αE-catenin is not a significant regulator of β-catenin signaling in the developing mammalian brain (original) (raw)
. Author manuscript; available in PMC: 2009 May 1.
Published in final edited form as: J Cell Sci. 2008 Apr 8;121(Pt 9):1357–1362. doi: 10.1242/jcs.020537
Abstract
β-catenin is a critical mediator of the canonical Wnt signaling pathway. α-catenin is a major β-catenin binding protein and overexpressed α-catenin can negatively regulate β-catenin activity. Thus, α-catenin may be an important modulator of Wnt pathway. We show here that endogenous α-catenin has little impact on transcriptional activity of β-catenin in developing mammalian organism. We analyzed β-catenin signaling in mice with conditional deletion of αE-catenin in the developing central nervous system. This mutation results in brain hyperplasia and we investigated whether activation of β-catenin signaling may be at least partially responsible for this phenotype. To reveal potential quantitative or spatial changes in β-catenin signaling, we utilized mice carrying a β-catenin-signaling reporter transgene. In addition, we analyzed the expression of known endogenous targets of the β-catenin pathway and the amount and localization of β-catenin in mutant progenitor cells. We found that while loss of αE-catenin resulted in disruption of intercellular adhesion and hyperplasia in the developing brain, β-catenin signaling was not altered. We conclude that endogenous _α_E-catenin has no significant impact on β-catenin transcriptional activities in the developing mammalian brain.
Keywords: brain development, β-catenin signaling, α-catenin
Introduction
β-catenin is an adherens junction (AJ) protein involved in both intercellular adhesion and regulation of the canonical Wnt signaling pathway (Clevers, 2006). In adhesion, β-catenin links cadherins with α-catenin and this interaction is critical for the assembly and maintenance of AJs (Perez-Moreno and Fuchs, 2006). In Wnt signaling, β-catenin binds to Lef/Tcf family of transcriptional factors and functions as a transcriptional co-activator (Clevers, 2006; Nelson and Nusse, 2004). Both the adhesion and signaling activities of β-catenin play a pivotal role in normal development and tissue homeostasis and it is often difficult to discern which of these functions is most critical at any given time of development or adult life of an organism. In fact, such a central position in both intercellular adhesion and signaling makes β-catenin a critical node that may be involved in orchestrating the behavior of individual cells assembled into a multicellular organism (Lien et al., 2006b).
In addition to its role in normal development and adult tissue homeostasis, the β-catenin signaling pathway also plays a causal role in a variety of human malignancies (Clevers, 2006; Perez-Moreno and Fuchs, 2006; Polakis, 2007). Elucidation of the mechanisms responsible for regulation of β-catenin signaling is necessary for understanding of the Wnt pathway and developing efficient tools for anti-cancer therapy. The canonical Wnt signal transduction pathway is the principal regulator of β-catenin-mediated transcription (Clevers, 2006). Wnt interacts with its receptor Frizzled and this activates a signaling cascade that ultimately results in attenuation of the activity of the β-catenin destruction complex and accumulation of cytoplasmic and nuclear β-catenin. Stabilized β-catenin associates with Lef/Tcf transcription factors and activates the transcription of multiple genes including the classic targets of β-catenin pathway c-myc, Cyclin D1 and Axin2 (He et al., 1998; Lustig et al., 2002; Tetsu and McCormick, 1999). Many β-catenin-binding proteins play a profound role in regulation of its signaling activities. Interaction of β-catenin with Lef/Tcf and BCL9/BCL9-2 is required for signaling (Behrens et al., 1996; Brembeck et al., 2004; Kramps et al., 2002; Molenaar et al., 1996). In contrast, adenomatous polyposis coli (APC) and Axin proteins are necessary for degradation of β-catenin and inactivating mutations in APC and Axin2 result in abnormal activation of the Wnt pathway and predisposition to colorectal cancer (Behrens et al., 1998; Korinek et al., 1997; Morin et al., 1997; Polakis, 2007).
In addition to known downstream effectors of classic Wnt pathway, AJ proteins can also influence β-catenin transcriptional activity. For example, cadherins negatively regulate β-catenin by competing for interaction with Lef/Tcf factors and perhaps, by tethering β-catenin to the cell plasma membrane away from the nucleus (Cox et al., 1996; Fagotto et al., 1996; Heasman et al., 1994; Orsulic et al., 1999; Sanson et al., 1996). It has been reported that another β-catenin-binding and AJ protein, α-catenin, is also a negative regulator of β-catenin signaling pathway (Giannini et al., 2000a; Hwang et al., 2005; Merdek et al., 2004; Sehgal et al., 1997; Simcha et al., 1998). Overexpression of α-catenin antagonized dorsalization effects of β-catenin in Xenopus embryos (Sehgal et al., 1997). In addition, overexpression of α-catenin in cell lines also resulted in attenuation of β-catenin transcriptional activity (Giannini et al., 2000b; Merdek et al., 2004; Simcha et al., 1998). Finally, siRNA-mediated depletion of α-catenin in cultured chondrocytes resulted in small, but statistically significant increase in β-catenin signaling (Hwang et al., 2005). Negative regulation of β-catenin transcriptional activity by α-catenin can be explained not only by sequestration of β-catenin away from the nucleus, but also by competition between α-catenin and DNA for binding to β-catenin/TCF complex (Giannini et al., 2000a). In contrast to these studies, abnormal activation of β-catenin was not detected in mouse keratinocytes lacking major epithelial α-catenin, αE-catenin (Vasioukhin et al., 2001) and it is still unclear whether endogenous α-catenin can regulate β-catenin signaling pathway in vivo.
