Single and Dual Amino Acid Substitutions in TCR CDRs Can Enhance Antigen-Specific T Cell Functions (original) (raw)
. Author manuscript; available in PMC: 2008 Jun 11.
Abstract
Single and dual amino acid substitution variants were generated in the TCR CDRs of three TCRs that recognize tumor-associated Ags. Substitutions that enhance the reactivity of TCR gene-modified T cells to the cognate Ag complex were identified using a rapid RNA-based transfection system. The screening of a panel of variants of the 1G4 TCR, that recognizes a peptide corresponding to amino acid residues 157-165 of the human cancer testis Ag NY-ESO-1 (SLLMWITQC) in the context of the HLA-A*02 class I allele, resulted in the identification of single and dual CDR3_α_ and CDR2_β_ amino acid substitutions that dramatically enhanced the specific recognition of NY-ESO-1+/HLA-A*02+ tumor cell lines by TCR gene-modified CD4+ T cells. Within this group of improved TCRs, a dual substitution in the 1G4 TCR CDR3_α_ chain was identified that enhanced Ag-specific reactivity in genemodified CD4+ and CD8+ T cells. Separate experiments on two distinct TCRs that recognize the MART-1 27-35 (AAGIGILTV) peptide/HLA-A*02 Ag complex characterized single amino acid substitutions in both TCRs that enhanced CD4+ T cell Ag-specific reactivity. These results indicate that simple TCR substitution variants that enhance T cell function can be identified by rapid transfection and assay techniques, providing the means for generating potent Ag complex-specific TCR genes for use in the study of T cell interactions and in T cell adoptive immunotherapy. The Journal of Immunology, 2008, 180: 6116-6131.
Multiple costimulatory and cellular adhesion molecules can influence the degree of T cell activation in response to antigenic stimulation; however, there is a substantial body of evidence indicating that TCR affinity represents the primary factor involved in determining T cell avidity and the outcome of antigenic stimulation (1-3). The affinity of native TCRs for cognate Ag complexes has been found to range between 1 and 50 _μ_M (4), a relatively low range that may reflect a balance between the need to effectively trigger T cell responses and the need to maintain self tolerance (5). In murine studies, immunization with a low dose of Ag elicited relatively high-avidity T cells whereas high Ag doses elicited cells of lower avidity (6). High-avidity T cells reactive against immunodominant epitopes derived either from tyrosinase-related protein 2 or the p15E endogenous retroviral envelope were more effective at reducing the number of lung metastases of the MCA-38 tumor cell line than low-avidity T cells (7). Chronic viral stimulation can also lead to the appearance of dominant clonotypes that possess higher avidities for Ag than subdominant clonotypes, suggesting that competition for Ag at the level of APCs may lead to the selective proliferation of T cells bearing high-affinity TCRs (8, 9).
Elucidation of the role of TCR amino acid residues in cognate Ag complex binding as well as T cell activation has primarily involved structural studies on the alteration of residues within the six CDRs of the _α_- and _β_-chains that form the heterodimeric TCR molecule. These regions are surface-exposed loops that play a primary role in determining TCR Ag recognition specificity due to their extensive contacts with the exposed surfaces of peptide/MHC complexes. A cell-based study of a murine class II-restricted TCR directed against arsonate-conjugated peptides resulted in the identification of positions in _α_- and _β_-chain CDR1, 2, and 3 where amino acid substitutions (AAS)2 of alanine for the residues present in the native TCR resulted in diminished T cell recognition, as well as residues where substitutions had no apparent effect, but failed to reveal alanine substitutions that led to enhanced T cell recognition (10). A murine class I-restricted TCR isolated from the alloreactive 2C clone that recognizes target cell lines pulsed with a self peptide in the context of the murine H-2Kb class I allele as well as peptides associated with the allo-MHC molecule H-2Ld has also been extensively studied to determine the role of individual CDR residues on TCR affinity and functional activity (11). The results of initial studies conducted by generating 2C-variant TCRs containing alanine substitutions of CDR residues indicated that _α_- and _β_-chain CDR1 and CDR2 regions were the primary contributors to the binding energy of the 2C TCR for the cognate QL9/Ld Ag complex (12, 13). Screening of libraries of yeast variants containing 2C CDR1, CDR2, or CDR3 substitutions resulted in the identification of high-affinity variants containing substitutions in each of these regions that at the same time appeared to maintain specificity for the cognate peptide (14). High-affinity variants possessing affinities that were >100-fold higher than the native 2C TCR greatly enhanced the response of transduced CD8- T cell hybridomas to peptide-pulsed target cell lines (15). Transduced CD8+ T cells expressing a high-affinity 2C TCR variant but not the wild-type (WT) 2C TCR also recognized normal non-peptide-pulsed H-2Kb+ murine target cell lines, indicating that this variant confers upon CD8+ T cells the ability to cross-recognize endogenously processed self ligands that presumably fail to trigger with the WT 2C TCR due to the lower avidity of their cellular interactions (16). Using a similar approach, AAS were also identified in the CDR1 and CDR3 regions of a murine class II-restricted TCR that resulted in a >100-fold increase in the affinity of the TCR for the cognate peptide/MHC complex that did not appear to alter the cognate Ag complex specificity of T cells transfected with these TCRs (17).
Additional studies conducted using bacteriophage display mutation and selection technology have resulted in the identification of high-affinity variants of the 1G4 TCR that recognize the HLA-A*02-restricted peptide 157-165 of the NY-ESO-1 cancer-testis Ag (18). Expression of the NY-ESO-1 molecule has been demonstrated in a wide variety of tumor types that include melanoma, breast, prostate, lung ovarian, thyroid, and bladder cancer, but is limited in normal tissues to the testis (19). These TCR variants contain multiple AAS in the α and β CDR2 and CDR3 regions and possess affinities that range 1 million-fold between the WT receptor (10-30 _μ_M) and that of the tightest-binding TCR with an Ag affinity of 20-50 pM. A soluble monomeric version of the mutant TCR that possesses a 1 million-fold higher affinity than the WT TCR bound in vitro to the Ag complex of the NY-ESO-1 peptide and HLA-A*02 without binding to a panel of 14 noncognate peptide/HLA-A*02 complexes. Tetrameric complexes prepared from this high-affinity TCR bound to HLA-A*02+ T2 cells that were pulsed with the cognate peptide but failed to bind to T2 cells pulsed with a control peptide, providing further evidence for the Ag specificity of a TCR with an affinity in the picomolar range. In addition, binding studies indicate that soluble monomeric complexes of this high-affinity 1G4 TCR can be used to probe cells that endogenously express the NY-ESO-1/HLA-A*02 Ag complex (20). Recent studies analyzing the function of T cells that express 1G4 variants have indicated that high-affinity (5-84 nM) variants that lead to the Ag-specific activation of TCR gene-modified CD4+ T cells result in nonspecific activation of TCR gene-modified CD8+ T cells, whereas TCR variants with affinities nearer to that of the WT 1G4 TCR show enhanced reactivity in CD4+T cells without any apparent loss of specificity in CD8+ T cells (21).
The results presented in this report demonstrate that single or dual AAS in the 1G4 TCR CDR2 or CDR3 regions can provide relatively modest increases in TCR affinity that nevertheless significantly enhance the ability of CD4+ T cells to recognize the NY-ES0-1/HLA-A*02 Ag complex while maintaining (and in one case enhancing) Ag-specific reactivity in CD8+ T cells. The current studies were guided by previous bacteriophage display studies that identified high-affinity variants of the 1G4 TCR that contained multiple combined substitutions principally in the CDR2 and CDR3 regions of both TCR chains (18, 22). Extending this approach, CDR2_β_ chain individual amino acid substitution variants were subsequently screened in two distinct TCRs (DMF4 and DMF5) directed against the immunodominant HLA-A2-restricted peptide epitope corresponding to amino acids 27-35 of the melanoma Ag MART-1 (23), a gene product expressed in 80-90% of fresh, uncultured melanomas as well as cultured melanoma cell lines (24) but not in additional histologies or normal tissues with the exception of melanocytes. These studies identified substituted TCRs with enhanced Ag-specific reactivity in TCR gene-modified CD4+ T cells, and in one case resulted in the identification of a TCR that also showed enhanced Ag-specific reactivity in CD8+ T cells. The TCR RNA cotransfection screening system used here represents an important complementary methodology that assists the rapid selection of TCRs that possess enhanced functional activity in T cells.
Materials and Methods
Cell lines
Melanoma cell lines (624.38, 397-A2, 1300, 526, 888-A2, 526) were isolated from surgically resected melanoma metastases, the A375 and SK23 cell lines were obtained from the American Type Culture Collection, and the renal carcinoma cell line 2661R, isolated from a surgically resected metastatic renal carcinoma lesion, was provided by Dr. K.-i. Hanada (National Institutes of Health, Bethesda, MD). The H1299 small cell lung carcinoma cell line was provided by Dr. D. Schrump (National Institutes of Health, Bethesda, MD). The H1299 lung carcinoma cell line, as well as the 397 melanoma cell line and the MDA-MB435S breast cancer cell line, were transduced with transient retroviral supernatants generated from a recombinant HLA-A*0201-expressing retrovirus constructed in the CLNCX vector and produced as previously described (25) and were designated H1299-A2, 397-A2, and MDA435S-A2, respectively. Tumor cell lines were grown in RPMI 1640 medium (Invitrogen) containing 5% FBS plus glutamine.
Analysis of NY-ESO-1 and MART-1 gene expression
Expression of NY-ESO-1 in tumor and normal cell lines was evaluated using quantitative RT-PCR conducted with an intron-spanning forward primer, 5′-CGGCAACATACTGACTATCCG-3′, and a reverse primer, 5′-CTGGAGACAGGAGCTGATGGA-3′, with a DNA probe, 5′-CTGCAGACCACCGCCAAC TGCAG-3′ (Integrated DNA Technologies). The 624.38, 397-A2, 1300, 526, 888-A2, and A375 melanoma cell lines and the MDA-MB435S breast cancer cell line expressed levels of the NY-ESO-1 transcript that ranged between 2,100 and 330,000 copies per million copies of the GAPDH transcript, whereas the SK23 and 526 melanoma cell lines, the renal carcinoma cell line 2661R, and activated PBMC expressed <100 copies per million copies of GAPDH, and were designated as NY-ESO-1-. Expression of the MART-1 Ag in melanoma cell lines was previously evaluated using a quantitative RT-PCR assay (26).
Generation of WT and variant TCR constructs used for in vitro RNA-screening assays
The 1G4, DMF4, and DMF5 TCR variants used to carry out RNA transfection assays were generated using an overlapping PCR method (27) using the general strategy outlined in Fig. 1_A_ and the primers detailed in Table I. The PCR products were generated using 1 U/50 _μ_l of Phusion high-fidelity DNA polymerase (New England Biolabs) 0.5 _μ_M primers, and 0.5 _μ_M dNTPs by incubation at 98°C for 30 s, followed by 35 amplification cycles of 98°C for 20 s, 58°C for 20 s, and 72°C for 20 s and sequences as previously described (28). Codon-optimized versions of the 1G4 _α_- and _β_-chains (GeneArt) generated in the pCR-Script vector (Stratagene) were used to carry out the initial screening of the TCR variants using RNA transfection methods that have been previously described (29). The _α_1.1F and α_2.1R oligonucleotide primers (Table I) were used to generate a single PCR product that encoded the N terminus of the 1G4 CDR3_α chain. Overlapping fragments corresponding to the C-terminal region of the 1G4 α_-chain that encoded substituted residues were generated using individual forward oligonucleotide primers that encoded the CDR3_α AAS, designated _α_3.1F to _α_3.20F, and the _α_4.1 reverse primer, which consisted of 66 d(T) residues at the 5′ end followed by sequences complementary to the 3′ end of the α_-chain C region. The two PCR products were combined and the full-length construct generated using the α_1.1F and the α_4.1R primers. The 1G4 CDR2_β and CDR3_β chain variants, as well as the DMF4 and DMF5 CDR2_β chain variants were generated using the same strategy using primers detailed in Table I. Upstream primers contained either T3 or T7 RNA polymerase binding sites to allow in vitro transcription of RNA with either T3 or T7 RNA polymerase.
FIGURE 1.
