RNA toxicity is a component of ataxin-3 degeneration in Drosophila (original) (raw)
. Author manuscript; available in PMC: 2008 Dec 19.
Published in final edited form as: Nature. 2008 Apr 30;453(7198):1107–1111. doi: 10.1038/nature06909
Abstract
Polyglutamine (polyQ) diseases are a class of dominantly-inherited neurodegenerative disorders caused by the expansion of a CAG repeat encoding glutamine within the coding region of the respective genes 1. The molecular and cellular pathways underlying polyQ-induced neurodegeneration are the focus of much research, and it is widely viewed that toxic activities of the protein, resulting from the abnormally long polyQ tract, cause pathogenesis 2, 3. We now provide evidence for a pathogenic role of the CAG repeat RNA in polyQ toxicity using Drosophila. In a Drosophila screen for modifiers of polyQ degeneration induced by the Spinocerebellar ataxia type 3 (SCA3) protein ataxin-3, we isolated an upregulation allele of muscleblind (mbl), a gene implicated in the RNA toxicity of CUG expansion diseases 4-6. Further analysis indicated that there may be a toxic role of the RNA in polyQ-induced degeneration. We tested the role of the RNA by altering the CAG repeat sequence to an interrupted CAACAG repeat within the polyQ-encoding region; this dramatically mitigated toxicity. In addition, expression of an untranslated CAG repeat of pathogenic length conferred neuronal degeneration. These studies reveal a role for the RNA in polyQ toxicity, highlighting common components in RNA-based and polyQ protein-based trinucleotide repeat expansion diseases.
To identify modifiers that add new insight into ataxin-3 pathogenesis, we performed an overexpression EP screen with a Drosophila model of ataxin-3 7 for modifiers of eye degeneration. Seven new _EP_-element insertional mutations were isolated; one of these (B2-E1) dramatically enhanced toxicity, causing severe pigmentation loss and striking collapse of the retina, but had no effect on its own when directed to the eye (Fig. 1a-d). Molecular analysis showed that the B2-E1 insertion was in the promoter of the mbl gene, and upregulated gene expression (Supplementary Fig. S2). As Mbl has been implicated as a modifier of CUG repeat RNA toxicity 4-6, these results suggested an unexpected role for mbl as a modulator of polyQ protein toxicity.
Figure 1. Upregulation of mbl enhances ataxin-3 toxicity.
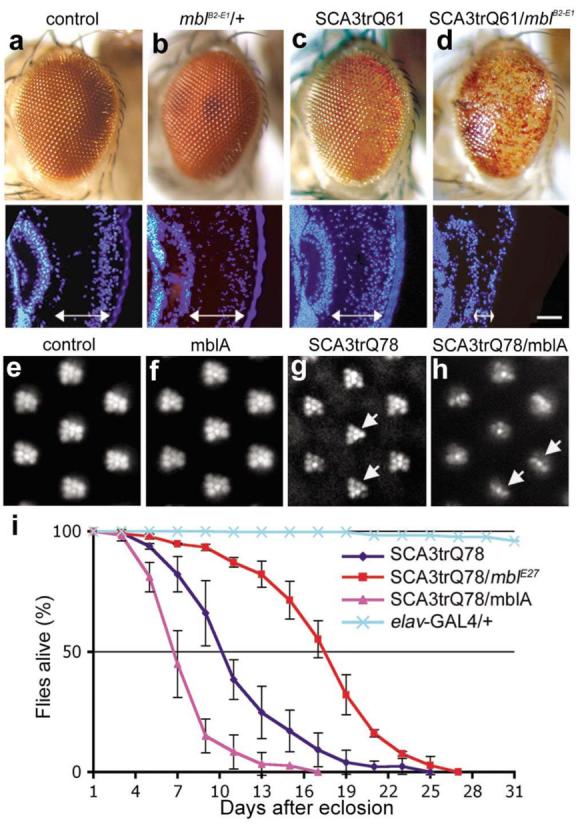
a-d, External eye of 1d flies. (a) Flies expressing gmr-GAL4 alone or (b) with mblB2-E1 have normal eye morphology. (c) Flies expressing SCA3trQ61 have a mild loss of pigmentation, and slightly disrupted internal retinal morphology. (d) Flies expressing SCA3trQ61 with mblB2-E1 show severe external degeneration and collapse of the retina. Genotypes w; gmr-GAL4 UAS-SCA3trQ61 in trans to (c) w (d) mblB2-E1. e-h, Retinal pseudopupils of 1d flies. (e) Flies expressing _elav_-GAL4 alone or (f) with MblA have a normal pattern of 7 photoreceptors (PR)/ommatidium. (g) Flies expressing SCA3trQ78 show mild loss of retinal integrity (arrows), with 5.8±0.4 SD PR/ommatidium (n=200 ommatidia). Genotype elav-GAL4/+; UAS-SCA3trQ78/+. (h) Co-expression of MblA with SCA3trQ78 enhances PR loss to 3.0±0.5 SD (n=200 ommatidia; significant difference from g, P< 0.0001 (two-tailed unpaired student _t_-test)). Genotype elav-GAL4/+;UAS-mblA/+; UAS-SCA3trQ78/+. (i) Neuronal toxicity by lifespan analysis. Upregulation of MblA further shortened the lifespan of SCA3trQ78 flies, whereas downregulation of mbl (flies heterozygous for allele mblE27) extended lifespan (P<0.001, SCA3trQ78 and SCA3trQ78/mblE27, SCA3trQ78 and SCA3trQ78/mblA, log-rank analysis). Mean ± SD; n=170-220 flies for each genotype. Genotypes for SCA3trQ78 and SCA3trQ78/mblA same as g and h, SCA3trQ78/mblE27 is elav-GAL4/+;mblE27/+;UAS-SCA3trQ78/+.