We have recently generated mice with conditional deletion of αE-catenin in the developing central nervous system and found that these animals display massive brain hyperplasia and dysplasia (Lien et al., 2006a). While we revealed that abnormal activation of the Hedgehog signaling pathway was playing an important role in hyperplasia in _αE-cateni_−/− brains, an abnormal increase in the total number of cells in the developing brain was also consistent with potential activation of β-catenin signaling. Since multiple studies demonstrated that α-catenin is a negative regulator of β-catenin transcriptional activity, we hypothesized that deletion of αE-catenin in developing mouse brain activates β-catenin and this activation may be at least partially responsible for hyperplasia in _αE-catenin_−/− brains. Surprisingly, our results revealed no significant changes in β-catenin signaling pathway in _αE-catenin_−/− brains. These data suggest that endogenous α-catenin may have very little if any role in regulation of β-catenin transcriptional activity in vivo.
Results and Discussion
αE-catenin is a prominent part of β-catenin protein complexes; however, depletion of αE-catenin has no effect on interaction between β-catenin, N-cadherin and other major β-catenin-binding proteins
Both α_E-_ and αN-catenins are expressed in the developing mammalian brain; however, these genes display striking cell type specificity, with αE-catenin primarily expressed in the dividing neural progenitors and αN-catenin in differentiated neurons (Lien et al., 2006a; Stocker and Chenn, 2006) Supplementary Materials Fig. S1). We previously reported that conditional deletion of αE-catenin in the developing central nervous system using Nestin-Cre driver results in disruption of cell-cell junctions, dysplasia and hyperplasia of neural progenitors (Lien et al., 2006a). The Nestin-Cre is activated in neural progenitors at embryonic day 10.5 (E10.5) of development (Graus-Porta et al., 2001). While no differences in cell numbers were detected between the wild-type and mutant brains in E12.5 embryos, E13.5 embryos displayed a 40% percent increase in the total number of cells in the mutant brains. The increase in cell numbers continued later in development; however, the most explosive hyperplasia in _αE-catenin_−/− brains took place early, between days E12.5 and E14.5 of development (Lien et al., 2006a). Since αE-catenin is a known binding partner of β-catenin and β-catenin plays an important role in regulating the proliferation of embryonic neural progenitor cells (Chenn and Walsh, 2002; Woodhead et al., 2006), we decided to analyze whether depletion of αE-catenin results in changes in transcriptional activity of β-catenin. Since the most drastic hyperplasia took place in αE-catenin−/− brains between E12.5 and E14.5 days of development, we concentrated on these developmental time points. We first analyzed potential changes in β-catenin protein complexes. For this purpose, β-catenin and its interacting proteins were immunoprecipitated from wild-type and αE-catenin−/− brains and analyzed by SDS-PAGE followed by Coomassie and Silver stainings and Western blotting with anti-β-catenin, anti-N-cadherin and anti-αE-catenin antibodies (Fig. 1A-B). Discrete protein bands corresponding to β-catenin, cadherins, α-catenin and few additional unidentified proteins were present in the immunoprecipitates from the wild-type brains (Fig. 1A). As expected, levels of α-catenin were significantly decreased in β-catenin protein complexes from _αE-catenin_−/− brains. Surprisingly, this was the only major change that was detected by Coomassie and Silver stainings of the proteins immunoprecipitated with anti-β-catenin antibodies (Fig. 1A). We conclude that α-catenin is a prominent part of β-catenin protein complexes; however, depletion of α-catenin has little effect on interaction between β-catenin and other major β-catenin-binding proteins. Small amounts of α-catenin remaining in the complexes from the mutant brains were likely to represent αN-catenin prominently expressed in the neurons of E14.5 brains (Fig. 1A), and the residual amounts of αE-catenin in the few brain cells (endothelial cells) that remained non-targeted by the Nestin-Cre transgene (Fig. 1B).
Fig. 1.
