Human Alkyladenine DNA Glycosylase Employs a Processive Search for DNA Damage (original) (raw)
. Author manuscript; available in PMC: 2009 Nov 4.
Published in final edited form as: Biochemistry. 2008 Oct 8;47(44):11434–11445. doi: 10.1021/bi801046y
Abstract
DNA repair proteins conduct a genome-wide search to detect and repair sites of DNA damage wherever they occur. Human alkyladenine DNA glycosylase (AAG) is responsible for recognizing a variety of base lesions, including alkylated and deaminated purines, and initiating their repair via the base excision repair pathway. We have investigated the mechanism by which AAG locates sites of damage using an oligonucleotide substrate containing two sites of DNA damage. This substrate was designed so that AAG randomly binds to either of the two lesions. AAG-catalyzed base excision creates a repair intermediate and the subsequent partitioning between dissociation and diffusion to the second site can be quantified from the rates of formation of the different products. Our results demonstrate that AAG has the ability to slide for short distances along DNA at physiological salt concentrations. The processivity of AAG decreases with increasing ionic strength to become fully distributive at high ionic strength, suggesting that electrostatic interactions between the negatively charged DNA and the positively charged DNA binding surface are important for nonspecific DNA binding. Although the amino terminus of the protein is dispensable for glycosylase activity at a single site, we find that deletion of the amino terminal 80 amino acids significantly decreases the processivity of AAG. These observations support the idea that diffusion on undamaged DNA contributes to the search for sites of DNA damage.
Although DNA is remarkably stable, it is nevertheless susceptible to spontaneous damage via reactions with cellular metabolites and environmental mutagens. Chemical reactions that alter the structure of the nucleobases within DNA are most commonly recognized and repaired by the base excision repair (BER)1 pathway. Some base lesions can block DNA replication and transcription with cytotoxic effects, and many more alter the base pairing properties so that replication leads to mispairing and mutation. The base excision repair pathway is initiated by a DNA repair glycosylase that must locate the site of damage within the genome. Once a damaged base is located, the glycosylase flips out the damaged nucleotide and catalyzes the hydrolysis of the _N_-glycosidic bond to release the lesioned base. The subsequent actions of an endonuclease, abasic site lyase, DNA polymerase and DNA ligase are required to complete the repair pathway.
It is estimated that ∼10,000 base lesions are formed in a typical human cell every day and that the vast majority of these are correctly repaired by BER or other DNA repair pathways (4). On the one hand, this is a large number of potential mutagenic events that must be corrected. On the other hand, these lesions are very rare considering the size of the human diploid genome (∼1010 nucleotides), with only one out of every million nucleotides sustaining damage on any given day. To underscore the magnitude of the damage recognition problem, this level of DNA damage requires a search of ∼100,000 nucleotides each day per enzyme molecule for an abundant protein of ∼105 copies per cell, and less abundant proteins would need to search a larger number of nucleotides.2 Despite its importance, there is still much to learn about the initial recognition of DNA damage and about the mechanisms that ensure a complete and continuous search of the genome.
Human alkyladenine DNA glycosylase (AAG) is a 33 kD monomeric protein that initiates repair of a diverse group of alkylated and deaminated purine nucleotides. These lesions include 3-methyladenine, 7-methylguanine, and 1,_N_6-ethenoadenine (εA), as well as the deaminated purines hypoxanthine and oxanine [(5, 6) and references therein]. A consequence of this broad specificity is that AAG removes normal bases at a low level (5, 7, 8). Substrate selection appears to be governed by a combination of selectivity filters. The first selectivity filter occurs at the nucleotide flipping step, since AAG preferentially selects lesions that are presented in unstable base pairs. The catalytic mechanism constitutes a second selectivity filter. Once bound, the use of general acid catalysis ensures that AAG only excises purine bases, even though smaller pyrimidines can fit into the active site (9, 10). The third selectivity filter consists of unfavorable steric clashes with the exocyclic amino groups of guanine and adenine, so that purine lesions lacking these functional groups are preferentially recognized. Finally, since alkylation of N3 and N7 of the purine ring leads to destabilization of the _N_-glycosidic bond, AAG is able to effectively excise _N_-alkyl lesions with a relatively modest rate enhancement.
In this study, we focus on understanding the mechanism by which AAG searches for sites of DNA damage. It is widely accepted that genome-wide searches for specific sites will be most efficient if a correlated search is used whereby each binding encounter with the DNA involves the search of multiple adjacent sites (11-14). This can be accomplished by diffusion along the DNA, which can be mediated by nonspecific binding interactions. A large body of work on restriction endonucleases and transcription factors has demonstrated the ability of these proteins to slide along DNA in search of their recognition sites [e.g., (15-17)]. Studies of several BER enzymes have also found evidence for processive action at adjacent sites on DNA, albeit with decreased processivity relative to the restriction endonucleases (18-24). A common criticism of many of these in vitro findings is that processive action occurs primarily at salt concentrations that are below physiological levels. Nevertheless, these studies provide compelling evidence that these proteins are capable of diffusion along DNA. In several cases, mutants have been used to directly correlate reductions in processivity with reduced biological function (25-27). This correlation between in vivo function and the ability to conduct a correlated search in vitro suggests that diffusion along DNA is important for lesion recognition in vivo.
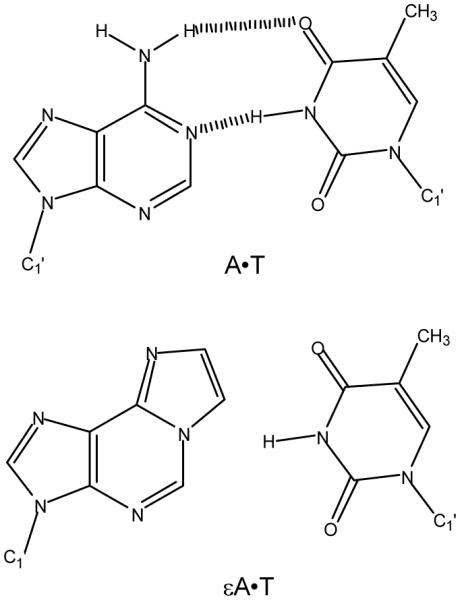
Our studies have focused on the repair of εA lesions. This lesion is thought to be the result of lipid peroxidation and it is found at low levels in human cells under normal growth conditions (28, 29). Since εA cannot hydrogen bond with any of the normal bases (Scheme 1), it is expected to present a relatively low barrier to nucleotide flipping. Indeed, it appears to bind more tightly than other substrates (5, 30). The crystal structure of AAG bound to an extrahelical εA lesion shows that it is readily accommodated in the active site pocket and the backbone amide of His136 donates a hydrogen bond to the N6 atom (31).
scheme 1.
We have developed a simple in vitro processivity assay and used it to characterize the ability of AAG to diffuse along DNA. We find that AAG is able to search at least 25 base pairs of DNA prior to dissociation at physiological ionic strength and pH. The ability of AAG to diffuse along DNA is eliminated at higher ionic strength, consistent with the importance of electrostatic interactions for DNA scanning. Deletion of the poorly conserved amino terminus of AAG results in decreased processivity, presumably by increasing the rate of dissociation from DNA. These observations demonstrate that AAG is capable of performing a correlated search of DNA in vitro and that this ability is expected to increase the efficiency of DNA damage recognition in vivo.
MATERIALS AND METHODS
Proteins
E. coli formamidopyrimidine DNA glycosylase (FPG) was obtained from New England Biolabs. Full-length and truncated recombinant human AAG was expressed in E. coli and purified as previously described (5, 9). We refer to the amino-terminally truncated protein as Δ80, but residues K80G81H82L83 have been replaced by the residues G80P81H82M83 that remain after proteolytic cleavage by human rhinovirus 3C protease. The concentrations of AAG proteins were determined from the absorbance at 280 nm and the calculated extinction coefficients. Under conditions of low ionic strength, the excision of hypoxanthine shows burst kinetics (see Supporting Information), with a rapid initial turnover followed by a slower steady state rate. We used this burst amplitude to calculate the concentration of active protein. The results from the burst analyses were in excellent agreement with the calculated concentration of AAG, indicating that greater than 90% of the recombinant proteins are active (see Supporting Information).
