Embryonic motor axon development in the severe SMA mouse (original) (raw)
Abstract
Spinal muscular atrophy (SMA) is caused by reduced levels of survival motor neuron (SMN) protein. Previously, cultured SMA motor neurons showed reduced growth cone size and axonal length. Furthermore, reduction of SMN in zebrafish resulted in truncation followed by branching of motor neuron axons. In this study, motor neurons labeled with green fluorescent protein (GFP) were examined in SMA mice from embryonic day 10.5 to postnatal day 2. SMA motor axons showed no defect in axonal formation or outgrowth at any stage of development. However, a significant increase in synapses lacking motor axon input was detected in embryonic SMA mice. Therefore, one of the earliest detectable morphological defects in the SMA mice is the loss of synapse occupation by motor axons. This indicates that in severe SMA mice there are no defects in motor axon formation however, we find evidence of denervation in embryogenesis.
INTRODUCTION
Proximal spinal muscular atrophy (SMA) is a motor neuron disease that is the leading genetic cause of infant mortality (1). SMA has a frequency of 1/10 000 live births and a carrier frequency of 1/50 (2–4). SMA is caused by loss or mutation of the SMN1 gene and retention of the SMN2 gene, which results in reduced levels of survival motor neuron (SMN) protein (5–8). The ubiquitously expressed SMN protein performs an essential function in small nuclear ribonucleoproteins (snRNP) assembly (9–11) thus complete elimination of SMN protein results in embryonic lethality (6,12). Indeed, loss of functional Smn in the mouse appears incompatible with survival of any tissue (12–15). Mouse models of SMA have been created by introducing human SMN2 into mice lacking functional mouse Smn (16–19). The most severe of these mice models lacks mouse Smn, has two copies of human SMN2 and survives for 5 days (16). Currently, it is unclear why low levels of SMN protein result specifically in a motor neuron disease.
The 38 kDa SMN protein is ubiquitously expressed and localizes to both the cytoplasm and nucleus of cells in culture and tissue. In the nucleus SMN localizes in dot like structures termed gems, which overlap, or are in close proximity to, coiled bodies (20–22). In cultured neurons, including motor neurons, SMN is found in granules in axons and growth cones (23–26). There have been reports of SMN identified in vivo in neuronal axons (27,28) however, recent reports indicate the difficulties in observing the axonal SMN complexes in vivo (29). Additionally, a truncated axonal form of SMN has been reported and is suggested to play a role in SMA (30). However, it is difficult to reconcile this finding with missense mutations that occur in the C-terminus of SMN in SMA patients that would not disrupt aSMN (31–35). Furthermore, correction of SMA in mice does not require aSMN (18,36). Thus although SMN protein is localized to axons, its activity in the axon is unknown.
Motor neurons cultured from severe SMA mice (SMN2 +/+ ; Smn −/−) show decreased axonal length and reduced growth cones size, as well as reduced levels of beta-actin message and protein at the growth cone (37,38). More recently, cultured sensory neurons have also been reported to have reduced axonal length and growth cone size (39). Knockdown of Smn in zebrafish to levels equivalent to that which occurs in SMA patients results in truncated motor neurons at early time points (40). These axons then branch excessively and reach the target muscle albeit with inappropriate trajectories. These zebrafish show no obvious defects in any other neurons including sensory neurons (40). The zebrafish phenotype can be rescued by full length SMN as well as certain mutant SMN forms predicted to be incapable of snRNP assembly (41). In addition, knockdown of gemin2 does not lead to a cell autonomous disruption of motor neuron axons (42). The SMN knockdown phenotype at this level of SMN reduction is clearly cell autonomous (40). However, in zebrafish and Xenopus strong knockdown of SMN causes morphological defects in body formation (43). snRNPs can rescue these defects suggesting that they are caused by loss of snRNPs. It is not clear, however, whether snRNPs can rescue the cell autonomous axonal phenotype resulting from SMN knockdown. The data from both zebrafish and cultured mouse SMA motor neurons clearly imply a developmental defect in motor neurons in SMA.
To investigate motor axon development and innervation in SMA mice we obtained SMA mice with GFP labeled motor neurons. The motor neurons innervating the intercostal muscles, brachial plexus, lumbosacral plexus and the diaphragm were examined in the SMA embryos and in neonates. Motor axons were examined for any abnormalities in outgrowth, branching, truncations or inappropriate connections. No defects in the development of motor axons were detected at any stage of development in the SMA animals. We conclude that SMA in the mouse does not result from incorrect development or outgrowth of motor axons. Furthermore, in embryonic SMA mice we observed a significant increase in unoccupied synapses in intercostal muscles and the presence of axonal varicosities in motor axons.
RESULTS
Motor axon development was analyzed in SMA, carrier and normal mice in the embryo at embryonic days e10.5–e18.5 of development as well as postnatal day 2 (PND02) mice. All animals described here have two copies of SMN2. In our studies of 32 SMA embryos examined (e10.5–e18.5) that could contain one or two copies of SMN2, only one embryo was identified with one copy of SMN2. That embryo was isolated at e10.5 and was severely dysmorphic. This indicates that one copy of SMN2 is seldom sufficient for the development of an SMA embryo and often results in early embryonic lethality in the severe mouse model of SMA. In this same colony, SMA animals containing two copies of SMN2 (SMN2 +/+ ; Smn −/−) were obtained from interbreeding SMA carrier mice (SMN2 +/+ ; Smn +/−). In this colony SMA animals comprised 17% of the total progeny obtained from these crosses and 73% of the SMA animals lived to 5 days (measured from birth at PND01 to PND05).
To investigate motor axon development and innervation in SMA mice, the HB9:GFP transgene, which is expressed at all stages of embryonic motor neuron development as well as postnatally (44), was crossed to severe SMA mice. HB9 is a transcription factor required for proper development of motor neurons (45). Thus we have obtained severe SMA mice with GFP labeled motor neurons throughout development. The motor neurons innervating the intercostal muscles, brachial plexus, lumbosacral plexus and the diaphragm were examined in the embryo from e10.5 to e18.5 days of development and in neonates at 2 days of development (PND02) in SMA, carrier, and normal littermates. Motor axons were examined for any defects in outgrowth, branching, truncations or inappropriate connections.