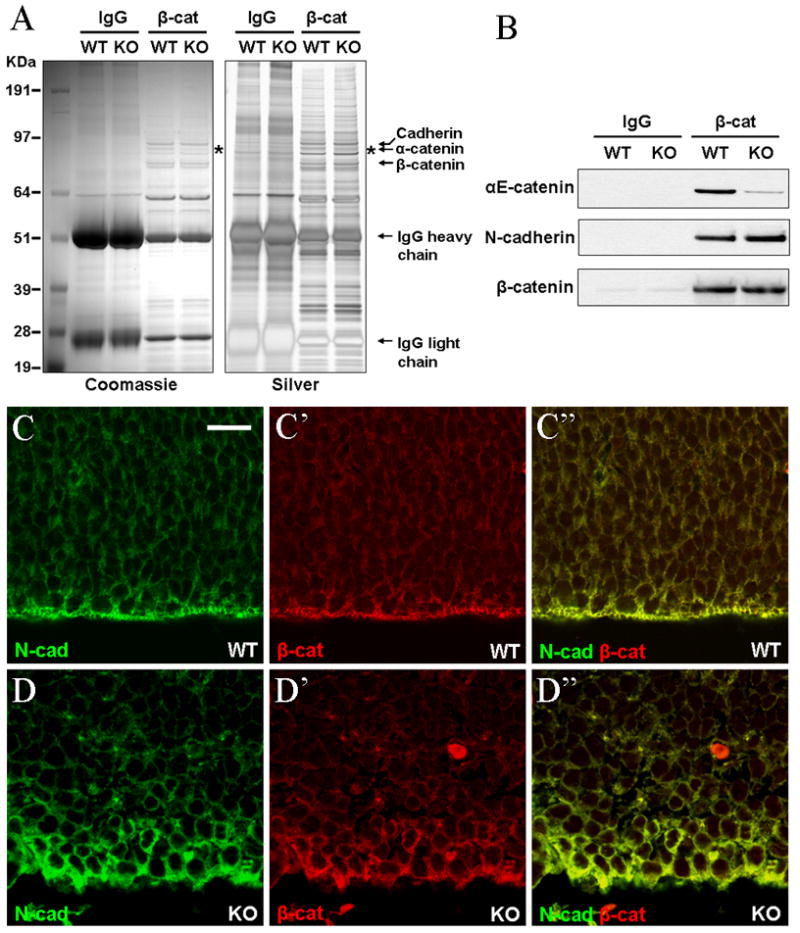
Depletion of αE-catenin has no major effect on interaction between β-catenin, N-cadherin and other β-catenin-binding proteins. (A-B) Total protein lysates from E14.5 wild-type (WT) and αE-catenin−/− (knockout, KO) brains were immunoprecipitated with control (IgG) or anti-β-catenin (β-cat) antibodies and resulting protein complexes were separated by SDS-PAGE and stained with Colloidal blue and Silver stain (A) or analyzed by Western blot with anti-αE-catenin, N-cadherin or β-catenin antibodies (B). Note that while αE-catenin becomes depleted from β-catenin protein complexes, composition or relative abundance of other proteins does not change. Western blotting reveals no significant changes in association between β-catenin and N-cadherin. (C-D″) Despite disruption of apical junctional complexes and loss of cell polarity, β-catenin continues to co-localize with N-cadherin at the periphery of αE-catenin−/− neural progenitor cells. Cortical sections from E13.5 wild-type (WT) and _αE-catenin_−/− (KO) embryos were stained with anti-N-cadherin (N-cad) and anti-β-catenin (β-cat) antibodies. Bar in C represents 15.9 μm.
To determine whether localization of β-catenin is changed in _αE-catenin_−/− brains, we performed immunofluorescent stainings of cortical sections from E13.5 embryos with anti-N-cadherin and anti-β-catenin antibodies. Analyses of sections using confocal microscope revealed disruption of apical junctional complexes and disorganization of _αE-catenin_−/− neural progenitors (Fig. 1C-D″). Nevertheless, β-catenin in these cells was still present at the cell periphery and co-localized with N-cadherin.
We conclude that despite the depletion of αE-catenin, composition of other major proteins in β-catenin protein complexes remains unchanged and β-catenin continues to co-localize with N-cadherin at the surface of _αE-catenin_−/− neural progenitor cells.
Depletion of αE-catenin has no impact on the total level of β-catenin and its nuclear localization
Wnt-mediated activation of β-catenin signaling usually results in inhibition of the β-catenin destruction complex and accumulation of β-catenin (Clevers, 2006). We analyzed potential changes in total levels of β-catenin in E12.5 and E13.5 wild-type and αE-catenin−/− brains using Western blot analysis (Fig. 2A). While αE-catenin was depleted in the mutant brains, the total levels of β-catenin remained unchanged (Fig. 2A′).
Fig. 2.
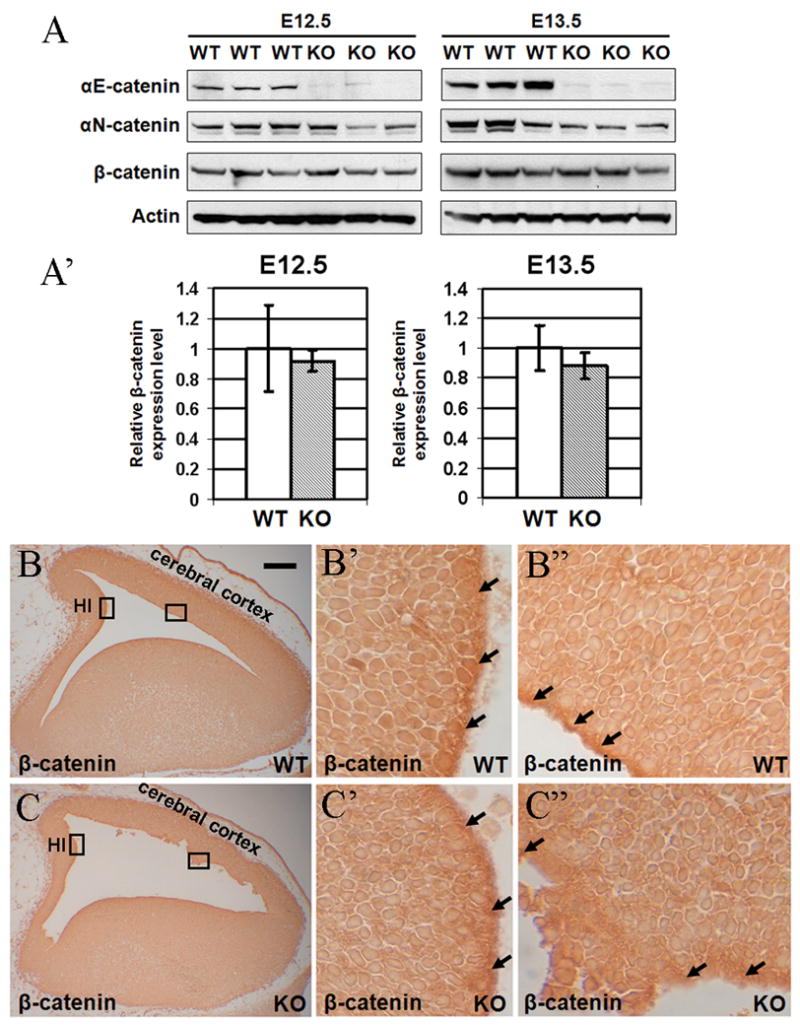
Depletion of αE-catenin has no effect on overall level of β-catenin or its nuclear localization. (A–A′) Total protein lysates from E12.5 and E13.5 wild-type (WT) and _αE-catenin_−/− (KO) brains were analyzed by Western blotting with anti-αE-catenin, anti-αN-catenin, β-catenin and β-actin antibodies. Quantitation of these results is shown in A′. Levels of β-catenin were normalized using β-actin and the results are shown as relative fold change. Data represent means ± SD (n=3). (B) Sagittal telencephalon sections from E13 embryos were immunostained for nuclear β-catenin. Sections were subjected to antigen retrieval, stained overnight with anti-β-catenin antibodies and processed using ABC MOM staining kit. β-catenin was present in the nuclei of progenitor cells, which were localized around the ventricles. Prominent staining was also seen in the AJs at the ventricular surface (black arrows). There were no significant differences in the nuclear β-catenin between the wild-type (WT) and knockout (KO) brains. HI – developing hippocampus. Bar in B represents 0.12 mm for B, C and 0.012 mm for B′–C″.