Synthesis and purification of Oligonucleotides
DNA substrates were synthesized by Integrated DNA Technologies or the Keck Center at Yale University. The εA-containing oligonucleotides were synthesized using ultra mild protecting groups and all other oligonucleotides were synthesized with standard protecting groups and deprotected according to the manufacturer’s recommendations (Glen Research). After desalting using Sephadex G-25, oligonucleotides were purified on denaturing polyacrylamide gels. DNA was extracted and desalted by binding to a C18 reverse phase column (Sep-pak, Waters) and eluting with 30% (v/v) acetonitrile. Concentrations were determined from the absorbance at 260 nm using the calculated extinction coefficients. For εA-containing strands we calculated the extinction coefficent for the identical sequence containing A in place of εA, and then subtracted a value of 9,400 M-1cm-1 per εA residue to correct for the weaker absorbance of εA relative to A. For 5′, 3′, or dual fluorescein-labeled oligonucleotides we assessed the labeling efficiency by comparing the absorbance at 260 nm with that at 495 nm and the calculated labeling efficiency was greater than 85% in all cases. For routine burst analysis to measure glycosylase activity and fraction of active enzyme, the deoxyinosine (I)-containing oligonucleotide 5′-(6-fam)-CGATAGCATCCTICCTTCTCTCCAT oligonucleotide was annealed to the complementary 5′-ATGGAGAGAAGGTAGGATGCTATCG with a 2-fold excess of the unlabeled strand. The sequence for the doubly-labeled εA-containing oligonucleotide duplex is given in Figure 1.
Figure 1.

Design of a simple substrate to monitor processivity of a DNA repair glycosylase. (A) A 47-mer oligonucleotide duplex was prepared that contains two εA lesions and the lesion-containing strand is labeled at both 3′ and 5′ ends by fluorescein (Fl). The immediate sequence contexts of the two lesions are identical (underlined). (B) The expected products after AAG-catalyzed base excision and alkaline cleavage of the abasic product. Only the lesion-containing strand is shown and the asterisk indicates a fluorescein label.
Glycosylase activity assay
Reactions were carried out at 37 °C. Unless otherwise indicated, the buffer consisted of 50 mM NaMES pH 6.1, 10% (v/v) glycerol, 0.1 mg/mL BSA, and sufficient NaCl to obtain the desired ionic strength. At pH 7.5, the NaMES was replaced with 50 mM NaHEPES. The enzyme concentration ranged from 20 nM to 10 μM. Control experiments demonstrated that both full-length and Δ80 AAG retained greater than 60% of their activity when incubated under these conditions without DNA for up to 15 hours, and greater than 90% of their activity when incubated with DNA substrate under turnover conditions (see Supporting Information). The reactions were initiated by adding a small volume of enzyme to a reaction volume of 20-60 μL that contained between 20 nM and 5 μM fluorescein-labeled DNA. Aliquots were withdrawn at various times and quenched with 2 volumes of 0.3 M sodium hydroxide to obtain 0.2 M final concentration. The quenched samples were stored at 4 °C until further processed to prevent base-catalyzed ring-opening and subsequent depurination of the εA lesions (32). Samples were heated at 70 °C for 15 minutes to quantitatively cleave abasic sites. Control reactions demonstrated that negligible εA sites were cleaved by this treatment (≤1 %, data not shown). After hydroxide-catalyzed cleavage of abasic sites, the samples were diluted with 3 volumes of formamide, containing 10 mM EDTA, and resolved on 20% (w/v) polyacrylamide sequencing gels containing 6.6 M Urea. Gels were scanned with a Typhoon fluorescence imager (GE Healthcare) to detect fluorescein (excitation at 488 nm and emission with a 520BP40 filter). The resulting fluorescence signal was quantified using ImageQuant and corrected for the amount of background signal. A standard curve was constructed by loading different amounts of fluorescently labeled oligo and this established the linearity of the assay up to 2 pmol of DNA per band (data not shown). The intensity of each DNA band was converted into a fraction by dividing its intensity by the sum of the intensities for all of the DNA species present.
Single turnover kinetics
The 47-mer processivity substrate (Figure 1) was incubated with excess enzyme to ensure single turnover conditions (this required >2-fold molar excess of enzyme). The reaction progress curve was plotted as the fraction of product versus time and was fit by a single exponential [F = e^(-kobs ×t)], in which F is the fraction of reaction, kobs is the observed rate constant, and t is the time. In all cases the nonlinear least squares fit was excellent (R2>0.95). At saturating concentration of enzyme the observed single turnover rate constant reaches a maximum that we will refer to as kmax [(33); we have previously referred to this value as kst, the single turnover rate constant (5, 9)]. The concentration of enzyme was varied by at least 3-fold to establish that the observed rate was independent of the concentration of enzyme, indicating that enzyme was in excess and at a saturating concentration (i.e., kobs = kmax).
Multiple turnover kinetics
Steady state kinetics for the dual lesion substrate were measured with a range of 10-1000-fold excess of substrate over enzyme. Unless otherwise indicated, the concentrations used were 2 μM DNA and 20 nM AAG (100-fold excess of substrate). Although reaction rates were linear to greater than 50% consumption of substrate, we used only the first 15-20% of the reactions to calculate initial rates. The initial rates for the disappearance of the substrate were calculated as a fractional change per unit of time. Values of kcat were calculated by multiplying this rate by the concentration of DNA and dividing by the concentration of enzyme (Vinit (nM/min) = Vinit(fract/min) × [DNA](nM) = kcat[E]). At pH 6.1 and low ionic strength, the rate-limiting step for multiple turnover is dissociation of the abasic product. We have fit the ionic strength dependence of kcat with a cooperative model [kcat = kmax(In/((Ka)n+In))], in which I is the ionic strength, n is the number of cation binding sites, Ka is the average affinity constant for binding of the cation, and kmax is the maximal single turnover rate constant.
Determination of the fraction processive (FP)
The ratio of processive events in which both damaged bases are excised in a single binding encounter divided by the total number of enzymatic events is defined as the fraction processive. Since only the ends of the DNA are labeled (Figure 1), an expression must be derived that depends only on the initial rates of formation of products and intermediates (15, 34). Accordingly, FP= (VA+VC-VAB-VBC)/(VA+VB+VAB+VBC); in which VA and VB are the initial velocities for the formation of the two products and VAB and VBC are the initial velocities for the formation of the two intermediates (Figure 1; see supporting information for the derivation). Averages and standard deviations are reported for 3-10 independent determinations. The fraction processive decreases as the rate of dissociation increases. We have fit this to a cooperative model in which cations can bind to multiple sites on the DNA and thereby affect the rate of AAG dissociation. This model is analogous to the Hill equation, that takes the following form. Fp=Fp,max-ΔFpIn/(Kan+In); in which Fp is the fraction processive, Fp, max is the maximal processivity observed, ΔFp is the difference between the maximal and minimal processivity observed, n is the number of cation binding sites, and the Ka is the average association rate constant for cation binding.