Overall axonal morphology in the embryonic SMA mouse
Motor axon development was analyzed in SMA (SMN2 +/+ , mSmn −/− , HB9:GFP +/−), carrier (SMN2 +/+ , mSmn +/− , HB9:GFP +/−) and normal mice (SMN2 +/+ , mSmn +/+ , HB9:GFP +/−) in the embryo at e10.5, e12.5, e13.5, e14.5, e15.5, and e18.5 days of development as well as PND02 and PND03. Motor axons were visualized by GFP labeling due to the expression of the HB9:GFP transgene. The HB9:GFP transgene expresses high levels of GFP in the cell bodies, axons and dendrites of spinal motor neurons from e9.5 to PND10 (44). Motor neuron development begins at e9.5 and is complete by e11.0 (46). Motor axon outgrowth also begins at e9.5 and by e12.5 motor axons have crossed the limb plexus region and passed the dorsal–ventral choice point (47). Motor axons were examined for any abnormalities in outgrowth, branching, truncations or inappropriate connections in the brachial plexus, lumbosacral plexus, intercostal muscles and the diaphragm. The overall morphology of motor axons in the brachial plexus, lumbosacral plexus and intercostal muscles at e12.5 days of development in normal, carrier and SMA embryos are shown in Figure 1A–P. The phrenic nerve, which innervates the diaphragm muscle, at e15.5 days of development is shown in Figure 1M–O. No defects in axonal formation in SMA embryos were observed in any tissue at any time in development. Although there is variation among the projections of individual motor axons the overall neuronal pattern of the plexuses, the phrenic nerve and the neurons innervating the intercostal muscles are highly stereotyped. No misrouted bundles of axons or aberrantly projecting motor axons were identified. All bundles of axons were tightly fasciculated in the intercostal muscles with no spreading of axonal bundles or crossing between main nerve branches as has been observed in mice with defects in motor axon development (45,47). Furthermore, staining with neurofilament heavy chain antibody at e12.5, e13.5, e14.4 and e15.5 revealed normal motor axon and sensory axon formation in the phrenic nerve of the diaphragm, the intercostal muscles and the brachial and lumbosacral plexus (unpublished data).
Figure 1.

Motor axon development in the severe SMA embryo. Overall morphology of motor axons at the brachial plexus of the forelimb (A–D), lumbosacral plexus of the hindlimb (E–H), and intercostal muscles in the thoracic region (I–L) of e12.5 embryos is normal in SMA embryos (C, G, K) as compared with normal (A, E, I) and carrier (B, F, J) littermate controls. The phrenic nerve of the diaphragm is also patterned correctly in e15.5 SMA embryos (O) when compared with normal (M) and carrier (N) littermate controls. In (A)–(L) anterior is to the top and proximal is to the left. The blue square in each of schematic drawing (B, H, L, P) indicates the location imaged in each tissue. Scale bar represents 300 µm for each micrograph.
To further determine if the diaphragm muscle of SMA mice was properly innervated, we labeled acetylcholine receptor (AChR) clusters in PND03 SMA and carrier diaphragm muscle with alpha-bungarotoxin. Excessive branching of motor axons as well as the absence of motor axon innervation can result in poorly formed AChR clusters and an increase in end plate bandwidth (45,48). As shown in Figure 2A and B, AChR clusters developed appropriately in SMA and carrier diaphragms with no increase in the width of the end plate band detected. Therefore, the alpha-bungarotoxin labeling of neuromuscular junctions (NMJ) in the SMA diaphragm confirms that the axonal development of the phrenic nerve is not affected in SMA mice. Furthermore, we found no increase in the end plate bandwidth of AChR clusters in the intercostal muscles of SMA animals at e18.5 (Fig. 2C and D) as well as at PND02 (Fig. 2E and F). This suggests the axonal development of the nerves innervating the intercostal muscles is not affected in embryonic or neonatal SMA mice.
Figure 2.
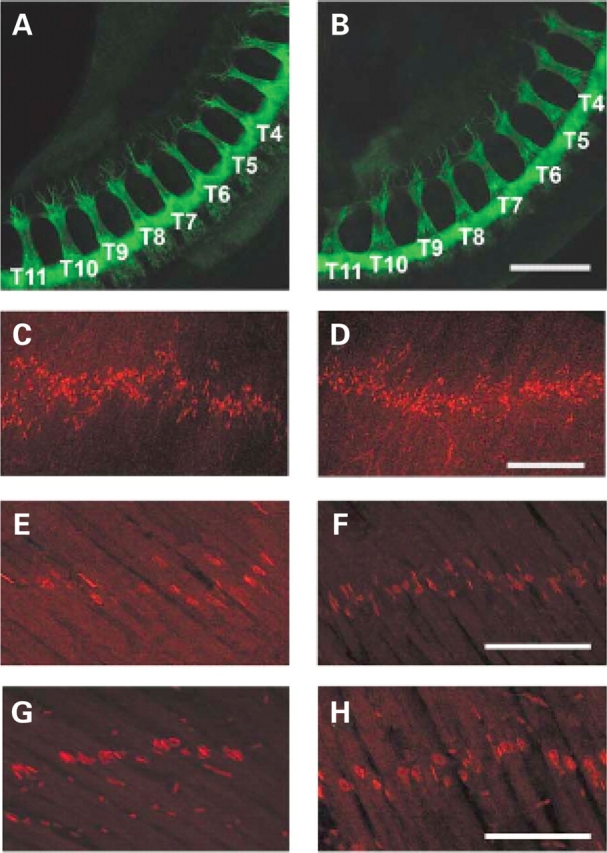
Axonal outgrowth and NMJ bandwidth in SMA mice. (A, B) No difference in the length of motor axon outgrowth was identified in the intercostal region (T4–T11) of e10.5 carrier (A) and SMA (B) littermates. No increase in the width of the end plate band was observed in PND03 diaphragm muscle between SMA (C) and normal (D) mice as assayed by alpha-bungarotoxin staining. Furthermore, the width of the end plate band was similar in carrier and SMA animals in the intercostal muscles at e18.5 (E, F respectively) and at PND02 (G, H respectively). Scale bar: (A–D) 300 µm; (E–H) 100 µm.