Activated β-catenin localizes to the cell nucleus and presence of cells with nuclear β-catenin is indicative of activation of β-catenin signaling pathway. Nuclear β-catenin is difficult to detect using regular immunofluorescent staining approach; however, it can be revealed using special antigen retrieval protocols (Merrill et al., 2001). We used this protocol to reveal potential changes in nuclear localization of β-catenin in _αE-catenin_−/− brains. No significant differences in the number or localization of cells with nuclear β-catenin were found between wild-type and αE-catenin −/− brains (Fig. 2B-C″).
We conclude that depletion of αE-catenin in the developing mouse brain does not alter the overall level or nuclear localization of β-catenin.
In vivo reporter for Lef/Tcf transcriptional activity reveals no changes in the spatial distribution or level of β-catenin signaling in αE-catenin−/− brains
While analyses of the total level of β-catenin or its nuclear localization can provide a general estimation of potential changes in β-catenin signaling pathway, these measurements may not be sufficiently sensitive and specific. The endogenous transcriptional reporter system has proven to be a very useful tool for quantitative and spatial analysis of β-catenin transcriptional activity. Several β-catenin signaling reporter mice have been developed and analyzed (DasGupta and Fuchs, 1999; Maretto et al., 2003; Moriyama et al., 2007). To determine potential changes in β-catenin transcriptional activity in vivo, we utilized TOPGAL mice (DasGupta and Fuchs, 1999). These animals carry the transgene containing 3 Lef/Tcf binding sites in front of a c-fos minimal promoter and LacZ reporter gene (Fig. 3A and (DasGupta and Fuchs, 1999)). We crossed our αE-cateninflox/flox/Nestin-Cre mice (Lien et al., 2006a) with TOPGAL animals and generated αE-cateninflox/flox/Nestin-Cre/TOPGAL mice. Staining the brains of TOPGAL αE-catenin+/+ E12.5 and E13.5 embryos for β-galactosidase activity revealed pattern of reporter expression that was similar to the pattern observed previously with other reporters of Lef/Tcf signaling (Maretto et al., 2003; Moriyama et al., 2007). β-catenin signaling in the developing mouse telencephalon was highly compartmentalized with the reporter displaying high levels of activity in E12.5 dorsal cortex, especially in the cingulate cortex area (Fig. 3B). In E13.5 cortexes, the area displaying active β-catenin signaling expands to encompass the developing hippocampus (Fig. 3C). Remarkably, nearly identical pattern of staining was observed in both E12.5 and E13.5 αE-cateninflox/flox/Nestin-Cre/TOPGAL mice (Fig. 3B′, C′). Therefore, we conclude that depletion of αE-catenin did not change the spatial pattern of active β-catenin signaling in the developing mouse telencephalon.
Fig. 3.