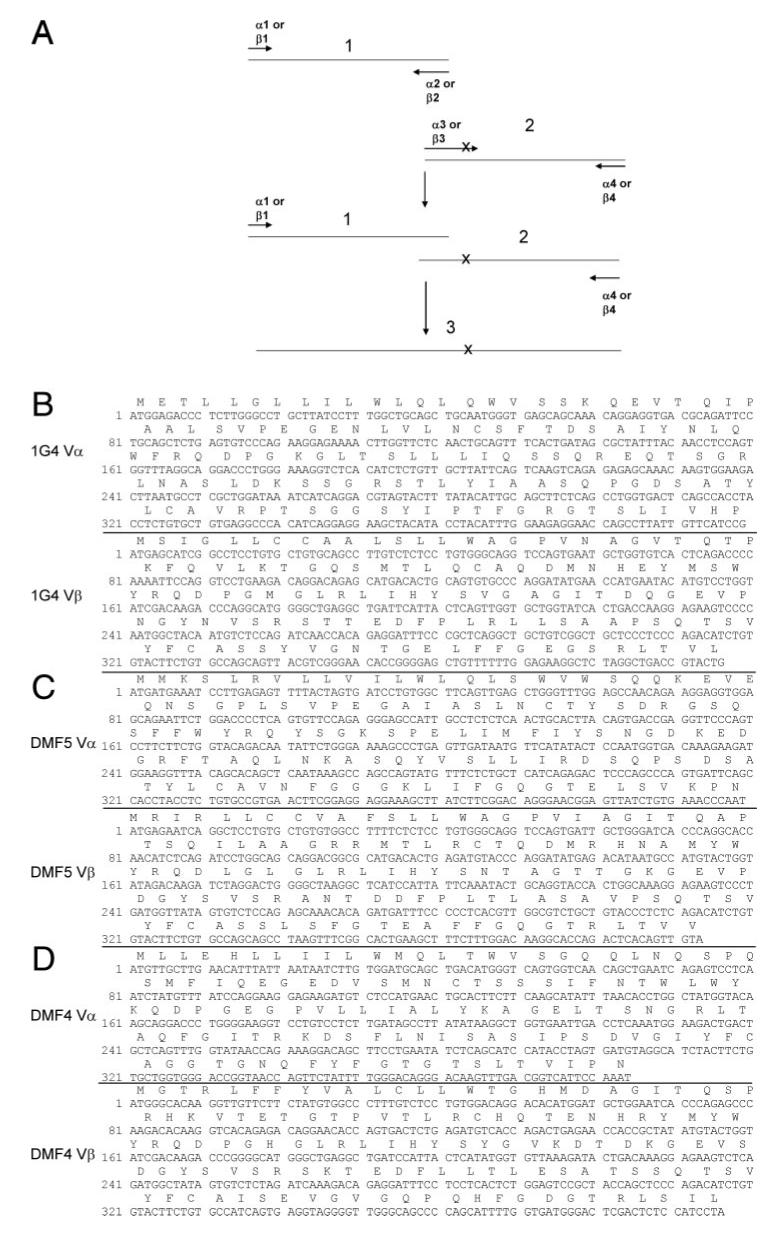
Generation of AAS variants of 1G4, DMF5, and DMF4 TCRs (A). Pairs of oligonucleotide primers designated _α_1 and _α_2 or _β_1 and _β_2 were used to amplify regions of TCR _α_-or _β_-chain sequences, respectively, that were upstream of the AAS. The AAS were encoded within a primer that partially overlapped with the _α_2 or _β_2 primer, designated _α_3 or _β_3, that was used to carry out a second PCR with primer _α_4 or _β_4. The products of the first two PCR were combined and amplified using primers _α_1 and _α_4 or _β_1 and _β_4. The nucleotide and amino acid sequences of the 1G4, DMF5, and DMF4 α and β TCR chains are presented in B, C, and D, respectively, and specific primers used to generate the AAS variants detailed in Table I.
Table I.
TCR variant primer sequencesa
| TCR Variant Primer Sequences | |
|---|---|
| 1G4 RNA constructs | |
| 1G4 CDR3_α_ | |
| _α_1.1F | 5′-ATATTAATACGACTCACTATAGGGCACCATGGAGACCCTGCTGGGC-3′ |
| _α_2.1R | 5′-CCGCACAGCGCACAGGTAGG-3′ |
| _α_3.1F | 5′-CCTACCTGTGCGCTGTGCGGGCCACCAGCGGCGGCAGCTAC-3′ |
| _α_3.2F | 5′-CCTACCTGTGCGCTGTGCGGCTGACCAGCGGCGGCAGCTAC-3′ |
| _α_3.3F | 5′-CCTACCTGTGCGCTGTGCGGCCTGCCAGCGGCGGCAGCTAC-3′ |
| _α_3.4F | 5′-CCTACCTGTGCGCTGTGCGGCCTCTGAGCGGCGGCAGCTAC-3′ |
| _α_3.5F | 5′-CCTACCTGTGCGCTGTGCGGCCTTACAGCGGCGGCAGCTAC-3′ |
| _α_3.6F | 5′-CCTACCTGTGCGCTGTGCGGCCTGAGAGCGGCGGCAGCTAC-3′ |
| _α_3.7F | 5′-CCTACCTGTGCGCTGTGCGGCCTAAGAGCGGCGGCAGCTAC-3′ |
| _α_3.8F | 5′-CCTACCTGTGCGCTGTGCGGCCTCCCAGCGGCGGCAGCTAC-3′ |
| _α_3.9F | 5′-CCTACCTGTGCGCTGTGCGGCCTACCGCCGGCGGCAGCTAC-3′ |
| _α_3.10F | 5′-CCTACCTGTGCGCTGTGCGGCCTACCCTGGGCGGCAGCTAC-3′ |
| _α_3.11F | 5′-CCTACCTGTGCGCTGTGCGGCCTACCTACGGCGGCAGCTAC-3′ |
| _α_3.12F | 5′-CCTACCTGTGCGCTGTGCGGCCTACCGAGGGCGGCAGCTAC-3′ |
| _α_3.13F | 5′-CCTACCTGTGCGCTGTGCGGCCTACCAAGGGCGGCAGCTAC-3′ |
| _α_3.14F | 5′-CCTACCTGTGCGCTGTGCGGCCTACCCCCGGCGGCAGCTAC-3′ |
| _α_3.15F | 5′-CCTACCTGTGCGCTGTGCGGCCTACCAGCGCCGGCAGCTAC-3′ |
| _α_3.16F | 5′-CCTACCTGTGCGCTGTGCGGCCTACCAGCGAGGGCAGCTAC-3′ |
| _α_3.17F | 5′-CCTACCTGTGCGCTGTGCGGCCTACCAGCAAGGGCAGCTAC-3′ |
| _α_3.18F | 5′-CCTACCTGTGCGCTGTGCGGCCTACCAGCCCCGGCAGCTAC-3′ |
| _α_3.19F | 5′-CCTACCTGTGCGCTGTGCGGCCCCTGCTGGGCGGCAGCTACATCC-3′ |
| _α_3.20F | 5′-CCTACCTGTGCGCTGTGCGGCCCCTGTACGGCGGCAGCTACATCC-3′ |
| _α_4.1R | 5′-T(66)AGCTGCTCCACAGCCGCAG-3′ |
| 1G4 CDR2_β_ | |
| _β_1.1F | 5′-ATATTAATACGACTCACTATAGGGATGAGCATCGGCCTGCTGTG-3′ |
| _β_2.1R | 5′-CACAGAGTAGTGGATCAGCCG-3′ |
| _β_3.1F | 5′-CGGCTGATCCACTACTCTGTGGCCGCCGGAATCACCGACCAGGGCGAG-3′ |
| _β_3.2F | 5′-CGGCTGATCCACTACTCTGTGGGAATCGGAATCACCGACCAGGGCGAG-3′ |
| _β_3.3F | 5′-CGGCTGATCCACTACTCTGTGGGAGCCCAGATCACCGACCAGGGCGAG-3′ |
| _β_3.4F | 5′-CGGCTGATCCACTACTCTGTGGGAGCCGGAACCACCGACCAGGGCGAG-3′ |
| _β_3.5F | 5′-CGGCTGATCCACTACTCTGTGGCCATCGGAATCACCGACCAGGGCGAG-3′ |
| _β_3.6F | 5′-CGGCTGATCCACTACTCTGTGGCCGCCGGAACCACCGACCAGGGCGAG-3′ |
| _β_3.7F | 5′-CGGCTGATCCACTACTCTGTGGGAATCGGAACCACCGACCAGGGCGAG-3′ |
| _β_3.8F | 5′-CGGCTGATCCACTACTCTGTGGCCATCGGAACCACCGACCAGGGCGAG-3′ |
| _β_4.1R | 5′-T(66)AGCCCCGGCTGTCCTTCC-3′ |
| 1G4 CDR3_β_ | |
| _β_1.1F | 5′-ATATTAATACGACTCACTATAGGGATGAGCATCGGCCTGCTGTG-3′ |
| _β_2.2R | 5′-ATAGCTGCTGGCGCAGAAGTAC-3′ |
| _β_3.9F | 5′-GTACTTCTGCGCCAGCAGCTATGCCGGCAACACCGGCGAGC-3′ |
| _β_3.10F | 5′-GTACTTCTGCGCCAGCAGCTATCTGGGCAACACCGGCGAGC-3′ |
| _β_3.11F | 5′-GTACTTCTGCGCCAGCAGCTATTACGGCAACACCGGCGAGC-3′ |
| _β_3.12F | 5′-GTACTTCTGCGCCAGCAGCTATGAGGGCAACACCGGCGAGC-3′ |
| _β_3.13F | 5′-GTACTTCTGCGCCAGCAGCTATGTGGCCAACACCGGCGAGC-3′ |
| _β_3.14F | 5′-GTACTTCTGCGCCAGCAGCTATGTGGGCGCCACCGGCGAGC-3′ |
| _β_3.15F | 5′-GTACTTCTGCGCCAGCAGCTATGTGGGCGACACCGGCGAGC-3′ |
| _β_3.16F | 5′-GTACTTCTGCGCCAGCAGCTATGTGGGCCTGACCGGCGAGC-3′ |
| _β_3.17F | 5′-GTACTTCTGCGCCAGCAGCTATGTGGGCAAGACCGGCGAGC-3′ |
| _β_3.18F | 5′-GTACTTCTGCGCCAGCAGCTATGTGGGCCCCACCGGCGAGC-3′ |
| _β_4.1R | 5′-T(66)AGCCCCGGCTGTCCTTCC-3′ |
| 1G4 retroviral constructs | |
| 1G4_α_ | |
| _α_1.2F | 5′-CACCATGGAGACCCTCTTGGGC-3′ |
| _α_1.3F | 5′-TACCATGGAGACCCTCTTGGGCCTGCTTATCCTTTG-3′ |
| _α_2.2R | 5′-CTGGTTCCTCTTCCAAATGTAGGTATGTAGCTTCCTCCTGATGTGGGCCTCACAGCACAGAGGTAGG-3′ |
| _α_2.3R | 5′-CTGCGGCTGTGGTCCAGCGGATCCGGAGCCACCAACTTCAGCCTGCTGAAGCAGGCCGGCG-3′ |
| _α_2.4R | 5′-CGCCGGCCTGCTTCAGCAGGCTGAAGTTGGTGGCTCCGGATCCGGACCGCTTGGCCCGGCTGGACCACAG\?\-3′ |
| _α_2.5R | 5′-CCTCACAGCACAGAGGTAGG-3′ |
| _α_3.21F | 5′-CTACATACCTACATTTGGAAGAGGAACCAGCCTTATTGTTCATCCGTATATCCAGAACCCTGACCC-3′ |
| _α_3.22F | 5′-CCTACCTCTGTGCTGTGAGGCCCCTGTACGGAGGAAGCTACATACCTACATTTGG-3′ |
| _α_4.2R | 5′-CGCCGGCCTGCTTCAGCAGGCTGAAGTTGGTGGCTCCGGATCCGCTTGGCCCGGCTGGACCACAGCCGCAG-3′ |
| _β_1.2F | 5′-CACCATGAGCATCGGCCTCCTGTG-3′ |
| _β_1.3F | 5′-CCTGCTGGAACAGGCCGGCGACGTGGAGGAGAACCCCGGCCCCATGAGCATCGGCCTCCTGTG-3′ |
| _β_2.2R | 5′-CTCCCCGGTGTTCCCGACGTAACTGCTGGCACAGAAGTAC-3′ |
| _β_2.3R | 5′-TGAGTAATGAATCAGCCTCAGC-3′ |
| _β_3.19R | 5′-CAGCAGTTACGTCGGGAACACCGGGGAGCTGTTTTTTGGAGAAG-3′ |
| _β_3.20F | 5′-GCTGAGGCTGATTCATTACTCAGTTGCCATCGGTATCACTGACCAAGGAGAAGTC-3′ |
| _β_3.21F | 5′-CCTGCTGAAGCAGGCCGGCGACGTGGAGGAGAACCCCGGCCCCATGAGCATCGGCCTCCTGTG-3′ |
| _β_4.2R | 5′-CTAGCCTCTGGAATCCTTTCTCTTG-3′ |
| _β_4.3R | 5′-TTGAATTCTAGCCTCTGGAATCCTTTCTCTTGACCATAGCCATC-3′ |
| DMF4 and DMF5 RNA constructs | |
| DMF4 CDR2_β_ | |
| _β_1.4F | 5′-GACTAATTAACCCTCACTAAAGGGACACCATGGGCACAAGGTTGTTCTTC-3′ |
| _β_2.4R | 5′-GTAATGGATCAGCCTCAGCC-3′ |
| _β_3.20F | 5′-GCTGAGGCTGATCCATTACTCATATGCCGTTAAAGATACTGACAAAGGAGAAGTC-3′ |
| _β_3.21F | 5′-GCTGAGGCTGATCCATTACTCATATGCCGCCAAAGATACTGACAAAGGAGAAGTC-3′ |
| _β_3.22F | 5′-GCTGAGGCTGATCCATTACTCATATGCCGTTGCCGATACTGACAAAGGAGAAGTC-3′ |
| _β_3.23F | 5′-GCTGAGGCTGATCCATTACTCATATGCCGTTAAAGCCACTGACAAAGGAGAAGTC-3′ |
| _β_4.4R | 5′-T(65)CAGAAATCCTTTCTCTTGACCATGGC-3′ |
| _β_1.5F | 5′-GACTAATTAACCCTCACTAAAGGGACACCATGAGAATCAGGCTCCTGTGCT-3′ |
| DMF5 CDR2_β_ | |
| _β_1.5F | 5′-GACTAATTAACCCTCACTAAAGGGACACCATGAGAATCAGGCTCCTGTGCT-3′ |
| _β_2.5R | 5′-TGAATAATGGATGAGCCTTAGC-3′ |
| _β_3.24F | 5′-GCTAAGGCTCATCCATTATTCAAATGCCGCAGGTACCACTGGCAAAGGAGAAGTCC-3′ |
| _β_3.25F | 5′-GCTAAGGCTCATCCATTATTCAAATATCGCAGGTACCACTGGCAAAGGAGAAGTCC-3′ |
| _β_3.26F | 5′-GCTAAGGCTCATCCATTATTCAAATACTATCGGTACCACTGGCAAAGGAGAAGTCC-3′ |
| _β_3.27F | 5′-GCTAAGGCTCATCCATTATTCAAATACTGTGGGTACCACTGGCAAAGGAGAAGTCC-3′ |
| _β_3.28F | 5′-GCTAAGGCTCATCCATTATTCAAATACTGCAGCCACCACTGGCAAAGGAGAAGTCC-3′ |
| _β_3.29F | 5′-GCTAAGGCTCATCCATTATTCAAATACTGCAGGTGCCACTGGCAAAGGAGAAGTCC-3′ |
| _β_3.30F | 5′-GCTAAGGCTCATCCATTATTCAAATACTGCAGGTATCACTGGCAAAGGAGAAGTCC-3′ |
| _β_4.3R | T(65)CAGAAATCCTTTCTCTTGACCATGGC |
Generation of retroviral constructs encoding 1G4 WT and variant TCRs
The 1G4 retroviral constructs were derived from native, noncodon-optimized α and β cDNA transcripts, as codon optimization did not enhance either the surface expression of the 1G4 TCR or the function of transfected T cells (data not shown). The native nucleotide sequences of the _α_- and _β_-chains expressed by the 1G4 T cell clone were provided by Dr. V. Cerundolo (John Radcliffe Hospital, Oxford, U.K.) (Fig. 1_B_). Noncodon optimized WT 1G4 α and β constructs were generated from TRAV-21 and TRBV6-5 cDNA clones identified within bulk tumor-infiltrating lymphocytes (28) by carrying out overlapping PCRs using primers as detailed in Table I containing sequences corresponding to the junctional regions of the WT 1G4 _α_- and _β_-chains. The 5′ end of the _α_-chain, which was generated by amplification of a TRAV-21 cDNA clone using the _α_1.2F primer and the _α_2.2R primer, and the 3′ end of the _α_-chain which was amplified using the _α_3.21F and _α_4.2R primers, were linked in a third PCR conducted with the _α_1.2F and _α_4.2R primers. Similarly, the 5′ end of the _β_-chain, which was generated by amplification of a TRBV-6-5 cDNA clone using the _β_1.2F and _β_2.2R primers, and the 3′ end of the _β_-chain which was amplified using the _β_3.19F and _β_4.2R primers, were linked in a third PCR conducted using the _β_1.2F and _β_4.2R primers. The resulting native 1G4 α and β products were cloned in the pCR4Blunt-TOPO vector using the Zero Blunt TOPO PCR Cloning kit (Invitrogen).