Normal axonal outgrowth at e10.5 in SMA mice
Primary motor neuron cultures from severe SMA mice (SMN2, Smn −/−) have been reported to have truncated axonal processes (37,38). Additionally, defects in motor axon outgrowth have been shown upon knockdown of Smn protein in a zebrafish model of SMA (40,41,43). To determine if motor axons are truncated in SMA mice, we measured the length of axonal bundles in both the intercostal muscles and the lumbosacral plexus of e10.5 SMA and carrier littermates. As shown in Figure 2G and H, the average length of axonal bundles in the intercostal region (T4–T11) was not different in SMA embryos from that of control littermates (SMA 378.5 ± 54.3 versus carrier 354.8 ± 37.9 µm). Additionally, there was no difference in the average length of axonal bundles in the lumbosacral region (L2–S2) in SMA embryos from that of control littermates (SMA 350.6 ± 22.4 versus 281.3 ± 45.2 µm).
Presence of swellings in SMA motor axons
Interestingly, while motor axon formation is normal in severe SMA mice, the axons of these mice did reveal swellings that are rarely observed in normal littermate controls (Fig. 3). These swellings are present in the motor neurons of all tissues examined in the SMA mice including the nerves of the intercostal, diaphragm, gastrocnemius and quadriceps muscles. The beads were also observed at all stages of development in the nerves of embryonic (e10.5–e18.5) and neonatal (PND02–PND03) SMA mice. Occasionally these axonal varicosities are found in the nerves of normal and carrier mice however they are much smaller in size and less numerous than in SMA mice. Previously, swellings have been reported in severe SMA patient muscle biopsies (49) and in other peripheral neuropathies (50,51). Ultrastructural analysis of axonal swellings revealed the presence of neurofilaments and organelles (50,51). Coërs and Woolf described the appearance of nerve fibers in SMA patient muscle biopsies that were ‘irregularly swollen’ and distal nerve fibers that contained ‘prominent lozenge-shaped swellings’ or ‘curious lozenge-shaped beads’ while ‘more proximal nerve bundles contain normal looking nerve fibers’. In our studies there was no difference observed in the proximal to distal distribution of beads in the motor axons. Although this work was under review others have reported the presence of neurofilament aggregates at the NMJ (52,53). The significance of these axonal varicosities is unknown however their presence in the motor axons of SMA mice illustrates how this mouse model recapitulates what is observed in type I SMA patients.
Figure 3.

Axonal swellings present in severe SMA mice. Motor axons of severe SMA mice (B, D, F) reveal swellings rarely seen in normal littermate controls (A, C, E). The phrenic nerve of PND03 diaphragm muscles (B, F) as well as the nerves innervating the intercostal muscles of e18.5 embryos (D) display several beads in the SMA tissue (arrowheads) that are seldom seen in normal controls (A, C, E). (E, F) Neurofilament antibody staining of PND02 diaphragm muscle also reveals the axonal swellings in SMA neurons (F) that are not often found in normal animals (E). Scale bar represents 50 µm for each micrograph.
Presence of aneural AChR clusters in SMA intercostal muscle at e18.5 and PND02
The observation that motor axons project to the muscle normally in the intercostal muscles of SMA mice yet the intercostal muscles of type I SMA patients are severely affected led us to examine NMJ more closely in this tissue (54). NMJs were visualized with alpha-bungarotoxin in SMA, carrier and normal embryos containing GFP labeled motor axons. At e18.5 and PND02 the number of occupied AChR clusters was determined by blinded evaluation of alpha-bungarotoxin positive AChR clusters juxtaposed to a nerve as visualized with GFP. At e18.5 51% of AChR clusters (n = 529) were occupied in the intercostal muscles of SMA mice as opposed to 92% occupied in normal littermates and 89% occupied in carrier littermate controls (P < 0.0001, n = 621 normal, n = 564 carrier) (Fig. 4, Table 1). At PND02 a similar number of AChR clusters (49%, n = 486) were occupied in the intercostal muscles of SMA mice as opposed to 92% occupied in normal littermates and 95% occupied in carrier littermate controls (P < 0.0001, n = 172 normal, n = 160 carrier) (Fig. 5, Table 1). Unoccupied synapses were also observed at PND02 using an additional presynaptic marker, synaptophysin along with GFP to label the axon (Fig. 6). This indicates that the unoccupied clusters observed in the SMA animals are not simply due to defective transport of GFP protein to the NMJ.
Figure 4.

Unoccupied AChR clusters are found in the SMA animal at e18.5. AChR clusters in the intercostal muscles of e18.5 normal (A–C) carrier (D–F) and SMA (G–I) littermates. GFP staining reveals the motor neuron axons innervating the intercostal muscles in (A), (D), (G), alpha-bungarotoxin labeling of AChR clusters in (B), (E), (H), and merged confocal images (C, F, I). All AChR clusters are fully innervated in normal and carrier animals while most AChR clusters are unoccupied in the SMA animal (arrowheads). One cluster is partially innervated as indicated by the arrow. Scale bar represents 50 µm in each micrograph.
Table 1.
Number of occupied synapses in embryonic and neonatal SMA mice
| Intercostal muscles | Diaphragm | ||
|---|---|---|---|
| e18.5 | PND02 | PND03 | |
| Normal | 571a, 92%b (n = 621c) | 158, 92% (n = 172) | 44, 100% (n = 44) |
| Carrier | 503, 89% (n = 564) | 152, 95% (n = 160) | 259, 98% (n = 264) |
| SMA | 271, 51%d (n = 529) | 240, 49%d (n = 486) | 322, 100% (n = 323) |
Figure 5.

Unoccupied AChR clusters are found in the SMA animal at PND02. AChR clusters in the intercostal muscles of PND02 normal (A–C) carrier (D–F) and SMA (G–I) littermates. GFP staining reveals the motor neuron axons innervating the intercostal muscles in (A), (D), (G), alpha-bungarotoxin labeling of AChR clusters in (B), (E), (H), and merged confocal images (C, F, I). All AChR clusters are fully innervated in normal and carrier animals while most AChR clusters are unoccupied in the SMA animal (arrowheads). Scale bar represents 50 µm in each micrograph.
Figure 6.