We found that a small percentage (∼4%) of the substrate contains a ring-opened form of εA, such that only a single εA lesion is available for AAG-catalyzed excision (see Supporting Information). This heterogeneity is expected to decrease the observed fraction processive. Assuming that AAG does not distinguish between oligonucleotides containing one or two lesions, it is predicted that single εA substrates will constitute ∼4% of the binding encounters during initial rate conditions (This has the effect of decreasing the initial rate of product formation by 4% and increasing the initial rate of intermediate formation by 4%). AAG-catalyzed excision of εA from a substrate that contains only a single site of damage will generate an intermediate length oligonucleotide of 37 or 34 nucleotides that cannot be distinguished from an intermediate that arose from distributive action (i.e., dissociation by AAG prior to engagement of the second εA site). Thus, even if AAG were 100% processive, our preparation of substrate sets an upper limit to the fraction processive of 0.92 [FP = (Vp-Vint)/(Vp+Vint) = (Vp-0.04Vp-0.04Vp) /(Vp-0.04Vp+0.04Vp) = 0.92; in which Vp and Vint are the sums of the initial rates for products and intermediates; Vp=VA+VC; Vint=VAB+VBC].
The processivity function predicts a minimum value of 0 for purely distributive action. In this case, the initial rates for formation of products and intermediates will be identical (Vp=Vint). However, for initial rates that proceed up to 10% reaction of substrate the probability of rebinding a released intermediate is not infinitely low. For example, at 10% reaction the probability of rebinding an intermediate is 10% that of binding a new substrate, if one assumes that the majority of binding encounters involve nonspecific interactions with DNA. The gradual accumulation of intermediates is expected to cause downward curvature in the reaction progress curve for the concentration of the intermediates and upward curvature in the reaction progress curve for the concentration of products. The effect of rebinding intermediate in a purely distributive mechanism can be roughly estimated by evaluating the processivity at both 10 and 20% reaction. When 10% product is formed, the predicted velocity for formation of product will not be affected because binding to either substrate or intermediates gives rise to the same products (Vp,corrected = 0.9Vp+0.1Vp = Vp). However, the corrected velocity for formation of the intermediates will be decreased because fewer enzymatic events form intermediates from substrate and because action on existing intermediates decreases the amount of intermediates (Vint, corrected = 0.9Vp-0.1Vp = 0.8Vp). This gives a predicted processivity value of 0.11 [FP = (Vp-Vint)/(Vp+Vint) = (Vp-0.8Vp)/(Vp+0.8Vp) = 0.11]. At 20% reaction this increases to Fp = 0.25. Therefore, the plateau of ∼0.1 that we observe for the fraction processive at high ionic strength is likely to reflect a fully distributive mechanism rather than a residual ionic strength independent processive mechanism.
RESULTS
Design of a quantitative processivity assay to characterize the ability of AAG to diffuse along DNA
We were interested in examining to what extent AAG can use linear diffusion to search multiple nucleotides during a single binding encounter. Previous studies have shown that other base excision repair enzymes are able to act processively at multiple sites on concatameric substrates (18-21). However, the previously employed assays have the limitation that most individual intermediates cannot be resolved. Therefore, we designed a synthetic oligonucleotide substrate that contains two sites of damage. By labeling both ends of the DNA, base excision at either site can be observed (Figure 1). Similar strategies have been used to study the processivity of restriction endonucleases [e.g., (15, 17, 34)] and a recent report described an internal labeling strategy to investigate the processivity of E. coli uracil DNA glycosylase (35).
The premise of the processivity assay is that an enzyme capable of diffusion along DNA will randomly bind to a site on the substrate, then diffuse along the DNA to locate one or the other lesion. Under multiple turnover conditions with excess substrate, most substrates will not have a protein bound. When the enzyme dissociates there is a low probability of rebinding the same DNA molecule. If the enzyme can diffuse along DNA and sample many binding sites prior to dissociation, then it may be able to excise both lesions before dissociating. We refer to the ability to remove multiple lesions in a single binding encounter as a processive mode of action. In the extreme case of 100% processive action, no intermediates corresponding to a single excision will be found. If AAG lacks the ability to diffuse along the DNA (i.e., dissociation into solution is much faster than translocation to the other site of damage), then intermediates corresponding to action at only a single site will accumulate at the same rate as the terminal fragments (enzymatic events E1 and E2; Figure 1). This extreme is referred to as distributive action. Depending on the distance between the target sites and the solution conditions, the behavior of the enzyme is expected to lie somewhere between these two extremes. We use the fraction processive (Fp) to define the fraction of enzymatic binding events that are processive versus the total number of processive and distributive events (Scheme 2; see Methods and Supporting Information). The average processivity can be determined by following many DNA binding events under steady state conditions (15).
scheme 2.

AAG is a monofunctional DNA glycosylase and its products are an abasic site and a free nucleobase. To monitor creation of the abasic site, enzymatic reactions were quenched in sodium hydroxide and heated to quantitatively convert the abasic sites into single strand breaks. Substrates, products and intermediates (resulting from base excision at one of the two sites of damage) were separated on a denaturing polyacrylamide gel and their relative intensities quantified with a fluorescence scanner (see Figures 1 & 2). The internal fragment (B; Figure 1) is unique to processive enzymatic events, but it is not labeled and cannot be detected directly in our experiments. Nevertheless, we can calculate the fraction processive because all of the other DNA species are observed and independently quantified (see Methods & Supporting Information).
Figure 2.

Characterization of the processivity substrate under single turnover conditions. A representative time course for AAG-catalyzed excision of εA with 35 nM oligonucleotide duplex and 350 nM Δ80 AAG at pH 6.1 and an ionic strength of 300 mM (see Methods for details). (A) Fluorescent scan of a 20% denaturing polyacrylamide gel showing increasing incubation times from left to right. The positions of the 47-mer substrate (ABC), 37-mer (BC), 34-mer (AB), 11-mer (C), and 9-mer (A) products are shown on the right (see Figure 1 for a schematic of the substrate and expected products resulting from _N_-glycosidic bond cleavage and hydroxide-catalyzed abasic site hydrolysis). (B) The amount of each labeled DNA is expressed as a fraction of the total fluorescence and the symbol legend is inset into the plot. Since the substrate contains two labels and the other species contain only a single label, the maximum for the product and intermediate bands is 0.5. The very similar maximal fractions observed for products and intermediates demonstrate that no correction is needed for either labeling efficiency or quantum yield of the 5′ and 3′-fluorescein labels. Furthermore, the almost identical rate constants indicate that both sites are recognized by AAG with equal efficiency.
Human AAG exists in at least two splice forms that differ slightly at their amino terminus and the larger splice variant is 298 amino acids in length (36-38). The amino terminus of AAG is poorly conserved even among mammals (see Supporting Information). In contrast, the carboxy-terminal glycosylase domain is highly conserved across vertebrates and sequence conservation can be detected in some prokaryotic DNA glycosylases (39, 40). The amino-terminal portion of human AAG is sensitive to proteolytic degradation and it was not included in the crystal structures of AAG bound to DNA substrate and inhibitor (31, 41). Our initial experiments used a truncated form of AAG (Δ80) that lacks the first 79 amino acids. This protein appears to be fully functional for _N_-glycosylase activity in a variety of in vitro assays (9, 36, 42). Other studies have implicated the amino terminus in interactions with other proteins, hHR23a/b and MBD1 (43, 44). Therefore, we have also examined the processivity of the full-length recombinant protein (longest splice variant).
Characterization of the processivity substrate
We performed single turnover reactions with excess Δ80 AAG over DNA as an initial characterization of the substrate (Figure 2). Under these conditions, both sites of damage can be simultaneously saturated (two AAG molecules per substrate oligonucleotide), so that any differences in reaction rate at the two sites can be monitored. As the two εA sites have very similar sequence contexts, it was expected that the two sites would have similar reactivity (Figure 1A). Indeed, the single turnover rate constant was essentially identical for both sites [kmax = 0.20 ± 0.01 min-1; Figures 2 and 3]. This rate constant is identical to the previously reported single turnover excision of a 25-mer εA-containing oligonucleotide that shares the same sequence context [kmax = 0.2 min-1;(5)]. The observed disappearance of substrate occurs at twice the rate (kobs = 0.4 min-1) since excision at either of the two sites depletes the concentration of substrate (Figure 2B). For two independent excision events, the intermediates, in which only a single εA is excised, build up and then decay as a function of the rate constants for excision at both sites, which are identical in this case. We let the AAG-catalyzed reaction go to completion in order to directly compare the fluorescence of the 5′- and 3′-labeled fragments (Figure 2). The almost identical fluorescence of the two bands indicates that the 5′-(6-amino)fluorescein and 3′-fluorescein labels that were used have very similar quantum yields. Therefore, no corrections need to be made to the raw fluorescence values obtained from scans of the gel.