The 1G4 _α_- and _β_-chains were expressed in retroviral constructs that contained the “self-cleaving” P2A sequence (30) between the two gene products. A retroviral construct encoding both the 1G4 WT α and β TCR was generated by carrying out a PCR using the _α_1.3F primer, which contained an _Nco_I restriction endonuclease site followed by sequences corresponding to the 5′ coding region of the AV21 gene, and the _α_2.3R primer that contained sequences complementary to those encoding a portion of the P2A sequence, followed by sequences encoding a three amino acid GSG spacer and the 3′ end of the _α_-chain C region coding sequence. The 1G4 _β_-chain TCR was amplified using the _β_1.3F primer that encoded a portion of the P2A sequence followed by sequences corresponding to the beginning of the BV6-5 coding region, and the _β_4.3R primer corresponding to the 3′ end of the C_β_2 C region along with an _Eco_RI restriction endonuclease site. The two PCR products were then linked in a third PCR conducted with the _α_1.3F and _β_4.3R primers, and the resulting product digested with _Nco_I and _Eco_RI and ligated to the MSGV-1 retroviral expression vector which had been digested with the same enzymes (31). For generating the 1G4 _α_95:LY/WT β retroviral variant, the 5′ _α_-chain fragment was amplified from the native 1G4 WT α construct using _α_1.2F and _α_2.5R primers, and the 3′ _α_-chain fragment was generated using the _α_3.23F and _α_4.2R primers. These fragments were then mixed and joined by carrying out a PCR using the _α_1.2 and _α_4.2R primers, and the resulting fragment was cloned in the pCR4Blunt-TOPO vector. The resulting fulllength 1G4 _α_-chain fragment was then used as a template for a PCR that was conducted with the _α_1.3F primer and a second primer, _α_2.4R, that encoded a furin cleavage site for the removal of additional amino acids at the C terminus of the TCR _α_-chain (32) followed by an SGSG spacer and a portion of the P2A sequence. The WT _β_-chain was generated using the _β_1.2F and _β_4.2R primers, and the α_95:LY and WT_β products were gel purified, mixed and amplified using the _α_1.3F and a second primer, _β_4.3R corresponding to the 3′ end of the 1G4 _β_-chain with a single nucleotide substitution designed to remove the _Nco_I site normally present near the end of the _β_-chain sequence. The linked _α_- and β_-chain product was cloned as described above in the MSGV-1 retroviral expression vector. To generate the 1G4 WT_α/_β_51:AI variant, the 5′end of the _β_51:AI variant that was amplified using the _β_1.2F and _β_2.3R primers and the 3′ end of the _β_51:AI variant that was amplified using the _β_3.20F and _β_4.2R primers were joined in a third PCR conducted using the _β_1.2F and _β_4.2R primers and the resulting product was cloned in the pCR4Blunt-TOPO vector. The WT _α_-chain was generated using the _α_1.3F and _α_2.4R primers, the _β_51:AI product was amplified using the _β_1.3F and _β_4.3R primers, and the WT α and _β_51:AI products were gel purified, mixed, and amplified using the _α_1.3F and _β_4.3R primers. The linked _α_- and _β_-chain product was then cloned as described above in the MSGV-1 retroviral expression vector. Stable retroviral producer clones were generated by transfected into Phoenix Eco cells and the vector supernatants were subsequently used to transduce PG13 packaging cells. Following limiting dilution cloning and selection, high-titer PG13 producer cell clones were identified for each construct.
Genetic modification of T cells
Transient RNA transfection was conducted as previously described (29) using PBMC that were stimulated for between 5 and 21 days using 30 ng/ml of the soluble anti-CD3 Ab OKT-3. Stimulated PBMC were coelectroporated with in vitro-transcribed TCR _α_- and _β_-chain RNA constructs at a concentration of 2 _μ_g of RNA per 106 T cells and, 2-4 h later, coculture assays were initiated. Additional experiments were conducted by transducing PBMC as previously described (31) with retroviral supernatants derived from stable packaging cell clones. Briefly, OKT-3 anti-CD3 Ab-activated PBMC were transduced on days 2 and 3 by overnight culture on retronectin (CH-296, GMP grade; Takara Bio) coated, vector-preloaded plates. For the initial transfection experiments, CD8+ T cells were isolated using anti-CD8 magnetic beads (Miltenyi Biotec). For subsequent experiments that involved transfection or transduction of CD4+ and CD8+ T cells, CD4+ T cells were initially isolated using anti-CD4 magnetic beads (Miltenyi Biotec) using MS- or LS-positive selections columns (Miltenyi Biotec), resulting in cell populations of >95% CD4 single-positive T cells. The cells that passed through the CD4 selection column were then passed through LD columns to remove residual CD4+ T cells (Miltenyi Biotec). For some experiments, CD4-depleted populations were incubated with anti-CD8 magnetic beads, followed by a positive selection on MS or LS columns.
FACS analysis
Analysis of the expression of cell surface markers was conducted using FITC- or PE-conjugated Abs directed against the V_β_13.1 TCR chain (Immunotech), CD3, CD4, and CD8 (BD Biosciences). The relative log fluorescence of live cells was measured using either a FACScan or a FACScanto flow cytometer (BD Biosciences). Fluorescent peptide/HLA-A*02 tetramers were produced by the National Institutes of Health Tetramer Core Facility (Atlanta, GA).
51Cr-release and cytokine assays
The ability of TCR-transduced PBMC to lyse target cell lines was measured in 51Cr-release assays, as described previously (33). Cytokine release was measured following the incubation of between 5 × 104 and 105 T cells with 105 target cells in 200 _μ_l for 18 h, as previously described (34). For measurement of IL-2 release, cocultures of T cells and target cells were incubated with 5 μ_g/ml of a blocking Ab directed against IL-2R_α that was added to inhibit the uptake of IL-2 by T cells, which was provided by Dr. T. Waldmann (National Institutes of Health, Bethesda, MD).
Soluble protein production and measurements of TCR affinity
Soluble monomeric TCR heterodimers were produced as previously described (35), and measurement of TCR affinities was conducted using Biacore surface plasmon resonance assays, as previously described (18).
Results
Evaluation of the function of 1G4 TCR CDR_α_ chain variants in CD8+ and CD4+ T cells
Initial attempts to enhance the function of TCR gene-modified CD8+ and CD4+ T cells focused on variants of the 1G4 TCR α_-chain containing single or dual AAS in either the CDR2_α or CDR3_α_ regions. These constructs were cotransfected as in vitro RNA transcripts along with a 1G4 WT TCR _β_-chain RNA construct and used to attempt to identify variant 1G4 TCR _α_-chains that lead to enhanced function in TCR gene-modified T cells. The results of cytokine release screening assays indicated that CD8+ PBMC transfected with variant TCRs containing individual alanine substitutions within the 1G4 CDR2 α region failed to recognize Ag-expressing target cell lines (data not shown).
The amino acid residues 94-97 (PTSG) of the 1G4 _α_-chain, which are found at the amino terminus of the CDR3 α loop (36), were then targeted for substitution as mutations in this region had previously been identified by bacteriophage display experiments as important for high-affinity Ag binding (18 ). Cotransfection of CD8+ PBMC with a WT 1G4 _β_-chain construct and individual _α_-chain 1G4 TCR CDR3 substitution constructs of alanine or leucine for proline at position 94 (designated 1G4 _α_94:A and 1G4 _α_94:L, respectively) resulted in diminished recognition of NY-ESO-1:157-165 peptide-pulsed T2 cells (Fig. 2_A_). Substitutions of alanine, leucine, or glutamic acid for the threonine residue at position 95 appeared to lead to enhanced recognition of target cells pulsed with cognate peptide, whereas substitutions of tyrosine, lysine, or proline at residue 95, as well as alanine at residue 97 led to decreased reactivity in transfected CD8+ T cells. Substitution of alanine, leucine, tyrosine, glutamic acid, or lysine for the serine residue at position 96 all appeared to modestly enhance Ag recognition by CD8+ T cells, whereas proline substitution abolished Ag recognition (Fig. 2_A_). Peptide titration studies indicated that individual substitutions of leucine at position 95 and leucine or tyrosine at position 96 as well as dual substitutions at both these positions had minimal effects on the strength and specificity of CD8+ T cell recognition (Fig. 3_A_). In marked contrast, while CD4+ T cells expressing the WT 1G4 TCR failed to secrete significant levels of cytokine in response to T2 target cells pulsed with <10 nM of the NY-ESO-1 peptide, CD4+ cells expressing the _α_95:L, _α_95:LL, and _α_95:LY variants recognized targets pulsed with between 1 and 0.1 nM peptide (Fig. 3_C_).
FIGURE 2.