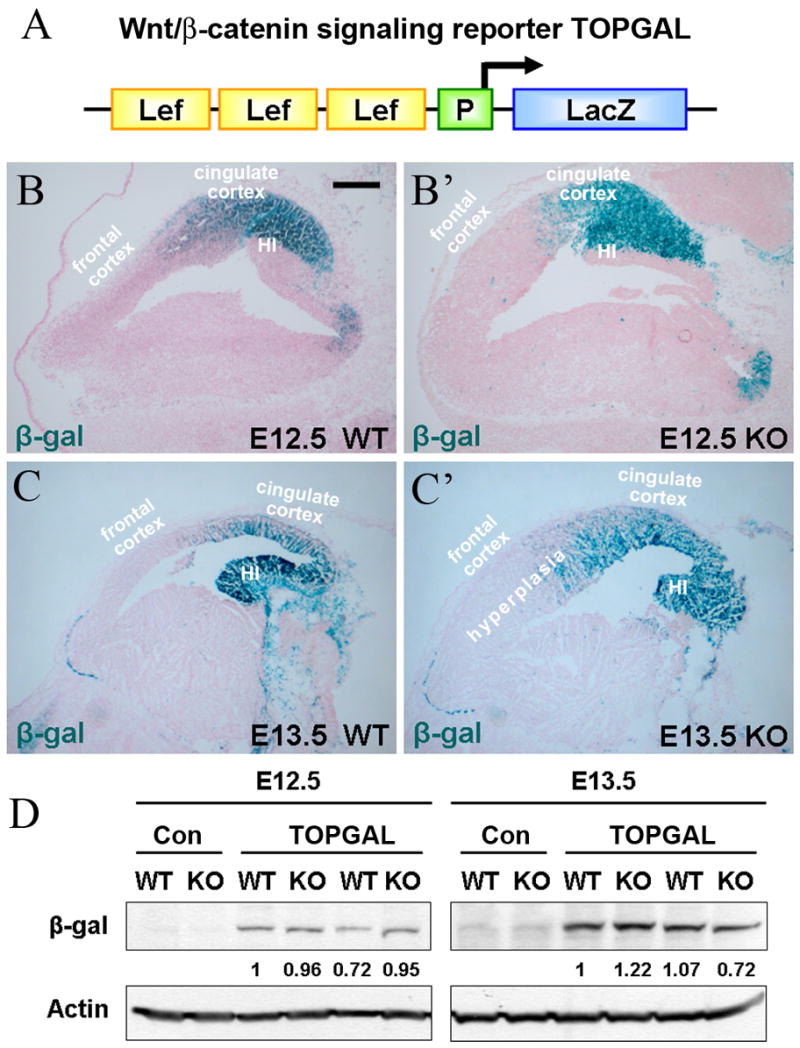
Endogenous reporter for β-catenin transcriptional activity reveals no changes in _αE-catenin_−/− brains. (A) Schematic representation of TOPGAL reporter (DasGupta and Fuchs, 1999). The reporter contains three consensus Lef/Tcf-binding motifs (Lef) and a minimal c-fos promoter (P) to drive transcription of the lacZ gene. (B-C′) Similar pattern of β-catenin reporter expression in the telencephalon of wild-type (WT) and _αE-catenin_−/− (KO) embryos. Sagital sections of brains from E12.5 (B–B′) and E13.5 (C–C′) embryos positive for TOPGAL transgene were stained for β-galactosidase (blue) and counterstained with nuclear fast red. HI – developing hippocampus. Bar in B represents 0.19 mm in BB′ and 0.26 mm in C-C′. (D) Similar levels of β-catenin reporter expression between wild-type (WT) and αE-catenin−/− (KO) brains. Total protein lysates from E12.5 and E13.5 telencephalons of TOPGAL positive animals were analyzed by Western blotting with anti-β-galactosidase (β-gal) and anti-beta;-actin antibodies. Con - control samples from TOPGAL-negative animals. The numbers indicate the relative amounts of β-gal adjusted by the levels of β-actin.
While staining of tissue sections for LacZ is useful for spatial localization of β-catenin signaling, quantitation of this enzymatic staining is challenging. To determine whether the overall levels of the reporter were different in _αE-catenin_−/− brains we performed western blot analyses of the total protein extracts from TOPGAL wild-type and _αE-catenin_−/− brains with anti-β-galactosidase antibodies. We found no significant differences in the level of the reporter between wild-type and mutant brains (Fig. 3D). Overall, we conclude that neither spatial distribution nor the level of expression of the Lef/Tcf reporter construct were significantly altered in _αE-catenin_−/− brain.
Depletion of α-catenin has no impact on the levels of endogenous transcriptional targets of β-catenin signaling
The synthetic TOPGAL promoter may not be able to faithfully reproduce the complexity of transcriptional regulation at the endogenous promoter sequences, which are controlled by β-catenin transcriptional activity. While β-catenin can control transcription of many genes in a tissue- and time-specific manner, c-myc, cyclin D1 and Axin2, are considered to be the classic endogenous transcriptional targets of β-catenin signaling (He et al., 1998; Lustig et al., 2002; Tetsu and McCormick, 1999). To examine whether depletion of αE-catenin results in changes in transcriptional levels of these genes, we performed real-time PCR analysis using total RNA extracted from E12.5 and E13.5 wild-type and _αE-catenin_−/− brains. While transition from E12.5 to E13.5 is associated with significant hyperplasia in _αE-catenin_−/− brains, we found no statistically significant changes in the levels of c-myc, cyclin D1 and Axin2 between the wild-type and _αE-catenin_−/− brains (Fig. 4). Thus, levels of known endogenous transcriptional targets of β-catenin signaling are not changed at the time of the most drastic increase in total cell number in _αE-catenin_−/− brains.
Fig. 4.