Unoccupied AChR clusters are also found in the SMA animal at PND02 with snyaptophysin presynaptic staining. AChR clusters in the intercostal muscles of PND02 normal (A–C) carrier (D–F) and SMA (G–I) littermates. GFP and synaptophysin double staining reveals the motor neuron axons innervating the intercostal muscles in (A), (D), (G). Alpha-bungarotoxin labels the AChR clusters in (B), (E), (H), and merged confocal images are shown in (C), (F), (I). AChR clusters are fully innervated in normal and carrier animals while several AChR clusters are unoccupied in the SMA animal (arrowheads). In this micrograph a fully occupied AchR cluster is also shown (arrow). Scale bar represents 50 µm in each micrograph.
At younger ages the AChR clusters were not well organized in the intercostal muscles (e15.5). At e16.5 and e17.5 we observed considerable numbers of unoccupied clusters in both normal and SMA animals. Therefore, the extent of innervation of AChR clusters in normal and SMA mice was similar during this period of synapse formation. There was no evidence of collateral sprouting at any time point examined in the SMA mice.
The denervation phenotype observed in e18.5 and PND02 in the intercostal muscles of SMA animals was not uniform. Although ∼50% of AChR clusters were unoccupied on an average in the SMA animals, the distribution of denervation was not equal throughout each section of tissue (Fig. 7). Although some AChR clusters were fully innervated, other areas of the same tissue section revealed several unoccupied clusters with little nerve present. This phenotype is never observed in carrier or normal littermates. There was no correlation between the levels of the spinal cord examined (T1–T12), or the proximal to distal location of the synapses in the intercostal muscles, to the degree of unoccupied AChR clusters. The lack of uniform denervation in SMA mice is consistent with results from Type I SMA patient muscle biopsies where extensive variation in the degree of muscle atrophy, thought to be caused by denervation, has been observed within the same muscle (54).
Figure 7.

AChR cluster occupation in the SMA animal. GFP staining reveals the motor neuron axons innervating the intercostal muscles of e18.5 embryos (A, C, E) while alpha-bungarotoxin identifies the AChR clusters in the merged image (B, D, F). In some areas of the intercostal muscles at e18.5, the AChR clusters are fully occupied in both the carrier (A, B) and SMA animals (C, D). Additionally, often all AChR clusters in a particular area of the intercostal muscles are unoccupied in the SMA animal and little of the motor neuron axon remains (E, F). Scale bar represents 50 µm in each micrograph.
Presence of fully occupied AChR clusters in SMA diaphragm muscle at PND03
The number of occupied AChR clusters was also measured in the diaphragm muscle of SMA, normal and carrier mice at PND03. As further evidence that the diaphragm is not affected in SMA mice, AChR clusters were fully occupied in the diaphragm of SMA animals (Fig. 8). On an average, 100% of AChR were occupied in the diaphragms of SMA mice (n = 323) as compared with 100% occupied in normal and 98% occupied in carrier diaphragms (normal n = 44, carrier n = 264). This result is consistent with what is seen in severe SMA patients where breathing is almost entirely diaphragmatic (55).
Figure 8.

All AChR clusters are occupied in the diaphragm of SMA animals at PND03. AChR clusters in the diaphragm of PND03 normal (A–C) carrier (D–F) and SMA (G–I) littermates. GFP staining of motor axons (A, D, G), alpha-bungarotoxin staining of AChR clusters (B, E, H), Merged images (C, F, I). Scale bar: (A–I) 100 µm. Insert in images (C), (F), (I) represents 50 µm in width.
DISCUSSION
In this study we have observed the motor axon development in severe SMA mice from axonal outgrowth (e10.5) until late embryogenesis (e18.5) as well as in neonates. The HB9:GFP transgene used in this study allows for the visualization of just the motor axons in the SMA animal throughout development. Additionally, the SMN protein was not overexpressed or modified with a tag, which may interfere with its normal function. We have shown that motor axon formation in the severe mouse model of SMA is normal. Motor axons develop and extend to reach the appropriate muscles in SMA animals at the same time as in carrier and normal littermates. No developmental delay, as has been previously suggested, was detected (56). Although SMN is essential for development of the organism, low levels of SMN present in SMA mice do not result in altered motor axon formation in the developing embryo.
The phenotype we see in the severe SMA mouse embryo is unlike the defects observed in other mouse models pertaining to motor axon and NMJ development. For example, in the HB9 knockout mouse bridging of the large nerve bundles in the intercostal muscles of e12.5 embryos has been reported (45). The HB9 transcription factor is required for proper development of motor neurons (45). In the SMA mouse no misrouted bundles of axons or aberrantly fasciculated motor axons were identified. All bundles of axons were tightly fasciculated in the intercostal muscles with no spreading of axonal bundles or crossing between main nerve branches. A second example examines the role of neuropilins and semaphorins, which have been implicated in the control of axon projections (47). In the neuropilin-1 Sema- knockin and neuropilin-2 knockout mice defasciculation and bridging of the intercostal nerves and ectopic nerve bundles were found (47). Additionally, neuropilin-1 Sema- knockin as well as Sema3A knockout mice revealed premature entry of motor axons into the plexus region at e10.5 and later defasciculation at e12.5 (47). Conversely, in our SMA mouse embryos no aberrant axonal outgrowth in the thoracic region or the lumbosacral plexus was detected with no difference in the overall average length of axons between carrier and SMA embryonic mice. Therefore, elimination of genes required for motor neuron development or axon guidance in the mouse can result in aberrant motor axon formation however this is not the case in the severe SMA mouse model.
In addition to the intercostal muscles and plexus regions we examined the diaphragm muscle in SMA embryos and showed no increase in branching of the phrenic nerve. Others have shown that mouse embryos lacking choline acetyltransferase (ChAT), the biosynthetic enzyme for acetylcholine, revealed an increase in branching and defasciculation of the intercostal nerves and the phrenic nerve in embryonic mice (48). ChAT is essential for proper formation of NMJs (48). In this case, increased branching of motor axons in the diaphragm results in ectopic AChR clusters and an increase in the width of the motor end plate band (48,57). However, in our SMA mice, all AChR clusters of the diaphragm were fully innervated and normal in size. The width of the motor end plate band was no different in SMA embryos as compared with normal and carrier littermates. Furthermore, when a motor axon fails to innervate muscle, as in the embryonic diaphragm of HB9 knockout mice, the AChR clusters are smaller in size and diffuse creating a wider motor end plate band (45). Yet, no change in the size of AChR clusters or increase in the motor end plate band was observed in the SMA mice in this study. Thus, these findings in the SMA mouse are consistent with observations of type I SMA patients where breathing is almost entirely diaphragmatic suggesting that the diaphragm is spared in SMA (55).