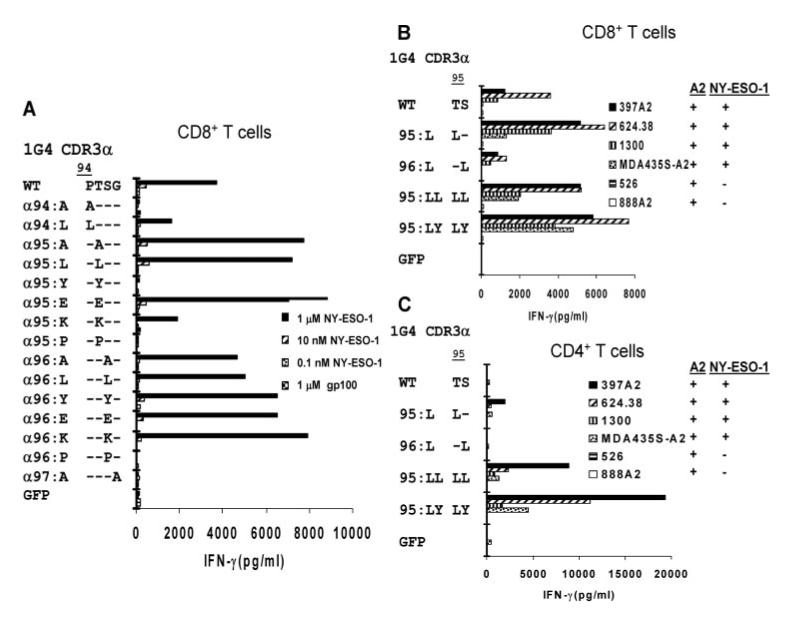
Rapid screening of NY-ESO-1/HLA-A*02-reactive TCR 1G4 CDR3_α_ variants in CD8+ and CD4+ T cells. T cells were activated with anti-CD3 Ab and isolated CD8+ (A and B) or CD4+ (C) T cells were cotransfected with RNA 1G4 TCR _α_-chain constructs encoding AAS within the CDR3 region along with a WT 1G4 TCR _β_-chain RNA construct. Transfected T cells were incubated overnight with T2 cells pulsed with the indicated concentrations of NY-ESO-1 peptide or with 1 _μ_M control peptide, gp100:154-162 (A), or with HLA-A*02+ melanoma and breast cancer target cell lines that did or did not also express the NY-ESO-1 transcript (B and C). The release of IFN-γ was measured the following day.
FIGURE 3.
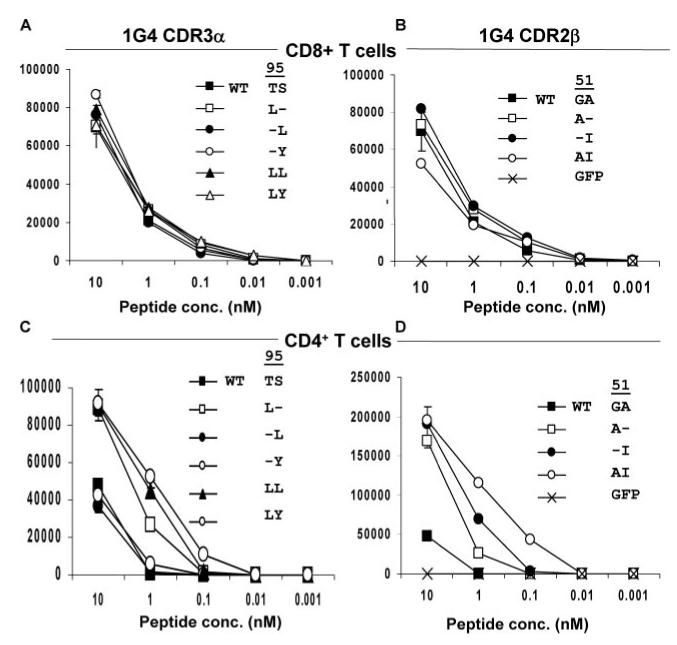
Peptide titration of responses of CD8+ and CD4+ T cells transfected with NY-ESO-1/HLA-A*02-reactive 1G4 WT, CDR2_β_, and CDR3_α_ TCR variants. T cells were activated with anti-CD3 Ab and isolated CD8+ (A and B) or CD4+ (C and D) T cells were cotransfected with RNA-encoding TCR constructs containing substitutions in the 1G4 _α_- or _β_-chains along with the complementary 1G4 WT TCR chain. The releases of IFN-γ in response to T2 cells pulsed with varying doses of the NY-ESO-1 peptide or with 10 nM control HLA-A*02-binding peptide from gp100 corresponding to amino acids 154-163 (KTWGQYWQV) were measured the following day.
The abilities of different tumor cell lines that naturally process and present the NY-ESO-1:157-165 epitope to stimulate CD8+ and CD4+ T cells expressing modified 1G4 TCRs containing single or dual AAS were then examined, and the results summarized in Table II. Cotransfection of CD8+ T cells with the WT 1G4 _β_-chain and either the CDR3 _α_95:L, _α_95:LL, or _α_95:LY constructs appeared to enhance the IFN-γ release response of transfected CD8+ T cells to NY-ESO-1 Ag-expressing HLA-A*02+ melanoma and breast cancer target cell lines, whereas the response of CD8+ T cells transfected with the _α_96:L was diminished in comparison with CD8+ T cells transfected with the WT α/β TCR (Fig. 2_B_). The CD4+ T cells transfected with the WT 1G4 TCR did not respond to NY-ESO-1+ and HLA-A*02+ tumor cell lines, and CD4+ T cells transfected with the _α_95:L and _α_96:L constructs also largely failed to recognize these same target cells. In marked contrast, CD4+ T cells transfected with the _α_95:LL and _α_95:LY constructs secreted highly elevated levels of IFN-γ in response to NY-ESO-1+/HLA-A*02+ tumor cell lines (Fig. 2_C_). At the same time, responses directed against NY-ESO-1- melanoma lines were not observed in CD8+ or CD4+ T cells transfected with either the WT or the variant TCRs, indicating that these TCR substitutions maintain Ag-specific tumor cell line recognition (Fig. 2, B and C).
Table II.
TCR amino acid substitutions that enhance tumor reactivity of gene-modified T cells
| CDR2a | CDR3 | ||||
|---|---|---|---|---|---|
| Ag | TCR | α | β | α | β |
| NY-ESO-1:157-165 | 1G4 | None | 51:A51:G to A52:I52:A to I51:AI51:G to A52:A to I | 95:LL95:T to L96: S to L95:LY95:T to L96:S to Y | None |
| MART-1:27-35 | DMF5 | None | 54:A54:T to A | None | NT |
| MART-1:27-35 | DMF4 | NTb | 51:A51:G to A | NT | NT |
Evaluation of the function of 1G4 TCR CDR _β_-chain variants in CD8+ and CD4+ T cells
An analysis of responses to peptide-pulsed T2 cells of CD8+ T cells transfected with 1G4 TCR variants failed to identify 1G4 β_-chain CDR3 substitution variants that greatly enhanced T cell function; most resulted in a diminished T cell responses (Fig. 4_A_). Previously, a TCR variant was identified using a bacteriophage display system that contained substitutions of AIQT for GAGI at the 1G4 WT CDR2_β positions 51-54. When combined with the WT 1G4 _α_-chain, this variant TCR possessed an ∼600-fold higher Ag affinity than the WT TCR (22). To probe the effects on T cell recognition of individual substitutions within this region, constructs containing single and double AAS were generated and evaluated in functional T cell assays. Cotransfection of CD8+ T cells with the WT 1G4 α construct along with _β_-chain constructs encoding single substitutions of either alanine for glycine at position 51 (_β_51:A), isoleucine for alanine at position 52 (_β_52:I), or threonine for isoleucine at position 54 (_β_54:T) led to enhanced recognition of peptide-pulsed target cells, whereas the _β_53:Q substitution led to diminished peptide recognition (Fig. 4_B_). Cotransfection of _β_-chain constructs encoding dual substitutions of alanine at position 51 plus isoleucine at position 52 (_β_51:AI), alanine at position 51 plus threonine at position 54 (_β_51:A-T) or isoleucine at position 52 plus threonine at position 54 (_β_52:I-T) along with the WT _α_-chain construct did not further enhance the activity of transfected CD8+ T cells when compared with cells transfected with the single AAS. Cotransfection of a 1G4 _β_-chain construct encoding three substitutions at positions 51, 52, and 54 (_β_51:AI-T) along with the WT _α_-chain, however, led to the nonspecific recognition of HLA-A*02+ T2 cells when pulsed with a noncognate control peptide (Fig. 4_B_).
FIGURE 4.
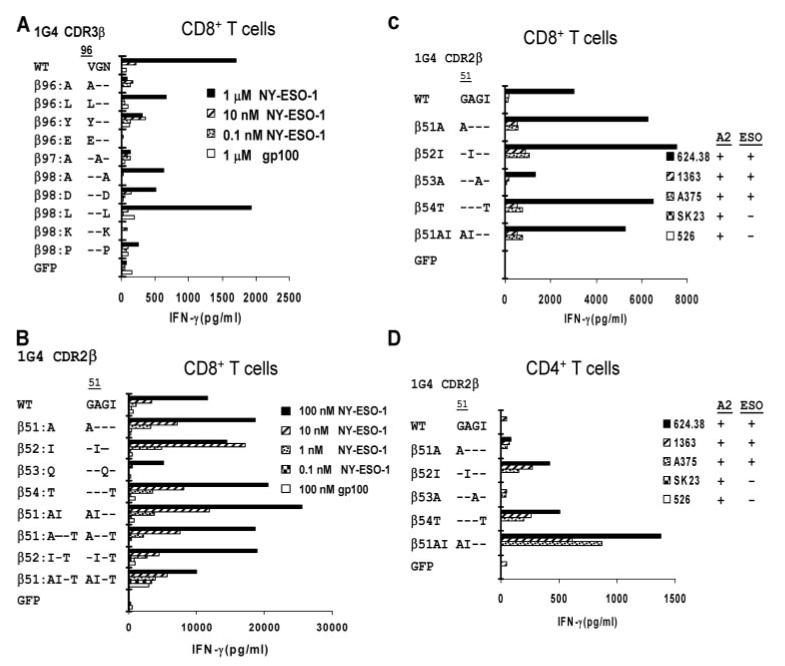
Rapid screening of NY-ESO-1/HLA-A*02-reactive TCR 1G4 CDR2 and CDR3 β variants in CD8+ and CD4+ T cells. T cells were activated with anti-CD3 Ab and isolated CD8+ (A-C) or CD4+ (D) T cells were cotransfected with RNA 1G4 TCR _β_-chain constructs encoding AAS within the CDR2 or CDR3 regions along with a WT 1G4 TCR _α_-chain RNA construct. Transfected T cells were incubated overnight with T2 cells pulsed with the indicated concentrations of NY-ESO-1 peptide or a control peptide, gp100:154-162 (A and B), or HLA-A*02+ melanoma target cell lines that did or did not also express NY-ESO-1 transcript (C and D). The release of IFN-γ was measured the following day.
The levels of IFN-γ released by CD8+ T cells that were transfected with 1G4 TCR CDR2_β_ chain variants containing single substitutions of alanine at position 51 or isoleucine at position 52 or both substitutions combined were then assessed. Responses to T2 cells pulsed with titered doses of the cognate peptide were not significantly different from those of CD8+ T cells transfected with the 1G4 WT TCR (Fig. 3_B_). The CD4+ T cells that were transfected with the WT 1G4 TCR responded only to the highest dose of peptide tested (10 nM) whereas CD4+ T cells transfected with the _β_51:A and _β_52:I variants responded to T2 target cells pulsed with 1 nM peptide (Fig. 3_D_), a 10-fold increase in sensitivity. Furthermore, CD4+ T cells transfected with the _β_51:AI variant responded to target cells pulsed with 0.1 nM peptide, equivalent to the lowest dose of peptide that resulted in significant cytokine release from CD8+ T cells and a 100-fold increase in the sensitivity of CD4+ T cells relative to that observed with 1G4 WT TCR.
Evaluation of the effects of CDR2_β_ substitutions on tumor cell line reactivity indicated that transfected CD8+ T cells expressing either the 1G4 TCR _β_51:A, _β_52:I, _β_54:T, or _β_51:AI variants, in conjunction with the WT 1G4 _α_-chain, enhanced the specific recognition of the HLA-A*02+ and NY-ESO-1+ melanoma cell lines (Fig. 4_C_). Transfection of CD4+ T cells with the WT 1G4 TCR did not lead to significant recognition of these melanoma cell lines, whereas CD4+ T cells cotransfected with either the _β_52:I or _β_54:T constructs, along with the 1G4 WT_α_-chain construct, released significant levels of IFN-γ in response to cognate tumor target cell lines (Fig. 4_D_). Transfection of CD4+ T cells with the _β_51:AI dual substitution construct further enhanced the levels of cytokine released in response to cognate tumor cell lines compared with cells that were transfected with TCRs containing single AAS (Fig. 4_D_).
Tumor cell line lysis mediated by T cells expressing 1G4 TCR variants
The effects of TCR substitutions on the lytic function of transfected CD8+ and CD4+ T cells were then evaluated. Cotransfection of CD8+ T cells with 1G4 _β_51:AI or _α_95:LL or _α_95:LY TCR chains in conjunction with the appropriate WT 1G4 TCR partner chain appeared to modestly enhance lysis of the two HLA-A*02+ and NY-ESO-1+ melanoma cell lines, 624.38 and A375 (Fig. 5_A_). The HLA-A*02+ but NY-ESO-1- renal carcinoma cell line 2661R was also lysed to a low but significant degree by CD8+ T cells transfected with the 1G4 _β_51:AI variant but not by those expressing the WT, _α_95:LL, or _α_95:LY constructs (Fig. 5_A_). None of the target cell lines were lysed by CD4+ T cells transfected with the WT 1G4 TCR, whereas CD4+ T cells transfected with the _β_51:AI, _α_95:LL, and _α_95:LY constructs demonstrated significant lysis of the 624.38 melanoma target cell line (Fig. 5_B_). A low level of lysis of the A375 melanoma cell line was also observed with CD4+ T cells transfected with the _β_51:AI or _α_95:LY constructs, while the NY-ESO-1- 2661R renal carcinoma cell line was not lysed by any of the CD4+ T cell TCR transfectants (Fig. 5_B_).
