Fragile X Premutation Disorders – Expanding the Psychiatric Perspective (original) (raw)
. Author manuscript; available in PMC: 2009 Jul 3.
Published in final edited form as: J Clin Psychiatry. 2009 May 5;70(6):852–862. doi: 10.4088/JCP.08m04476
Abstract
Objective:
Fragile X premutation conditions are associated with a significant degree of psychopathology and thus are of interest to the psychiatrist. Remarkable advances at the molecular level have enhanced our understanding of fragile X premutation disorders.
Methods:
The authors review the genetic, molecular, neuroimaging, and clinical (systemic, neurologic, and psychiatric) manifestations of the premutation carrier state (55-200 CGG repeats) of the fragile X mental retardation 1 (FMR1) gene.
Results:
Clinical manifestations of psychiatric illness in premutation carriers include cognitive, mood, anxiety, and other psychiatric disorders. Fragile X premutation-associated conditions are part of the clinical differential diagnosis of several psychiatric syndromes, particularly in pedigrees with known fragile X syndrome (FXS) cases.
Conclusions:
Fragile X-associated psychiatric manifestations serve as a useful model for a molecular genesis of neuropsychiatric illness. Because of the multigenerational expression of fragile X-associated neuropsychiatric illness, there is a prominent role for genetic testing and genetic counseling of patients and their relatives. Genetic testing is confirmatory of the FMR1 premutation and is an essential component of the clinical evaluation. Psychopharmacological and psychotherapeutic treatment of fragile X-associated psychiatric illnesses may improve patient function and assist in adaptation to the burden of a genetic neuropsychiatric illness.
Keywords: fragile X syndrome, fragile X-associated tremor/ataxia syndrome (FXTAS), Primary ovarian insufficiency, Psychiatric co-morbidity, Dementia
Introduction
Fragile X syndrome (FXS), the most common inherited form of intellectual disability and the most common single gene cause of autism,1 is a trinucleotide repeat disorder caused by an expansion of greater than 200 CGG repeats in the 5′ untranslated region of the fragile X mental retardation 1 (FMR1) gene on the X chromosome. The inheritance pattern of fragile X is complex and based upon a progressive generational expansion of the repeat size in transmission from mother to child. Based on the size of the CGG repeat expansion, individuals are classified as having normal alleles (5-44 CGG repeats), premutation alleles (55-200 CGG repeats), or full mutation alleles (>200 CGG repeats). Alleles that have 45-54 CGG repeats are labeled as the “gray zone” because instability in transmission to the next generation is common.2
The FMR1 gene codes for FMR1 protein (FMRP), which is thought to be important for dendritic transport of mRNAs, regulation of translation, and synaptic plasticity. This protein is deficient or absent in FXS, due to silencing of the FMR1 gene. FMR1 mRNA and FMRP levels were initially thought to be normal in the premutation range; however, it was recently demonstrated that FMR1 mRNA levels in peripheral blood leukocytes are two- to eight-fold higher than normal, despite a normal or slightly low level of FMRP.3 The excess FMR1 mRNA in premutation carriers is hypothesized to exert a toxic gain of function effect leading to dysregulation of several proteins, including lamin A/C and several heat shock (stress response) proteins, with subsequent accumulation of these and other proteins, along with the _FMR1_-mRNA, in the intranuclear inclusions found in FXTAS cases examined post-mortem.4, 5 These inclusions are a pathologic hallmark of FXTAS.6, 7
Fragile X premutation-associated conditions are associated with a significant degree of psychopathology and thus represent psychiatric illnesses of defined molecular/genetic origin that are of interest to the practicing clinician. Cases of fragile X premutation-related psychiatric illness may be identified in family members through analysis of genetic pedigrees of probands with FXS (often children presenting with developmental delay or intellectual disability). Table 1 summarizes fragile X-associated terminology.
TABLE 1.
Definition of Terms
| Fragile X syndrome | Most common inherited form of intellectual disability, fragile X syndrome is a trinucleotide repeat disorder caused by an expansion of greater than 200 CGG repeats in the 5′ untranslated region of the fragile X mental retardation 1 (FMR1) gene on the X chromosome. This is also called the full mutation of fragile X. Full mutation alleles result in hypermethylation and silencing of the FMR1 gene and consequent deficiency or absence of FMR1 protein. Physical signs including a long face and prominent ears are present in many individuals with FXS, and seizures occur in about 15%, most commonly in young children. Due to the presence of a presumably normal FMR1 allele on the second X chromosome, females with a full mutation are more mildly affected than males, but can have a range of associated features, both physical and developmental. |
|---|---|
| FMR1 premutation | Premutation alleles range from 55-200 CGG repeats of the FMR1 gene. Clinical phenotypes specific to premutation carriers have emerged, fragile X-associated primary ovarian insufficiency (FXPOI) in female premutation carriers and fragile X-associated tremor/ataxia syndrome (FXTAS) in both male and female premutation carriers. |
| Fragile X- associated tremor/ataxia syndrome (FXTAS) | Fragile X–associated tremor/ataxia syndrome (FXTAS) is a late onset (> 50 years) neurodegenerative disorder, occurring in carriers of a premutation CGG-repeat expansion in the fragile X mental retardation 1 (FMR1) gene. The disorder consists of intention tremor, ataxia, parkinsonism, cognitive decline, neuropathy and psychiatric features along with MRI findings of increased signal intensity in the middle cerebellar peduncles (MCP) sign on T2-weighted images or FLAIR sequences in many affected persons. |
| Fragile X- associated primary ovarian insufficiency | Fragile X-associated primary ovarian insufficiency (FXPOI; previously, premature ovarian failure, POF) is defined as menopause occurring before age 40. It is a unique phenotype among women who are premutation carriers, and is not seen in females with the full mutation. Carriers of the FMR1 premutation (55-200 CGG repeats) are at risk for FXPOI, early menopause and ovarian dysfunction (decreased fertility) in general. Approximately 20% of females with the premutation experience FXPOI. The premutation is the most common single gene mutation associated with POI in the general population. Sullivan et al.14 demonstrated a significant, non-linear association between CGG repeat number and prevalence of POI. |
Premutation expansions are frequent in the general population, with estimates of 1 per 113-259 females and 1 per 260-800 males.8-12 Recent clinical and molecular studies have changed the view that there are no clinical manifestations associated with the premutation allele condition. It is now apparent that individuals with the premutation can present with a late-onset neurodegenerative disorder, fragile X-associated tremor/ataxia syndrome (FXTAS), which affects nearly 40% of premutation males and 8% of premutation females over 50 years of age who are ascertained through families with fragile X syndrome probands.13 In addition, primary ovarian insufficiency (POI) affects approximately 16-25% of females with the premutation.14 Co-morbid psychiatric illness is also seen in a subgroup of premutation carriers, both males and females, children and adults, and these problems will be reviewed here.15-18
It is important to recognize disorders of premutation carriers for several reasons. Recognition of a genetic disorder in a family has important implications for multiple members within the family tree. For example, a premutation carrier male will pass the premutation to all of his daughters. His daughters, depending upon their CGG repeat size, will have up to 50% risk of having a child with fragile X syndrome (which is caused by expansion of the CGG repeat into the full mutation range). New targeted treatments are becoming available for fragile X syndrome19 and emerging treatment information is available for premutation involvement as described in more detail below,20 making diagnosis imperative.
Brain Structure and Function in the Fragile X Premutation: A Substrate for Psychiatric Symptoms?