In our study we find that while the overall motor axon morphology and pattern of innervation is normal, the presence of nerve occupied AChR receptors is decreased by 50% in the intercostal muscles of SMA animals at e18.5 as compared with littermate controls. A similar number of unoccupied AChR clusters are also observed at in SMA animals at PND02. At earlier time points (e16 and e17) there is an equal distribution of occupied and unoccupied AChR clusters in normal verses SMA animals. Thus we are not able to fully determine if motor neuron phenotype we observe in the severe SMA mouse is denervation or simply failure of the motor neuron to innervate the muscle in the first place. This question can be better addressed using a mild SMA mouse model with a later phenotypic onset. However, since there is no increase in end plate bandwidth or diffuse alpha-bungarotoxin staining, as has been observed in other mice when a nerve fails to innervate, we suggest that denervation is occurring in the severe SMA mouse embryo. Consistent with this, a recent study has shown denervation of the NMJ in the transversus abdominis and levator auris longus muscles of the postnatal severe SMA mouse has been reported (53).
During embryonic and neonatal synapse development AChR clusters are maintained for less than a day and plaques rapidly disassemble (58). Thus the similar number of unoccupied clusters at both e18.5 and PND02 likely indicates that denervation and reinnervation of the muscle are ongoing in the SMA animals during development. Collateral sprouting of neurons, which is often suggestive of renervation and has been observed in the mild SMA mouse model, was not observed here in the severe SMA mice (18). Although notable this finding is not unexpected for three reasons. First, the muscle is multiply innervated in embryonic and neonatal mice. At this early stage of development, even though denervation is occurring the muscle may still be innervated as polyneuronal innervation of muscle is still present. Thus the signal for renervation may not be sent from the muscle. Collateral sprouting seems to occur when the muscle has no nerve input. Secondly, fiber type grouping, as has been reported in the nmd mouse, is not observed before 3 weeks of age (59). Thirdly, these finding in the SMA mice are consistent with observed muscle pathology in SMA patients. In the most severe Type 0 Electromyogram patients muscle fibers are of uniform small size with no fiber type grouping or indication of reinnervation (54). Electromyogram studies reveal clear evidence of collateral sprouting in Type III SMA but not in Type I SMA patients (60).
Interestingly, in this study the motor axons of the severe SMA mice reveal axonal swellings present in the neurons of all tissues examined at all stages of development. Occasionally the beads are found in the nerves of normal and carrier mice however they are smaller and less abundant than in SMA mice. These axonal varicosities were found to be co-localized with neurofilament in the motor nerves of SMA embryos. It is important to note that these beads were identified in all motor nerves of the SMA mouse including the phrenic nerve of the diaphragm muscle that does not display unoccupied AChRs. Previously, ‘irregularly swollen’ nerve fibers have been reported in severe SMA patient muscle biopsies (49). Although the significance of these swellings to SMA pathology is unknown, their presence in the motor axons of SMA mice correlates with observations made in human SMA patients.
Contrary to previous studies in cell culture and in zebrafish, we find no defects in motor axon outgrowth. How does one reconcile these findings? In vitro assays in cell culture allow for examination of individual axons with tightly controlled environmental conditions. However, even primary motor neuron cultures from SMA mice, which most closely represent the in vivo situation, are still grown outside of the context of the muscle environment the motor axon normally innervates (38). Therefore, it is likely that cell culture cannot fully recapitulate the complex environment motor axons normally develop in. Axonal defects found in cultured motor axons may not be identified in the whole embryo due to extrinsic cues in the nerve environment that compensate for low levels of SMN in vivo. Studies in zebrafish have shown aberrant motor axon truncation and branching when levels of SMN are reduced in vivo (40). It is possible that the more complex pattern of motor axon development in a mammal, such as the mouse, includes redundancy of developmental cues that allows the motor axon to reach the correct muscle. Therefore, while in the zebrafish defects in axonal outgrowth and branching are observed, the first defect observed in the mice is at the NMJ.
It is interesting to note that in these severe SMA mice denervation of the NMJ is detected at e18.5 before weakness and motor neuron loss is detected (PND03) in the mouse (16). Indeed, at birth severe SMA mice are phenotypically indistinguishable from carrier mice. In SMA patients the period of time before the loss of motor neuron function is observed is known as the ‘pre-pathological’ state. Studying the pre-pathological stage of SMA in mouse models might allow us to determine when the administration of therapeutics will be most effective in treating SMA in humans. Electrophysiological studies have shown in prenatally identified severe SMA infants a significant decrease in function shortly after birth (61). If the disease is progressing before clinical symptoms are observed then early detection through newborn screening and administration of therapeutics in the pre-pathological state may yield the greatest benefit in preserving motor neuron function.
MATERIALS AND METHODS
Mouse strains and breeding
HB9:GFP transgenic mice were obtained from T. Jessell (44). Transgenic mice containing human SMN and the mouse Smn knockout are as previously described (12,16). Genotyping and SMN2 copy number determination was performed as previously described (19). Mouse genotypes are as follows: SMA (SMN2 +/+ , mSmn −/− , HB9:GFP +/−), carrier (SMN2 +/+ , mSmn +/− , HB9:GFP +/−) and normal mice (SMN2 +/+ , mSmn +/+ , HB9:GFP +/−). For normal controls SMN2 +/+ , mSmn +/+ , HB9:GFP +/− mice are phenotypically no different from mSmn +/+ , HB9:GFP +/− mice that do not carry the human SMN2 transgene. Mice were maintained in accordance with The Ohio State University Institutional Laboratory Animal Care and Use Committee regulations.
Mouse embryo preparation
Timed matings were used to obtain GFP SMA embryos. The presence of a coital plug after mating as well as the morphology of the dissected embryo were used to determine embryonic stages (62). Pregnant females were sacrificed, the embryos removed and fixed in 4% paraformaldehyde in PBS overnight. DNA was isolated from either the embryo sac or the tail for genotyping (Puregene). Genotyping of embryos was performed as previously described (16). A total of 169 embryos and neonates were examined in this study. The intercostal muscles, brachial plexus and lumbosacral plexus on both the right and left sides of the animals were examined.