FIGURE 5.
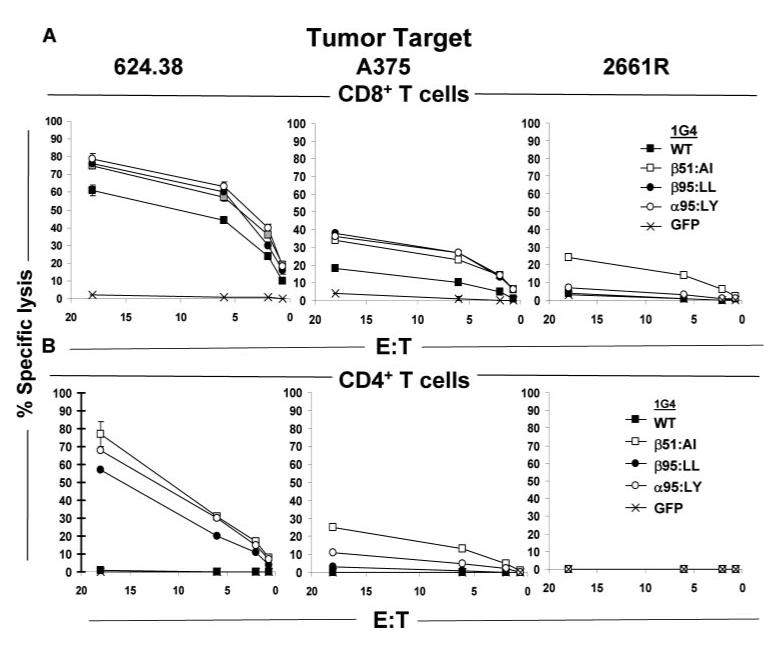
Effects of NY-ESO-1/HLA-A*02-reactive TCR 1G4 CDR3_α_ and CDR2_β_ variants on the cytolytic activity of CD8+ and CD4+ T cells. The cytolytic activities of anti-CD3 Ab stimulated CD8+ (A) and CD4+ (B) T cells cotransfected with 1G4 WT and variant TCRs to the HLA-A*02+ and NY-ESO-1+ melanoma cell lines 624.38 and A375 as well as the response to the HLA-A*02+, but the NY-ESO-1- renal cell carcinoma line 2661R was measured in standard 4-h 51Cr-release cell lysis assays.
IFN-γ release by T cells stably transduced with 1G4 TCR dual substitution variants
To further examine the effects of 1G4 TCR dual AAS on T cell recognition, recombinant retroviruses coexpressing either the WT α plus β 1G4, the _α_95:LY plus WT β, or the WT α plus _β_51:AI TCR chains were used to stably transduce populations of CD8+ and CD4+ T cells. The percentage level of TCR expressed on the surface of CD8+ T cells transduced with the WT 1G4 TCR chains, as determined by anti-V_β_13 (also known as VB6-5; IMGT) Ab staining of the 1G4 _β_-chain V region, appeared to be somewhat lower than the levels obtained with the _α_95:LY and _β_51:AI TCR constructs (Fig. 6_A_), a difference that was not, however, consistently observed in subsequent experiments (data not shown). In contrast, nearly identical levels of anti-V_β_13 Ab staining were observed with all the TCR-transduced CD4+ T cells (Fig. 6_B_). Further analysis indicated that while similar percentage levels of NY-ESO-1/HLA-A*02 tetramer binding were observed on CD8+ T cells transduced with the WT, _α_95:LY, or _β_51:AI TCRs, the levels of tetramer staining were significantly lower on CD4+ T cells that were transduced with the WT 1G4 TCR than on CD4+ T cells that were transduced with either the _α_95:LY or _β_51:AI TCRs (Fig. 6, A and B).
FIGURE 6.
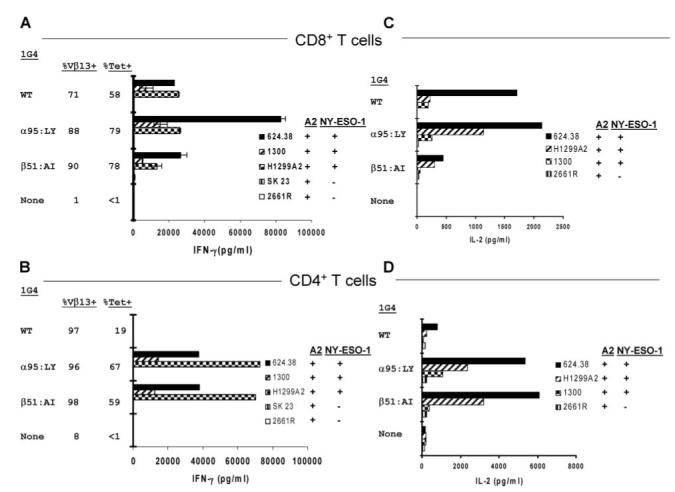
Effects of NY-ESO-1/HLA-A*02-reactive 1G4 WT and variant TCRs on CD8+ and CD4+ T cell activity following retroviral transduction. Anti-CD3 Ab stimulated PBMC were transduced on days 2 and 3 with retroviral supernatants that coexpressed either the 1G4 WT α and β, α_95:LY and WT β, or WT_α and _β_51:AI TCR chains. Ten days following anti-CD3 Ab stimulation, CD8+ (A and C) and CD4+ (B and D) T cells were separated and were tested 4 days later for their ability to release IFN-γ (A and B) or IL-2 (C and D) in response to melanoma, lung carcinoma, and renal cell carcinoma cell lines. The results presented are representative of those obtained following the transduction of three samples of PBMC. Cell surface TCR expression was evaluated by FACS analysis of V_β_13 as well as NY-ESO-1/HLA-*02 tetramer staining 10 days following anti-CD3 Ab stimulation, and coculture assays were initiated on day 13.
The CD8+ T cells that were transduced with retroviruses encoding either the 1G4 WT, _α_95:LY, or _β_51:AI TCRs generated high levels of IFN-γ when stimulated by the NY-ESO-1+/HLA-A*02+ melanoma cell lines 624.38 and 1300, as well as by the NY-ESO-1+/HLA-A*02+ small cell lung carcinoma cell line, H1299-A2 (Fig. 6_A_). In marked contrast, the CD4+ T cells that were transduced with the _α_95:LY and _β_51:AI TCR constructs generated high levels of IFN-γ in response to all three HLA-A*02+/NY-ESO-1+ tumor cell lines, whereas no response was seen with CD4+ T cells transduced with the 1G4 WT TCR retrovirus (Fig. 6_B_).
The results of multiple repeated retroviral TCR transduction experiments were then evaluated. The observation that CD8+ T cells transduced with the _α_95:LY/WT β TCR released higher levels of IFN-γ in response to the NY-ESO-1+/HLA-A*02+ tumor target cell lines than those expressing the WT α/β TCR (Fig. 5_A_) was confirmed in the overall analysis of the results of multiple experiments (ratio of 2.2 ± 0.34, mean ± SEM), a statistically significant level of enhancement (p < 0.01 using a paired _t_ test, 36 pairstested). At the same time, the multiply measured responses of CD8+ T cells transduced with the WT _α_/_β_51:AI TCR pair did not differ significantly from the WT _α_/_β_ TCR (Fig. 6_A_; ratio of 1.2 ± 0.24, _p_ > 0.4, 34 pairs tested). The IFN-γ responses of CD4+ T cells transduced with either _α_95:LY/WT β or WT α/_β_51:AI TCRs, however, were greatly enhanced relative to CD4+ T cells transduced with the WT 1G4 α/β TCR (Fig. 6_B_; ratio of 140 ± 50, p < 0.01, and 150 ± 52, _p_ < 0.02, respectively, 33 pairs tested). A direct comparison of responses induced by these two dual substitution TCR variants revealed that the responses of CD8+ T cells transduced with the _α_95:LY/WT _β_ TCR was significantly enhanced relative to those of cells transduced with the WT _α_/_β_51:AI variant (Fig. 6_A_; ratio of 3.1 ± 0.67, _p_ < 0.01, 34 pairs tested). In contrast, significant differences were not observed between the responses of CD4+ T cell transduced with the _α_95:LY/WT _β_ and WT _α_/_β_51:AI TCR constructs (Fig. 6_B_; ratio of 1.2 ± 0.24, _p_ > 0.1, 33 pairs tested).
Expression of the WT α/_β_51:AI TCR in CD8+ T cells also resulted in a low but significant level of cross-reactivity with Ag-negative target cells. The CD8+ T cells that expressed the WT α/_β_51:AI TCR released 730 pg/ml IFN-γ in response to the HLA-A*02+ but NY-ESO-1- 2661R renal cancer cell line (Fig. 6_A_), whereas control untransduced CD8+ T cells and the WT and _α_95:LY TCR transductants released only background levels of <30 pg/ml IFN-γ in response to this cell line. These results were consistent with the low but measurable levels of nonspecific lysis of 2661R cells mediated by CD8+ T cells that were cotransfected with the WT α and β_51:AI RNA constructs (Fig. 5_A_), as well as experiments in which CD8+ T cells expressing the WT_α/_β_51:AI variant recognized, to a variable extent, this as well as additional HLA-A*02+ but NY-ESO-1- tumor cell lines (data not shown).
IL-2 release by T cells stably transduced with 1G4 TCR dual substitution variants
The ability of stable TCR-transduced T cells to produce IL-2 in response to tumor cell line targets was then evaluated. The results demonstrate that similar levels of IL-2 were secreted by CD8+ T cells retrovirally transduced with the 1G4 WT TCR and the 1G4 _α_95:LY variant in response to the NY-ESO-1+ and HLA-A*02+ melanoma cell lines 624.38 and 1300, whereas IL-2 secretion in response to the NY-ESO-1+ and HLA-A*02+ small cell lung carcinoma cell line, H1299-A2, appeared to be enhanced in CD8+ T cells transduced with the 1G4 _α_95:LY variant (Fig. 6_C_). Lower levels of IL-2 were released from T cells transduced with the _β_51:AI variant in response to two of the three NY-ESO-1+ and HLA-A2+ target cell lines, which is consistent with the relatively lower levels of IFN-γ released by CD8+ T cells transduced with the _β_51:AI variant (Fig. 6_A_).
The levels of IL-2 released by CD4+ T cells that were transduced with the _α_95:LY/WT β and WT α/_β_51:AI constructs in response to the NY-ESO-1+ and HLA-A2+ tumor targets were similar, both being significantly higher than those observed with CD4+ T cells transduced with the WT 1G4 α/β TCR (Fig. 6_D_). Again, this was similar to the IFN-γ responses of CD4+ T cells transduced with these constructs (Fig. 6_B_).
Correlation of Ag-binding affinity of 1G4 TCR variants with T cell activation
The observations that one or two amino acid substitutions in the 1G4 _α_- or _β_-chain can enhance the function and tetramer staining of TCR gene-modified T cells indicates that the Ag affinities of these modified TCRs may be higher than the 1G4 WT TCR. These differences may be particular marked in CD4+ T cells where the increase in T cell avidity is not obscured by the simultaneous binding of the CD8 α/β coreceptor to the HLA class I protein.
Direct binding measurements of the Ag affinities of soluble TCR proteins, which were conducted by surface plasmon resonance, demonstrated that the monomeric WT 1G4 TCR heterodimer possesses an Ag complex affinity of 9.3 _μ_M (Table III). The _α_95: LY/WT β 1G4 TCR variant possesses an affinity of 730 nM, and the WT α/β_52:I and WT_α/_β_51:AI variants possess Ag affinities of 1.1 _μ_M and 280 nM, respectively (Table III). These increased affinities are primarily due to decreased Ag dissociation rates as all of the WT and variant TCRs possess similar rates of association with NY-ESO-1/HLA-A*02 Ag complex (Table III). These findings are consistent with IFN-γ response results of peptide titration assays conducted in TCR-transfected CD4+ T cells (Fig. 3, C and D) as well as with the increased levels of tetramer staining of CD4+ T cells expressing either the _α_95:LY or _β_51:AI variant TCRs in comparison with cells expressing the WT 1G4 TCR (Fig. 6_B_).
Table III.