Because of the previous long-standing belief that no neuropsychiatric abnormalities were associated with the FMR1 premutation, few studies have been published on brain structure in carriers of the premutation. One such study reported that, in comparison to matched controls, males and females with the premutation had significantly smaller hippocampal volumes than controls, and that this decrease was associated with memory deficits in the premutation carriers.21 Similarly, Murphy et al showed that premutation female carriers had significant decreases in whole brain volume as well as caudate and thalamic nuclei bilaterally.22
More recently, male premutation carriers were reported to have significantly reduced grey matter density, as compared to IQ matched controls, in a number of brain regions including the cerebellum, caudate, insula, amygdalo-hippocampal complex, brainstem, and thalamus.23 Additionally, within this group, increased age, increased CGG repeat size and decreases in the percentage of blood lymphocytes expressing FMRP were associated with decreased grey matter density in the amygdalo-hippocampal complex.
In a recent paper investigating amygdala function in adult male carriers of the FMR1 premutation, fMRI was used to measure brain responses to viewing fearful faces, stimuli that reliably activate this brain region in normative studies. Compared to controls, men with the premutation showed diminished brain activation in the amygdala and several brain areas that mediate social cognition (i.e., bilateral superior temporal sulcus (STS), bilateral orbital gyrus, and bilateral insula) while viewing fearful faces, as compared to age and IQ-matched controls.24 The reduced amygdala activation in the premutation group was significantly associated with self-report of psychological symptoms on the Symptom Checklist-90-Revised (SCL-90-R). In the psychophysiology laboratory, these men also displayed a lack of startle potentiation while viewing fearful faces and showed reduced skin conductance response when greeting an unfamiliar experimenter in comparison to the control group. In a subsequent study with the same participants, it was found that the men with the premutation had reduced activity of the hippocampus during a memory recall task that was also correlated with abnormal elevation of the FMR1 mRNA and psychological symptom severity.25
FXTAS
FXTAS is a late onset (generally > 50 years) neuropsychiatric degenerative disorder, occurring predominantly in males with the premutation, although approximately 8% of females with the premutation are affected.26-31 Because the premutation is relatively common in the general population, FXTAS may be one of the most common late-onset, progressive neurological diseases associated with a single gene mutation.32 The major motor features of FXTAS are kinetic, intention or postural tremor, cerebellar gait and limb ataxia, and parkinsonism.33, 34 Age of onset of both tremor and ataxia and degree of brain atrophy have been found to correlate with CGG repeat length.35-37 The major features of FXTAS constitute the diagnostic criteria in Table 2). FXTAS clinical staging is outlined in Table 3.
TABLE 2.
Diagnostic criteria for FXTAS*
| MOLECULAR | CGG repeat 55 – 200 | |
|---|---|---|
| CLINICAL Major Minor | Intention tremor Cerebellar gait ataxia | |
| Parkinsonism Moderate to severe short term memory deficit Executive function deficit | ||
| RADIOLOGICAL Major Minor | MRI white matter lesions involving middle cerebellar peduncles | |
| MRI lesions involving cerebral white matter Moderate to severe generalized brain atrophy | ||
| Diagnostic Categories | ||
| DEFINITE One major clinical, and One major radiological, or Presence of FXTAS inclusions | PROBABLE Two major clinical, or One minor clinical, and One major radiological | POSSIBLE One major clinical, and One minor radiological |
TABLE 3.
FXTAS Stage Description
| STAGE | DESCRIPTION |
|---|---|
| 0 | Normal function |
| 1 | Subtle or questionable signs such as subtle tremor or mild balance problems, with no interference in ADLs |
| 2 | Minor, but clear, tremor and/or balance problems with minor interference with ADLs |
| 3 | Moderate tremor and/or balance problems and occasional falls with significant interference in ADLs |
| 4 | Severe tremor and/or balance problems; uses cane or walker |
| 5 | Uses wheelchair on a daily basis |
| 6 | Bedridden |
Patients may have substantial autonomic dysfunction, including orthostatic hypotension, impotence, and loss of bowel and bladder control.29, 38, 39 Signs of peripheral neuropathy, including decreased deep tendon reflexes and impaired vibration sense in the distal lower extremities, are present in many persons affected with FXTAS.7,35,39-42 As such, patients may have received a broad range of other clinical diagnoses including cerebellar ataxia, essential tremor, olivopontocerebellar atrophy (OPCA), multi-system atrophy (MSA), Parkinson's disease, dementia, stroke and/or multiple sclerosis before the diagnosis of FXTAS is validated by clinical evaluation and genetic testing.40
A family-based, retrospective, longitudinal study29 reported the progression of motor deficits related to tremor and ataxia and the length of survival after onset of first report of tremor or ataxia in 55 males with the premutation. Tremor generally occurred first, with median onset at about age 60. Median onset of ataxia was two years after the onset of the first other motor sign (usually tremor); followed sequentially by onset of falls at six years; dependence on a walking aid at 15 years; and, finally, death at 21 years. Early data on life expectancy were variable, ranging from five to 25 years after onset of motor symptoms. In the months before death, patients tended to be bedridden, dysarthric, dysphagic, without bladder or bowel control, and had severe Parkinsonism (rigidity, rest tremor and bradykinesia). Goncalves et al43 reported a case of FXTAS with rapidly progressive dementia with minimal motor symptoms.
MRI findings in patients with FXTAS include cerebellar and cerebral atrophy and bilateral regions of increased T2 signal intensity of the middle cerebellar peduncles (MCPs) and subcortical regions (Figure 1).5, 33, 41 The MCP hyperintensity is seen in approximately 60% of males with FXTAS and 13% of females with FXTAS.34, 37 Another study using structural MRI compared male premutation carriers who were affected with FXTAS with unaffected carriers and age-matched controls.36 Significant volume loss in the whole brain, cerebrum, and cerebellum, as well as increases in whole-brain white matter hyperintensity volume was found in the FXTAS group, correlating with CGG repeat number and becoming more severe with age.
FIGURE 1.
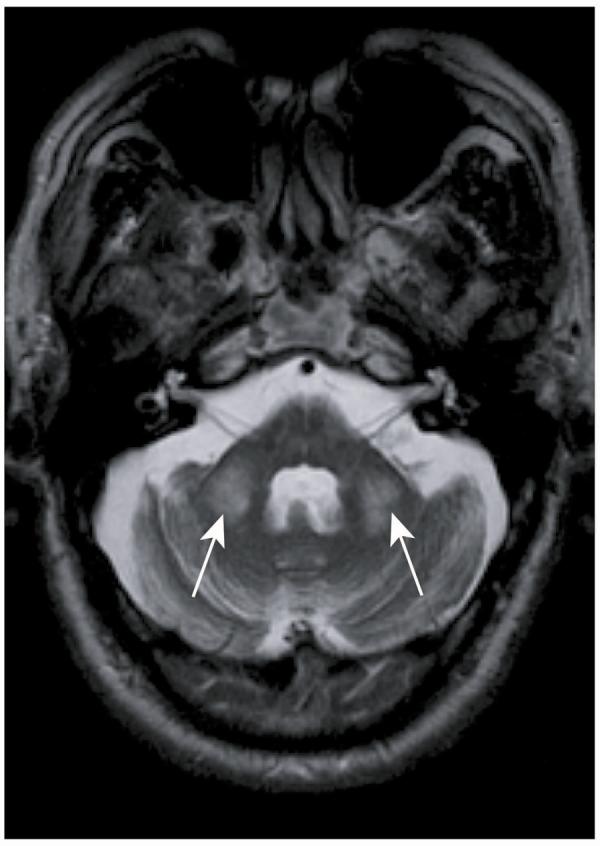
MRI image with an axial cut through the cerebellum demonstrates the middle cerebellar peduncle sign bilaterally denoted by the arrows
The pattern of features associated with FXTAS seems to differ somewhat between female and male premutation carriers. Compared with controls, female carriers with definite or probable FXTAS had greater medical co-morbidity, with increased prevalence of thyroid disease, hypertension, seizures, peripheral neuropathy, and fibromyalgia, in addition to the typical symptoms of FXTAS, tremor and ataxia.42 The non-FXTAS group of females with the premutation had more complaints of chronic muscle pain, persistent paresthesias in the extremities, and history of tremor than controls.42
Pathology of FXTAS
The distinctive neuropathological feature of FXTAS is the presence of intranuclear inclusions in neurons and astrocytes that are broadly distributed throughout the central nervous system.4-6, 44, 45 The peripheral nervous system, including the autonomic nervous system are also known to be involved, as evidenced by the presence of similar-appearing intranuclear inclusions in ganglion cells. 4, 5, 45 Studies of inclusion formation in individuals with FXTAS have demonstrated inclusions in the anterior and posterior pituitary, in addition to the hypothalamus.39, 46 Inclusions are also present in the Leydig and smooth muscle cells of the testicles, a finding that may relate to low testosterone levels observed in a small number of males with FXTAS45 and presence of impotence as a common finding in males with FXTAS. 39 Symptoms of impotence often occur prior to the onset of tremor and ataxia.39, 46
Psychopathology in FXTAS: Cognitive Disorders and Psychiatric Comorbidity
Until recent years there was relatively little literature on the psychiatric aspects of FXTAS. As is common in neurodegenerative conditions (especially those with a molecular basis; e.g., Huntington's disease), either concurrent or sequential clinical expression of psychiatric illnesses may occur in FXTAS. Indeed, the conceptualization for CNS-derived symptoms and signs in such illnesses is that of “neuropsychiatric” illness.