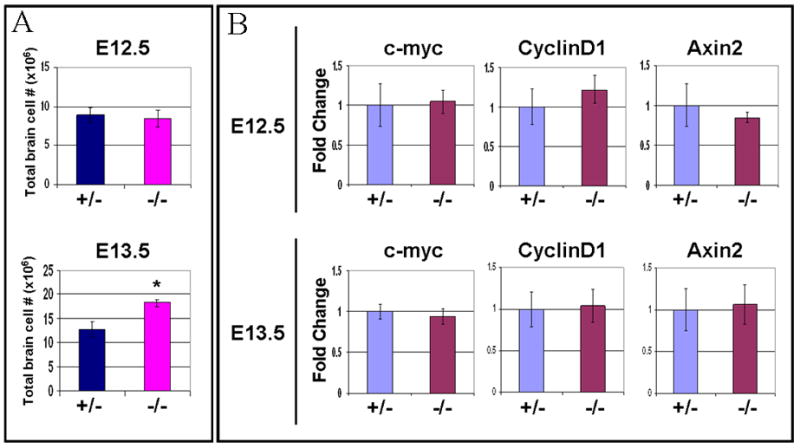
Transition from E12.5 to E13.5 results in extensive hyperplasia without significant changes in expression of endogenous transcriptional targets of β-catenin signaling pathway. (A) Hyperplasia in E13.5 αE-catenin−/− brains. Total cells were isolated from E12.5 and E13.5 wild-type (WT) and _αE-catenin_−/− brains and counted using Coulter Counter. Data represent means ± SD. n=3 to 5. Asterisk indicates statistically significant difference with P<0.0001. (B) qPCR analysis of β-catenin pathway transcripts c-myc, Cyclin D1, Axin2 in E12.5 and E13.5 heterozygous and αE-catenin−/− brains. The levels of expression are shown in arbitrary units with mean of levels in heterozygous embryos adjusted to one. Data represent means ± SD. n=4.
In summary, we used a loss of function approach to determine the potential role of endogenous αE-catenin in the regulation of β-catenin signaling in the developing brain. For this purpose, we analyzed the level and localization of β-catenin, activity of β-catenin signaling via a TOPGAL reporter construct and the levels of known endogenous transcriptional targets of the β-catenin signaling pathway. We did not find significant changes in β-catenin-mediated transcriptional activity in _αE-catenin_−/− neural progenitor cells. While it is possible that our analysis was not sensitive enough to detect potential minor changes in β-catenin signaling, our results indicate that α-catenin has very little impact on β-catenin signaling in vivo. This is different from the results obtained using overexpression or knockdown of α-catenin in cultured cell lines (Giannini et al., 2000a; Hwang et al., 2005; Merdek et al., 2004; Sehgal et al., 1997; Simcha et al., 1998). It is possible that small amounts of αN-catenin in neural progenitors compensate for the loss of αE-catenin, however, this is unlikely, because _αE-catenin_−/− progenitors display prominent cell-cell adhesion defects. Both αE- and αN-catenins are completely competent in AJs formation (Hirano et al., 1992). Therefore, disruption of AJs in _αE-catenin_−/− progenitors indicates absence of compensation by αN-catenin. The most likely reason for differences between our results and previously published studies are the different model systems that were utilized. We believe that our in vivo approach may be more relevant for the analysis of α-catenin, because absence of 3-dimentional tissue organization in cells in culture may produce major changes, which are simply not pertinent to the situation in the live organism. This is especially critical for studies on catenins, because these proteins are directly involved in tissue organization via their role in AJs formation.
Materials and Methods
Mice
Mice with _Nestin-Cre_-mediated conditional deletion of αE-catenin in the developing central nervous system were generated as described (Lien et al., 2006a). TOPGAL Lef/Tcf reporter mice were obtained from Dr. Elaine Fuchs (DasGupta and Fuchs, 1999). To obtain TOPGAL/Nestin-Cre/αE-cateninflox/flox mice, we crossed TOPGAL females with Nestin-Cre/αE-cateninflox/+ males, and the resulting TOPGAL/Nestin-Cre/αE-cateninflox/+ males were crossed with αE-cateninflox/flox females. All mice were on C57BL/6J genetic background.
Immunoprecipation and Western blotting
Total proteins were extracted from embryonic brains with IP buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.5% Brij, 10% glycerol, 0.1 mM EDTA, 0.5 mM MgCl2, 20 mM sodium fluoride, 10 mM sodium pyrophosphate, 1 mM sodium vanadate and a cocktail of protease inhibitors). Extracts were precleared by centrifugation at 25,000 × g for 15 min at 4°C and supernatants were incubated with 30 μl of 50% slurry of ProteinA-Sepharose (Amersham) for 30 minutes. Resulting extracts were rotated at 4°C for 1 hour with anti-b-catenin antibody (Sigma) and then for 1 hour with 50 μl of 50% slurry of Protein A-Sepharose conjugated to rabbit anti-mouse antibody. Sepharose beads were washed 4 times with IP buffer and bound proteins were released by addition of LDS loading buffer and heating at 100°C for 5 min. Immunoprecipated proteins or total protein extracts were resolved by NuPAGE electrophoresis (Invitrogen) and transferred to Immobilon P membrane (Millipore) or stained with Colloidal Blue or Silver stain (Invitrogen). The membranes were incubated with anti-αE-catenin (1:500, gift from Dr. Tsukita), anti-αN-catenin (NCAT2, 1:500, University of Iowa Hybridoma Bank) anti-β-catenin (1:2000, Sigma), anti-β-actin (1:10,000, Sigma), anti-N-cadherin (1:2000, Zymed), or anti-β-galactosidase (1:2000, Rockland) antibodies at 4°C overnight. The membranes were washed and incubated with anti-mouse or anti-rabbit IgG coupled to horseradish peroxidase (Jackson Laboratories) at a dilution of 1:10,000. The blots were developed using ECL chemiluminescence detection reagent (Pierce).