Affinity of 1G4 WT TCR and TCR variants
| α | β | Kon (M-1s-1)a | Koff (s-1) | KD | Monomer _t_1/2 (seconds) |
|---|---|---|---|---|---|
| WT | WT | 3.4 × 104 | 0.31 | 9.3 _μ_M | 2.2 |
| WT | 52:I | 6.4 × 104 | 7.2 × 10-2 | 1.1 _μ_M | 9.6 |
| 95:LY | WT | 4.9 × 104 | 3.6 × 10-2 | 730 nM | 19 |
| WT | 51:AI | 5.8 × 104 | 1.7 × 10-2 | 280 nM | 41 |
These results, taken together with those presented in an earlier study of modified 1G4 TCR variants (21), demonstrate that the TCR variants that are capable of mediating enhanced tumor recognition by TCR gene-modified CD4+ T cells possess higher affinities for the NY-ESO-1/HLA-A*02 complex than the WT TCR but suggest that TCRs with affinities equal to or greater than the WT α/_β_51:AI TCR (280 nM) can lead to impaired function in CD8+ T cells.
Identification of single substitution variants in two MART-1/HLA*02 reactive TCRs, DMF4 and DMF5, that enhance T cell function
Attempts were then made to determine whether CDR substitutions that enhance function could be replicated in TCRs directed against a different T cell tumor Ag without prior knowledge of key residues gained from mutated TCR display or from atomic structure. To this end, CDR substitutions were introduced into two distinct WT TCRs (DMF4 and DMF5), both of which recognize the immunodominant MART-1:27-35 HLA-A*02-restricted epitope. The AAS variants generated in these TCRs are detailed in Table II and Fig. 7. These TCRs were isolated from two prevalent clonotypes that were present within a population of tumor infiltrating lymphocytes derived from a single patient. Administration of this lymphocyte population was associated with a positive response to clinical melanoma following T cell adoptive immunotherapy, and one of these clonotypes (DMF4) demonstrated a high level of persistence following adoptive transfer (37). Previously, the DMF5 TCR was identified as CD8 coreceptor independent and the most potent TCR among a panel of 24 MART-1 Ag-specific TCRs (including the DMF4 TCR) based upon their abilities to recognize Ag complex in both TCR gene-modified CD8+ and CD4+ T cells (38). In contrast to the 1G4 TCR which contains the AV21 and BV6-5 TCR variable chains, the DMF5 TCR is comprised of the AV12-2 and BV6-4 variable chains (IMGT TCR nomenclature).
FIGURE 7.
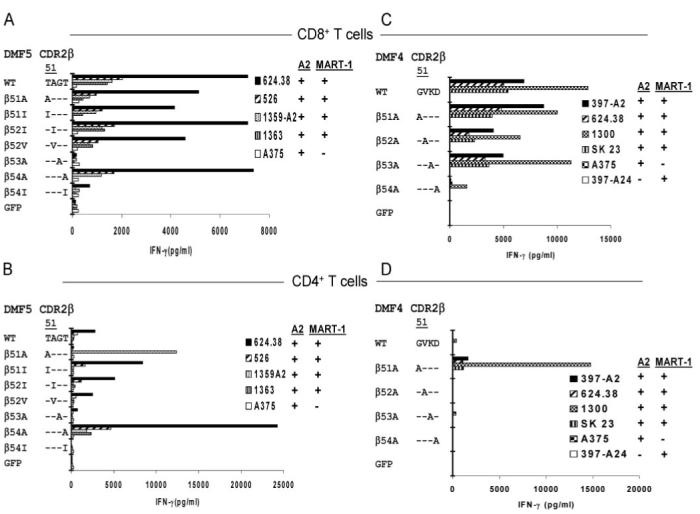
Rapid screening of MART-1/HLA-A*02-reactive TCR DMF5 and DMF4 WT and CDR2_β_ variants. T cells were activated with anti-CD3 Ab and isolated CD8+ (A and C) and CD4+ (B and D) T cells were cotransfected with RNA constructs encoding the WT α, along with either the WT β_-chain or CDR2_β variant chains generated from either the DMF5 TCR (A and B) or the DMF4 TCR (C and D). Transfected T cells were evaluated the following day for their ability to generate IFN-γ in response to melanoma target cell lines.
Variants of the DMF5 TCR (Fig. 1_C_) that were initially generated contained single AAS of the CDR2_β_ residues 51-54 corresponding to the sequence TAGT in the WT TCR. Populations of CD4+ T cells that were transfected with the WT DMF5 TCR recognized two of the four MART-1+ and HLA-A*02+ melanoma cell lines that were tested, although these responses were substantially lower than those observed in CD8+ T cells also transfected with the WT TCR (Fig. 7, A and B). Transfection of CD4+ T cells with the DMF5 β_54:A variant substantially enhanced the levels of IFN-γ released in response to the four cognate melanoma cell lines while the response of CD8+ T cells transfected with the same variant was not significantly altered (Fig. 7, A and B). The results of subsequent assays conducted with a wide variety of melanoma cell lines that either did or did not express HLA-A*02 or MART-1 provided further evidence that the recognition mediated by the DMF5 β_54:A variant was both HLA-A*02 and MART-1 specific in both CD8+ and CD4+ T cells (Table IV). A substitution of alanine for the threonine residue at DMF5 CDR2_β chain position 51 significantly enhanced the CD4+ T cell response to one of the four MART-1+ and HLA-A*02+ melanoma cell lines, 1359-A2, while at the same time leading to diminished recognition of additional MART-1+ and HLA-A*02+ tumor cell lines (Fig. 7, A and B). The CD8+ T cells that were transfected with the DMF5 WT_α/_β_51:A variant appeared to recognize the appropriate target cells, although these responses appeared to be somewhat diminished relative to those of cells transfected with the DMF5 WT TCR. The analysis of responses directed against additional tumor targets, however, indicated that the _β_51:A substitution may have lead to the generation of an alloreactive TCR, as both CD8+ as well as CD4+ T cells expressing this TCR variant were also stimulated by the HLA-A*02-negative 1359 melanoma cell line (Table IV). Thus, the ability of CD8+ T cells transfected with the DMF5 _β_51:A variant to recognize the cognate peptide Ag may have obscured any alloreactivity directed against the 1359-A2 tumor cell line, which was only revealed when the HLA-A2- 1359 tumor cell line was tested.
Table IV.
Effects of DMF5 CDR2β AAS on the function of gene-modified CD8+ and CD4+ T cells
| DMF5 CDR2_β_ Variant | ||||||
|---|---|---|---|---|---|---|
| Stimulator | WT | _β_51:A | β:54A | |||
| T2+: | IFN-γ (pg/ml) | GFP | ||||
| CD8+ T cells | ||||||
| gp100:154-162 1 _μ_M | <30 | <30 | <30 | <30 | ||
| MART-1 1 _μ_M | 4740 | 7770 | 5180 | <30 | ||
| MART-1 0.1 _μ_M | 1420 | 680 | 1320 | <30 | ||
| MART-1 0.01 _μ_M | 53 | <30 | 56 | <30 | ||
| MART-1 1 nM | <30 | <30 | <30 | <30 | ||
| Melanomas | HLA-A2 | MART-1 | ||||
| 397-A2 | + | + | 2030 | 1340 | 2160 | 27 |
| 526 | + | + | 1083 | 680 | 671 | <30 |
| 624.38 | + | + | 3660 | 2650 | 3470 | 21 |
| 888-A2 | + | + | 9570 | 7360 | 7020 | 45 |
| 1300 | + | + | 6170 | 4180 | 5380 | 55 |
| 1359-A2 | + | + | 175 | 157 | 207 | 57 |
| 1363 | + | + | 496 | 209 | 595 | <30 |
| SK23 | + | + | 4090 | 2830 | 3790 | 14 |
| 397-A24 | - | + | <30 | <30 | <30 | <30 |
| 624.28 | - | + | <30 | <30 | <30 | <30 |
| 888 | - | + | <30 | <30 | <30 | <30 |
| 1359 | - | + | <30 | 270 | <30 | <30 |
| A375 | + | - | <30 | 68 | 32 | 30 |
| T alone | <30 | <30 | <30 | <30 | ||
| CD4+ T cells | ||||||
| gp100:154-162 1 _μ_M | <30 | <30 | <30 | <30 | ||
| MART-1 1 _μ_M | 1180 | <30 | 4580 | <30 | ||
| MART-1 0.1 _μ_M | 5 | <30 | 399 | <30 | ||
| MART-1 0.01 _μ_M | <30 | <30 | <30 | <30 | ||
| MART-1 1 nM | <30 | <30 | <30 | <30 | ||
| Melanomas | HLA-A2 | MART-1 | ||||
| 397-A2 | + | + | 206 | <30 | 1028 | <30 |
| 526 | + | + | 35 | <30 | 351 | <30 |
| 624.38 | + | + | 250 | <30 | 1287 | <30 |
| 888-A2 | + | + | 2799 | 650 | 4285 | <30 |
| 1300 | + | + | 827 | <30 | 2370 | <30 |
| 1359-A2 | + | + | <30 | 862 | 32 | <30 |
| 1363 | + | + | <30 | <30 | <30 | <30 |
| SK23 | + | + | 187 | <30 | 732 | <30 |
| 397-A24 | - | + | <30 | <30 | <30 | <30 |
| 624.28 | - | + | <30 | <30 | <30 | <30 |
| 888 | - | + | <30 | <30 | <30 | <30 |
| 1359 | - | + | <30 | 3690 | <30 | <30 |
| A375 | + | - | 53 | 20 | 55 | 64 |
| T alone | <30 | <30 | <30 | <30 |
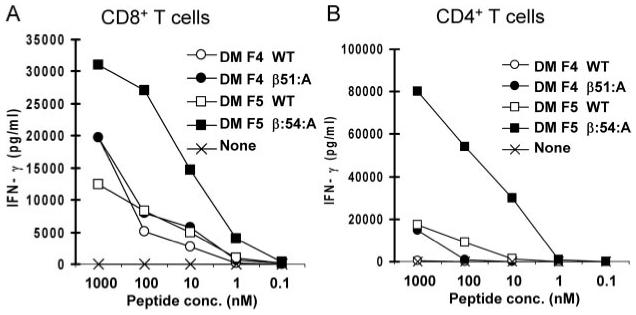
Further studies were undertaken to evaluate the function of TCR variants containing substitutions of DMF5 CDR2_α_ residues 50 to 54 as well as the CDR3_α_ residues 93 and 94; however, none of these variants enhanced the function of TCR gene-modified T cells (data not shown). Taken together, these results indicate that the DMF5 _β_-chain residue 54 represents a critical residue that can be altered to enhance the specific function of this TCR in CD4+ T cells.
In an attempt to evaluate the generality of this approach, individual CDR AAS were generated in a second MART-1/HLA-A*02-reactive TCR, DMF4, that contained the AV35 and BV10-3 variable chain regions (Fig. 1_D_), both of which differ from the DMF5 clone. Single alanine AAS were generated across the CDR2 _β_-chain residues 51-54, which corresponds to GVKD in the DMF4 WT _β_-chain. Responses directed against HLA-A*02+ and MART-1+ melanoma cell lines were not significantly altered by transfection of CD8+ T cells with the WT DMF4 _α_-chain along with variants containing single substitutions of alanine at either residue positions 51, 52, or 53 of the DMF4 _β_-chain (Fig. 7_C_). In contrast, these responses were greatly reduced in CD8+ T cells transfected with a DMF4 β_-chain construct containing a substitution of alanine for aspartic acid residue at position 54. Relatively high levels of IFN-γ were released from CD4+ T cells that were transfected with the DMF4 WT_α/β_51:A substitution in response to four MART-1+ and HLA-A2+ tumor cell lines relative to those released from CD4+ T cells transfected with the WT DMF4 TCR (Fig. 7_D_), although the dramatically higher levels of IFN-γ from CD4+ T cells expressing the DMF4 WT_α/_β_51:A TCR in response to the 1300 melanoma cell line than those directed against the additional MART-1+ and HLA-A*02+ tumor cell lines were not observed in additional experiments (data not shown). The observation that CD8+ and CD4+ T cells that were transfected with the DMF4 _β_51:A construct failed to respond to any of the MART-1- or HLA-A*02- target cell lines that were tested (Fig. 7, C and D and data not shown) indicated that these responses are both peptide and HLA specific.
Lysis of tumor cell lines mediated by DMF4 and DMF5 TCR variants
The abilities of CD8+ and CD4+ T cells expressing the DMF4 or DMF5 WT TCRs to mediate specific tumor cell lysis were then compared with T cells that express the corresponding TCR variants. The levels of Ag-specific lysis mediated by CD8+ T cells expressing either the DMF4 _β_51:A or the DMF5 _β_54:A variants directed against the MART-1+ and HLA-A*02+ melanoma cell line 624.38 were not appreciably higher than those seen with the corresponding DMF4 or DMF5 WT TCRs (Fig. 8_A_). CD4+ T cells transfected with the DMF5 WT TCR showed a low level lysis of the 624.38 tumor cell line confirming CD8 coreceptor independence of this TCR (23). In contrast, 624.38 cells were not significantly lysed by CD4+ T cells transfected with the DMF4 WT TCR (Fig. 8_C_). The level of lysis of 624.38 cells mediated by CD4+ T cells expressing the DMF5 _β_54:A TCR variant was enhanced in comparison to that observed with CD4+ T cells expressing the DMF5 WT TCR, and a low but significant level of lysis of 624.38 cells was also observed following incubation with CD4+ T cells expressing the DMF4 _β_51:A variant (Fig. 8_C_). Expression of these modified TCRs in CD8+ or CD4+ T cells, however, did not enhance the lysis of the MART-1- but HLA-A*02+ renal carcinoma cell line 2661R indicating that these AAS did not substantially alter the lytic specificity of T cells that expressed these TCRs (Fig. 8, B and D). Although there appeared to be a low level of lysis of the 2661R tumor cell line mediated by CD4+ T cells transfected with the DMF4 and DMF5 WT TCRs, this was not observed in additional experiments (data not shown).