We confirmed that mbl upregulation enhanced polyQ toxicity by generating transgenic flies bearing a mbl cDNA (MblA, which is implicated in eye and photoreceptor (PR) neuron development 8). As with the original EP insertion, flies expressing MblA showed strongly enhanced SCA3trQ61 toxicity, as well as enhanced photoreceptor (PR) degeneration when expressed with rh1-GAL4, but MblA had no effect on its own (Fig. 1e-h, Supplementary Fig. S3). B2-E1 and MblA enhanced toxicity of full-length pathogenic forms of ataxin-3 as well as truncated versions, and enhanced the toxicity of pathogenic forms of the Huntington's disease protein (Supplementary Figs. S3, S4). Expression of Mbl, however, did not enhance deleterious eye phenotypes due to compromised chaperone activity or tau (Supplementary Fig. S3d-g). Upregulation of MblA also enhanced the shortened lifespan of flies expressing SCA3trQ78 globally in the nervous system (elav-GAL4). Heterozygosity for a mbl null allele (mblE27 or mblE22) 8, 9 suppressed lifespan reduction (Fig. 1i, Supplementary Table S1), indicating that mbl modulated SCA3 toxicity upon both upregulation and with reduced activity.
To reveal insight into potential mechanisms of enhancement, we examined effects of Mbl on polyQ protein and RNA. PolyQ proteins undergo abnormal aggregation, accumulating into nuclear inclusions (NI) 2, 3. With the late-onset rh1-GAL4 driver, polyQ protein accumulates slowly over adult lifespan, allowing sensitive analysis of protein levels and NI formation. MblA increased the level of the polyQ protein, accelerating inclusion formation (Supplementary Fig. S5j, k). Notably, Mbl increased the level of the polyQ RNA as well as protein (Supplementary Fig. S5i), suggesting that Mbl may act on the RNA to enhance polyQ toxicity.
To test whether the interaction with polyQ toxicity was conserved, we determined whether human muscleblind (MBNL1) could also modulate polyQ toxicity in the fly. Flies co-expressing human MBNL140 with pathogenic SCA3trQ78 had dramatically enhanced toxicity with striking loss of pigmentation and accelerating PR neurodegeneration (Supplementary Fig. S6d, g, i). Previous studies indicate that functional depletion of muscleblind contributes to CUG repeat toxicity, with upregulation of MBNL140 suppressing CUG RNA toxicity 10. To further determine whether muscleblind modulation of polyQ toxicity shared features with CUG RNA modulation, we asked whether an isoform of MBNL1 that fails to bind CUG repeats in binding assays in vitro 6, 11 was altered in ability to modulate polyQ toxicity. Transgenics were generated that expressed MBNL135, a form of MBNL1 that lacks exon 4 encoding a domain required for optimal CUG repeat binding 6, 11 and conserved in Drosophila Mbl (Supplementary Fig. S6a). Flies expressing MBNL135 at similar levels to MBNL140 with SCA3trQ78 disease gene had mitigated enhancement, with only mild pigmentation loss and modest loss of PR neurons (Supplementary Fig. S6). These results indicated that a MBNL1 isoform which is compromised in ability to bind CUG repeat RNA, is also compromised in ability to enhance CAG-encoded polyQ toxicity. This suggested that muscleblind may modulate polyQ toxicity at least in part by acting at the level of the RNA, although mechanisms with CUG RNA modulation are likely distinct given the opposite effects in these two disease situations.