Immunofluorescence
Whole mount tissue
Whole mount embryo tissues were blocked in 10% Triton X-100 (Sigma), 4% goat serum (Sigma), PBS overnight. Whole mount embryo tissue was incubated with rabbit anti-GFP antibody (1:1000, Molecular Probes, Invitrogen, Carlsbad, CA, USA) or chicken anti-mouse neurofilament heavy chain, (1:1000, EnCor Biotechnology Inc., Gainesville, FL, USA) in 10% Triton X-100, 0.4% goat serum, PBS overnight and incubated with Alexa Fluor-488 or Alexa Fluor-594 secondary antibody (1:1000, Molecular Probes) for 2 h. Tissues were mounted in Vectashield (Vector Labs, Burlingame, CA, USA).
Tissue sections
Thick cryostat sections (34 µm) were obtained from 4% paraformaldehyde fixed tissue frozen in liquid nitrogen cooled isopentane. Muscle sections were stained with rabbit anti-GFP antibody (1:1000, Molecular Probes) and/or rabbit anti-synaptophysin antibody (1:100, Zymed, Invitrogen, Carlsbad, CA, USA) in 10% Triton X-100, 0.4% goat serum, PBS for 2 h and incubated with Alexa Fluor 488 secondary antibody (1:100, Molecular Probes) and alpha-bungarotoxin Alexa Fluor 594 (1:400, Molecular probes) for 30 min. Tissue sections were mounted in Vectashield (Vector Labs).
Confocal microscopy
All images were captured with the Leica TCS_SL scanning confocal microscope system using an inverted Leica DMIRE2 microscope and photomultiplier tube detectors. Images were captured at room temperature with the following objectives: 63× HCX Plan Apo CS oil, NA = 1.4; 40× HCX Plan Apo CS oil, NA = 1.25; and 10× HC Plan Fluotar Phl, NA = 0.30. A Z-Galvo stage was used to obtain Z-series stacks of ∼30 images each. Image acquisition, overlays, scale bars, and measurements were produced with the Leica Confocal Software v2.61 and subsequential image processing was performed with Adobe Photoshop CS2. Confocal images of axonal morphology in whole mount tissue were composed of 30 individual images taken at 10× magnification spanning on an average to 100 µm. Confocal images of AChR clusters in 30 µm sections of intercostal muscles were 30 individual images taken at 40× magnification spanning on an average to 19 µm.
Measurement of axonal outgrowth
Axonal outgrowth was measured in e10.5 SMA and carrier embryos at the thoracic (T2–T10) and lumbosacral region (L2–S2) levels of the spinal cord. Measurements of the extension of each axonal bundle were made with the Leica Confocal Software v2.61 from confocal stacks of 30 images totaling ∼70 µm. One single linear measurement from the point at which the axonal bundle exited the spinal cord to the end of the axon of greatest extension from the spinal cord was made at each level of the spinal cord. The overall lengths of extension of the bundles of axons at the thoracic and lumbosacral levels were then averaged and the standard deviation determined for each sample.
Quantification of AChR clusters
The number of occupied AChR clusters was determined by blinded evaluation of alpha-bungarotoxin positive AChR clusters with or without the presence of GFP labeled neurons. Thick tissue sections (34 µm) were obtained from intercostal muscles and diaphragms of GFP SMA, carrier and normal e17.5 and e18.5 embryos as well as PND02 and PND03 mice. AChR clusters were counted in the same regions in SMA and control animals. All AChR clusters found in between the ribs in the intercostal muscles at the thoracic level of the spinal cord were counted. The intercostal muscles located both left and right of the spinal cord were assayed for each animal in serial sections. Statistical significance was measured using a 2 × 2 χ2 contingency table with Yates correction of continuity.
FUNDING
This study was funded by Families of SMA postdoctoral research awards MCG03, MCG04, MCG05-06 to V.L.M., NIH NINDS grant 5R01NS050414-02 to C.E.B. and additional support is provided by NIH grant P30NS045758.
ACKNOWLEDGEMENT
We would like to thank Dr T. Jessell for the HB9:GFP mice.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Roberts D.F., Chavez J., Court S.D. The genetic component in child mortality. Arch. Dis. Child. 1970;45:33–38. doi: 10.1136/adc.45.239.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J. Med. Genet. 1978;15:409–413. doi: 10.1136/jmg.15.6.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melki J. Spinal muscular atrophy. Curr. Opin. Neurol. 1997;10:381–385. doi: 10.1097/00019052-199710000-00005. [DOI] [PubMed] [Google Scholar]
- 4.McAndrew P.E., Parsons D.W., Simard L.R., Rochette C., Ray P.N., Mendell J.R., Prior T.W., Burghes A.H. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 1997;60:1411–1422. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 6.Burghes A.H. When is a deletion not a deletion? When it is converted. Am. J. Hum. Genet. 1997;61:9–15. doi: 10.1086/513913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lefebvre S., Burlet P., Liu Q., Bertrandy S., Clermont O., Munnich A., Dreyfuss G., Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 8.Coovert D.D., Le T.T., McAndrew P.E., Strasswimmer J., Crawford T.O., Mendell J.R., Coulson S.E., Androphy E.J., Prior T.W., Burghes A.H. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 9.Eggert C., Chari A., Laggerbauer B., Fischer U. Spinal muscular atrophy: the RNP connection. Trends Mol. Med. 2006;12:113–121. doi: 10.1016/j.molmed.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Gubitz A.K., Feng W., Dreyfuss G. The SMN complex. Exp. Cell Res. 2004;296:51–56. doi: 10.1016/j.yexcr.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 11.Pellizzoni L., Yong J., Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–1779. doi: 10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- 12.Schrank B., Gotz R., Gunnersen J.M., Ure J.M., Toyka K.V., Smith A.G., Sendtner M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl Acad. Sci. USA. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frugier T., Tiziano F.D., Cifuentes-Diaz C., Miniou P., Roblot N., Dierich A., Le Meur M., Melki J. Nuclear targeting defect of SMN lacking the C-terminus in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2000;9:849–858. doi: 10.1093/hmg/9.5.849. [DOI] [PubMed] [Google Scholar]