FIGURE 8.
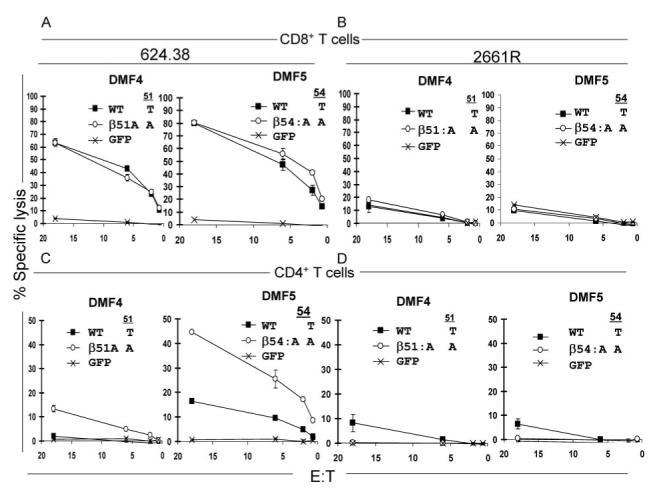
Effects of MART-1/HLA-A*02-reactive TCR DMF4 and DMF5 CDR2_β_ variants on the cytolytic activity of CD8+ and CD4+ T cells. The cytolytic activities of CD8+ (A and B) and CD4+ (C and D) T cells transfected with RNA encoding the DMF4 and DMF5 WT and variant TCRs to the HLA-A*02+ and MART-1+ melanoma cell line 624.38 (A and C) and to the HLA-A*02+ but MART-1- tumor cell line renal cell carcinoma 2661R (B and D) were evaluated in standard 4-h 51Cr-release cell lysis assays.
Similar levels of IFN-γ were released by CD8+ T cells cotransfected with either the DMF4 WT TCR or the DMF4 WT α/_β_51:A variant in response to T2 target cells pulsed with the cognate MART-1:27-35 peptide (Fig. 9_A_). In contrast, CD8+ T cells cotransfected with the DMF5 WT α/β_54:A TCR variant recognized target cells pulsed with between 10- and 100-fold lower levels of the MART-1 peptide than CD8+ T cells transfected with the DMF5 WT TCR (Fig. 9_A_). Additionally, transfected CD4+ T cells expressing the DMF5 WT_α/β_54:A and DMF4 WT_α/_β_51:A TCR variants recognized between 10- and 100-fold lower concentrations of the peptide than CD4+ cells transfected with the corresponding WT TCRs (Fig. 9_B_). These observations, taken together with the results of studies conducted with the 1G4 TCR variants, suggest that the evaluation of T cells that have been genetically modified to express TCRs containing as few as one or two CDR AAS may represent a general strategy for identifying TCRs with enhanced functional avidities.
FIGURE 9.
Peptide titration of responses of CD8+ and CD4+ T cells transfected with MART-1/HLA-A*02-reactive DMF4 and DMF5 WT TCRs and their CDR2_β_ variants. T cells were activated with anti-CD3 Ab and isolated CD8+ (A) or CD4+ (B) cells were cotransfected with RNA encoding TCR constructs containing substitution of DMF4 or DMF5 _β_-chains along with the complementary DMF4 or DMF5 WT _α_-chains. The releases of IFN-γ in response to T2 cells pulsed with varying doses of the MART-1 peptide or with a 1 _μ_M control HLA-A*02-binding peptide corresponding to tyrosinase residues 369-377 (YMDTMSQV) were measured the following day.
Discussion
In this report, single and dual AAS variants were created in both chains of the 1G4 TCR that recognizes the HLA-A*02-restricted NY-ESO-1:157-165 peptide Ag, to identify substituted TCRs with enhanced levels of Ag-specific reactivity in TCR gene-modified T cells. As exemplified here, mutant CDR motifs selected from large mutational libraries can provide an effective starting point for this process. In addition, the effects of single AAS on two TCRs (DMF4 and DMF5) both reactive with the HLA-A*02-restricted MART-1:27-35 epitope, but with distinct variable chain usage, were evaluated to determine whether the cellular properties of these TCRs could also be enhanced without the prior knowledge of key TCR residues derived from affinity improvement selection or structural studies.
Although increases in T cell reactivity in TCR-transfected CD4+ and/or CD8+ cells are desired, this should not come at the expense of a loss of Ag specificity. Ag cross-reactivity is of particular concern for T cell adoptive immunotherapy and may be more likely if CD8-independent TCRs are used in a CD8+ T cell environment. Improved TCRs with responses that remain Ag-specific in both CD4+ and CD8+ T cells can potentially enhance T cell adoptive immunotherapy.
This study used an RNA-transfection/screening assay method which facilitated the rapid evaluation of TCR AAS variants in T cells. TCR variants which showed potentially improved properties in both CD4+ and CD8+ T cells were then further characterized in T cells stably transduced by retroviral TCR gene expression constructs. Among the substituted 1G4 TCRs that were evaluated were two dual AAS variants; one in the CDR3 α region (_α_95:LY) and another in CDR2 β (_β_51:AI). Both variant TCRs significantly increased the CD4+ T cell sensitivity both to peptide-pulsed target cells and to cognate tumor cell lines. The high levels of recognition of cognate cells by CD4+ T cells transfected with these and a third single substitution (_β_52:I) indicates that increased CD8-independent T cell responsiveness to physiologically relevant levels of Ag is associated with increased TCR affinity. A 10- to 30-fold increase in affinity is seen, ranging from the WT 1G4 TCR (9.3 _μ_M), across _β_52:I (1.1 _μ_M) and _α_95:LY variants (730 nM) to the _β_51:AI 1G4 TCR (280 nM). These responses by substituted 1G4 TCRs concur with those seen previously with the more complex 1G4 TCR wt/c59 and c12/c2 mutants, having respective Ag complex affinities of 4 _μ_M and 450 nM (21). Taken together, an elevated response by TCR-transfected CD4+ T cells is seen over a TCR affinity range of ∼5 _μ_M to ∼500 nM, a 2- to 20-fold higher affinity than the 1G4 WT TCR. The findings of an earlier study on the murine 2C class I TCR are consistent with this as peptide/MHC complex affinities of ∼3 _μ_M or stronger were associated with CD8-independent T cell triggering (39).
These elevated 1G4 TCR affinities result mainly from an increase in the _t_1/2 of the binding of the 1G4 TCR to the NY-ESO-1 peptide/HLA-A*02 complex (Table III) and suggest elevated TCR dwell times. Other studies suggest that efficient T cell activation may require an optimal dwell time of interaction between the TCR and Ag complex (40). Earlier studies have identified a correlation between the strength of the T cell response and the _t_1/2 of TCR binding (41), although exceptions to this rule have been noted which may relate to TCR structural flexibility (42).
The Ag-specific responses to tumor cell lines of CD8+ T cells transduced with the _α_95:LY/WT β variant 1G4 TCRs (730 nM) were enhanced ∼2-fold relative to the WT α/β TCR (∼10 _μ_M), while at the same time the responses of CD8+ T cells transduced with the WT α/_β_51:AI variant (280 nM) to the appropriate target cells were not significantly enhanced. The increased responses observed from CD8+ as well as CD4+ T cells transduced with the 1G4 _α_95:LY variant were observed in the absence of any apparent cross-reactivity with NY-ESO-1- or HLA-A2-negative target cells, indicating that the affinity of the _α_95:LY/WT β TCR (730 nM) TCR variant may be close to the affinity that is close to the optimum for the maximal triggering of CD8+ T cell antitumor responses.
In parallel to these observations on TCR-transfected T cell responses, the T cell Ag specificities were also measured and compared. Previously, a multiple substitution variant of the 1G4 TCR was shown to possess an affinity ∼1 million-fold higher than the WT 1G4 TCR. As a soluble monomeric protein, this TCR was shown to bind specifically to NY-ESO-1 peptide-pulsed T2 cells but not to a panel of 16 other unrelated control peptide/HLA-A*02 Ags (18). When this TCR and other mutant1G4 TCRs were transfected into CD8+ T cells, Ag cross-reactivity with NY-ESO-1-negative target cell lines was observed with the highest affinity TCR and with those with Ag affinities ranging from 20 to 50 pM to 84 nM. However, when CD8+ T cells were transfected with mutant 1G4 TCRs with affinities 450 nM and weaker were Ag specific (21). In agreement with these findings, the _β_51:AI 1G4 TCR variant (280 nM affinity) was also associated with a loss of Ag specificity in CD8+ T cells, as these cells were stimulated by the NY-ESO-1-negative 2661R tumor cell line. Therefore, the upper affinity limit of the 1G4 TCR in CD8+ T cells compatible with specific Ag recognition may lie between 450 and 280 nM.
These results are compatible with the hypothesis that both the strength and the specificity of the T cell response are closely related to TCR affinity. As noted earlier, the principal kinetic parameter change responsible for these observed mutational increases in 1G4 TCR affinity is the increase seen in the Ag dissociation half-life. Hence, in defining the optimal properties of TCR variants suitable for Ag-specific tumor cell recognition, it may be more appropriate to describe a range of Ag-dissociation rates rather than the product of the association and dissociation rates that together constitute the overall affinity.
Crystal structure studies on soluble WT 1G4 TCR protein have demonstrated that amino acid residues 95 and 96 (TS) in the 1G4 CDR3 α_-chain interact with the NY-ESO-1 peptide in the HLA-A*02-binding groove, whereas CDR2_β residues interact with the HLA-A*02 α_2 helix and do not interact at all with the NY-ESO-1 peptide (36). Previously, a crystal structure of a high-affinity 1G4 TCR containing substitutions in both the CDR2_α and CDR2_β_ regions revealed that the orientation of the native CDR3 loops and their specific contacts with bound peptide remained essentially unchanged from the parental wild type (22). A recent study indicates that dual substitutions of two leucine residues for the threonine plus serine residues at CDR3 positions 95 and 96 in the 1G4 WT α_-chain are unfavorable in the unbound TCR due to the solvent accessibility of the hydrophobic leucine side chains (43). When this variant TCR binds to cognate peptide/HLA-A*02 complex, however, these leucine residues form favorable interactions with CDR1_α and CDR2_α_ residues that enhanced the overall stability of this complex (43), interactions that may similarly stabilize the Ag binding of the _α_95:LY/WT β variant TCR analyzed in the current report.
The effects of single AAS in the CDR2_β_ regions of two TCRs (DMF4 and DMF5) both reactive with the HLA-A*02-restricted MART:27-35 peptide epitope but with distinct variable chain usages were evaluated to determine whether the results obtained using the 1G4 TCR could be extended to include other receptors without prior knowledge from affinity improvement or structural studies. The results of screening assays indicated that separate alanine substitutions of a single CDR2_β_ residue in both TCRs could significantly enhance the Ag-specific tumor cell line responses of TCR gene-modified CD4+ T cells. One of these mutant TCRs, DMF5 _β_54:A, also demonstrated increased CD8+ T cell reactivity without loss of Ag specificity. The in vitro affinity binding kinetics and atomic structures of the native and modified DMF4 and DMF5 TCRs have not been determined, but the results of peptide titration assays indicate that these modifications may have led to enhancements in TCR affinity that are similar to those observed with the 1G4 _α_95:LY variant (Fig. 9).
Studies conducted with an additional DMF5 TCR variant containing an alanine substitution at position 51 of the _β_-chain indicate that this AAS substitution results in the generation of an alloreactive TCR parallel and emphasizes the need for caution in evaluating the effect of AAS on TCR function. The use of wide panels of target cells lacking expression of the tumor marker and/or the HLA restriction element is critical to insure that the chosen AAS do not significantly alter the specificity of the TCR for cognate peptide/HLA Ag, especially in CD8+ T cells.
The results of modifications of the 1G4 TCR, taken together with those obtained with the DMF4 and DMF5 TCRs, provide guidelines that can be used in studies aimed at enhancing the function of additional TCRs. The function of altered TCRs containing alanine substitutions of individual CDR residues can be evaluated to identify alterations that either enhance or diminish T cell function. Conservative substitutions at critical residues identified in the initial alanine substitution scan, as well as combinations of substituted residues, can then be evaluated for the effects of these changes on the function of gene-modified T cells.