The data discussed so far suggested that enhancement by Mbl was associated with an increased level of the RNA with consequent increased level of polyQ protein. Consistent with an effect on the RNA, MBNL1 has been shown to interact with CAG repeat RNA in vitro and co-localize with CAG repeat RNA foci in cell culture, suggesting that it can interact with CAG repeat RNA11, 12. We therefore considered whether the enhanced toxicity observed when polyQ RNA levels were increased by Mbl was due simply to an increase in the level of the protein, or whether the CAG repeat RNA itself possessed an inherent toxicity contributing to polyQ disease. The ataxin-3 transgenes used thus far were composed of pure CAG repeat sequences within the glutamine-encoding domain; we therefore tested whether the CAG repeat sequence was important for Mbl enhancement. SCA3 transgenes were generated with a CAG repeat sequence interrupted by CAA codons (SCA3trQ78CAA/G). These transgenes are predicted to express a protein of identical amino acid sequence to the CAG-repeat encoding transgenic lines, differing only in the sequence of the RNA within the polyQ-encoding region (Supplementary Fig. S7a). Lines were selected that expressed the protein at levels similar to those of CAG-repeat transgenic lines (Supplementary Fig. S7b, c; Supplementary Table S2).
Analysis of the toxicity in flies expressing similar levels of SCA3trQ78CAA/G and the pure CAG-encoded protein (SCA3trQ78CAG) showed that the two transgenes conferred strikingly different degrees of degeneration. Whereas SCA3trQ78(s)CAG flies had strong loss of pigmentation and PR neurons (4.6±0.6 PR/ommatidium), SCA3trQ78CAA/G flies expressing identical levels of protein had only mild pigmentation loss and minimal PR degeneration (6.5±0.2 PR/ommatidium) (Fig. 2e-f, Supplementary Fig. S8). Mitigation of toxicity also occurred with expression globally in the nervous system with elav-GAL4: SCA3trQ78CAA/G flies lived longer, with milder progression of neuronal dysfunction (Fig. 2g, h). These findings were confirmed with multiple independent SCA3trQ78CAA/G and SCA3trQ78CAG transgenic lines (Supplementary Table S2). We also analyzed flies expressing full-length ataxin-3. These studies showed similar mitigation of toxicity when the CAG repeat sequence was replaced by an interrupted CAA/G sequence within ataxin-3 encoding transgene (Supplementary Fig. S9). These results indicated that toxicity of pathogenic ataxin-3 is mitigated upon interruption of the CAG repeat.
Figure 2. Interruptions of the CAG repeat mitigate SCA3 protein pathogenesis.
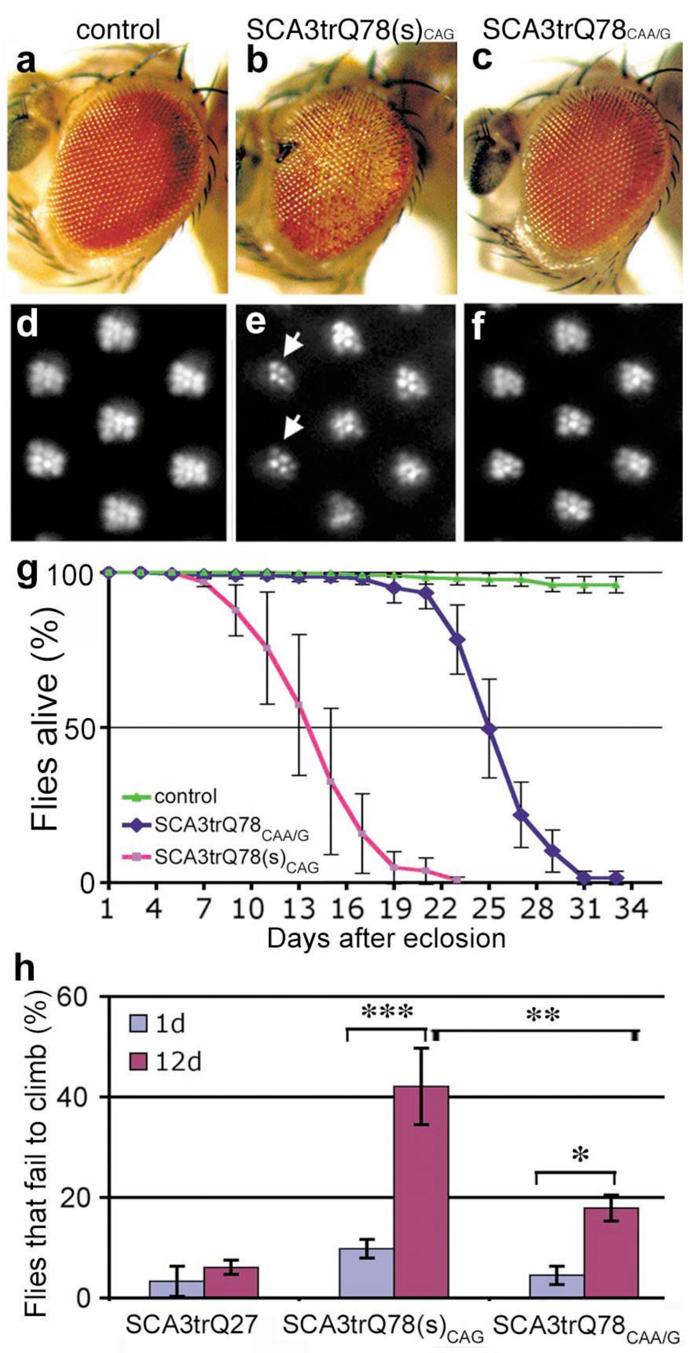