Immunofluorescence and immunohistochemistry
Immunofluorescence staining was performed as described (Klezovitch et al., 2004). Stained sections were analyzed using the Zeiss LSM510 confocal two photon microscope. For nuclear b-catenin staining brain tissues were first fixed in 4% paraformaldehyde for 15 mins on ice, washed from formaldehyde in PBS, processed, embedded in paraffin, sectioned and resulting sections were deparafinized, hydrated, subjected to antigen retrieval by autoclaving for 15 min in the antigen unmasking solution (Vector Laboratories, H3300) and incubated with primary anti-β-catenin antibodies (1:2000, Sigma, C-7082) overnight at 4°C, as described above. The ABC (mouse on mouse) MOM kit (Vector Laboratories) was used for immunohistochemical detection of primary antibodies (Jackson Laboratories). Secondary antibodies were detected with DAB peroxidase substrate kit (Vector Laboratories).
X-gal staining
E12.5 to E13.5 mouse embryos were pre-fixed for 20 min in 4% paraformaldehyde in PBS on ice, washed 4 times in cold PBS and incubated in 30% sucrose in PBS at 4°C, overnight. Subsequently, mouse heads were embedded in OCT (Tissue-Tek) and cryosectioned at 7 μm. Sections were post-fixed in 0.5% glutaraldehyde in PBS for 2 min at room temperature, rinsed 7 times with PBS and then stained overnight in the dark, at room temperature with X-gal solution (100 mM Na phosphate pH 7.3, 1.3 mM MgCl2, 3 mM K3Fe(CN)6, 3 mM K4Fe(CN)6,, 1 mg/ml X-gal). Sections were counterstained with Nuclear Fast Red (Vector Labs).
RNA isolation and quantitive RT-PCR
Total brain RNA was extracted with TRIZOL and reverse transcribed using SuperScript III First-strand synthesis system kit (Invitrogen). Quantitative-PCR was performed using Prism 7900HT instrument (Applied Biosystems), platinum qPCR mix (Invitrogen) and Universal ProbeLibrary kit utilizing the primers, probes and PCR conditions recommended by Universal ProbeLibrary assay center (www.roche-applied-science.com/sis/rtpcr/upl/adc.jsp). PCR for ribosomal protein Rsp16 was used for normalization.
Total brain cell number counting
To determine the total brain cell numbers, brains were dissected, incubated in DMEM media containing 0.6 mg/ml papain (Worthington) and 20 μg/ml DNAse (Sigma) for 20 min at room temperature, and dissociated to a single cell suspension by trituration. Cells were counted using Z1 Coulter particle counter (Beckman Coulter).
Supplementary Material
SFig.1
Fig. S1. Expression patterns of αE- and αN-catenins in E12.5 cerebral cortexes from wild-type (WT) and αE-cateninfl/fl/Nestin-Cre (KO) embryos. (A-B″) Frozen cortical sections were stained with anti-αE-catenin (αE-cat, red) and anti-N-cadherin (N-cad, green) antibodies. (C-D″) Frozen cortical sections were stained with anti-αN-catenin (αN-cat, red) and anti-N-cadherin (N-cad, green) antibodies. Note prominent staining for αN-catenin in developing neuronal cortical plate (C, C″), and staining for αE-catenin in the area containing neural progenitors and throughout the entire cortex. Bar in A represents 0.1 mm.
Acknowledgments
We thank all members of Dr. Vasioukhin’s laboratory for help and critical reading of the manuscript. Drs. Nagafuchi, Tsukita and University of Iowa Developmental Studies Hybridoma Bank for generous gift of antibodies. Dr. Elaine Fuchs for the gift of TOPGAL mice. Vi Nguyen for help with mouse genotyping. This work was supported by the NCI grant 1R01CA098161 to VV.
References
- Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280:596–9. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–42. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Brembeck FH, Schwarz-Romond T, Bakkers J, Wilhelm S, Hammerschmidt M, Birchmeier W. Essential role of BCL9-2 in the switch between beta-catenin’s adhesive and transcriptional functions. Genes Dev. 2004;18:2225–30. doi: 10.1101/gad.317604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–9. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Cox RT, Kirkpatrick C, Peifer M. Armadillo is required for adherens junction assembly, cell polarity, and morphogenesis during Drosophila embryogenesis. J Cell Biol. 1996;134:133–48. doi: 10.1083/jcb.134.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–68. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- Fagotto F, Funayama N, Gluck U, Gumbiner BM. Binding to cadherins antagonizes the signaling activity of beta-catenin during axis formation in Xenopus. J Cell Biol. 1996;132:1105–14. doi: 10.1083/jcb.132.6.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini AL, Vivanco M, Kypta RM. alpha-catenin inhibits beta-catenin signaling by preventing formation of a beta-catenin*T-cell factor*DNA complex. J Biol Chem. 2000a;275:21883–8. doi: 10.1074/jbc.M001929200. [DOI] [PubMed] [Google Scholar]
- Giannini AL, Vivanco MM, Kypta RM. Analysis of beta-catenin aggregation and localization using GFP fusion proteins: nuclear import of alpha-catenin by the beta-catenin/Tcf complex. Exp Cell Res. 2000b;255:207–20. doi: 10.1006/excr.1999.4785. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–79. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Heasman J, Crawford A, Goldstone K, Garner-Hamrick P, Gumbiner B, McCrea P, Kintner C, Noro CY, Wylie C. Overexpression of cadherins and underexpression of beta-catenin inhibit dorsal mesoderm induction in early Xenopus embryos. Cell. 1994;79:791–803. doi: 10.1016/0092-8674(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Hirano S, Kimoto N, Shimoyama Y, Hirohashi S, Takeichi M. Identification of a neural alpha-catenin as a key regulator of cadherin function and multicellular organization. Cell. 1992;70:293–301. doi: 10.1016/0092-8674(92)90103-j. [DOI] [PubMed] [Google Scholar]
