiol. 1998;32:492–498. doi: 10.1016/s0735-1097(98)00259-9. [DOI] [PubMed] [Google Scholar]
- 36.Iserin L., de Lonlay P., Viot G., Sidi D., Kachaner J., Munnich A., Lyonnet S., Vekemans M., Bonnet D. Prevalence of the microdeletion 22q11 in newborn infants with congenital conotruncal cardiac anomalies. Eur. J. Pediatr. 1998;157:881–884. doi: 10.1007/s004310050959. [DOI] [PubMed] [Google Scholar]
- 37.Lee J.A., Madrid R.E., Sperle K., Ritterson C.M., Hobson G.M., Garbern J., Lupski J.R., Inoue K. Spastic paraplegia type 2 associated with axonal neuropathy and apparent PLP1 position effect. Ann. Neurol. 2006;59:398–403. doi: 10.1002/ana.20732. [DOI] [PubMed] [Google Scholar]
- 38.Smyk M., Berg J.S., Pursley A., Curtis F.K., Fernandez B.A., Bien-Willner G.A., Lupski J.R., Cheung S.W., Stankiewicz P. Male-to-female sex reversal associated with an approximately 250 kb deletion upstream of NR0B1 (DAX1) Hum. Genet. 2007;122:63–70. doi: 10.1007/s00439-007-0373-8. [DOI] [PubMed] [Google Scholar]
- 39.Lupski J.R. Genome structural variation and sporadic disease traits. Nat. Genet. 2006;38:974–976. doi: 10.1038/ng0906-974. [DOI] [PubMed] [Google Scholar]