a-f, Flies expressing similar levels of SCA3trQ78CAG and SCA3trQ78CAA/G protein show strikingly different degeneration. (a-c) Flies expressing SCA3 with gmr-GAL4. (d-f) Adult-onset PR retinal degeneration, with expression by rh1-GAL4 (25d). (a, d) Controls expressing non-pathogenic SCA3trQ27 protein have normal eye structure. Genotype UAS-SCA3trQ27 in trans to (a) gmr-GAL4 or (d) rh1-GAL4. (b, e) Flies expressing SCA3trQ78(s)CAG show (b) severe retinal degeneration and (e, arrows) striking loss of rhabdomeres (4.6±0.6 PR/ommatidium). UAS-SCA3trQ78(s)CAG in trans to (b) gmr-GAL4 and (e) rh1-GAL4. (c, f) Flies expressing SCA3trQ78CAA/G at the same level show mild degeneration, with (c) normal external eye morphology and (f) mild PR loss (6.5±0.2 PR/ommatidium, statistically significant from e, P<0.001, two-tailed unpaired student _t_-test). Genotype UAS-SCA3trQ78CAA/G in trans to (c) gmr-GAL4 or (f) rh1-GAL4. (g) Neuronal toxicity by lifespan analysis. Flies expressing SCA3trQ78(s)CAG have a strikingly shorter lifespan than flies expressing SCA3trQ78CAA/G at the same level (P< 0.001, log-rank test). Mean ± SD, n=150-200 flies for each. Genotypes: elav-GAL4 in trans to UAS-SCA3trQ78(s)CAG or UAS-SCA3trQ78CAA/G. (h) Climbing behavior with age. At 1d, both SCA3trQ78CAG and SCA3trQ78CAA/G flies show normal climbing compared to SCA3trQ27 control flies, with only ∼5% failing to climb with agitation (mean ± SD, n=120-200 flies total). SCA3trQ78CAG flies show more progressive climbing defects, such that at 12d, 42.1±7.6% of the flies fail to climb (mean ± SD, n=180). In contrast, only 17.9±2.6 % of SCA3trQ78CAA/G flies fail to climb at 12d (mean ± SD, n=200). Genotypes as in h. *, P<0.05; **, P<0.01; ***, P<0.001 (two-way ANOVA).
Western blots and immunohistochemistry confirmed that flies expressing pure CAG or CAA/G interrupted transgenes for truncated or full-length ataxin-3 expressed similar levels of polyQ protein. In both, protein was initially diffuse, then accumulated into inclusions of similar size and intensity over time, despite the differential toxicity (Supplementary Fig. S7c, S9b, c). The SCA3trQ78CAA/G flies, however, had lower steady-state levels of the transgene-encoding RNA (50±10% of that of SCA3trQ78(s)CAG flies), indicating that interruption of the CAG repeat sequence affected the ratio of SCA3trQ78 transcript to protein (Supplementary Fig. S7b). We also tested whether interrupting the RNA sequence affected the interaction with Mbl. Whereas MblA normally dramatically enhanced polyQ toxicity, MblA had only a modest effect on flies expressing SCA3trQ78CAA/G, with minimal pigmentation and PR loss (Supplementary Fig. S10). Results were similar with co-expression of human MBNL1 (data not shown). Moreover, whereas upregulation of MblA increased the level of the CAG repeat RNA by 1.6±0.2 fold, it did not affect the level of SCA3trQ78CAA/G RNA (Supplementary Fig. S10i). This result suggested that enhancement of ataxin-3 toxicity by MblA correlated with increased CAG repeat RNA.