- 14.Cifuentes-Diaz C., Frugier T., Tiziano F.D., Lacene E., Roblot N., Joshi V., Moreau M.H., Melki J. Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J. Cell Biol. 2001;152:1107–1114. doi: 10.1083/jcb.152.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vitte J.M., Davoult B., Roblot N., Mayer M., Joshi V., Courageot S., Tronche F., Vadrot J., Moreau M.H., Kemeny F., et al. Deletion of murine Smn exon 7 directed to liver leads to severe defect of liver development associated with iron overload. Am. J. Pathol. 2004;165:1731–1741. doi: 10.1016/S0002-9440(10)63428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monani U.R., Sendtner M., Coovert D.D., Parsons D.W., Andreassi C., Le T.T., Jablonka S., Schrank B., Rossol W., Prior T.W., et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 17.Hsieh-Li H.M., Chang J.G., Jong Y.J., Wu M.H., Wang N.M., Tsai C.H., Li H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 18.Monani U.R., Pastore M.T., Gavrilina T.O., Jablonka S., Le T.T., Andreassi C., DiCocco J.M., Lorson C., Androphy E.J., Sendtner M., et al. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J. Cell Biol. 2003;160:41–52. doi: 10.1083/jcb.200208079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le T.T., Pham L.T., Butchbach M.E., Zhang H.L., Monani U.R., Coovert D.D., Gavrilina T.O., Xing L., Bassell G.J., Burghes A.H. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 20.Liu Q., Dreyfuss G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 1996;15:3555–3565. [PMC free article] [PubMed] [Google Scholar]
- 21.Young P.J., Le T.T., thi Man N., Burghes A.H., Morris G.E. The relationship between SMN, the spinal muscular atrophy protein, and nuclear coiled bodies in differentiated tissues and cultured cells. Exp. Cell Res. 2000;256:365–374. doi: 10.1006/excr.2000.4858. [DOI] [PubMed] [Google Scholar]
- 22.Young P.J., Le T.T., Dunckley M., Nguyen T.M., Burghes A.H., Morris G.E. Nuclear gems and Cajal (coiled) bodies in fetal tissues: nucleolar distribution of the spinal muscular atrophy protein, SMN. Exp. Cell Res. 2001;265:252–261. doi: 10.1006/excr.2001.5186. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H.L., Pan F., Hong D., Shenoy S.M., Singer R.H., Bassell G.J. Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J. Neurosci. 2003;23:6627–6637. doi: 10.1523/JNEUROSCI.23-16-06627.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan L., Simard L.R. Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum. Mol. Genet. 2002;11:1605–1614. doi: 10.1093/hmg/11.14.1605. [DOI] [PubMed] [Google Scholar]
- 25.Sharma A., Lambrechts A., Hao le T., Le T.T., Sewry C.A., Ampe C., Burghes A.H., Morris G.E. A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp. Cell Res. 2005;309:185–197. doi: 10.1016/j.yexcr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H., Xing L., Rossoll W., Wichterle H., Singer R.H., Bassell G.J. Multiprotein complexes of the survival of motor neuron protein SMN with Gemins traffic to neuronal processes and growth cones of motor neurons. J. Neurosci. 2006;26:8622–8632. doi: 10.1523/JNEUROSCI.3967-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tizzano E.F., Cabot C., Baiget M. Cell-specific survival motor neuron gene expression during human development of the central nervous system: implications for the pathogenesis of spinal muscular atrophy. Am. J. Pathol. 1998;153:355–361. doi: 10.1016/S0002-9440(10)65578-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pagliardini S., Giavazzi A., Setola V., Lizier C., Di Luca M., DeBiasi S., Battaglia G. Subcellular localization and axonal transport of the survival motor neuron (SMN) protein in the developing rat spinal cord. Hum. Mol. Genet. 2000;9:47–56. doi: 10.1093/hmg/9.1.47. [DOI] [PubMed] [Google Scholar]
- 29.Briese M., Esmaeili B., Sattelle D.B. Is spinal muscular atrophy the result of defects in motor neuron processes? Bioessays. 2005;27:946–957. doi: 10.1002/bies.20283. [DOI] [PubMed] [Google Scholar]
- 30.Setola V., Terao M., Locatelli D., Bassanini S., Garattini E., Battaglia G. Axonal-SMN (a-SMN), a protein isoform of the survival motor neuron gene, is specifically involved in axonogenesis. Proc. Natl Acad. Sci. USA. 2007;104:1959–1964. doi: 10.1073/pnas.0610660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parsons D.W., McAndrew P.E., Iannaccone S.T., Mendell J.R., Burghes A.H., Prior T.W. Intragenic telSMN mutations: frequency, distribution, evidence of a founder effect, and modification of the spinal muscular atrophy phenotype by cenSMN copy number. Am. J. Hum. Genet. 1998;63:1712–1723. doi: 10.1086/302160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clermont O., Burlet P., Benit P., Chanterau D., Saugier-Veber P., Munnich A., Cusin V. Molecular analysis of SMA patients without homozygous SMN1 deletions using a new strategy for identification of SMN1 subtle mutations. Hum. Mutat. 2004;24:417–427. doi: 10.1002/humu.20092. [DOI] [PubMed] [Google Scholar]
- 33.Rochette C.F., Surh L.C., Ray P.N., McAndrew P.E., Prior T.W., Burghes A.H., Vanasse M., Simard L.R. Molecular diagnosis of non-deletion SMA patients using quantitative PCR of SMN exon 7. Neurogenetics. 1997;1:141–147. doi: 10.1007/s100480050021. [DOI] [PubMed] [Google Scholar]
- 34.Hahnen E., Schonling J., Rudnik-Schoneborn S., Raschke H., Zerres K., Wirth B. Missense mutations in exon 6 of the survival motor neuron gene in patients with spinal muscular atrophy (SMA) Hum. Mol. Genet. 1997;6:821–825. doi: 10.1093/hmg/6.5.821. [DOI] [PubMed] [Google Scholar]
- 35.Wang C.H., Papendick B.D., Bruinsma P., Day J.K. Identification of a novel missense mutation of the SMN(T) gene in two siblings with spinal muscular atrophy. Neurogenetics. 1998;1:273–276. doi: 10.1007/s100480050040. [DOI] [PubMed] [Google Scholar]
- 36.Gavrilina T.O., McGovern V.L., Workman E., Crawford T.O., Gogliotti R.G., DiDonato C.J., Monani U.R., Morris G.E., Burghes A.H. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum. Mol. Genet. 2008;17:1063–1075. doi: 10.1093/hmg/ddm379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ting C.H., Lin C.W., Wen S.L., Hsieh-Li H.M., Li H. Stat5 constitutive activation rescues defects in spinal muscular atrophy. Hum. Mol. Genet. 2007;16:499–514. doi: 10.1093/hmg/ddl482. [DOI] [PubMed] [Google Scholar]
- 38.Rossoll W., Jablonka S., Andreassi C., Kroning A.K., Karle K., Monani U.R., Sendtner M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jablonka S., Karle K., Sandner B., Andreassi C., von Au K., Sendtner M. Distinct and overlapping alterations in motor and sensory neurons in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2006;15:511–518. doi: 10.1093/hmg/ddi467. [DOI] [PubMed] [Google Scholar]
- 40.McWhorter M.L., Monani U.R., Burghes A.H., Beattie C.E. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 2003;162:919–932. doi: 10.1083/jcb.200303168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carrel T.L., McWhorter M.L., Workman E., Zhang H., Wolstencroft E.C., Lorson C., Bassell G.J., Burghes A.H., Beattie C.E. Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J. Neurosci. 2006;26:11014–11022. doi: 10.1523/JNEUROSCI.1637-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McWhorter M.L., Boon K.L., Horan E.S., Burghes A.H., Beattie C.E. The SMN binding protein Gemin2 is not involved in motor axon outgrowth. Dev. Neurobiol. 2008;68:182–194. doi: 10.1002/dneu.20582. [DOI] [PubMed] [Google Scholar]
- 43.Winkler C., Eggert C., Gradl D., Meister G., Giegerich M., Wedlich D., Laggerbauer B., Fischer U. Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes. Dev. 2005;19:2320–2330. doi: 10.1101/gad.342005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wichterle H., Lieberam I., Porter J.A., Jessell T.M. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 45.Thaler J., Harrison K., Sharma K., Lettieri K., Kehrl J., Pfaff S.L. Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron. 1999;23:675–687. doi: 10.1016/s0896-6273(01)80027-1. [DOI] [PubMed] [Google Scholar]
- 46.Arber S., Han B., Mendelsohn M., Smith M., Jessell T.M., Sockanathan S. Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron. 1999;23:659–674. doi: 10.1016/s0896-6273(01)80026-x. [DOI] [PubMed] [Google Scholar]
- 47.Huber A.B., Kania A., Tran T.S., Gu C., De Marco Garcia N., Lieberam I., Johnson D., Jessell T.M., Ginty D.D., Kolodkin A.L. Distinct roles for secreted semaphorin signaling in spinal motor axon guidance. Neuron. 2005;48:949–964. doi: 10.1016/j.neuron.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 48.Brandon E.P., Lin W., D’Amour K.A., Pizzo D.P., Dominguez B., Sugiura Y., Thode S., Ko C.P., Thal L.J., Gage F.H., et al. Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J. Neurosci. 2003;23:539–549. doi: 10.1523/JNEUROSCI.23-02-00539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coërs C., Woolf A.L. The Innervation of Muscle: A Biopsy Study. Springfield, IL: Thomas; 1959. [Google Scholar]
- 50.Alderson K. Axonal swellings in human intramuscular nerves. Muscle Nerve. 1992;15:1284–1289. doi: 10.1002/mus.880151111. [DOI] [PubMed] [Google Scholar]
- 51.Tarrade A., Fassier C., Courageot S., Charvin D., Vitte J., Peris L., Thorel A., Mouisel E., Fonknechten N., Roblot N., et al. A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum. Mol. Genet. 2006;15:3544–3558. doi: 10.1093/hmg/ddl431. [DOI] [PubMed] [Google Scholar]
- 52.Kariya S., Park G., Maeno-Hikichi Y., Leykekhman O., Lutz C., Arkovitz M.S., Landmesser L.T., Monani U.R. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008 doi: 10.1093/hmg/ddn156. advance access published online on May 20, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murray L.M., Comley L.H., Thomson D., Parkinson N., Talbot K., Gillingwater T.H. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- 54.Dubowitz V., Sewry C.A., Fitzsimons R.B. Muscle Biopsy: A Practical Approach. 2nd edn. Eastbourne: Baillière Tindall; 1985. [Google Scholar]
- 55.Dubowitz V. Muscle Disorders in Childhood. Philadelphia: Saunders; 1978. [Google Scholar]
- 56.Hausmanowa-Petrusewicz I., Vrbova G. Spinal muscular atrophy: a delayed development hypothesis. Neuroreport. 2005;16:657–661. doi: 10.1097/00001756-200505120-00001. [DOI] [PubMed] [Google Scholar]
- 57.Lin W., Burgess R.W., Dominguez B., Pfaff S.L., Sanes J.R., Lee K.F. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–1064. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- 58.Sanes J.R., Lichtman J.W. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- 59.Cox G.A., Mahaffey C.L., Frankel W.N. Identification of the mouse neuromuscular degeneration gene and mapping of a second site suppressor allele. Neuron. 1998;21:1327–1337. doi: 10.1016/s0896-6273(00)80652-2. [DOI] [PubMed] [Google Scholar]
- 60.Hausmanowa-Petrusewicz I., Karwanska A. Electromyographic findings in different forms of infantile and juvenile proximal spinal muscular atrophy. Muscle Nerve. 1986;9:37–46. doi: 10.1002/mus.880090106. [DOI] [PubMed] [Google Scholar]
- 61.Swoboda K.J., Prior T.W., Scott C.B., McNaught T.P., Wride M.C., Reyna S.P., Bromberg M.B. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann. Neurol. 2005;57:704–712. doi: 10.1002/ana.20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bard J.L., Kaufman M.H., Dubreuil C., Brune R.M., Burger A., Baldock R.A., Davidson D.R. An internet-accessible database of mouse developmental anatomy based on a systematic nomenclature. Mech. Dev. 1998;74:111–120. doi: 10.1016/s0925-4773(98)00069-0. [DOI] [PubMed] [Google Scholar]