At this time, the treatment effects of tumor-reactive, class I-restricted CD4+ T cells containing CD8-independent TCRs on in vivo or clinical responses are unknown; however, multiple observations suggest that the presence of CD4+ T cells enhances the function of the CD8+ T cell population. The persistence of CMV-reactive CD4+ T cells is correlated with enhanced control of disease by CMV-reactive CD8+ T cells (44, 45). Cooperative interactions between CD8+ and CD4+ T cells expressing a high-affinity CD8-independent TCR directed against a human p53 epitope have been observed, as assessed by their abilities both to secrete IFN-γ in response to cognate Ag complex expressing target cell lines and to activate dendritic cells (46). The adoptive transfer of CD4+ T cells expressing a CD8-independent TCR has been shown to enhance tumor protection mediated by CD8+ tumor-reactive T cells (47). In addition, the priming of CD8+ T cells directed against a viral epitope was enhanced by the adoptive transfer of CD4+ T cells that had been genetically modified to coexpress the CD8 α/CD8 β coreceptor along with a CD8-dependent class I-specific TCR (48).
The findings presented above indicate that the substitutions of one to two TCR _α_- or _β_-chain CDR residues can dramatically enhance the function of gene-modified T cells. The development of rapid screening assays to identify enhanced TCR substitution variants may provide valuable assistance to studies on T cell functions and to the understanding of the physical nature of interactions between TCRs and peptide/MHC complexes as well as generating TCR reagents that lead to improved T cell immunotherapy.
Acknowledgments
We thank Arnold Mixon and Shawn Farid for their expert assistance with flow cytometry analysis.
Footnotes
2
Abbreviations used in this paper:
AAS
amino acid substitution
WT
wild type
Disclosures
The authors have no financial conflict of interest.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, Chien Y. Ligand recognition by αβ T cell receptors. Annu. Rev. Immunol. 1998;16:523–544. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 2.Manning TC, Kranz DM. Binding energetics of T-cell receptors: correlation with immunological consequences. Immunol. Today. 1999;20:417–422. doi: 10.1016/s0167-5699(99)01508-x. [DOI] [PubMed] [Google Scholar]
- 3.van der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annu. Rev. Immunol. 2003;21:659–684. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- 4.Rudolph MG, Luz JG, Wilson IA. Structural and thermodynamic correlates of T cell signaling. Annu. Rev. Biophys. Biomol. Struct. 2002;31:121–149. doi: 10.1146/annurev.biophys.31.082901.134423. [DOI] [PubMed] [Google Scholar]
- 5.Savage PA, Davis MM. A kinetic window constricts the T cell receptor repertoire in the thymus. Immunity. 2001;14:243–252. doi: 10.1016/s1074-7613(01)00106-6. [DOI] [PubMed] [Google Scholar]
- 6.Alexander-Miller MA, Leggatt GR, Berzofsky JA. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc. Natl. Acad. Sci. USA. 1996;93:4102–4107. doi: 10.1073/pnas.93.9.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeh HJ, 3rd, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J. Immunol. 1999;162:989–994. [PubMed] [Google Scholar]
- 8.Trautmann L, Rimbert M, Echasserieau K, Saulquin X, Neveu B, Dechanet J, Cerundolo V, Bonneville M. Selection of T cell clones expressing high-affinity public TCRs within human cytomegalovirus-specific CD8 T cell responses. J. Immunol. 2005;175:6123–6132. doi: 10.4049/jimmunol.175.9.6123. [DOI] [PubMed] [Google Scholar]
- 9.Price DA, Brenchley JM, Ruff LE, Betts MR, Hill BJ, Roederer M, Koup RA, Migueles SA, Gostick E, Wooldridge L, et al. Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J. Exp. Med. 2005;202:1349–1361. doi: 10.1084/jem.20051357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kasibhatla S, Nalefski EA, Rao A. Simultaneous involvement of all six predicted antigen binding loops of the T cell receptor in recognition of the MHC/antigenic peptide complex. J. Immunol. 1993;151:3140–3151. [PubMed] [Google Scholar]
- 11.Eisen HN. Specificity and degeneracy in antigen recognition: yin and yang in the immune system. Annu. Rev. Immunol. 2001;19:1–21. doi: 10.1146/annurev.immunol.19.1.1. [DOI] [PubMed] [Google Scholar]
- 12.Manning TC, Schlueter CJ, Brodnicki TC, Parke EA, Speir JA, Garcia KC, Teyton L, Wilson IA, Kranz DM. Alanine scanning mutagenesis of an αβ T cell receptor: mapping the energy of antigen recognition. Immunity. 1998;8:413–425. doi: 10.1016/s1074-7613(00)80547-6. [DOI] [PubMed] [Google Scholar]
- 13.Manning TC, Parke EA, Teyton L, Kranz DM. Effects of complementarity determining region mutations on the affinity of an α/β T cell receptor: measuring the energy associated with CD4/CD8 repertoire skewing. J. Exp. Med. 1999;189:461–470. doi: 10.1084/jem.189.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chlewicki LK, Holler PD, Monti BC, Clutter MR, Kranz DM. High-affinity, peptide-specific T cell receptors can be generated by mutations in CDR1, CDR2 or CDR3. J. Mol. Biol. 2005;346:223–239. doi: 10.1016/j.jmb.2004.11.057. [DOI] [PubMed] [Google Scholar]
- 15.Holler PD, Lim AR, Cho BK, Rund LA, Kranz DM. CD8- T cell transfectants that express a high affinity T cell receptor exhibit enhanced peptide-dependent activation. J. Exp. Med. 2001;194:1043–1052. doi: 10.1084/jem.194.8.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holler PD, Chlewicki LK, Kranz DM. TCRs with high affinity for foreign pMHC show self-reactivity. Nat. Immunol. 2003;4:55–62. doi: 10.1038/ni863. [DOI] [PubMed] [Google Scholar]
- 17.Weber KS, Donermeyer DL, Allen PM, Kranz DM. Class II-restricted T cell receptor engineered in vitro for higher affinity retains peptide specificity and function. Proc. Natl. Acad. Sci. USA. 2005;102:19033–19038. doi: 10.1073/pnas.0507554102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, Dunn S, Liddy N, Jacob J, Jakobsen BK, Boulter JM. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 19.Chen YT, Scanlan MJ, Sahin U, Tureci O, Gure AO, Tsang S, Williamson B, Stockert E, Pfreundschuh M, Old LJ. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc. Natl. Acad. Sci. USA. 1997;94:1914–1918. doi: 10.1073/pnas.94.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purbhoo MA, Sutton DH, Brewer JE, Mullings RE, Hill ME, Mahon TM, Karbach J, Jager E, Cameron BJ, Lissin N, et al. Quantifying and imaging NY-ESO-1/LAGE-1-derived epitopes on tumor cells using high affinity T cell receptors. J. Immunol. 2006;176:7308–7316. doi: 10.4049/jimmunol.176.12.7308. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y, Bennett AD, Zheng Z, Wang QJ, Robbins PF, Yu LY, Li Y, Molloy PE, Dunn SM, Jakobsen BK, et al. High-Affinity TCRs Generated by Phage Display Provide CD4+ T Cells with the Ability to Recognize and Kill Tumor Cell Lines. J. Immunol. 2007;179:5845–5854. doi: 10.4049/jimmunol.179.9.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dunn SM, Rizkallah PJ, Baston E, Mahon T, Cameron B, Moysey R, Gao F, Sami M, Boulter J, Li Y, Jakobsen BK. Directed evolution of human T cell receptor CDR2 residues by phage display dramatically enhances affinity for cognate peptide-MHC without increasing apparent cross-reactivity. Protein Sci. 2006;15:710–721. doi: 10.1110/ps.051936406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawakami Y, Eliyahu S, Sakaguchi K, Robbins PF, Rivoltini L, Yannelli JR, Appella E, Rosenberg SA. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J. Exp. Med. 1994;180:347–352. doi: 10.1084/jem.180.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marincola FM, Hijazi YM, Fetsch P, Salgaller ML, Rivoltini L, Cormier J, Simonis TB, Duray PH, Herlyn M, Kawakami Y, Rosenberg SA. Analysis of expression of the melanoma-associated antigens MART-1 and gp100 in metastatic melanoma cell lines and in in situ lesions. J. Immunother. Emphasis Tumor Immunol. 1996;19:192–205. doi: 10.1097/00002371-199605000-00004. [DOI] [PubMed] [Google Scholar]
- 25.Naviaux RK, Costanzi E, Haas M, Verma IM. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 1996;70:5701–5705. doi: 10.1128/jvi.70.8.5701-5705.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riker AI, Kammula US, Panelli MC, Wang E, Ohnmacht GA, Steinberg SM, Rosenberg SA, Marincola FM. Threshold levels of gene expression of the melanoma antigen gp100 correlate with tumor cell recognition by cytotoxic T lymphocytes. Int. J. Cancer. 2000;86:818–826. doi: 10.1002/(sici)1097-0215(20000615)86:6<818::aid-ijc10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 27.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 28.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, Powell DJ, Jr., Rosenberg SA. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol. 2004;173:7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, Rosenberg SA, Morgan RA. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol. Ther. 2006;13:151–159. doi: 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single "self-cleaving" 2A peptide-based retroviral vector. Nat. Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 31.Hughes MS, Yu YY, Dudley ME, Zheng Z, Robbins PF, Li Y, Wunderlich J, Hawley RG, Moayeri M, Rosenberg SA, Morgan RA. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum. Gene Ther. 2005;16:457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Topalian SL, Solomon D, Rosenberg SA. Tumor-specific cytolysis by lymphocytes infiltrating human melanomas. J. Immunol. 1989;142:3714–3725. [PubMed] [Google Scholar]
- 34.Robbins PF, El-Gamil M, Kawakami Y, Stevens E, Yannelli J, Rosenberg SA. Recognition of tyrosinase by tumor infiltrating lymphocytes from a patient responding to immunotherapy. Cancer Res. 1994;54:3124–3126. [PubMed] [Google Scholar]
- 35.Boulter JM, Glick M, Todorov PT, Baston E, Sami M, Rizkallah P, Jakobsen BK. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 2003;16:707–711. doi: 10.1093/protein/gzg087. [DOI] [PubMed] [Google Scholar]
- 36.Chen JL, Stewart-Jones G, Bossi G, Lissin NM, Wooldridge L, Choi EM, Held G, Dunbar PR, Esnouf RM, Sami M, et al. Structural and kinetic basis for heightened immunogenicity of T cell vaccines. J. Exp. Med. 2005;201:1243–1255. doi: 10.1084/jem.20042323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson LA, Heemskerk B, Powell DJ, Jr., Cohen CJ, Morgan RA, Dudley ME, Robbins PF, Rosenberg SA. Gene transfer of tumor reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J. Immunol. 2006;177:6548–6559. doi: 10.4049/jimmunol.177.9.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holler PD, Kranz DM. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity. 2003;18:255–264. doi: 10.1016/s1074-7613(03)00019-0. [DOI] [PubMed] [Google Scholar]
- 40.Kalergis AM, Boucheron N, Doucey MA, Palmieri E, Goyarts EC, Vegh Z, Luescher IF, Nathenson SG. Efficient T cell activation requires an optimal dwell-time of interaction between the TCR and the pMHC complex. Nat. Immunol. 2001;2:229–234. doi: 10.1038/85286. [DOI] [PubMed] [Google Scholar]
- 41.Kersh GJ, Kersh EN, Fremont DH, Allen PM. High- and low-potency ligands with similar affinities for the TCR: the importance of kinetics in TCR signaling. Immunity. 1998;9:817–826. doi: 10.1016/s1074-7613(00)80647-0. [DOI] [PubMed] [Google Scholar]
- 42.Krogsgaard M, Prado N, Adams EJ, He XL, Chow DC, Wilson DB, Garcia KC, Davis MM. Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol. Cell. 2003;12:1367–1378. doi: 10.1016/s1097-2765(03)00474-x. [DOI] [PubMed] [Google Scholar]
- 43.Sami M, Rizkallah PJ, Dunn S, Molloy P, Moysey R, Vuidepot A, Baston E, Todorov P, Li Y, Gao F, et al. Crystal structures of high affinity human T-cell receptors bound to peptide major histocompatibility complex reveal native diagonal binding geometry. Protein Eng. Des. Sel. 2007;20:397–403. doi: 10.1093/protein/gzm033. [DOI] [PubMed] [Google Scholar]
- 44.Peggs KS, Verfuerth S, Pizzey A, Khan N, Guiver M, Moss PA, Mackinnon S. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet. 2003;362:1375–1377. doi: 10.1016/S0140-6736(03)14634-X. [DOI] [PubMed] [Google Scholar]
- 45.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 46.Kuball J, Schmitz FW, Voss RH, Ferreira EA, Engel R, Guillaume P, Strand S, Romero P, Huber C, Sherman LA, Theobald M. Cooperation of human tumor-reactive CD4+ and CD8+ T cells after redirection of their specificity by a high-affinity p53A2.1-specific TCR. Immunity. 2005;22:117–129. doi: 10.1016/j.immuni.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 47.Morris EC, Tsallios A, Bendle GM, Xue SA, Stauss HJ. A critical role of T cell antigen receptor-transduced MHC class I-restricted helper T cells in tumor protection. Proc. Natl. Acad. Sci. USA. 2005;102:7934–7939. doi: 10.1073/pnas.0500357102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kessels HW, Schepers K, van den Boom MD, Topham DJ, Schumacher TN. Generation of T cell help through a MHC class I-restricted TCR. J. Immunol. 2006;177:976–982. doi: 10.4049/jimmunol.177.2.976. [DOI] [PubMed] [Google Scholar]