These results suggested that a pathogenic length CAG repeat RNA may contribute to polyQ toxicity beyond coding for a pathogenic polyQ protein. This raised the possibility that expression of RNAs containing non-coding CAG repeats of pathogenic length may, on their own, be deleterious. We therefore generated flies bearing a pathogenic length CAG repeat within the 3'UTR of the DsRed open reading frame (Fig. 3a). Repeats of ∼100 and ∼250 CAGs were examined, the latter being within the upper limit of expansions in polyQ diseases like SCA2 and SCA7 2. Transgenic lines were selected or combined that expressed the RNA at levels similar to that of SCA3trQ78CAG protein-encoding transgenes (Supplementary Fig. S11a). We confirmed that the non-coding CAG repeat transgenics did not express a CAG-repeat containing protein or generate anti-sense CUG transcripts (Supplementary Fig. S11, and data not shown).
Figure 3. Untranslated CAG repeats induce progressive neural dysfunction.
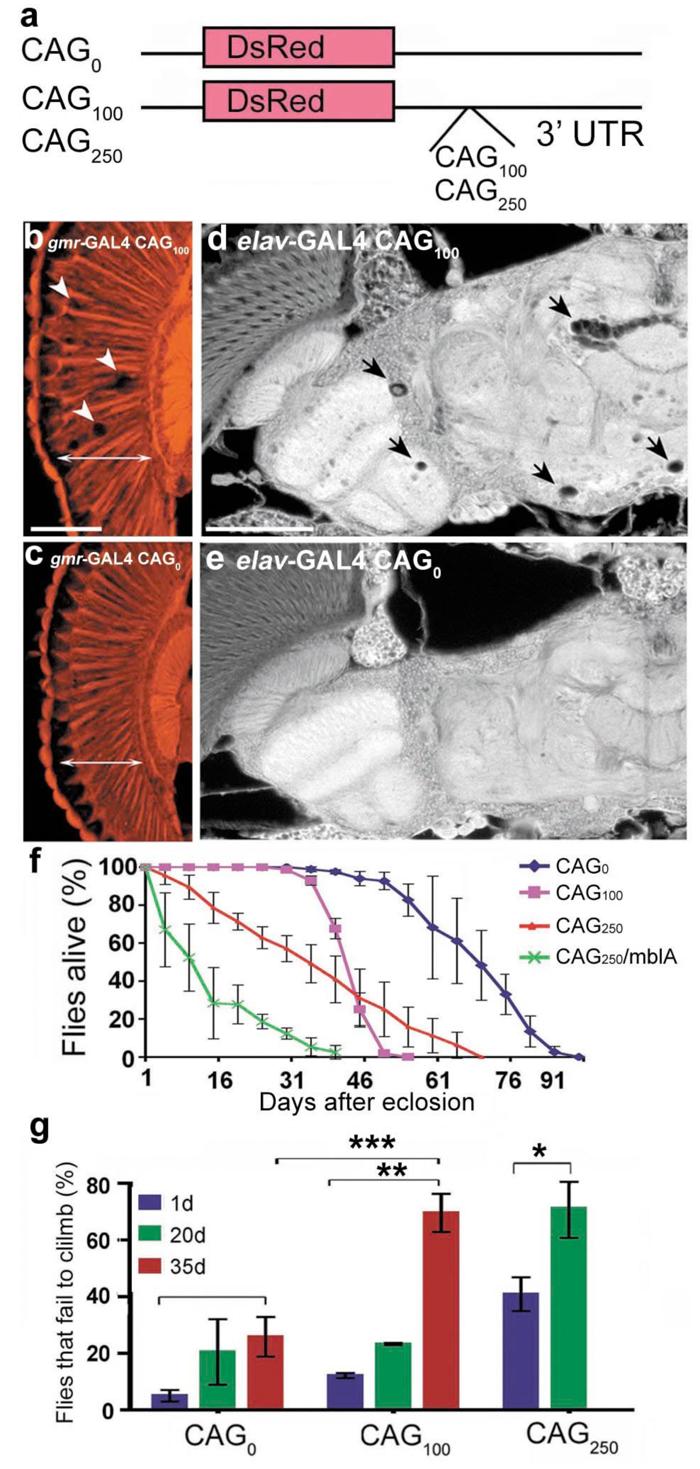
a, Constructs with untranslated CAG repeats within the 3' UTR of a transgene encoding a control protein DsRed. b-e, Flies expressing untranslated CAG repeats show neuronal degeneration. (b, c) Flies expressing CAG100 in the eye with gmr-GAL4 showed loss of retinal integrity (arrows). Paraffin sections of 1d flies, genotypes: (b) gmr-GAL4/ UAS-CAG100 (4x), (c) gmr-GAL4/ UAS-CAG0. (d, e) Flies expressing CAG100 showed progressive brain degeneration with vacuoles in the brain (arrows). Paraffin sections of 35d flies, genotypes: (c) elav-GAL4/UAS-CAG100 (5x) and (e) elav-GAL4/UAS-CAG0. Bar in b, 5 μm for b, c. Bar in d, 10 μm for d, e. f-g, Expression of untranslated CAG repeats induces length-dependent, progressive neural dysfunction. (f) Neurotoxicity by lifespan analysis. Flies expressing untranslated CAG repeats show length-dependent reduced lifespan. Differences in lifespan of flies expressing CAG0, CAG100, and CAG250, and CAG250/mblA are significantly different at P<0.001 (log-rank analysis). Mean ± SD, n= 250, 260, 300, 100 flies, respectively. elav-GAL4 in trans to (CAG0) UAS-CAG0, (CAG100) UAS-CAG100 (five UAS-trangenes were combined to match the RNA expression level to that of SCA3trQ78(s)CAG), (CAG250) UAS-CAG250. (g) Climbing ability with age. Flies expressing CAG0 showed normal climbing defects with age (* P<0.05). Flies expressing CAG100 had mild climbing defects at 20d, which strikingly degenerated by 35d (*** P<0.001 compared to 1d; ** P<0.01 compared to 35d CAG0). CAG250 flies had moderate climbing defects at 1d, which were strikingly worse by 20d (* P<0.05). Mean ± SD, 100-200 flies per time point for each genotype in each experiment, two-way ANOVA. Genotypes as in (f).