Patients may experience symptoms on several psychiatric dimensions either simultaneously or sequentially, adding further conceptual and diagnostic imprecision. In FXTAS, this dispersion of psychological symptoms that bridge DSM-IV-TR categories was illustrated by Hessl et al.16 in a study of subjects with the premutation both with and without FXTAS. On the SCL-90-R, elevated scores on the somatization, obsessive compulsive, interpersonal sensitivity, psychoticism, and global severity index were found for premutation carriers of both genders, and increased scores on depression and phobic anxiety in male carriers.
The psychopathology of FXTAS is characterized in many cases by dementia, which presents as memory loss with both frontal lobe features (disinhibition, inappropriate social behavior, poor executive functioning, perseveration, irritability, mood disturbances) and subcortical features (psychomotor slowing, bradyphrenia, attention and concentration difficulties).33, 47, 48 The onset of the cognitive impairment often follows the onset of movement disorder symptoms, and is infrequently seen before age 50. 33, 49 With formal cognitive assessment, patients with FXTAS are found to suffer a greater decrement in performance IQ than verbal IQ, probably in part because of the movement disorder.33 As patients may present with movement disorder and subsequent dementia, some cases are may be misclassified as Parkinson's disease or multiple sclerosis.40 A recent paper of 15 subjects with FXTAS revealed dementia in 7 cases; 7 were also diagnosed with mood or anxiety disorders. Twelve had cognitive impairment on formal neuropsychological testing.50
The rate of progression of cognitive impairment in FXTAS dementia awaits prospective clarification. As this is a neurodegenerative condition, it appears likely that the dementia in FXTAS is insidiously progressive. The behavioral disturbances in FXTAS dementia have been demonstrated on the Neuropsychiatric Inventory (NPI) with apathy, depression, and agitation as key features compared to age-matched controls.48
The cognitive phenotype of FXTAS is characterized by impairment of executive cognitive functioning (ECF),51, 52 very similar to that observed in various subcortical or frontal dementias. These include such disorders as the frontal variant of frontotemporal dementia (FTD), many of the spinocerebellar ataxias, corticobasal degeneration, progressive supranuclear palsy, Parkinson's disease, and the cerebellar type of multiple system atrophy (MSA-C). 53-56 The existence of deficits in ECF was apparent in the early reports of FXTAS,57 and it since has been confirmed in a series of research studies and case reports.58, 59
The phenotype appears to differ somewhat for males and females.26 Moreover, women are less likely to be affected than men. Consequently, systematic study of the cognitive phenotype of FXTAS has been undertaken only among males, among whom it has been found that individuals with FXTAS typically performed worse than control subjects on measures of general intelligence (Wechsler Verbal and Performance IQ scores), with especially deficient performance on the nonverbal subtests of the Wechsler Adult Intelligence Scale-Third Edition (WAIS-III). 51, 60 Certain areas of functioning appear to be relatively unaffected. These include language, visuospatial perception, and verbal reasoning—all abilities that are, in contrast, affected relatively early in the course of Alzheimer's disease.61 The overall cognitive status of men with FXTAS, measured by the Folstein MiniMental State Examination,62 differs significantly from that of control subjects.
Most noteworthy is a striking impairment of executive functions, including working memory,51, 60 especially on a measure of the capacity for behavioral self-regulation (the Behavioral Dyscontrol Scale63) and the Controlled Oral Word Association Test (COWAT) (a measure of verbal fluency.) Deficits have been noted as well in declarative (semantic) learning and memory, information processing speed, and temporal sequencing as well as some measures of verbal comprehension and visuospatial functioning.52 When test scores on most WAIS subtests and measures of memory are adjusted for age, education, and ECF performance, the majority of group differences disappear.52 This suggests that many of the observed problems are mediated by the executive abilities, and are not primarily deficits of such capacities as memory or visuospatial functioning, at least in the early and intermediate stages of the disorder. Careful study of one case over time, until a few months prior to his death, suggests that eventually such cognitive disorders as ideomotor apraxia may be observed.60 Moreover, given the high burden of inclusion bodies in the hippocampus,5 and the hippocampal atrophy observed among persons with FXTAS,36 it is likely that the memory disorder shifts over time from being dysexecutive in nature to one in which consolidation of memory per se becomes problematic.
At this time, little is known regarding the timing of onset of cognitive difficulties in FXTAS, although research on unaffected carriers of the premutation23, 51, 64 suggests that they may have a neurodevelopmental deficit affecting executive cognitive functioning that may predate the appearance of cognitive symptoms in FXTAS. It also is unclear whether all persons with FXTAS will eventually develop this cognitive phenotype, and if so, how severe it will be. Longitudinal research currently underway will provide insight into the progressive nature of the disorder.
Psychopathology in Premutation Carriers without FXTAS
Initial studies of psychiatric status in premutation carriers focused on women because they were more available in the clinical evaluation of their children with FXS. Females may be more buffered from RNA toxicity, depending on activation ratio, since they have a normal X chromosome in addition to the premutation X chromosome.16 Initial reports did not find significant psychopathology in female carriers compared to controls.65 However, several later reports demonstrated that a subgroup of women with the premutation had symptoms of depression and/or anxiety. Although the stress of raising a child with FXS could certainly add to the risk for psychopathology, many female carriers report the presence of anxiety and low level depression prior to giving birth to children who have FXS. Studies in knock-in mice expressing a premutation range allele have demonstrated an enhanced release of cortisone, the stress hormone equivalent to cortisol in humans.66 Therefore, individuals with the premutation may have an increased stress response.