- Hwang SG, Yu SS, Ryu JH, Jeon HB, Yoo YJ, Eom SH, Chun JS. Regulation of beta-catenin signaling and maintenance of chondrocyte differentiation by ubiquitin-independent proteasomal degradation of alpha-catenin. J Biol Chem. 2005;280:12758–65. doi: 10.1074/jbc.M413367200. [DOI] [PubMed] [Google Scholar]
- Klezovitch O, Fernandez TE, Tapscott SJ, Vasioukhin V. Loss of cell polarity causes severe brain dysplasia in Lgl1 knockout mice. Genes Dev. 2004;18:559–71. doi: 10.1101/gad.1178004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–7. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S, Murone M, Zullig S, Basler K. Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell. 2002;109:47–60. doi: 10.1016/s0092-8674(02)00679-7. [DOI] [PubMed] [Google Scholar]
- Lien WH, Klezovitch O, Fernandez TE, Delrow J, Vasioukhin V. alphaE-catenin controls cerebral cortical size by regulating the hedgehog signaling pathway. Science. 2006a;311:1609–12. doi: 10.1126/science.1121449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien WH, Klezovitch O, Vasioukhin V. Cadherin-catenin proteins in vertebrate development. Curr Opin Cell Biol. 2006b;18:499–506. doi: 10.1016/j.ceb.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22:1184–93. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci U S A. 2003;100:3299–304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merdek KD, Nguyen NT, Toksoz D. Distinct activities of the alpha-catenin family, alpha-catulin and alpha-catenin, on beta-catenin-mediated signaling. Mol Cell Biol. 2004;24:2410–22. doi: 10.1128/MCB.24.6.2410-2422.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill BJ, Gat U, DasGupta R, Fuchs E. Tcf3 and Lef1 regulate lineage differentiation of multipotent stem cells in skin. Genes Dev. 2001;15:1688–705. doi: 10.1101/gad.891401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–9. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–90. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Moriyama A, Kii I, Sunabori T, Kurihara S, Takayama I, Shimazaki M, Tanabe H, Oginuma M, Fukayama M, Matsuzaki Y, et al. GFP transgenic mice reveal active canonical Wnt signal in neonatal brain and in adult liver and spleen. Genesis. 2007;45:90–100. doi: 10.1002/dvg.20268. [DOI] [PubMed] [Google Scholar]
- Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–7. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsulic S, Huber O, Aberle H, Arnold S, Kemler R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci. 1999;112 (Pt 8):1237–45. doi: 10.1242/jcs.112.8.1237. [DOI] [PubMed] [Google Scholar]
- Perez-Moreno M, Fuchs E. Catenins: keeping cells from getting their signals crossed. Dev Cell. 2006;11:601–12. doi: 10.1016/j.devcel.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Sanson B, White P, Vincent JP. Uncoupling cadherin-based adhesion from wingless signalling in Drosophila. Nature. 1996;383:627–30. doi: 10.1038/383627a0. [DOI] [PubMed] [Google Scholar]
- Sehgal RN, Gumbiner BM, Reichardt LF. Antagonism of cell adhesion by an alpha-catenin mutant, and of the Wnt-signaling pathway by alpha-catenin in Xenopus embryos. J Cell Biol. 1997;139:1033–46. doi: 10.1083/jcb.139.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simcha I, Shtutman M, Salomon D, Zhurinsky J, Sadot E, Geiger B, Ben-Ze’ev A. Differential nuclear translocation and transactivation potential of beta-catenin and plakoglobin. J Cell Biol. 1998;141:1433–48. doi: 10.1083/jcb.141.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker AM, Chenn A. Differential expression of alpha-E-catenin and alpha-N-catenin in the developing cerebral cortex. Brain Res. 2006:1073–1074. 151–8. doi: 10.1016/j.brainres.2005.12.057. [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Bauer C, Degenstein L, Wise B, Fuchs E. Hyperproliferation and defects in epithelial polarity upon conditional ablation of alpha-catenin in skin. Cell. 2001;104:605–17. doi: 10.1016/s0092-8674(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Woodhead GJ, Mutch CA, Olson EC, Chenn A. Cell-autonomous beta-catenin signaling regulates cortical precursor proliferation. J Neurosci. 2006;26:12620–30. doi: 10.1523/JNEUROSCI.3180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SFig.1
Fig. S1. Expression patterns of αE- and αN-catenins in E12.5 cerebral cortexes from wild-type (WT) and αE-cateninfl/fl/Nestin-Cre (KO) embryos. (A-B″) Frozen cortical sections were stained with anti-αE-catenin (αE-cat, red) and anti-N-cadherin (N-cad, green) antibodies. (C-D″) Frozen cortical sections were stained with anti-αN-catenin (αN-cat, red) and anti-N-cadherin (N-cad, green) antibodies. Note prominent staining for αN-catenin in developing neuronal cortical plate (C, C″), and staining for αE-catenin in the area containing neural progenitors and throughout the entire cortex. Bar in A represents 0.1 mm.