Our study revealed that expression of untranslated CAG repeats caused late-onset loss of neuronal integrity when directed to the eye and nervous system. Flies expressing CAG repeat (CAG100 or CAG250) in the eye showed internal loss of retinal tissue, with holes within the retina (Fig. 3b, c, and data not shown). When expressed globally in neurons with _elav_-GAL4, flies bearing CAG100 had normal brain morphology at 1d (data not shown), but underwent striking degeneration of the brain with time, and died early with progressive loss of climbing compared to controls with CAG0 (Fig. 3 d-g). CAG repeat-induced neuronal dysfunction was repeat-length dependent, with flies expressing CAG250 at lower levels than CAG100 showing more severe defects (Fig. 3f, g). MblA also enhanced toxicity of the untranslated CAG RNA (Fig. 3f), and could increase the level of the transcript (Supplementary Fig. S12), indicating that Mbl enhancement of neurotoxicity does not require polyQ protein. We further determined that a long, untranslated, but interrupted RNA construct (CAA/G100 or 262), when expressed at levels comparable to CAG250 lines was not toxic (Supplementary Fig. S13). These results confirmed that pathogenic length CAG repeat RNAs can induce neuronal degeneration independent of coding for a toxic polyQ protein.
We next examined whether untranslated CAG repeats induce toxicity through similar mechanisms as other non-coding trinucleotide repeat diseases; namely like CTG repeat sequences. We examined whether CAG repeat RNA formed RNA foci or affected RNA splicing, which are hallmarks of CUG repeat diseases 13, 14. We found that the CAG repeat RNA (CAG250) could form RNA foci, although the foci were present in a limited number of cells and were small compared to those formed by control CUG repeat RNA (Supplementary Fig. S14). There was no effect on the splicing pattern of a reporter construct 15 when expressed in the fly (Supplementary Fig. S15). These results suggest that untranslated CAG repeat RNA induces neurodegeneration through mechanisms different from those of CUG repeat RNA.