Franke et al.17 reported a lifetime diagnosis of unipolar affective disorder of 21.3%, major depression of 19.7%, and bipolar affective disorder of 11.5% in premutation carrier mothers of children with FXS and an overall rate of affective disorders diagnosis of 55.7% in this group. Thompson et al.67 studied 14 women with the premutation (and 5 with the full mutation) and found 78% of these patients (in the combined group) to have had a lifetime history of major depression; 33% of the subjects had had recurrent depression. Johnston et al.68 found higher scores on the depression and interpersonal sensitivity subscales of the SCL-90-R in premutation carriers with greater than 100 CGG repeats. A more recent study examining the association between FMR1 CGG repeat size, FMRP, and mRNA in men and women with the premutation documented that the abnormal elevation of mRNA was correlated with severity of psychological symptoms in male premutation carriers without FXTAS.16
A recently published study by Roberts et al.69 studied 93 premutation females compared to 2,159 female controls from the National Comorbidity Survey Replication (NCS-R) data set. The methodology included the Structured Clinical Interview for the DSM-IV (SCID-I) to assess for lifetime and current history of mood and anxiety disorders. This study found a significantly elevated risk of lifetime major depressive disorder (43.0% vs. 31.9%), lifetime panic disorder without agoraphobia (8.6% vs. 2.3%), and current agoraphobia without panic disorder (3.2% vs. 0.7%) in the premutation population compared to controls. Strikingly, the risk of mood disorder in premutation subjects was associated with decreased CGG repeat length (a counterintuitive finding) and was not associated with variables pertinent to number of children with FXS or child behavioral problems. Anxiety disorders in the premutation group were correlated with number of children with FXS and problematic child behaviors.
Anxiety, social avoidance, interpersonal sensitivity, shyness, and avoidance of eye contact have been described in premutation carriers, although prevalence rates and psychiatric diagnostic specificity (e.g., Axis I social phobia and/or Axis II avoidant personality disorder) are as of yet unclear.15, 49, 70 Franke et al.17 reported a lifetime rate of diagnosis of an anxiety disorder on 41.0% in premutation carrier mothers of children with FXS; among these patients, the lifetime risk of social phobia was 18.0% and the lifetime risk of panic disorder was 11.5%. This higher rate of social phobia was also seen in female siblings of the women, who also carried the premutaion allele but did not have children with FXS. In retrospective examination, many patients with FXTAS have been shown to have chronic anxiety and obsessionality since early adulthood, although these were not always formally diagnosed as anxiety disorders.15 Franke et al.17 reported a higher rate of social phobia in premutation carriers who were the mothers of full mutation children; this higher rate of social phobia was also seen in siblings of the women, who also carried the premutation allele, but who did not have children with FXS.
The rate of clearly distinct psychotic disorder in premutation carriers appears to be low.17 Individual cases reported in the literature include an 18-year old female with hallucinations, delusions, cognitive disorganization, and mania, followed by social and intellectual dysfunction. She was diagnosed as schizoaffective disorder.71 Khin et al.72 described a case of an adult male with FMR1 methyation mosaicism who developed paranoid delusions, vague thought content, and inappropriate laughter and was diagnosed with paranoid schizophrenia and schizoid personality disorder.
Premutation carriers have been reported to have increased prevalence of learning disabilities, ADHD, intellectual disability, and autism.18, 73 Schizotypal personality features (not necessarily a full personality disorder) have also been reported in premutation carriers.74 The Franke et al. study17 found a higher rate of avoidant personality disorder (8.2%) and schizotypal personality disorder (4.9%) in premutation carriers, a finding which, along with their other finding of more social phobia in premutation carriers, is consistent with higher rates of social avoidance in these patients.68 This is substantiated by Thompson et al.67, who found schizotypal features in 17% of a fragile X group which consisted of both premutation and full mutation. Dorn et al.75 found high rates of adult ADHD, alcohol abuse/dependence, and OCD-related behaviors in premutation carrier males.
There have been several case reports of children with the premutation and psychopathology including autism spectrum disorders (ASD) or ADHD, sometimes related to lowered FMRP.76 Although lowered FMRP levels are typically seen in those with a full mutation, the protein levels may fall in premutation carriers, particularly those in the upper range of the premutation, therefore causing fragile X syndrome features in some individuals with a large premutation.3 Whether autism in premutation carriers is related to the RNA toxic effect of elevated mRNA or also to lowered FMRP is not known. In Australia, ASD has been reported in 1 of 7 males with a premutation (14%) and in 2 of 43 (5%) females with a premutation. In a family study of carrier brothers there was a significant group difference in ASD and ADHD between premutation carrier males presenting clinically (proband; n = 14), non-proband carrier males (n = 13) identified through cascade testing of the family (siblings of the proband), and control siblings without the premutation (n = 16).18 The rate of ASD as confirmed by the ADOS was found to be significantly higher (79%) in probands compared to control siblings without the premutation (0%). It is important to note that the number of autistic symptoms reported on the Social Communication Questionnaire (SCQ) in the non-proband premutation carriers was also statistically higher compared to control siblings without the premutation. This study demonstrated a significant group difference between the probands (93%) and the controls (13%) with ADHD diagnoses. The ASD symptoms and ADHD problems in young male carriers are reminiscent of the social deficits reported in adult male carriers without FXTAS by Cornish et al.,77 and previously demonstrated in a retrospective study by Dorn et al.75 Therefore, significant psychopathology may be relatively common in male carriers and may be developmentally based and present well before onset of FXTAS neurological symptoms. Currently available studies, however, involve small numbers of subjects, and are subject to ascertainment bias, and definition of the true frequency of these executive and social deficits in premutation carriers will await large population-based epidemiological studies.
Genetic Testing
Laboratory testing for fragile X mutations is usually straightforward and highly reliable in both clinically affected patients and asymptomatic carriers. Testing involves a single blood sample on which direct DNA analysis of the FMR1 gene is performed to determine both the number of CGG repeats and the methylation status of the gene using both Southern blot and PCR testing. Fragile X mutation testing is widely available and covered by most major insurance companies in the U.S., including Medicare and state Medicaid plans. The cost of the genetic test is generally between 200and200 and 200and400 per test. Results typically distinguish among normal, premutation, and full mutation allele sizes, although it is sometimes difficult to interpret the significance of “gray zone” alleles that overlap the junction between the normal and premutation ranges (approximately 45 –54 CGG repeats). Such “gray zone” results should be interpreted in the context of the family and clinical history and in genetic counseling.78
Psychiatric Clinical Indications for Genetic Testing
The increasing appreciation of the varied, if somewhat phenomenologically nonspecific, psychiatric manifestations of fragile X premutation-associated conditions leads to consideration of which psychiatric patients should receive fragile X genetic testing. For a “maximally inclusive” approach, the psychiatrist may consider genetic analysis in cases of anxiety disorders, mood disorders, patients with tremor, ataxia, apparent Parkinson's disease, and/or dementia. It is likely that testing “all” anxiety or mood disorder patients for a fragile X premutation may yield relatively few cases a priori (although a large scale assessment of anxiety and mood disorder patients in this fashion would address the question of the frequency of the premutation in anxiety and mood disorder populations).
In cases of mood and anxiety disorders, testing of probands for fragile X may be more targeted in some specific circumstances. Adult patients with mood or anxiety disorders who are biological parents of developmentally delayed, autistic or intellectually disabled children should be tested. The phenotype of FXS in children may be variable, thus testing for fragile X in all such children is clearly indicated. The occasional case of an adult patient with these conditions not already attributed to another known genetic or environmental cause may benefit from testing as well.
Adult patients with mood or anxiety disorders who also have family history of dementia (especially of dementia with a movement disorder) may also benefit from testing; in such cases, the adult patient may not have had children with FXS but can be phenomenologically identified by the dementia with a movement disorder in a parent.
Adult women who present with a history of anxiety and/or mood disorders and a history of premature ovarian insufficiency may benefit from testing. Some of these women may have been childless until their late 30s and in pursuing reproductive options are diagnosed with primary ovarian insufficiency. The loss of reproductive capacity may need to be processed and mourned, but the clinical context is a good place for fragile X testing of the patient offers the opportunity to rule out the premutation as the cause of the infertility problems.