Figure 3.

Single turnover excision of εA is independent of ionic strength and the two sites are equivalent under all of the conditions tested. Single turnover excision of εA at site 1 (open circles) and site 2 (open squares) were measured with 1 μM DNA and 3, 6, and 9 μM Δ80 AAG. The observed rate constants were independent of the concentration of AAG, so the average and standard deviation are shown (each data point represents at least 9 independent determinations of the rate constant). The rates of excision at both sites are the same within error and independent of ionic strength (kmax = 0.2 min-1). These results are the same as was previously reported for excision of a single εA lesion from a similar sequence context using a 32P-based glycosylase assay, suggesting that the activity of AAG is not affected by either of the fluorescein labels (5).
Since εA is susceptible to ring-opening, especially at alkaline pH (32), we were concerned that some of the εA sites might be damaged during solid phase synthesis, deprotection, and purification. Indeed, this could explain the persistence of the intermediate fragments AB and BC over long reaction times (e.g., Figure 2). Since AAG has little or no activity against the ring-opened from of εA (45), such damage would lead to an underestimate of the degree of processivity because the enzyme could only act on these substrate molecules once, regardless of residence time. We allowed the single turnover reaction with AAG to proceed for more than 10 half-lives and determined that greater than 99% of the fluorescein-labeled substrate contains at least one εA that could be recognized by AAG and ∼95% contains two εA lesions. This is consistent with a ∼2% chance that a given εA nucleotide undergoes spontaneous degradation during synthesis, deprotection, and purification. Control reactions with E. coli FPG, an enzyme known to be active on the ring-opened form of εA (45), confirmed that the majority of the AAG-resistant lesions could be excised by FPG and are likely to be ring-opened εA bases (see Supporting Information). This small percentage of substrate that is refractory to dual excision by AAG (∼4%) leads to a slight underestimate of the processivity. We calculate a theoretical maximum of Fp = 0.92 for a fully processive enzyme acting on this substrate (see Methods). Since none of our conclusions rely on the exact value of the fraction processive, we have not corrected any of the observed values of Fp for the heterogeneity existing in the DNA substrate.
To ensure that the AAG·DNA complex is fully saturated at high ionic strength, we investigated the ionic strength dependence of the single turnover reaction at several concentrations of AAG. At saturating concentrations, the single turnover rate constant reports on steps that occur subsequent to DNA binding up to and including _N_-glycosidic bond cleavage (Scheme 2). Since base flipping is expected to be fast, _N_-glycosidic bond cleavage is likely to be rate-limiting under these conditions (5). In the range from 30 to 300 mM ionic strength, the single turnover rate constant is independent of ionic strength and identical for both sites (kmax = 0.20 ± 0.01; Figure 3). These data are for Δ80 AAG, but the single turnover rate constant for full-length AAG under these conditions was the same within error (kmax = 0.23 ± 0.04; data not shown). These data confirm that the AAG·DNA complex is saturated at both sites even at high salt concentrations, and suggest that AAG-catalyzed _N_-glycosidic bond cleavage is insensitive to ionic strength in this range.
Processivity of AAG at low ionic strength
Previous studies of processive action of enzymes on DNA have consistently found that processivity is greatest at low ionic strength and that it decreases with increasing ionic strength, presumably because dissociation from DNA is accelerated at increased ionic strength (15, 18, 46). Therefore, we performed multiple turnover experiments under low ionic strength conditions (I = 50 mM) to address whether AAG exhibits a processive searching mechanism. AAG has a slightly acidic pH-rate optimum for excision of neutral lesions, including εA, so reactions were initially carried out at pH 6.1 (5, 9). Under these conditions, the multiple turnover reaction was very slow, requiring incubation times of up to 12 hours. This corresponds to multiple turnover rate constants that are 10-20-fold lower than the single turnover rate constant. Stability controls indicated that both full-length and Δ80 AAG retain at least 50% activity over 12 hours when incubated without DNA, but are considerably more stable when incubated with DNA substrate so that no loss of AAG activity was detected during the course of the assay (see Supporting Information). Figure 4 shows a representative gel from a multiple turnover processivity experiment. The left-most lanes show time courses for reactions with no enzyme, Δ80 AAG, or full-length AAG at 50 mM ionic strength (Figure 4A). For both enzymes it is apparent that the products resulting from processive action build up much more quickly than the intermediates that result from distributive action, as expected for a processive mode of action (Figure 4B & 4C). Surprisingly, Δ80 AAG was ∼2-fold faster than full-length AAG for multiple-turnover excision of εA under these conditions (see below). The fraction processive was calculated as described in the Methods and gave values of 0.76 ± 0.09 and 0.88 ± 0.04 for Δ80 and full-length AAG, respectively, indicating that both enzymes are highly processive at low ionic strength.
Figure 4.

The multiple turnover processivity assay. (A) This representative gel compares reactions containing full-length or Δ80 AAG at pH 6.1 with 50-150 mM ionic strength. All reactions contained 2 μM oligonucleotide substrate and 20 nM enzyme. Three time points between 5 and 20% reaction were chosen for each reaction condition (the time varies between 0.1 and 12 hours, since the steady state rate is dependent upon ionic strength). (B-G) Reaction progress curves for products (fragments A & C) and intermediates (fragments AB & BC) are shown for each set of reactions. In this experiment each reaction was performed in triplicate and additional time points analyzed on additional gels are included. The average values for each condition are plotted and the error bars indicate the standard deviation. Panels B, D, & F show results obtained with Δ80 AAG and Panels C, E, & G show results obtained with full-length AAG. The ionic strength was 50 mM (Panels A & B), 150 mM (Panels C & D), and 300 mM (Panels E & G). The fraction processive was calculated from these data and from additional experiments to obtain the average Fp values that are shown in Figure 6A.
Under multiple turnover conditions, there is very low probability of multiple proteins binding to the same DNA molecule so any intrinsic difference in binding or base excision at the two sites can be detected. Both products (fragment A and C) are formed at essentially the same rate and therefore AAG does not have a preference for either the 5′- or 3′-εA lesion (Figure 4A and data not shown). Although much lower amounts of the two intermediate fragments (AB and BC) were formed under these conditions, the rates for their formation were also the same within error (Figure 4A and data not shown). In principle, it is possible for an enzyme to exhibit different processivity depending upon which site it acts at first (i.e., E1,2 ≠ E2,1; Figure 1). We considered this possibility, but did not find a significant difference between the two possible pathways [(15, 17); See Supporting Information]. This suggests that AAG recognizes and excises the lesions from the two sites with identical efficiency despite the different polarity and distance from the two DNA ends. Therefore, we routinely determined initial rates for the sum of the two products and for the sum of the two intermediates (e.g., Figure 4B & C). This facilitated our ability to measure extremely slow rates of formation of intermediates.
Effect of ionic strength on multiple turnover reaction and processivity
The ionic strength dependence of the steady state reaction was characterized in order to better understand the slow steady state rate at low ionic strength and the modest, but reproducible, increase in the reaction rate for the truncated form of AAG relative to the full-length protein. Since the DNA binding surface of AAG contains many charged groups and because linear diffusion along DNA by other enzymes is very sensitive to ionic strength, we anticipated that increased ionic strength would cause a switch to a distributive mechanism. Comparison of processivity of full-length and truncated proteins at increased ionic strength would allow even a modest change in processivity to be detected.