Taken together, our results suggest that CAG-repeat RNAs may contribute a toxic component in polyQ disease, inducing neuronal dysfunction and progressive degeneration (Supplementary Fig. S1). In support of a role of the RNA in polyQ disease are our findings that interruptions of the CAG repeat within protein coding transgenes mitigate toxicity, and that an untranslated CAG repeat RNA can cause toxicity on its own. Although previous studies suggest a minimal role of the RNA in polyQ toxicity 16, raw polyQ domains used in such studies induce strikingly more severe toxicity than polyQ domains within host protein context 17; this may mask toxic contributions of the RNA. We find that flies bearing pathogenic SCA3 polyQ proteins encoded by CAA/G repeat transgenes can still develop degenerative phenotypes, but they are less severe and progress more slowly than those of SCA3tr Q78CAG flies with similar levels of protein. We therefore suggest that the RNA contributes to a shift of the toxicity curve, rather than being solely responsible for toxicity. Host protein context clearly plays a crucial role in polyQ diseases: indeed, a change of a single amino acid outside the polyQ stretch can substantially diminish pathogenesis, despite expression of an expanded CAG repeat RNA 18-20. Within the protein context of ataxin-3, including truncated forms of the protein which occur in disease 21, effects of the CAG-repeat RNA may be particularly apparent. Despite the striking neural dysfunction and degeneration caused by CAG-repeat RNA, we did not observe effects on external eye morphology. This is consistent with a previous study that examined only the external eye 16, and is reminiscent of toxicity of full-length ataxin-3 disease gene in Drosophila 22; these findings suggest a selective sensitivity of neurons to the RNA toxicity.
The pathogenic effects of the CAG repeat RNA may extend to interactions with other cellular factors, as revealed by Mbl. Human MBNL1 was initially identified as a CUG RNA-binding protein 6, although it has been shown to also interact with CAG repeat RNA 11, 12. Our studies reveal that mbl modulation of polyQ toxicity is affected by the polyQ-encoding CAG sequence of ataxin-3, as well as by the domain of MBNL1 for optimal CUG repeat RNA binding. Comparison of flies expressing the same steady-state level of polyQ RNA in the presence or absence of added Mbl indicated that polyQ toxicity with Mbl was more severe (Supplementary Fig. S16), indicating that Mbl enhancement is not exclusively due to an increase in the level of the RNA transcript. These results suggest that muscleblind may act at least in part at the level of the CAG repeat RNA in the animal in vivo. Additional studies are required to determine the role of muscleblind in the human disease situation.
A large number of human degenerative diseases are caused by nucleotide repeat expansions 2. Although the polyQ diseases have typically been viewed as a separate class due to a protein toxicity, other repeat expansion diseases like DM1, fragile X-associated ataxia and SCA8 are associated with deleterious actions of an expanded repeat RNA 23-25. Data with DM1 and SCA8—both CUG repeat RNA expansion diseases—also indicates more complex mechanisms that include the production of anti-sense CAG repeat RNAs that may act at the level of the RNA and/or generation of polyQ protein 26, 27. Our findings that the CAG repeat RNA in the ataxin-3 situation may confer pathogenicity highlights additional aspects of these two classes of disease that may be shared.
Methods summary
General fly lines from the Bloomington Drosophila Stock Center. SCA3trQ78 fly lines are described 7. The SCA3trQ78CAA/G and SCA3nQ81CAA/G constructs were generated from MJDQ82CAA/G 28, using the QuikChange Site-Directed Mutagenesis kit (Stratagene). The MblA cDNA was generated by RT-PCR from wildtype flies. MBNL135 was generated from MBNL140 29 using site-directed mutagenesis. CAG100 and CAG250 repeats (sequences kindly provided by C. Thornton) were inserted into the 3'UTR of DsRed2 gene (Clontech) in the pUAST vector. Non-coding CAG repeats were sized by genescan analysis 30 with primers: forward 5'-cgtggagcagtacgagcgcac-3'; reverse 5'-aggttccttcacaaagatcctc-3'. This analysis showed that the CAG100 lines had repeats that were moderately unstable over time, and at the end of the analysis, had repeats of ∼102-106 (although several lines had flies with repeats of ∼85-90, and one line contracted to 17). Multiple lines were combined to achieve levels of the RNA comparable to that of the SCA3trQ78 lines, and is noted (e.g., 4x means 4 independent transgenic lines were combined for expression). The CAG250 lines had repeat lengths ∼240 to 270, and an expression level of ∼25% that of the SCA3trQ78 lines. Techniques for westerns and northerns were standard; details are described in Supplementary Information. PR counts were performed using the corneal optical neutralization technique. For each data point, 10 ommatidia/eye and 15-20 eyes/genotype were scored; for at least one experiment of each type, genotypes were scored in a blinded manner. Fly climbing ability was analyzed by negative geotaxis. Groups of ∼20 females were anesthetized and placed in a vertical plastic column. After 30 min recovery, flies were banged to the bottom, then scored for climbing ability as the percentage of flies remaining at the bottom (< 2cm) at 25s. Three trials were performed at 3 min intervals in each experiment. 100-200 flies were tested per genotype. Additional details in Supplementary Information.