In cases with a primary presentation of cognitive impairment, the clinician is advised to actively include the possibility of FXTAS dementia when evaluating patients for cognitive impairment. This is especially true if the patient (especially a male patient) presents with dementia in the setting of a movement disorder, although in some cases, FXTAS may present with cognitive impairment in advance of a movement disorder. The differential diagnosis of “dementia with a movement disorder” typically includes other neurodegenerative illnesses such as Parkinson's disease, Huntington's disease, the spinocerebellar ataxias, and multiple sclerosis. Cases of dementia with a movement disorder with a family history of fragile X-associated symptoms and diagnoses including intellectual disability or autism would specifically benefit from genetic testing, as the true incidence of FXTAS dementia among patients with dementia and a movement disorder, although currently unknown, may be relatively common, based on the rate of the fragile X premutation in population samples.79
The use of MRI in dementia evaluations may identify the middle cerebellar peduncle T2 hyperintensity characteristic of FXTAS.80 Due to the apparent relative specificity of the cerebellar hyperintensity for FXTAS, based on the current literature, it appears reasonable to obtain fragile X testing on cases with this radiological sign, especially when cognitive impairment is clinically evident.
Genetic Counseling
Once the diagnosis of a fragile X-associated disorder is made, the clinician must not only consider the patient's clinical course, but also the implications of the diagnosis for the extended family. Fragile X inheritance is complex and there are few absolutes, apart from the fact that X-linked inheritance precludes male-to-male transmission of fragile X mutations. None of the sons of a man with FXTAS, for example, will inherit his premutation, while all of his daughters will be carriers of the premutation. Females with the premutation or full mutation are at an increased risk to have sons and/or daughters affected by FXS. As such, it is not unusual to identify families with multiple relatives across several generations who are affected by a wide range of fragile X-associated effects, from intellectual disability and psychiatric illness to infertility and ataxia.78
Genetic counseling for fragile X-associated disorders is challenging because of the complex multigenerational inheritance, variable phenotype, as well as the emotional impact of the diagnoses on families. Newly diagnosed patients should be provided with information about high quality fragile X resources, such as the National Fragile X Foundation (www.nfxf.org or www.fragilex.org) which has parent support groups available in most states, and the FRAXA Research Foundation (www.fraxa.org). They should also be encouraged to share information about the diagnosis with their extended family members. Using the family pedigree, special attention should be given to identifying relatives who are pregnant or contemplating pregnancy as timely information about the presence of a familial fragile X mutation maximizes reproductive options, including carrier testing and prenatal diagnosis. Anger, denial, anxiety, guilt, blame, lowered self-esteem, and depression are common reactions among diagnosed carriers of any genetic disorder. Further complicating the situation in fragile X is the presence of high incidence of psychiatric disorders among individuals with premutations and full mutations. Referral to a genetic counselor or geneticist is recommended, as these professionals can provide support with an emphasis on anticipatory guidance for families throughout the life cycle — from newborn screening, pediatric evaluations, reproductive counseling, to evaluations of individuals for FXTAS and FXPOI.
Clinical Management, Psychotherapy, and Psychopharmacology
Premutation carriers, even those without FXTAS or fragile X-associated POI, are likely at greater risk for psychiatric illness based on emerging understanding of these conditions and the limited literature to date. Caution is advised against attributing psychiatric symptoms in carriers of the premutation as nonspecific consequences of the stress of raising a child with FXS. Empirical study has shown that psychiatric symptoms in carriers also may be a neuropsychiatric consequence of the premutation condition itself. Because of the risk of eventual FXTAS in some carriers of the premutation, baseline cognitive status should be assessed with a standardized cognitive instrument (e.g., the MMSE) and consideration should be given to formal neuropsychological testing of these patients if cognitive function is of concern.
Women with the premutation experiencing fragile X-associated POI might need to mourn the premature loss of reproductive capacity. If these women have given birth to children diagnosed with FXS, they may benefit from psychotherapy approaches to deal with the sense of guilt and responsibility associated with having been the carrier of a genetic disorder. While not formally studied, women with fragile X-associated POI may be at risk for menopause-associated onset and/or exacerbation of psychiatric symptoms.
Newly identified carriers who are identified as part of a pedigree analysis generated following the clinical assessment of a FXS affected child may experience guilt at being the “source” of the genetically mediated illness that afflicts their family member. Relatively common scenarios where this may happen include the mother of a newly diagnosed child with FXS (herself a carrier), where obvious concern for the child and the burdens of raising an impaired child are compounded by guilt at having transmitted the genetic disorder. Similarly, a newly diagnosed elder who is the grandparent of a child with FXS in such a pedigree may have a significant psychological burden. Beyond generational guilt at having introduced fragile X to descendants, the older patient may simultaneously need to confront his or her own illness characterized by cognitive decline and progressing motor impairment. Supportive psychotherapy models focusing on gentle confrontation of distortions and a supportive and/or cognitive—behavioral approach may be of benefit. Couples and family psychotherapy models may also be considered due the “family” nature of genetic illness.
There are a number of treatments that help both tremor and balance problems in addition to Parkinsonian symptoms.20 Treatment of hypertension is essential, because it can worsen white matter disease and cause further cardiovascular problems.81 Identification of hypothyroidism is also essential because if left untreated it can exacerbate psychiatric symptoms, especially anxiety and depression, which are also common in carriers16, 82 and may be associated with cognitive dysfunction.
The psychopharmacologic treatment of premutation-associated psychiatric conditions is not specific to these conditions and borrows from conventional psychopharmacological interventions for various psychiatric disorders. For FXTAS-associated mood, anxiety, and psychotic disorders, conventional psychopharmacologic interventions should be considered. Antidepressants are often used for mood disorders in these patients, and when prescribing SSRIs the physician should take into account the drug-drug interactions common with paroxetine, fluoxetine, and fluvoxamine, since many of these patients will be receiving other systemic medications. The use of SNRI agents such as venlafaxine and duloxetine and the other dual action agent mirtazapine (which are largely devoid of drug-drug interaction problems) may be useful. TCAs and MAOIs should generally be avoided, as the side effects of these agents (especially the orthostatic hypotension) may be problematic in older patients with neurologic illness.
For anxiety disorders, clinicians should use antidepressants and buspirone, rather than benzodiazepines, due to the risks of tolerance with chronic use, and the well-known difficulties in using benzodiazepines in patients with risk for cognitive impairment or balance and postural problems. For psychotic disorders, atypical antipsychotics should used cautiously; because of the possibility of medication-induced movement disorders. If antipsychotics are necessary, quetiapine is a good first choice because it is associated with less risk of extrapyramidal side effects.
Empirical treatment of FXTAS dementia with off-label use of cholinesterase inhibitors and/or memantine appears warranted in selected cases, with regular close monitoring of cognitive status with a standardized cognitive assessment instrument such as the MMSE.47. 49 Avoidance of benzodiazepines and anticholinergic psychopharmacological agents is also indicated in dementia cases, to preserve vulnerable cognition and to minimize the risk of episodes of delirium. Further research into the newly identified molecular physiological mechanisms of involvement in FXTAS will likely lead to targeted treatments in the future.20
Conclusions
From a psychiatric perspective, fragile X premutation-associated conditions are a group of illnesses at high risk for psychiatric co-morbidity. The premutation carriers without clinical FXTAS appear to have an increased risk of mood and anxiety disorders, and males in particular may have an increased risk for disorders characterized by social avoidance (schizotypal personality disorder, avoidant personality disorder, and social phobia). Psychiatrists must be cautious not to solely attribute psychiatric distress in premutation carriers to the stress of raising an impaired child (although this may clearly be profound), but should conceptualize the psychiatric symptoms of these patients as a primary manifestation of the neuropsychiatric vulnerability represented by the carrier/premutation state. There is a role for genetic testing and counseling to establish the genetic pedigree, as well as to ascertain the number of CGG repeats, which may ultimately have prognostic significance. However, clinicians need to remain cautious and recall that a patient's psychiatric disorder could be mistakenly attributed to the premutation when it is in fact related to something else, like other life circumstances/stresses or a predisposition to a psychiatric disorder that was inherited in another way. This may be the case when a psychiatrically symptomatic member of a fragile X pedigree is found to not have the premutation or full mutation on genetic testing.
At the present time, it is impossible to predict with certitude which premutation carriers will develop FXTAS; furthermore, within the cohort of established FXTAS cases, it is not presently possible to predict which patient are at risk for FXTAS-associated mood, anxiety, psychotic, or cognitive disorders. There is a role for genetic testing and counseling to establish the genetic pedigree as well as to ascertain the number of CGG repeats, which may ultimately have prognostic significance. As with routine dementia workups, early use of MRI is highly recommended; cases with the characteristic MCP sign appear to be most specific for FXTAS. At the very least, FXTAS dementia needs to be on the differential diagnosis of dementia in certain clinical contexts, such as dementia with a movement disorder, or dementia with a family history of intellectual disability/autism/other childhood developmental disorders. In concert with genetic testing of the proband, a pedigree analysis of available family members, and genetic counseling for this “multigenerational family neuropsychiatric illness” needs to be made routine.
In a broader sense, wider clinician appreciation of fragile X-associated conditions as molecularly-based psychiatric illness may serve as a useful model for conceptualization of psychiatric illness, and may eventually lead to consideration of a genetic basis for other molecular illnesses that may present primarily with psychopathology.
ACKNOWLEDGEMENT
This work was supported by the National Fragile X Foundation and the National Institute of Health (NIH) grants HD036071, HD056031, NS044299, HD02274, MH77554, MH078041, RL1 AG032115, RL1NS062412, and RL1 AG032119, Centers for Disease Control and Prevention collaborative agreement U10/CCU925123, the National Fragile X Foundation, and the M.I.N.D. Institute. This publication was also made possible by a grant (UL1 RR024992) administered by the National Center of Dental and Craniofacial Research (NCRCR), a component of the NIH, and NIH Roadmap for Medical Research. The work was also supported in part by the Intramural Research Program, NIH.
REFERENCES
- 1.Hagerman RJ, Rivera SM, Hagerman PJ. The fragile X family of disorders: A model for autism and targeted treatments. Current Pediatric Reviews. 2008;4:40–52. [Google Scholar]
- 2.Nolin SL, Brown WT, Glicksman A, et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet. 2003;72:454–464. doi: 10.1086/367713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tassone F, Hagerman RJ, Taylor AK, et al. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in fragile X syndrome. Am J Hum Genet. 2000;66:6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greco CM, Hagerman RJ, Tassone F, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125:1760–1771. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- 5.Greco CM, Berman RF, Martin RM, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS) Brain. 2006;129(Pt 1):243–255. doi: 10.1093/brain/awh683. [DOI] [PubMed] [Google Scholar]
- 6.Iwahashi CK, Yasui DH, An HJ, et al. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129(Pt 1):256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- 7.Arocena DG, Iwahashi CK, Won N, et al. Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum Mol Genet. 2005;14:3661–3671. doi: 10.1093/hmg/ddi394. [DOI] [PubMed] [Google Scholar]
- 8.Dawson AJ, Chodirker BN, Chudley AE. Frequency of FMR1 premutations in a consecutive newborn population by PCR screening of Guthrie blood spots. Biochem Mol Med. 1995;56:63–69. doi: 10.1006/bmme.1995.1057. [DOI] [PubMed] [Google Scholar]
- 9.Dombrowski C, Levesque ML, Morel ML, et al. Premutation and intermediate-size FMR1 alleles in 10 572 males from the general population: loss of an AGG interruption is a late event in the generation of fragile X syndrome alleles. Hum Mol Genet. 2002;11:371–378. doi: 10.1093/hmg/11.4.371. [DOI] [PubMed] [Google Scholar]
- 10.Rousseau F, Rouillard P, Morel ML, et al. Prevalence of carriers of premutation-size alleles of the FMRI gene--and implications for the population genetics of the fragile X syndrome. Am J Hum Genet. 1995;57:1006–1018. [PMC free article] [PubMed] [Google Scholar]
- 11.Toledano-Alhadef H, Basel-Vanagaite L, Magal N, et al. Fragile-X Carrier Screening and the Prevalence of Premutation and Full-Mutation Carriers in Israel. Am J Hum Genet. 2001;69:351–360. doi: 10.1086/321974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagerman PJ. The Fragile X Prevalence Paradox. J Med Gen. 2008 April 15; doi: 10.1136/jmg.2008.059055. [epub ahead of print](PMID: 18413371) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacquemont S, Hagerman RJ, Leehey MA, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291:460–469. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- 14.Sullivan AK, Marcus M, Epstein MP, et al. Association of FMR1 repeat size with ovarian dysfunction. Hum Reprod. 2005;20:402–412. doi: 10.1093/humrep/deh635. [DOI] [PubMed] [Google Scholar]
- 15.Hagerman RJ. Lessons from fragile X regarding neurobiology, autism, and neurodegeneration. J Dev Behav Pediatr. 2006;27:63–74. doi: 10.1097/00004703-200602000-00012. [DOI] [PubMed] [Google Scholar]
- 16.Hessl D, Tassone F, Loesch DZ, et al. Abnormal elevation of FMR1 mRNA is associated with psychological symptoms in individuals with the fragile X premutation. Am J Med Genet B Neuropsychiatr Genet. 2005;139:115–121. doi: 10.1002/ajmg.b.30241. [DOI] [PubMed] [Google Scholar]
- 17.Franke P, Leboyer M, Gansicke M, et al. Genotype-phenotype relationship in female carriers of the premutation and full mutation of FMR-1. Psychiatry Res. 1998;80:113–127. doi: 10.1016/s0165-1781(98)00055-9. [DOI] [PubMed] [Google Scholar]
- 18.Farzin F, Perry H, Hessl D, et al. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27(2 Suppl):S137–144. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- 19.Hagerman RJ, Berry-Kravis E, Kaufmann WE, et al. Advances in the Treatment of Fragile X Syndrome. Pediatrics. doi: 10.1542/peds.2008-0317. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagerman RJ, Hall DA, Coffey S, et al. Treatment of fragile X-associated tremor ataxia syndrome (FXTAS) and related neurological problems. Clin Interv Aging. 2008;3:251–262. doi: 10.2147/cia.s1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jäkälä P, Hanninen T, Ryynanen M, et al. Fragile-X: neuropsychological test performance, CGG triplet repeat lengths, and hippocampal volumes. J Clin Invest. 1997;100:331–338. doi: 10.1172/JCI119538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy DG, Mentis MJ, Pietrini P, et al. Premutation female carriers of fragile X syndrome: a pilot study on brain anatomy and metabolism. J Am Acad Child Adolesc Psychiatry. 1999;38:1294–1301. doi: 10.1097/00004583-199910000-00019. [DOI] [PubMed] [Google Scholar]
- 23.Moore CJ, Daly EM, Tassone F, et al. The effect of pre-mutation of X chromosome CGG trinucleotide repeats on brain anatomy. Brain. 2004;127:2672–2681. doi: 10.1093/brain/awh256. [DOI] [PubMed] [Google Scholar]
- 24.Hessl D, Rivera S, Koldewyn K, et al. Amygdala dysfunction in men with the fragile X premutation. Brain. 2007;130(Pt 2):404–416. doi: 10.1093/brain/awl338. [DOI] [PubMed] [Google Scholar]
- 25.Koldewyn K, Hessl D, Adams J, et al. Reduced hippocampal activation during recall is associated with elevated FMR1 mRNA and psychiatric symptoms in men with the fragile X premutation. Brain Imaging Behav. 2008;2:105–116. doi: 10.1007/s11682-008-9020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagerman RJ, Leavitt BR, Farzin F, et al. Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am J Hum Genet. 2004;74:1051–1056. doi: 10.1086/420700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacquemont S, Hagerman RJ, Hagerman PJ, Leehey MA. Fragile-X syndrome and fragile X-associated tremor/ataxia syndrome: two faces of FMR1. Lancet Neurol. 2007;6:45–55. doi: 10.1016/S1474-4422(06)70676-7. [DOI] [PubMed] [Google Scholar]
- 28.Berry-Kravis E, Sumis A, Hervey C, et al. Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr. doi: 10.1097/DBP.0b013e31817dc447. in press. [DOI] [PubMed] [Google Scholar]
- 29.Leehey MA, Berry-Kravis E, Min SJ, et al. Progression of tremor and ataxia in male carriers of the FMR1 premutation. Mov Disord. 2007;22:203–206. doi: 10.1002/mds.21252. [DOI] [PubMed] [Google Scholar]
- 30.Willemsen R, Mientjes E, Oostra BA. FXTAS: a progressive neurologic syndrome associated with Fragile X premutation. Curr Neurol Neurosci Rep. 2005;5:405–410. doi: 10.1007/s11910-005-0065-5. [DOI] [PubMed] [Google Scholar]
- 31.Hagerman PJ, Hagerman RJ. Fragile X-associated tremor/ataxia syndrome-an older face of the fragile X gene. Nat Clin Pract Neurol. 2007;3:107–112. doi: 10.1038/ncpneuro0373. [DOI] [PubMed] [Google Scholar]
- 32.Jacquemont S, Leehey MA, Hagerman RJ, et al. Size bias of fragile X premutation alleles in late-onset movement disorders. J Med Genet. 2006;43:804–809. doi: 10.1136/jmg.2006.042374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacquemont S, Hagerman RJ, Leehey M, et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72:869–878. doi: 10.1086/374321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacquemont S, Farzin F, Hall D, et al. Aging in individuals with the FMR1 mutation. Am J Ment Retard. 2004;109:154–164. doi: 10.1352/0895-8017(2004)109<154:AIIWTF>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tassone F, Adams J, Berry-Kravis EM, et al. CGG repeat length correlates with age of onset of motor signs of the fragile X-associated tremor/ataxia syndrome (FXTAS) Am J Med Genet B Neuropsychiatr Genet. 2007;144:566–569. doi: 10.1002/ajmg.b.30482. [DOI] [PubMed] [Google Scholar]
- 36.Cohen S, Masyn K, Adams J, et al. Molecular and imaging correlates of the fragile X-associated tremor/ataxia syndrome. Neurology. 2006;67:1426–1431. doi: 10.1212/01.wnl.0000239837.57475.3a. [DOI] [PubMed] [Google Scholar]
- 37.Adams JS, Adams PE, Nguyen D, et al. Volumetric brain changes in females with fragile X-associated tremor/ataxia syndrome (FXTAS) Neurology. 2007;69:851–859. doi: 10.1212/01.wnl.0000269781.10417.7b. [DOI] [PubMed] [Google Scholar]
- 38.Pugliese P, Annesi G, Cutuli N, et al. The fragile X premutation presenting as postprandial hypotension. Neurology. 2004;63:2188–2189. doi: 10.1212/01.wnl.0000145709.61117.08. [DOI] [PubMed] [Google Scholar]
- 39.Greco CM, Soontarapornchai K, Wirojanan J, et al. Testicular and pituitary inclusion formation in fragile X associated tremor/ataxia syndrome. J Urol. 2007;177:1434–1437. doi: 10.1016/j.juro.2006.11.097. [DOI] [PubMed] [Google Scholar]
- 40.Hall DA, Berry-Kravis E, Jacquemont S, et al. Initial diagnoses given to persons with the fragile X associated tremor/ataxia Syndrome (FXTAS) Neurology. 2005;65:299–301. doi: 10.1212/01.wnl.0000168900.86323.9c. [DOI] [PubMed] [Google Scholar]
- 41.Brunberg J, Jacquemont S, Hagerman RJ, et al. Fragile X premutation carriers: characteristic MR imaging findings in adult male patients with progressive cerebellar and cognitive dysfunction. Am J Neuroradiol. 2002;23:1757–1766. [PMC free article] [PubMed] [Google Scholar]
- 42.Coffey SM, Cook K, Tartaglia N, et al. Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A. 2008;146:1009–1016. doi: 10.1002/ajmg.a.32060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goncalves MRR, Capelli LP, Nitrini R, et al. Atypical clinical course of FXTAS: rapidly progressive dementia as the major symtpom. Neurology. 2007;68:1864–1866. doi: 10.1212/01.wnl.0000262058.68100.ea. [DOI] [PubMed] [Google Scholar]
- 44.Tassone F, Hagerman RJ, Garcia-Arocena D, et al. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet. 2004;41:e43. doi: 10.1136/jmg.2003.012518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gokden M, Al-Hinti JT, Harik SI. Peripheral nervous system pathology in fragile X tremor/ataxia syndrome (FXTAS) Neuropathology. 2008 Jun 30; doi: 10.1111/j.1440-1789.2008.00948.x. ePub ahead of print. [DOI] [PubMed] [Google Scholar]
- 46.Louis E, Moskowitz C, Friez M, et al. Parkinsonism, dysautonomia, and intranuclear inclusions in a fragile X carrier: a clinical-pathological study. Mov Disord. 2006;21:420–425. doi: 10.1002/mds.20753. [DOI] [PubMed] [Google Scholar]
- 47.Bourgeois JA, Farzin F, Brunberg JA, et al. Dementia with mood symptoms in a fragile X premutation carrier with the fragile X-associated tremor/ataxia syndrome: clinical intervention with donepezil and venlafaxine. J Neuropsychiatry Clin Neurosci. 2006;18:171–177. doi: 10.1176/jnp.2006.18.2.171. [DOI] [PubMed] [Google Scholar]
- 48.Bacalman S, Farzin F, Bourgeois JA, et al. Psychiatric phenotype of the fragile X-associated tremor/ataxia syndrome (FXTAS) in males: newly described fronto-subcortical dementia. J Clin Psychiatry. 2006;67:87–94. doi: 10.4088/jcp.v67n0112. [DOI] [PubMed] [Google Scholar]
- 49.Hagerman RJ, Hagerman PJ. The fragile X premutation: into the phenotypic fold. Curr Opin Genet Dev. 2002;12:278–283. doi: 10.1016/s0959-437x(02)00299-x. [DOI] [PubMed] [Google Scholar]
- 50.Bourgeois JA, Cogswell JB, Hessl D, et al. Cognitive, anxiety and mood disorders in the fragile X-associated tremor/ataxia syndrome. Gen Hosp Psychiatry. 2007;29:349–356. doi: 10.1016/j.genhosppsych.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grigsby J, Brega AG, Jacquemont S, et al. Impairment in the cognitive functioning of men with fragile X-associated tremor/ataxia syndrome (FXTAS) J Neurol Sci. 2006;248:227–233. doi: 10.1016/j.jns.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 52.Grigsby J, Brega AG, Leehey MA, et al. Impairment of executive cognitive functioning in males with fragile X-associated tremor/ataxia syndrome. Mov Disord. 2007;22:645–650. doi: 10.1002/mds.21359. [DOI] [PubMed] [Google Scholar]
- 53.Bak T, Crawford L, Hearn V, et al. Subcortical dementia revisited: Similarities and differences in cognitive function between progressive supranuclear palsy (PSP), corticobasal degeneration (CBD) and multiple system atrophy (MSA) Neurocase. 2005;11:268–273. doi: 10.1080/13554790590962997. [DOI] [PubMed] [Google Scholar]
- 54.Burk K, Globas C, Bosch S. Cognitive deficits in spinocerebellar ataxia type 1, 2, and 3. J Neurol. 2003;250:207–211. doi: 10.1007/s00415-003-0976-5. [DOI] [PubMed] [Google Scholar]
- 55.Schelhaas HJ, van de Warrenburg BP. Clinical, psychological, and genetic characteristics of spinocerebellar ataxia type 19 (SCA19) Cerebellum. 2005;4:51–54. doi: 10.1080/14734220510007888. [DOI] [PubMed] [Google Scholar]
- 56.Kertesz A, McMonagle P, Blair M, et al. The evolution and pathology of frontotemporal dementia. Brain. 2005;128:1996–2005. doi: 10.1093/brain/awh598. [DOI] [PubMed] [Google Scholar]
- 57.Hagerman RJ, Leehey M, Heinrichs W, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- 58.Grigsby J, Kaye K, Shetterly SM, et al. Prevalence of disorders of executive cognitive functioning among the elderly: Findings from the San Luis Valley Health and Aging Study. Neuroepidemiology. 2002;21:213–220. doi: 10.1159/000065638. [DOI] [PubMed] [Google Scholar]
- 59.Peters N, Kamm C, Asmus F, et al. Intrafamilial variability in fragile X-associated tremor/ataxia syndrome. Mov Disord. 2006;21:98–102. doi: 10.1002/mds.20673. [DOI] [PubMed] [Google Scholar]
- 60.Grigsby J, Leehey MA, Jacquemont S, et al. Cognitive impairment in a 65-year-old male with the fragile X-associated tremor-ataxia syndrome (FXTAS) Cogn Behav Neurol. 2006;19:165–171. doi: 10.1097/01.wnn.0000213906.57148.01. [DOI] [PubMed] [Google Scholar]
- 61.Rosen HJ, Hartikainen KM, Jagust W, et al. Utility of clinical criteria in differentiating frontotemporal lobar degeneration (FTLD) from AD. Neurology. 2002;58:1608–1615. doi: 10.1212/wnl.58.11.1608. [DOI] [PubMed] [Google Scholar]
- 62.Folstein MF, Folstein SW, McHugh PR. “Mini Mental State”: A practical method of grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 63.Kaye K, Grigsby J, Robbins LJ, Korzun B. Prediction of independent functioning and behavior problems in geriatric patients. J Am Geriatr Soc. 1990;38:1304–1310. doi: 10.1111/j.1532-5415.1990.tb03452.x. [DOI] [PubMed] [Google Scholar]
- 64.Cornish KM, Li L, Kogan CS, et al. Age-dependent cognitive changes in carriers of the fragile X syndrome. Cortex. 2008;44:628–636. doi: 10.1016/j.cortex.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reiss AL, Freund L, Abrams MT, et al. Neurobehavioral effects of the fragile X premutation in adult women: a controlled study. Am J Hum Genet. 1993;52:884–894. [PMC free article] [PubMed] [Google Scholar]
- 66.Brouwer JR, Severijnen E, de Jong FH, et al. Altered hypothalamus-pituitary-adrenal gland axis regulation in the expanded CGG-repeat mouse model for fragile X-associated tremor/ataxia syndrome. Psychoneuroendocrinology. 2008;33:863–873. doi: 10.1016/j.psyneuen.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thompson NM, Rogeness GA, McClure E, et al. Psychiatry Res. 1996;64:97–104. doi: 10.1016/0165-1781(96)02785-0. [DOI] [PubMed] [Google Scholar]
- 68.Johnston C, Eliez S, Dyer-Friedman J, et al. Neurobehavioral phenotype in carriers of the fragile X premutation. Am J Med Genet. 2001;103:314–319. [PubMed] [Google Scholar]
- 69.Roberts JE, Bailey DB, Jr., Mankowski J, et al. Mood and anxiety disorders in females with the FMR1 premutation. Am J Med Genet B Neuropsychiatr Genet. 2008 Jun 13; doi: 10.1002/ajmg.b.30786. ePub ahead of print. [DOI] [PubMed] [Google Scholar]
- 70.Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective. Am J Hum Genet. 2004;74:805–816. doi: 10.1086/386296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Al-Semaan Y, Malla AK, Lazosky A. Schizoaffective disorder in a fragile-X carrier. Aust N Z J Psychiatry. 1999;33:436–440. doi: 10.1046/j.1440-1614.1999.00564.x. [DOI] [PubMed] [Google Scholar]
- 72.Khin NA, Tarleton J, Raghu B, Park SK. Clinical description of an adult male with psychosis who showed FMR1 gene methylation mosaicism. Am J Med Genet. 1998;81:222–224. doi: 10.1002/(sici)1096-8628(19980508)81:3<222::aid-ajmg3>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 73.Goodlin-Jones B, Tassone F, Gane LW, Hagerman RJ. Autistic spectrum disorder and the fragile X premutation. J Dev Behav Pediatr. 2004;25:392–398. doi: 10.1097/00004703-200412000-00002. [DOI] [PubMed] [Google Scholar]
- 74.Hagerman RJ, Hagerman PJ. Fragile X Syndrome: Diagnosis, Treatment, and Research. 3rd Edition The Johns Hopkins University Press; Baltimore: 2002. [Google Scholar]
- 75.Dorn MB, Mazzocco MM, Hagerman RJ. Behavioral and psychiatric disorders in adult male carriers of fragile X. J Am Acad Child Adolesc Psychiatry. 1994;33:256–264. doi: 10.1097/00004583-199402000-00015. [DOI] [PubMed] [Google Scholar]
- 76.Clifford S, Dissanayake C, Bui QM, et al. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. 2007;37:738–747. doi: 10.1007/s10803-006-0205-z. [DOI] [PubMed] [Google Scholar]
- 77.Cornish KM, Kogan C, Turk J, et al. The emerging fragile X premutation phenotype: Evidence from the domain of social cognition. Brain Cogn. 2005;57:53–60. doi: 10.1016/j.bandc.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 78.McConkie-Rosell A, Abrams L, Finucane B, et al. Recommendations from multi-disciplinary focus groups on cascade testing and genetic counseling for fragile X-associated disorders. J Genet Couns. 2007;16:593–606. doi: 10.1007/s10897-007-9099-y. [DOI] [PubMed] [Google Scholar]
- 79.Hagerman PJ. The fragile X prevalence paradox. J Med Genet. 2008;45:498–499. doi: 10.1136/jmg.2008.059055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jagust WJ, Convit A, Frisoni GB, et al. Structural and functional brain imaging and dementia diagnosis. NeuroScience News. 2000;3:54–61. [Google Scholar]
- 81.Sierra C, Coca A. White matter lesions and cognitive impairment as silent cerebral disease in hypertension. TheScientificWorldJournal. 2006;6:494–501. doi: 10.1100/tsw.2006.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Franke P, Leboyer M, Hardt J, et al. Neuropsychological profiles of FMR-1 premutation and full-mutation carrier females. Psychiatry Res. 1999;87:223–231. doi: 10.1016/s0165-1781(99)00067-0. [DOI] [PubMed] [Google Scholar]