First, we determined the effect of added sodium chloride on steady state reaction rate. The kcat values were obtained from linear fits to the initial rates for disappearance of substrate for reactions in which the ionic strength was adjusted between 50 mM and 300 mM by the addition of sodium chloride (see Supporting Information for a representative plot). The results for both full-length and Δ80 AAG are summarized in Figure 5A. For Δ80 AAG, the multiple turnover rate constant increases with increasing ionic strength until ∼200 mM at which point it reaches the single turnover rate constant. The full-length AAG follows a very similar ionic strength dependence, but does not reach the single turnover rate constant until ∼250 mM ionic strength. Across the range between 50 and 200 mM ionic strength the truncated enzyme maintains ∼2-fold faster rate of reaction. Although this difference between full-length and Δ80 AAG is modest, there are several reasons to believe that it is real. To control for possible differences in protein concentration, we performed active site titrations of both enzymes on the same substrate to determine the active concentration of enzyme (see Supporting Information). To control for possible differences in DNA substrate concentration, since the kcat value in a gel-based assay is also dependent upon this concentration, the steady state kinetics were carried out side-by-side with the same stock of DNA. Furthermore, both enzymes gave the same kcat value at high ionic strength and this rate constant was in very close agreement with the kmax values for single turnover excision.
Figure 5.

Ionic strength affects multiple turnover excision of εA at pH 6.1, but not at pH 7.5. The multiple turnover rate constants for Δ80 (open circles) and full-length AAG (open squares) were measured at the indicated ionic strength. The average of 3-8 replicates is shown and the error bars indicate one standard deviation from the mean. The solid lines shows the best fits to a cooperative model in which multiple sodium ions cause an increased rate of dissociation up to the threshold at which the rate of dissociation is greater than the rate constant for _N_-glycosidic bond cleavage (see Methods for details).
The ionic strength dependence of the steady state excision by AAG strongly suggests that a different step is rate-limiting at low and high ionic strength. The rate-limiting step at low ionic strength increases in response to added salt up to the point at which an ionic strength independent step becomes rate limiting. At high ionic strength the excellent agreement between the single turnover and multiple turnover rate constants indicates that the rate-limiting step is the same; hydrolysis of the _N_-glycosidic bond. The rate-limiting step at low ionic strength for multiple turnover excision must occur subsequent to hydrolysis of the _N_-glycosidic bond and is most likely to be dissociation of the DNA product. An increased dissociation rate constant at higher concentration of salt is consistent with weakened electrostatic interactions between the positively charged protein and the negatively charged DNA.
We compared the ionic strength dependence of the fraction processive for both full-length and Δ80 AAG and the results are shown in Figure 6A. Both proteins show a steep decrease in processivity with increasing ionic strength, however the full-length protein shows significantly higher processivity at intermediate ionic strength (100-200 mM). We believe that the lower limit of the processivity of ∼0.1 that both proteins approach at high ionic strength is reflective of a fully distributive mechanism, since this value is expected for distributive mechanisms at 10-20% reaction due to the low, but finite, probability of rebinding an intermediate containing a single εA lesion (see Methods). The observation of decreased fraction processivity under certain conditions provides an important validation of the processivity assay by addressing a trivial alternative interpretation of the low levels of intermediates observed in the steady state assay. For example, if AAG preferentially rebinds a substrate with an abasic site, then the pattern of products would appear to be processive even though the mechanism of base excision was distributive. Further evidence against this alternative model is that linear initial rates are observed for greater than 40% of the reaction, indicating that even when abasic product and εA-containing substrate are present in roughly equal amounts, AAG preferentially binds to the substrate (see Supporting Information for a representative time course).
Figure 6.

Ionic strength affects the processivity of AAG. Processivity at the optimal pH of 6.1 (A) and at pH 7.5 (B) was determined at increasing ionic strength, as described in the methods. Both Δ80 (open symbols) and full-length AAG (closed symbols) were examined. The average value of 3-8 independent determinations is shown and the error bars indicate the standard deviation for each condition.
Multiple turnover and Processivity of AAG at physiological pH
The initial processivity experiments were carried out at pH 6.1 because this is the optimal pH for AAG-catalyzed excision of εA in vitro. However, since εA base excision is slower at higher pH, we also performed processivity experiments at pH 7.5. The kcat values for both enzymes are significantly lower at pH 7.5 than at pH 6.1 (Figure 5). However, in contrast to the ionic strength-dependent multiple turnover reaction observed at lower pH, the multiple turnover reaction at higher pH was independent of ionic strength between 50 and 300 mM (Figure 5B). The change in the ionic strength dependence suggests that multiple turnover excision is limited by _N_-glycosidic bond cleavage at pH 7.5, and that dissociation of the abasic product is relatively fast.
As described above for the pH 6.1 conditions, the fraction processive was calculated for full-length and truncated AAG as a function of ionic strength at pH 7.5 (Figure 6B). The results closely resemble the results at the lower pH, with maximal processivity at low ionic strength (Fp = 0.87 ± 0.04 for both full-length and Δ80 AAG) and with decreasing processivity at higher ionic strength. The midpoint of the ionic strength dependence is shifted to lower ionic strength for both full-length and truncated proteins, relative to the midpoint observed at pH 6.1. For intermediate ionic strength at both pH 6.1 and pH 7.5, the full-length protein exhibits higher processivity than the Δ80 truncated protein. Although the contribution of the amino terminus to a processive search is modest, it is interesting to note that this effect is greatest at physiological ionic strength. This observation is consistent with the idea that residues in the amino terminus fine-tune the nonspecific DNA binding activity of AAG to allow for short-range correlated searching of adjacent sites.
DISCUSSION
We have investigated the mechanism by which human AAG, a DNA repair glycosylase that recognizes a wide variety of alkylated and deaminated purines, locates sites of DNA damage. We describe a processivity assay that allows quantitative measurement of the ability of AAG to remove multiple base lesions from a simple oligonucleotide substrate in a single binding encounter. This assay has provided evidence that AAG employs a processive searching mechanism that makes use of nonspecific DNA binding interactions to carry out a highly redundant search of adjacent sites. By comparing an amino-terminally truncated enzyme to the full-length enzyme, we found that the amino terminus plays a role in nonspecific DNA binding and increases the probability of a correlated search at physiological ionic strength. These results are similar to those obtained for a variety of DNA binding enzymes and further supports the idea that nonspecific DNA binding is an important feature of enzymes that must carry out genome-wide searches for specific sites [e.g., (11, 14, 15, 23, 24, 47-49)].
The relatively short, dual-lesion substrate that we have utilized allows a simple and quantitative measure of the ability of a DNA repair glycosylase to translocate between nearby sites. This assay has several advantages over more commonly employed assays involving concatameric substrates. Most importantly, the small substrate size allows each possible product to be quantified and any inherent directionality of the scanning process to be detected. In addition, these substrates can be directly synthesized to allow for a wide-range of site-specific modifications, such as the incorporation of fluorescent labels. However, the dual-lesion assay has the disadvantage that long sliding distances cannot be measured, because solid phase synthesis is limited to relatively short oligonucleotides. We have overcome this limitation by increasing the ionic strength to weaken the protein-DNA interaction. Beyond allowing quantitative comparison of mutants or alternative substrates, the ionic strength dependence provides mechanistic insight into the DNA damage recognition process.
At low ionic strength, both full-length and Δ80 AAG exhibit a high degree of processivity at both pH 6.1 and pH 7.5 (Figure 6). This demonstrates that nonspecific DNA binding by AAG enables a correlated search over a distance of at least several turns of the DNA helix. Since the εA lesions are separated by 25 base pairs and the pitch of B-form DNA is 10.4 base pairs per turn, these lesions are predicted to be on opposite sides of the DNA duplex. This rules out a hand-over-hand model for diffusion along one face of the duplex that would be analogous to the models that have been proposed for the movement of motor proteins such as myosin and kinesin along either actin or microtubule filaments (50, 51). These data are consistent with either a one-dimensional mode of diffusion along one strand of the duplex ((52, 53)), or a two-dimensional mode of diffusion in which both strands can be simultaneously searched (34, 54). We have fit the ionic strength dependence of the fraction processive with a cooperative model whereby multiple cations affect the probability of finding a second site of damage. In principle, ionic strength dependent changes in dissociation rate, scanning rate, or excision rate could be responsible for the ionic strength dependence of the processivity. However, the base excision step is insensitive to ionic strength because the single turnover reaction does not change between 50 and 300 mM ionic strength (Figure 3). Therefore, the simplest interpretation of the ionic strength dependence of the processivity is that the dissociation rate is dependent on ionic strength and that the rate of scanning is not. Consistent with this, the ionic strength at which half of the maximal kcat is obtained is similar to the ionic strength at which half of the maximal processivity is observed. Nevertheless, these experimental observations cannot rule out the possibility that the rate of scanning is also dependent upon the ionic strength.
At physiological pH and ionic strength (pH 7.5, I=150 mM), AAG exhibits a processivity of 0.45 which is near the midpoint of the range of processivity values that we observe (0.1-0.9; Figure 6B). Under these conditions, AAG has a roughly equal probability of finding the second site of damage or dissociating. It is possible that interactions with other proteins or covalent modifications of AAG increase its processivity in vivo. However, the observed in vitro processivity is consistent with the requirements for a genome-wide search in vivo. The dissociative extreme in which only a single base is sampled per binding encounter would be inefficient because the search for damage would involve the entire three-dimensional space of the nucleus. The associative extreme would be inefficient because the repair protein would be restricted to distinct domains of DNA and movement between accessible regions of DNA would be limited. Therefore, an intermediate level of processivity, in which short sections of DNA are exhaustively searched prior to dissociation, is expected to balance the requirements of covering every single base of the genome (14, 16).
At the optimal pH of 6.1, the kcat value for both Δ80 and full-length AAG is dependent upon the ionic strength. Since the single turnover rate constant is independent of ionic strength, this suggests that dissociation from the abasic DNA product is rate-limiting for multiple turnover base excision. The AAG DNA binding surface has a high density of positive charge, so it is not surprising that high ionic strength weakens binding (Figure 7). We fit this ionic strength dependence with a simple model in which the rate of dissociation is dependent upon the number of sodium ions bound. At high ionic strength, dissociation becomes sufficiently fast that an earlier step, presumably _N_-glycosidic bond hydrolysis, becomes rate limiting. Consistent with this interpretation, the multiple turnover rate constant reaches the same value as the kmax value for single turnover (Figure 5A). In contrast, at pH 7.5 the value of kcat is independent of ionic strength, suggesting that _N_-glycosidic bond hydrolysis step is rate-limiting for multiple turnover at even the lowest ionic strength tested. This is consistent with the observation that the single turnover rate constant for excision of εA is ∼8-fold lower at pH 7.5 relative to pH 6.1 (9).
Figure 7.

Electrostatic surface potential of the AAG catalytic domain reveals a positively charged DNA binding surface. The crystal structure of the AAG·εA-DNA complex was used to generate this figure (1F4R; (31)). Electrostatic calculations were performed with Pymol on the protein alone (W.L. DeLano; http://www.pymol.org) and APBS ((58); using a plug-in written by M. Lerner; http://wwwpersonal.umich.edu/~mlerner/PyMOL/). A continuum from -2 (red) to +2 (blue) is shown. A view of the active site and bound DNA is on the left, illustrating the positively charged DNA binding surface, and on the right the molecule is rotated horizontally by 180° to show that the positively charged surface continues around the protein.
The slow rate constants that are observed for dissociation of the abasic product, reflected by slow multiple turnover excision at low ionic strength, suggest a relatively long-lived AAG·DNA complex. For example, at pH 6.1 and 150 mM ionic strength the full-length protein has a half-life of 7 minutes [t1/2 = ln 2/kdissociation = ln (2/0.1 min-1) = 7 min] for its dissociation from DNA containing an abasic DNA product. This can be compared to the half-life of 3 minutes for the Δ80 truncated protein. At 50 mM ionic strength, the half-life of the AAG·DNA complexes increases to 46 and 23 minutes for the full-length and truncated proteins, respectively. Presumably dissociation would be faster from undamaged DNA, but even 100-fold weaker binding to undamaged DNA would imply a dissociation half-time of many seconds. A single molecule study of 8-oxoguanosine DNA glycosylase provided an estimate of as many as 3000 base pairs sampled per second on undamaged DNA (55). If AAG exhibits similar fast sliding, then this implies a massively redundant search of adjacent sites in this long-lived AAG·DNA complex, in which case our lower estimate of >25 base pairs is an underestimate of the distance traveled during a binding encounter.
The importance of electrostatic interactions for the stabilization of the AAG·DNA complex is apparent from the increasing rate of dissociation from DNA and from the decreasing processivity that are observed upon increasing the ionic strength. These observations are consistent with the positively charged DNA binding groove that is observed in crystal structures of the AAG·εA-DNA complex [Figure 7; (31, 41)]. The DNA binding groove contains 4 positively charged residues that directly contact the phosphate backbone (Arg 141, Arg197, Arg182, K229). It is not known whether there are additional contacts between the amino terminus of AAG, which was not present in the crystal structure, and the DNA. However, there are 13 arginine and lysine residues present in the first 80 amino acids, one or more of which could provide a positive electrostatic interaction. The increase in the multiple turnover rate constant and the decreased processivity at higher ionic strength that was observed upon deletion of the amino terminal 80 amino acids suggests that the amino terminus either contacts DNA directly or alters the conformation of AAG to slow dissociation. However, the amino terminus does not appear to have any effect on the rate of base excision since the rate constant for the full-length protein reaches the rate constant of the Δ80 truncated protein at high ionic strength (Figure 5).
Although it is difficult to quantitatively compare the processivity results from AAG with the two-lesion substrate with the results from concatameric multiple-lesion substrates that have been reported for other base excision repair enzymes, there are some obvious qualitative similarities. Whereas several enzymes exhibit processive excision of adjacent lesions at low ionic strength (≤70 mM), including E. coli and human UNG (19, 20), M. luteus (56) and T4 endoV (46, 57), human APE1 (18), E. coli MutY (21), and E. coli FPG (21), their behavior becomes distributive when the ionic strength is greater than 70 mM. Only E. coli FPG acting on an 8-oxoguanosine·cytosine-containing concatameric DNA showed processive behavior with ionic strength greater than 100 mM (21). It appears that AAG is more processive than these other enzymes that have been previously studied, because it shows a bias towards processive action up to 200 mM ionic strength at optimal pH and up to 150 mM at physiological pH. Taken together, the results from a variety of eukaryotic and prokaryotic base excision repair enzymes are consistent with the idea that a coordinated search is important over relatively short distances along DNA.
In summary, our results reveal that AAG searches many adjacent sites on a DNA molecule in a single binding event prior to dissociation. This observation suggests that the majority of lesion recognition events involve initial nonspecific binding to undamaged sites followed by diffusion along the DNA. This searching process is expected to be highly redundant given the long lifetime of the AAG·DNA complex, providing ample opportunity for the enzyme to recognize and excise lesions that minimally perturb the structure of DNA. As ionic strength is increased above physiological levels, the rate of dissociation from DNA increases and AAG switches to a distributive searching mechanism. In addition, deletion of the amino terminal 80 amino acids, a region dispensable for catalytic activity, results in significantly decreased processivity at physiological ionic strength. These observations suggest that the nonspecific binding affinity of AAG is tuned to allow for correlated searches of a local DNA domain, while still allowing freedom for a long-range three-dimensional search.
Supplementary Material
1_si_001
ACKNOWLEDGMENT
We thank Abby Wolfe for providing comments on the manuscript.
Footnotes
†
This work was supported by a grant from the NIH to P.O. (CA122254).
1
Abbreviations:
AAG
Alkyladenine DNA glycosylase, also known as methylpurine DNA glycosylase (MPG) and 3-methyladenine DNA glycosylase
BER
base excision repair
BSA
bovine serum albumin
εA
1,_N_6-ethenoadenine
Fp
fraction processive
FPG
formamidopyrimidine DNA glycosylase
I
ionic strength
NaHEPES
sodium _N_-(2-hydroxyethyl)piperazine-_N_′-(2-ethanesulfonate)
NaMES
sodium 2-(_N_-morpholino)ethanesulfonate
PAGE
polyacrylamide gel electrophoresis
2
The abundance of AAG in human fibroblasts was determined to be 2 ×105 molecules per cell from glycosylase activity in cell extracts (1). This is similar to the abundance of other base excision repair enzymes, which have been reported to be between 0.5-3×105 molecules per cell (2, 3).
REFERENCES
- (1).Ye N, Holmquist GP, O’Connor TR. Heterogeneous repair of N-methylpurines at the nucleotide level in normal human cells. J Mol Biol. 1998;284:269–85. doi: 10.1006/jmbi.1998.2138. [DOI] [PubMed] [Google Scholar]
- (2).Chen DS, Herman T, Demple B. Two distinct human DNA diesterases that hydrolyze 3′-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res. 1991;19:5907–14. doi: 10.1093/nar/19.21.5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cappelli E, Hazra T, Hill JW, Slupphaug G, Bogliolo M, Frosina G. Rates of base excision repair are not solely dependent on levels of initiating enzymes. Carcinogenesis. 2001;22:387–93. doi: 10.1093/carcin/22.3.387. [DOI] [PubMed] [Google Scholar]
- (4).Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–15. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- (5).O’Brien PJ, Ellenberger T. Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J Biol Chem. 2004;279:9750–7. doi: 10.1074/jbc.M312232200. [DOI] [PubMed] [Google Scholar]
- (6).Hitchcock TM, Dong L, Connor EE, Meira LB, Samson LD, Wyatt MD, Cao W. Oxanine DNA glycosylase activity from Mammalian alkyladenine glycosylase. J Biol Chem. 2004;279:38177–83. doi: 10.1074/jbc.M405882200. [DOI] [PubMed] [Google Scholar]
- (7).Berdal KG, Johansen RF, Seeberg E. Release of normal bases from intact DNA by a native DNA repair enzyme. Embo J. 1998;17:363–7. doi: 10.1093/emboj/17.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Connor EE, Wyatt MD. Active-site clashes prevent the human 3-methyladenine DNA glycosylase from improperly removing bases. Chem Biol. 2002;9:1033–41. doi: 10.1016/s1074-5521(02)00215-6. [DOI] [PubMed] [Google Scholar]
- (9).O’Brien PJ, Ellenberger T. Human alkyladenine DNA glycosylase uses acid-base catalysis for selective excision of damaged purines. Biochemistry. 2003;42:12418–29. doi: 10.1021/bi035177v. [DOI] [PubMed] [Google Scholar]
- (10).Biswas T, Clos LJ, 2nd, SantaLucia J, Jr., Mitra S, Roy R. Binding of specific DNA base-pair mismatches by N-methylpurine-DNA glycosylase and its implication in initial damage recognition. J Mol Biol. 2002;320:503–13. doi: 10.1016/S0022-2836(02)00519-3. [DOI] [PubMed] [Google Scholar]
- (11).Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–48. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- (12).Winter RB, Berg OG, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 3. The Escherichia coli lac repressor--operator interaction: kinetic measurements and conclusions. Biochemistry. 1981;20:6961–77. doi: 10.1021/bi00527a030. [DOI] [PubMed] [Google Scholar]
- (13).Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 2. The Escherichia coli repressor--operator interaction: equilibrium measurements. Biochemistry. 1981;20:6948–60. doi: 10.1021/bi00527a029. [DOI] [PubMed] [Google Scholar]
- (14).Zharkov DO, Grollman AP. The DNA trackwalkers: principles of lesion search and recognition by DNA glycosylases. Mutat Res. 2005;577:24–54. doi: 10.1016/j.mrfmmm.2005.03.011. [DOI] [PubMed] [Google Scholar]
- (15).Terry BJ, Jack WE, Modrich P. Facilitated diffusion during catalysis by EcoRI endonuclease. Nonspecific interactions in EcoRI catalysis. J Biol Chem. 1985;260:13130–7. [PubMed] [Google Scholar]
- (16).Halford SE, Marko JF. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004;32:3040–52. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Stanford NP, Szczelkun MD, Marko JF, Halford SE. One- and three-dimensional pathways for proteins to reach specific DNA sites. Embo J. 2000;19:6546–57. doi: 10.1093/emboj/19.23.6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Carey DC, Strauss PR. Human apurinic/apyrimidinic endonuclease is processive. Biochemistry. 1999;38:16553–60. doi: 10.1021/bi9907429. [DOI] [PubMed] [Google Scholar]
- (19).Higley M, Lloyd RS. Processivity of uracil DNA glycosylase. Mutat Res. 1993;294:109–16. doi: 10.1016/0921-8777(93)90019-d. [DOI] [PubMed] [Google Scholar]
- (20).Bennett SE, Sanderson RJ, Mosbaugh DW. Processivity of Escherichia coli and rat liver mitochondrial uracil-DNA glycosylase is affected by NaCl concentration. Biochemistry. 1995;34:6109–19. doi: 10.1021/bi00018a014. [DOI] [PubMed] [Google Scholar]
- (21).Francis AW, David SS. Escherichia coli MutY and Fpg utilize a processive mechanism for target location. Biochemistry. 2003;42:801–10. doi: 10.1021/bi026375+. [DOI] [PubMed] [Google Scholar]
- (22).Zharkov DO, Shoham G, Grollman AP. Structural characterization of the Fpg family of DNA glycosylases. DNA Repair (Amst) 2003;2:839–62. doi: 10.1016/s1568-7864(03)00084-3. [DOI] [PubMed] [Google Scholar]
- (23).Lloyd RS, Hanawalt PC, Dodson ML. Processive action of T4 endonuclease V on ultraviolet-irradiated DNA. Nucleic Acids Res. 1980;8:5113–27. doi: 10.1093/nar/8.21.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ganesan AK, Seawell PC, Lewis RJ, Hanawalt PC. Processivity of T4 endonuclease V is sensitive to NaCl concentration. Biochemistry. 1986;25:5751–5. doi: 10.1021/bi00367a060. [DOI] [PubMed] [Google Scholar]
- (25).Dowd DR, Lloyd RS. Biological significance of facilitated diffusion in protein-DNA interactions. Applications to T4 endonuclease V-initiated DNA repair. J Biol Chem. 1990;265:3424–31. [PubMed] [Google Scholar]
- (26).Jeltsch A, Wenz C, Stahl F, Pingoud A. Linear diffusion of the restriction endonuclease EcoRV on DNA is essential for the in vivo function of the enzyme. Embo J. 1996;15:5104–11. [PMC free article] [PubMed] [Google Scholar]
- (27).McKinney K, Mattia M, Gottifredi V, Prives C. p53 linear diffusion along DNA requires its C terminus. Mol Cell. 2004;16:413–24. doi: 10.1016/j.molcel.2004.09.032. [DOI] [PubMed] [Google Scholar]
- (28).Barbin A. Etheno-adduct-forming chemicals: from mutagenicity testing to tumor mutation spectra. Mutat Res. 2000;462:55–69. doi: 10.1016/s1383-5742(00)00014-4. [DOI] [PubMed] [Google Scholar]
- (29).Gros L, Ishchenko AA, Saparbaev M. Enzymology of repair of etheno-adducts. Mutat Res. 2003;531:219–29. doi: 10.1016/j.mrfmmm.2003.07.008. [DOI] [PubMed] [Google Scholar]
- (30).Abner CW, Lau AY, Ellenberger T, Bloom LB. Base excision and DNA binding activities of human alkyladenine DNA glycosylase are sensitive to the base paired with a lesion. J Biol Chem. 2001;276:13379–87. doi: 10.1074/jbc.M010641200. [DOI] [PubMed] [Google Scholar]
- (31).Lau AY, Wyatt MD, Glassner BJ, Samson LD, Ellenberger T. Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc Natl Acad Sci U S A. 2000;97:13573–8. doi: 10.1073/pnas.97.25.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Speina E, Ciesla JM, Wojcik J, Bajek M, Kusmierek JT, Tudek B. The pyrimidine ring-opened derivative of 1,N6-ethenoadenine is excised from DNA by the Escherichia coli Fpg and Nth proteins. J Biol Chem. 2001;276:21821–7. doi: 10.1074/jbc.M100998200. [DOI] [PubMed] [Google Scholar]
- (33).Bennett MT, Rodgers MT, Hebert AS, Ruslander LE, Eisele L, Drohat AC. Specificity of human thymine DNA glycosylase depends on N-glycosidic bond stability. J Am Chem Soc. 2006;128:12510–9. doi: 10.1021/ja0634829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kampmann M. Obstacle bypass in protein motion along DNA by two-dimensional rather than one-dimensional sliding. J Biol Chem. 2004;279:38715–20. doi: 10.1074/jbc.M404504200. [DOI] [PubMed] [Google Scholar]
- (35).Sidorenko VS, Mechetin GV, Nevinsky GA, Zharkov DO. Correlated cleavage of single- and double-stranded substrates by uracil-DNA glycosylase. FEBS Lett. 2008;582:410–4. doi: 10.1016/j.febslet.2008.01.002. [DOI] [PubMed] [Google Scholar]
- (36).O’Connor TR. Purification and characterization of human 3-methyladenine-DNA glycosylase. Nucleic Acids Res. 1993;21:5561–9. doi: 10.1093/nar/21.24.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Vickers MA, Vyas P, Harris PC, Simmons DL, Higgs DR. Structure of the human 3-methyladenine DNA glycosylase gene and localization close to the 16p telomere. Proc Natl Acad Sci U S A. 1993;90:3437–41. doi: 10.1073/pnas.90.8.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bonanno K, Wyrzykowski J, Chong W, Matijasevic Z, Volkert MR. Alkylation resistance of E. coli cells expressing different isoforms of human alkyladenine DNA glycosylase (hAAG) DNA Repair (Amst) 2002;1:507–16. doi: 10.1016/s1568-7864(02)00051-4. [DOI] [PubMed] [Google Scholar]
- (39).Alseth I, Rognes T, Lindback T, Solberg I, Robertsen K, Kristiansen KI, Mainieri D, Lillehagen L, Kolsto AB, Bjoras M. A new protein superfamily includes two novel 3-methyladenine DNA glycosylases from Bacillus cereus, AlkC and AlkD. Mol Microbiol. 2006;59:1602–9. doi: 10.1111/j.1365-2958.2006.05044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Aamodt RM, Falnes PO, Johansen RF, Seeberg E, Bjoras M. The Bacillus subtilis counterpart of the mammalian 3-methyladenine DNA glycosylase has hypoxanthine and 1,N6-ethenoadenine as preferred substrates. J Biol Chem. 2004;279:13601–6. doi: 10.1074/jbc.M314277200. [DOI] [PubMed] [Google Scholar]
- (41).Lau AY, Scharer OD, Samson L, Verdine GL, Ellenberger T. Crystal structure of a human alkylbase-DNA repair enzyme complexed to DNA: mechanisms for nucleotide flipping and base excision. Cell. 1998;95:249–58. doi: 10.1016/s0092-8674(00)81755-9. [DOI] [PubMed] [Google Scholar]
- (42).Roy R, Biswas T, Hazra TK, Roy G, Grabowski DT, Izumi T, Srinivasan G, Mitra S. Specific interaction of wild-type and truncated mouse N-methylpurine-DNA glycosylase with ethenoadenine-containing DNA. Biochemistry. 1998;37:580–9. doi: 10.1021/bi972313l. [DOI] [PubMed] [Google Scholar]
- (43).Miao F, Bouziane M, Dammann R, Masutani C, Hanaoka F, Pfeifer G, O’Connor TR. 3-Methyladenine-DNA glycosylase (MPG protein) interacts with human RAD23 proteins. J Biol Chem. 2000;275:28433–8. doi: 10.1074/jbc.M001064200. [DOI] [PubMed] [Google Scholar]
- (44).Watanabe S, Ichimura T, Fujita N, Tsuruzoe S, Ohki I, Shirakawa M, Kawasuji M, Nakao M. Methylated DNA-binding domain 1 and methylpurine-DNA glycosylase link transcriptional repression and DNA repair in chromatin. Proc Natl Acad Sci U S A. 2003;100:12859–64. doi: 10.1073/pnas.2131819100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Speina E, Zielinska M, Barbin A, Gackowski D, Kowalewski J, Graziewicz MA, Siedlecki JA, Olinski R, Tudek B. Decreased repair activities of 1,N(6)-ethenoadenine and 3,N(4)-ethenocytosine in lung adenocarcinoma patients. Cancer Res. 2003;63:4351–7. [PubMed] [Google Scholar]
- (46).Gruskin EA, Lloyd RS. The DNA scanning mechanism of T4 endonuclease V. Effect of NaCl concentration on processive nicking activity. J Biol Chem. 1986;261:9607–13. [PubMed] [Google Scholar]
- (47).von Hippel PH, Berg OG. Facilitated target location in biological systems. J Biol Chem. 1989;264:675–8. [PubMed] [Google Scholar]
- (48).Jack WE, Terry BJ, Modrich P. Involvement of outside DNA sequences in the major kinetic path by which EcoRI endonuclease locates and leaves its recognition sequence. Proc Natl Acad Sci U S A. 1982;79:4010–4. doi: 10.1073/pnas.79.13.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Gowers DM, Halford SE. Protein motion from non-specific to specific DNA by three-dimensional routes aided by supercoiling. Embo J. 2003;22:1410–8. doi: 10.1093/emboj/cdg125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Yildiz A, Tomishige M, Vale RD, Selvin PR. Kinesin walks hand-over-hand. Science. 2004;303:676–8. doi: 10.1126/science.1093753. [DOI] [PubMed] [Google Scholar]
- (51).Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–5. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- (52).Verdine GL, Bruner SD. How do DNA repair proteins locate damaged bases in the genome? Chem Biol. 1997;4:329–34. doi: 10.1016/s1074-5521(97)90123-x. [DOI] [PubMed] [Google Scholar]
- (53).Sun J, Viadiu H, Aggarwal AK, Weinstein H. Energetic and structural considerations for the mechanism of protein sliding along DNA in the nonspecific BamHI-DNA complex. Biophys J. 2003;84:3317–25. doi: 10.1016/S0006-3495(03)70056-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Gowers DM, Wilson GG, Halford SE. Measurement of the contributions of 1D and 3D pathways to the translocation of a protein along DNA. Proc Natl Acad Sci U S A. 2005;102:15883–8. doi: 10.1073/pnas.0505378102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc Natl Acad Sci U S A. 2006;103:5752–7. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hamilton RW, Lloyd RS. Modulation of the DNA scanning activity of the Micrococcus luteus UV endonuclease. J Biol Chem. 1989;264:17422–7. [PubMed] [Google Scholar]
- (57).Nickell C, Lloyd RS. Mutations in endonuclease V that affect both protein-protein association and target site location. Biochemistry. 1991;30:8638–48. doi: 10.1021/bi00099a021. [DOI] [PubMed] [Google Scholar]
- (58).Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–41. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1_si_001