Supplementary Material
Supplementary
ACKNOWLEDGMENTS
We thank A. Cashmore, D. Lessing and R. Pittman for comments, J. Weissman, C. Thornton, M. Baylies, R. Artero, and the Developmental Studies Hybridoma Bank (supported by NICHD and University of Iowa) for reagents. These studies were supported by the NINDS. NMB is an Investigator of the Howard Hughes Medical Institute.
REFERENCES
- 1.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 2.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–55. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 3.Ross CA, Poirier MA. Opinion: What is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol. 2005 doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- 4.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13:3079–88. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 5.Mankodi A, et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet. 2001;10:2165–70. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 6.Miller JW, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. Embo J. 2000;19:4439–48. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warrick JM, et al. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–49. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 8.Begemann G, et al. muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins. Development. 1997;124:4321–31. doi: 10.1242/dev.124.21.4321. [DOI] [PubMed] [Google Scholar]
- 9.Artero R, et al. The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev Biol. 1998;195:131–43. doi: 10.1006/dbio.1997.8833. [DOI] [PubMed] [Google Scholar]
- 10.Kanadia RN, et al. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci U S A. 2006;103:11748–53. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kino Y, et al. Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum Mol Genet. 2004;13:495–507. doi: 10.1093/hmg/ddh056. [DOI] [PubMed] [Google Scholar]
- 12.Ho TH, et al. Colocalization of muscleblind with RNA foci is separable from misregulation of alternative splicing in myotonic dystrophy. J Cell Sci. 2005;118:2923–33. doi: 10.1242/jcs.02404. [DOI] [PubMed] [Google Scholar]
- 13.Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci. 2006;29:259–77. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 14.Wheeler TM, Thornton CA. Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol. 2007;20:572–6. doi: 10.1097/WCO.0b013e3282ef6064. [DOI] [PubMed] [Google Scholar]
- 15.Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–41. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 16.McLeod CJ, O'Keefe LV, Richards RI. The pathogenic agent in Drosophila models of ‘polyglutamine’ diseases. Hum Mol Genet. 2005;14:1041–8. doi: 10.1093/hmg/ddi096. [DOI] [PubMed] [Google Scholar]
- 17.Marsh JL, et al. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Hum Mol Genet. 2000;9:13–25. doi: 10.1093/hmg/9.1.13. [DOI] [PubMed] [Google Scholar]
- 18.Emamian ES, et al. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38:375–87. doi: 10.1016/s0896-6273(03)00258-7. [DOI] [PubMed] [Google Scholar]
- 19.Graham RK, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–91. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 20.Klement IA, et al. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 21.Goti D, et al. A mutant ataxin-3 putative-cleavage fragment in brains of Machado-Joseph disease patients and transgenic mice is cytotoxic above a critical concentration. J Neurosci. 2004;24:10266–79. doi: 10.1523/JNEUROSCI.2734-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warrick JM, et al. Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol Cell. 2005;18:37–48. doi: 10.1016/j.molcel.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 23.Jin P, et al. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39:739–47. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- 24.Ranum LP, Day JW. Pathogenic RNA repeats: an expanding role in genetic disease. Trends Genet. 2004;20:506–12. doi: 10.1016/j.tig.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Tapscott SJ, Thornton CA. Biomedicine. Reconstructing myotonic dystrophy. Science. 2001;293:816–7. doi: 10.1126/science.1063517. [DOI] [PubMed] [Google Scholar]
- 26.Cho DH, et al. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol Cell. 2005;20:483–9. doi: 10.1016/j.molcel.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Moseley ML, et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38:758–69. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 28.Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI(+)] prion. Cell. 2001;106:183–94. doi: 10.1016/s0092-8674(01)00440-8. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Casado MZ, Artero RD, Paricio N, Terol J, Perez-Alonso M. Generation of GAL4-responsive muscleblind constructs. Genesis. 2002;34:111–4. doi: 10.1002/gene.10147. [DOI] [PubMed] [Google Scholar]
- 30.Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315:1857–9. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary