Ribosomal Protein Genes RPS10 and RPS26 Are Commonly Mutated in Diamond-Blackfan Anemia (original) (raw)
Abstract
Diamond-Blackfan anemia (DBA), an inherited bone marrow failure syndrome characterized by anemia that usually presents before the first birthday or in early childhood, is associated with birth defects and an increased risk of cancer. Although anemia is the most prominent feature of DBA, the disease is also characterized by growth retardation and congenital malformations, in particular craniofacial, upper limb, heart, and urinary system defects that are present in ∼30%–50% of patients. DBA has been associated with mutations in seven ribosomal protein (RP) genes, RPS19, RPS24, RPS17, RPL35A, RPL5, RPL11, and RPS7, in about 43% of patients. To continue our large-scale screen of RP genes in a DBA population, we sequenced 35 ribosomal protein genes, RPL15, RPL24, RPL29, RPL32, RPL34, RPL9, RPL37, RPS14, RPS23, RPL10A, RPS10, RPS12, RPS18, RPL30, RPS20, RPL12, RPL7A, RPS6, RPL27A, RPLP2, RPS25, RPS3, RPL41, RPL6, RPLP0, RPS26, RPL21, RPL36AL, RPS29, RPL4, RPLP1, RPL13, RPS15A, RPS2, and RPL38, in our DBA patient cohort of 117 probands. We identified three distinct mutations of RPS10 in five probands and nine distinct mutations of RPS26 in 12 probands. Pre-rRNA analysis in lymphoblastoid cells from patients bearing mutations in RPS10 and RPS26 showed elevated levels of 18S-E pre-rRNA. This accumulation is consistent with the phenotype observed in HeLa cells after knockdown of RPS10 or RPS26 expression with siRNAs, which indicates that mutations in the RPS10 and RPS26 genes in DBA patients affect the function of the proteins in rRNA processing.
Main Text
Diamond-Blackfan anemia (DBA) (MIM 105650) is an inherited bone marrow failure syndrome characterized by anemia that usually presents during infancy or early childhood.1 Red cell aplasia is the most prominent feature of DBA; however, the disease is also characterized by growth retardation and congenital malformations, in particular craniofacial, upper limb, heart, and urinary system defects that are present in ∼30%–50% of patients.2–4 In addition to anemia and birth defects, the disease is associated with predisposition to cancer, in particular acute myeloid leukemia (AML) and osteogenic sarcoma.5,6 DBA is a clinically heterogeneous disease: laboratory findings such as increased mean corpuscular volume (MCV), elevated erythrocyte adenosine deaminase activity (eADA), and hemoglobin F are observed in a majority but not in all DBA patients.7–9 Furthermore, within affected families, some individuals may exhibit mild or absent anemia with only subtle indications of erythroid abnormalities such as macrocytosis, elevated eADA, and/or hemoglobin F. The incidence of DBA is estimated to be between 1:100,000 and 1:200,000 live births9 or 5–7 per million live births,2,10,11 which remains consistent across ethnicities and equal in both genders.9 Approximately 45% of patients are familial cases, with disease inherited in an autosomal-dominant pattern; the remaining 55% of patients are sporadic cases.3
DBA is genetically heterogeneous: heterozygous mutations in four small subunit ribosomal protein (RP) genes, RPS19 (MIM 603474), RPS24 (MIM 602412), RPS17 (MIM 180472), and RPS7 (MIM 612563), and in three large subunit ribosomal protein genes, RPL35A (MIM 180468), RPL5 (MIM 612561), and RPL11 (MIM 612562), have been reported in about 43% of DBA patients, indicating that DBA is a disorder of ribosomal biogenesis and/or function.12–16
Several studies showed that RPS19 protein plays an important role in 18S rRNA maturation in yeast and in human cells.17–20 Other studies demonstrated alterations of pre-RNA processing and small or large ribosomal subunit synthesis in human cells with RPS24, RPS7, RPL35A, RPL5, and RPL11 deficiency.15,16,21 These observations further indicate that DBA is a disorder of the ribosome. Increased apoptosis has been demonstrated by Perdahl22 and also in hematopoietic cell lines and in bone marrow cells with deficiency of RPS19 and RPL35A,15,23 and imbalance of the p53 family proteins has been suggested as a mechanism of abnormal embryogenesis and anemia in zebrafish upon perturbation of RPS19 expression.24 Furthermore, the DBA phenotype in the mouse is ameliorated by the knockdown of p53.25
Here we report the results of a large-scale screen of 35 additional RP genes in a cohort of 117 DBA probands. Remarkably, we identified probable pathogenic mutations in two of these genes, RPS10 (MIM 603632) and RPS26 (MIM 603701). In addition, we found a possible missense mutation of RPL9 (MIM 603686) in one family.
One hundred and seventeen DBA families participated in the study. The diagnosis of DBA in all probands and their family members was based on normochromic, often macrocytic, anemia; reticulocytopenia; a low number or lack of erythroid precursors in bone marrow; and, in some patients, congenital malformations and elevated eADA. Among these families, 14 were multiplex families and 103 comprised only one clinically affected individual. One hundred and three probands were negative for mutations in seven known DBA genes, RPS19, RPS24, RPL35A, RPS17, RPS7, RPL5, and RPL11, and did not have any sequence change in RPS15, RPS27A, and RPL36, previously described as variants of unknown significance.16 Twelve probands were mutated in one of the known genes, three with mutations in RPS19, one with RPS24, five with mutations in RPL5, and three with RPS11 mutation. Two probands had sequence changes in RPS15 and RPS27A, previously described as variants of unknown significance (Table 1).16 Informed consent was obtained from all patients and their family members participating in the study under a protocol at Children's Hospital Boston.
Table 1.
One Hundred and Seventeen Probands Studied for Mutations in 35 Ribosomal Protein Genes
| All Studied Probands | Sporadic Cases | Familial Cases | Probands Negative for Mutations in Ribosomal Protein (RP) Genes S19, S24, L35A, S17, S7, L5, L11, S15, S27A, and L36 | Probands with Sequence Change in RP Genes S19, S24, L5, L11, S15, and S27A |
|---|---|---|---|---|
| 117 | 103 | 14 | 103 | 14 |
Genomic DNA was isolated from blood samples with nucleic acid isolation system QuickGene-610L (AutoGen) according to the manufacturer's instructions. To continue our large-scale screen of RP genes in a DBA population, we amplified genomic DNA samples from 96 unrelated DBA probands enrolled in the study by PCR and sequenced for mutations in 35 RP genes, RPL15, RPL24, RPL29, RPL32, RPL34, RPL9, RPL37, RPS14, RPS23, RPL10A, RPS10, RPS12, RPS18, RPL30, RPS20, RPL12, RPL7A, RPS6, RPL27A, RPLP2, RPS25, RPS3, RPL41, RPL6, RPLP0, RPS26, RPL21, RPL36AL, RPS29, RPL4, RPLP1, RPL13, RPS15A, RPS2, and RPL38 (chromosomes 3, 4, 5, 6, 8, 9, 11, 12, 14, 15, 16, and 17), in our DBA patient cohort of 117 families (the majority of the patient cohort is the same as in previous studies).13,15,16 In total, we screened 64 RP genes, in addition to the 29 RP genes sequenced previously.13,15,16,26 Primers (see Table S1 available online) were designed with Primer3 software to amplify the coding exons and intron/exon boundaries of the above genes. PCR products, between 200 and 600 base pairs long, were robotically prepared with a Hamilton MicroLab STARplus (Hamilton Robotics), purified with ExoSAP enzyme (USB), and sequenced on both strands with an Applied Biosystems 3730 DNA Analyzer. The chromatograms were analyzed with Sequencher software version 4.7 (Gene Codes). When a sequence change was identified in a given gene, we sequenced an additional 21 samples (for a total of 117 samples) from unrelated probands for mutations in this gene. Duplicate, independent PCR products were sequenced to confirm the observed nucleotide changes in the probands. DNA samples from at least 260 control individuals, i.e., 520 chromosomes, were sequenced to determine whether the observed sequence variations were nonpathogenic variants. Subsequently, DNA samples from available family members were sequenced to determine whether the mutation cosegregated with the DBA phenotype within the pedigree.
Interestingly, we identified heterozygous sequence changes in RPS10 in 5 of 117 probands. One sequence change is a missense mutation 3G>A that changes Met1Ile and eliminates the start codon. The next downstream start codon is located at nucleotide position 61–63 (codon 21) and is predicted to start translation of a truncated protein of 144 amino acids. Another mutation is a c.260_261insC causing a frameshift at codon 87 and a stop at codon 97. Three other probands have a common nonsense mutation, c.337C>T, causing Arg113 stop (Table 2). Proband P3 has a de novo mutation, whereas SNP analysis of 14 polymorphisms spanning an ∼20 kb region surrounding the RPS10 gene demonstrated that probands P3 and P4 do not share the same background haplotype (data not shown) (Table 2). All of these changes are unique (i.e., not previously described in the literature or databases), and two of them were found in only a single kindred, whereas one was seen in three unrelated families.
Table 2.
Sequence Changes in RPS10 in DBA Patients
| Proband's ID (Gender), Inheritance | Family Members | DNA Mutation | Exon or Intron | Predicted Amino Acid Change | Age at Diagnosis | Malformation Status | Response at First Steroid Therapy | Present Therapy |
|---|---|---|---|---|---|---|---|---|
| P1 (M), sporadic | - | c.3G>A | exon 1 | Met1Ile | NA | NA | NA | NA |
| P2 (F), sporadic | f – normal sequence | c.260_261insC | exon 3 | frameshift at codon 87; stop at 97 | 2 mo | none | responsive | steroid therapy |
| P3 (M), sporadic | - | c.337C>T | exon 4 | Arg113stop | 7 mo | none | responsive | RBC trx |
| P4 (F), sporadic | - | c.337C>T | exon 4 | Arg113stop | 12 yrs | none | responsive | steroid therapy |
| P5 (F), de novo | f, m – normal sequence | c.337C>T | exon 4 | Arg113stop | birth | webbed neck | unresponsive | RBC trx |
We also identified nine distinct mutations in 12 probands in RPS26 gene located on chromosome 12. Interestingly, we found mutation of the first codon, methionine change to leucine, valine, or arginine, in six probands and in two family members (Table 3), which suggests that the translation-initiation codon may be a hot spot in RPS26. These sequence changes eliminate a start codon; the next downstream start codon is located at nucleotide position 343–345. This is the last codon, codon 115, and the protein is predicted to not be translated. Probands P7–P10 share the same M1V mutation. Probands P7 and P8 have de novo mutations, whereas SNP analysis of 15 polymorphisms spanning an ∼20 kb region surrounding the RPS26 gene demonstrated that probands P9 and P10 do not share the same background haplotype (data not shown) (Table 3). Other identified mutations are two missense mutations (c.97G>A changing Asp33Asn and c.334T>C resulting in Met115Thr), insertion G in exon 2 causing frameshift at codon 11 and stop at codon 25, and three donor splice-site mutations in introns 1 and 2. All of these changes are also unique, and eight of them were found in only a single kindred, whereas one was seen in four apparently unrelated families (Table 3). Parental DNA was available in two probands with RPS10 mutations and in seven with RPS26 mutations. De novo sequence changes of RPS10 and RPS26 were identified in one and five probands, respectively, with sporadic disease, further supporting our belief that these sequence changes are likely pathogenic mutations. In addition, six mutations in RPS26 among 75 samples from DBA probands were identified by a separate ongoing RP gene resequencing project (unpublished data).
Table 3.
Sequence Changes in RPS26 in DBA Patients
| Mutation Type | Proband's ID (Gender), Inheritance | Family Members | DNA Mutation | Exon or Intron | Predicted Amino Acid Change | Age at Diagnosis | Malformation Status | Response at First Steroid Therapy | Present Therapy |
|---|---|---|---|---|---|---|---|---|---|
| Missense mutation | P6 (M), de novo | f, m, s – normal sequence | c.1A>T | exon 1 | Met1Leu | NA | none | unresponsive | RBC trx |
| - | P7 (M), familial | f, m – normal sequence | c.1A>G | exon 1 | Met1Val | 2 mo | none | responsive | steroid therapy |
| - | d of P7 | c.1A>G | exon 1 | Met1Val | 6 wks | none | unresponsive | RBC trx | |
| - | P8 (M), de novo | f, m – normal sequence | c.1A>G | exon 1 | Met1Val | NA | none | unresponsive | RBC trx |
| - | P9 (M), familial | - | c.1A>G | exon 1 | Met1Val | NA | none | responsive | steroid therapy |
| - | d of P9 | c.1A>G | exon 1 | Met1Val | NA | none | responsive | no therapy | |
| - | P10 (M), sporadic | - | c.1A>G | exon 1 | Met1Val | 7 wks | duplicated pelvocalicon on right kidney | unresponsive | RBC trx |
| - | P11 (M), familial | d – normal sequence | c.1T>G | exon 1 | Met1Arg | NA | NA | NA | NA |
| - | P12 (M), de novo | f, m, s – normal sequence | c.97G>A | exon 2 | Asp33Asn | 8 mo | inguinal hernia, missing vas deferens (unilateral), slightly abnormal epidymis, pronounced boney prominence of a cervical spinous process | unresponsive | RBC trx |
| - | P13 (M), de novo | f, m, s – normal sequence | c.344T > C | exon 4 | Met115Thr | 8 mo | none | responsive | no therapy |
| Insertion | P14 (F), sporadic | - | c.31_32insG | exon 2 | frameshift at codon 11; stop at 25 | NA | none | unresponsive | deceased |
| Splice-site mutations | P15 (M), sporadic | s – normal sequence | donor splice site IVS1+1g>c | intron 1 | - | NA | NA | NA | NA |
| - | P16 (M) | f, m, s, b – normal sequence | donor splice site IVS1+1g>a | intron 1 | - | NA | none | unresponsive | RBC trx |
| - | P17 (F), de novo | f, m – normal sequence | donor splice site IVS1+t>g | intron 2 | - | NA | NA | NA | NA |
We also found a heterozygous missense mutation, c.375G>C, resulting in Arg125Ser substitution in RPL9 in one proband and his affected mother. None of the identified sequence changes were found on the NCBI SNP lists, and none were identified in at least 520 control chromosomes from a control population of similar, largely European origin.
Review of medical records of DBA probands and data from the DBA Registry of North America (DBAR)4,6 revealed that among four patients with RPS10 mutations and available medical data, one had a physical malformation of a webbed neck and three patients were without malformations, whereas among 11 RPS26 mutated patients whose clinical data were available, only three patients had malformations. These physical abnormalities were similar to those observed in other RP gene mutations (Table 2; Table 3). One patient with RPS26 mutation has cleft lip and cleft palate. Interestingly, to date this malformation has only been found in DBA patients with RPL5 and RPL11 mutations.16,27 Thus, the RPL26 gene is the third DBA gene whose mutation is associated with clefting in DBA. In general, physical malformations are present in about 30%–50% of patients in the overall study cohort.2–4 Although the sample size is small, physical malformations seem to be rare events in patients with RPS26 mutations.
Interestingly, 5 of 11 patients with RPS26 mutations whose treatment data were available responded well to the steroid treatment and six were unresponsive. Among four patients with RPS10 mutations and available medical data, three patients were responsive and one unresponsive to steroid treatment. According to data from DBAR, 79% of DBA patients have been initially responsive to steroids, 17% unresponsive, and 4% were never treated with steroids.4 At present, four _RPS26_-mutated patients are dependent on red blood transfusions, three are steroid dependent, two do not need any treatment, and two are deceased (Table 3). Two _RPS10_-mutated patients are red-blood-cell-transfusion dependent and two are dependent on steroid treatment (Table 2).
The role of RPS10 and RPS26 in pre-rRNA processing was assessed by knocking down expression of these proteins with siRNAs in HeLa cells. Pre-rRNA processing analysis was performed as previously described.21 Depletion of both proteins led to decreased levels of 18S rRNA, indicating that they are necessary for production of the small subunit. Accordingly, RNA blot analysis of the pre-rRNAs revealed that these proteins are both required for proper processing of the pre-rRNA (Figure 1A). Depletion of RPS26 led to the accumulation of 43S, 26S, and 18S-E pre-rRNAs, which indicates defects in cleavages at both ends of the 18S rRNA. Interestingly, knockdown of RPS10 expression yielded a very similar phenotype. This phenotype differed from what we observed previously upon depletion of RPS19, RPS24, or RPS7, three other RPS proteins mutated in DBA21 for which the processing was blocked upstream of 18S-E pre-rRNA formation. Pre-rRNAs were next detected by RNA blot analysis in RNAs extracted from lymphoblastoid cells derived from patients with mutations in RPS10 and RPS26 (Figure 1B). In the case of the RPS26+/mut patients, we compared their profiles to those of unaffected relatives. Quantitative analysis by phosphorimager indicated that four out of five patients had elevated levels of 18S-E RNA when compared to their relatives (Figures 1B and 1C). The remaining RPS26+/mut patient instead showed a higher amount of 21S RNA, a species just upstream the 18S-E RNA in the pre-rRNA maturation pathway (Figures 1B and 1C, patient D). Higher levels of 18S-E pre-rRNA were also detected in RPS10+/mut patients when compared to cells from unaffected individuals (Figure 1B). Interestingly, this phenotype was not detected in an individual bearing a missense mutation in RPS10 (Figure 1B, last lane). Higher levels of 18S-E pre-rRNA in RPS26+/mut and RPS10+/mut DBA patients translated into high 18S-E/30S or 18S-E/21S pre-RNA ratios (Figure 1D), whereas the 21S/30S ratio remained in the same range as in controls, unlike in RPS19+/mut patient cells. The similarity of the pre-rRNA pattern modifications in patient cells and in cells depleted of the corresponding ribosomal proteins strongly suggests that the mutations in RPS10 and RPS26 identified in DBA patients alter the function of ribosomal proteins in ribosome biogenesis, as shown before for other ribosomal protein genes affected in DBA.
Figure 1.
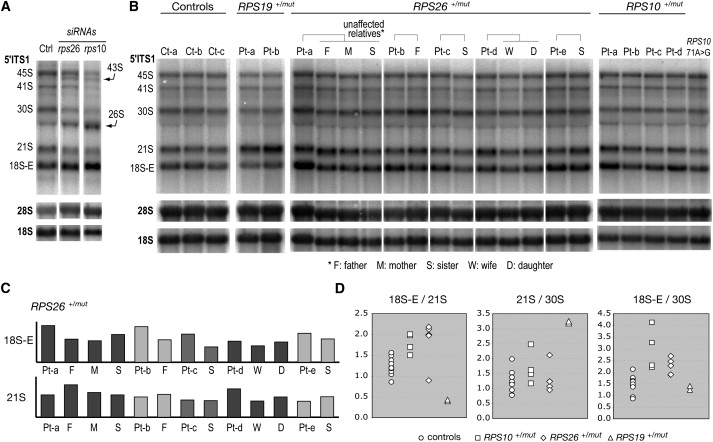
RNA Blot Analysis of the Effects of RPS10 and RPS26 Mutation on Pre-rRNA Processing
(A) HeLa cells were transfected with siRNAs to knock down RPS10 and RPS26 expression. RNAs extracted 48 hr posttransfection were probed with oligonucleotides complementary to the 5′ end of the ITS1 (5′ ITS1), which detects the precursors to the 18S rRNA or to the mature 18S and 28S rRNAs. Transfected cells show similar phenotypes with high levels of 43S, 26S, and 18S-E RNA, together with low levels of 18S rRNA. Similar results were obtained with two independent siRNAs for each gene.
(B) Total RNAs from lymphoblastoid cells derived from DBA patients (Pt) and unaffected relatives were analyzed as in (A). Controls (Ct) correspond to cells from unaffected individuals unrelated to patients. RNAs from patients mutated in RPS19 are also shown for comparison. The last lane on the right corresponds to an individual bearing a missense change in RPS10, most likely a rare normal variant.
(C) Phosphorimager analysis of the 18S-E and 21S pre-rRNA levels in RPS26+/mut patients shows that four out of five patients have elevated levels of 18S-E rRNA when compared to their unaffected relatives. In each lane, measurements were normalized to the amount of 28S rRNA and divided by the mean of the corresponding value in the three controls.
(D) Relative abundance of the 18S RNA precursors was quantified by phosphorimaging in DBA patients mutated in RPS10, RPS26, and RPS19. Controls in this panel correspond to all the unaffected individuals shown in (B), both patient relatives and unrelated persons.
In summary, we found that mutations in RPS10 and RPS26 are present in about 4.8% and 11.6% of our DBA proband cohort, respectively. After correcting for the fact that our study population was prescreened for RPS19, RPS24, RPL35A, RPS17, RPL5, RPL11, and RPS7 mutations, we estimate that RPS10 and RPS26 mutations are present in about 2.6% and 6.4% of the overall DBA population. The RPS10 gene is located on chromosome 6 and contains six exons, with the start codon in exon 2. RPS10 encodes a 165-amino-acid-long RPS10 protein, a component of the 40S ribosomal subunit. In the present work, we identified three sequence changes in RPS10 in five DBA probands. In addition, we found one missense change, c.71A>G, resulting in Lys24Arg in the patient with transfusion-dependent anemia and in her unaffected brother. Although this sequence change was not found in 285 control individuals, we strongly suspect that it may be a rare normal variant because there is no phenotype in the pre-RNA maturation assay in the sample of the brother carrying the sequence change (Figure 1B, last lane). Because the substituted amino acid is also basic, we hypothesize that this change affects only mildly the overall RPS10 conformation and/or function.
We also identified nine distinct mutations in the RPS26 gene in 12 patients. The RPS26 gene is located on chromosome 12 and contains four exons, with the start codon in exon 1. This gene encodes a 115-amino-acid-long RPS26 protein, which is also a component of the 40S ribosomal subunit. The translation-initiation codon may be a hot spot for mutation in RPS26, because we found mutation of the first codon, methionine change to leucine, valine, or arginine, in six probands and in two affected family members (Table 3). One missense sequence change, c.344T>C, resulting in substitution of methionine by threonine at the last codon 115, may be a rare or private nonpathogenic genetic variant; therefore, the DNA sample carrying this sequence change is currently being screened for mutations in the remaining 15 RP genes.
The RPL9 gene is located on chromosome 4 and comprises two isoforms, which contain seven and eight exons, respectively. In the seven-exon-long isoform the start codon is located in exon 1, and in the eight-exon-long isoform the start codon is located in exon 2. Both isoforms encode a 192-amino-acid-long RPL9 protein, a component of the large ribosomal subunit. The only RPL9 missense change, c.375G>C, causing Arg125Ser, was found in one of the probands and his affected mother. Unlike for mutations RPS10 and RPS26, lymphoblastoid cells established from these persons do not display a pre-rRNA processing defect similar to that observed upon knockdown of RPL9 with siRNAs (data not shown). Our data do not allow definitive determination of pathogenicity for this mutation. Therefore, we consider this change to be a rare variant of unknown significance, and DNA samples carrying these sequence changes are currently being screened for mutations in the remaining 15 RP genes.
Thus, this report brings the total number of known DBA-associated genes to nine and includes RPS19 (∼25%), RPS24 (∼2%), RPS17 (∼1%), RPL35A (∼3.5%), RPL5 (∼6.6%), RPL11 (∼4.8%), RPS7 (∼1%), RPS10 (∼6.4%), and RPS26 (∼2.6%), in total accounting for approximately ∼52.9% of patients with DBA. In addition, there were four rare variants of unknown significance identified in RPS15, RPS27A, RPL36, and RPL9.12–16
Our data indicate that RPS10 and RPS26 contribute differently to pre-rRNA processing when compared to other RPS proteins involved in DBA, like RPS19 and RPS24.18,21 It remains to be understood how mutations in proteins having different functions in ribosome biogenesis all lead to the same disease. The most direct consequence of these mutations is a defect in ribosome synthesis. This may elicit a ribosomal stress response, to which erythrocytic progenitors would be especially sensitive at some stage of their differentiation. A defect of ribosome biogenesis may also alter regulation and efficacy of translation by affecting the rate of ribosome production and ribosome quality. Such translation disorders could disturb the synthesis timing of important regulators in the erythrocyte differentiation pathway or impede high-rate synthesis of globin. The effects of ribosomal protein gene mutations could be further exacerbated in erythrocytic progenitors when compared to other cell lineages because of the dramatic changes in gene expression that result from the massive chromatin condensation preceding nuclear extrusion. Generation of animal models reproducing the mutations in RPS10 and RPS26 described here could help to answer these questions.
Acknowledgments
This work was supported by a generous gift from The Manton Foundation and a grant from the French National Research Agency (Agence Nationale de la Recherche, RIBODBA project), as well as grants from the Daniella Maria Arturi Foundation, the Diamond Blackfan Anemia Foundation, the Pediatric Cancer Foundation (J.M.L.), the National Institutes of Health (NIH) (R01 HL 079571) (J.M.L., A.V., E.A.), and the Feinstein Institute for Medical Research at the North Shore Long Island Jewish General Clinical Research Center (M01 RR018535) (J.M.L., A.V., E.A.). DNA sequencing was performed by the Children's Hospital Boston Program in Genomics and the Molecular Genetics Core Facility supported by the Developmental Disabilities Research Center (NIH P30 HD18655) and the Harvard Neuromuscular Disease Project (NIH P50 NS040828), by the Children's Hospital Boston SNP Genotyping Facilities, and by the Diamond Blackfan Anemia Gene Discovery and Resequencing Project (National Heart, Lung, and Blood Institute Resequencing & Genotyping Service) (R109 MOHLKE). We thank Alan Beggs for many helpful discussions, advice, and support; Marie Arturi for stimulating DBA research collaboration; and all of the physicians and DBA patients for participating in the study.
Supplemental Data
Document S1. Table S1
Web Resources
The URLs for data presented herein are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
- Primer 3, http://frodo.wi.mit.edu/primer3/
- UCSC Genome Browser, http://genome.ucsc.edu/
- Entrez Gene, http://www.ncbi.nlm.nih.gov/gene
- Single Nucleotide Polymorphism, http://www.ncbi.nlm.nih.gov/SNP/
Accession Numbers
The GenBank accession numbers for human RPS10, RPS26, and RPL9 are NM_001014; NM_001029; and NM_000661 and NM_001024921, respectively.
References
- 1.Alter B.P., Young N.S. The bone marrow failure syndromes. In: Nathan D.G., Orkin H.S., editors. Hematology of Infancy and Childhood. Volume 1. Saunders; Philadelphia: 1998. pp. 237–335. [Google Scholar]
- 2.Willig T.N., Niemeyer C.M., Leblanc T., Tiemann C., Robert A., Budde J., Lambiliotte A., Kohne E., Souillet G., Eber S. Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Société d'Hématologie et d'Immunologie Pédiatrique (SHIP), Gesellshaft für Pädiatrische Onkologie und Hämatologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI) Pediatr. Res. 1999;46:553–561. doi: 10.1203/00006450-199911000-00011. [DOI] [PubMed] [Google Scholar]
- 3.Orfali K.A., Ohene-Abuakwa Y., Ball S.E. Diamond Blackfan anaemia in the UK: Clinical and genetic heterogeneity. Br. J. Haematol. 2004;125:243–252. doi: 10.1111/j.1365-2141.2004.04890.x. [DOI] [PubMed] [Google Scholar]
- 4.Lipton J.M., Atsidaftos E., Zyskind I., Vlachos A. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: An update from the Diamond Blackfan Anemia Registry. Pediatr. Blood Cancer. 2006;46:558–564. doi: 10.1002/pbc.20642. [DOI] [PubMed] [Google Scholar]
- 5.Janov A.J., Leong T., Nathan D.G., Guinan E.C. Diamond-Blackfan anemia. Natural history and sequelae of treatment. Medicine (Baltimore) 1996;75:77–78. doi: 10.1097/00005792-199603000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Vlachos A., Klein G.W., Lipton J.M. The Diamond Blackfan Anemia Registry: Tool for investigating the epidemiology and biology of Diamond-Blackfan anemia. J. Pediatr. Hematol. Oncol. 2001;23:377–382. doi: 10.1097/00043426-200108000-00015. [DOI] [PubMed] [Google Scholar]
- 7.Glader B.E., Backer K. Elevated red cell adenosine deaminase activity: A marker of disordered erythropoiesis in Diamond-Blackfan anaemia and other haematologic diseases. Br. J. Haematol. 1988;68:165–168. doi: 10.1111/j.1365-2141.1988.tb06184.x. [DOI] [PubMed] [Google Scholar]
- 8.Willig T.N., Draptchinskaia N., Dianzani I., Ball S., Niemeyer C., Ramenghi U., Orfali K., Gustavsson P., Garelli E., Brusco A. Mutations in ribosomal protein S19 gene and diamond blackfan anemia: Wide variations in phenotypic expression. Blood. 1999;94:4294–4306. [PubMed] [Google Scholar]
- 9.Vlachos A., Ball S., Dahl N., Alter B.P., Sheth S., Ramenghi U., Meerpohl J., Karlsson S., Liu J.M., Leblanc T., Participants of Sixth Annual Daniella Maria Arturi International Consensus Conference Diagnosing and treating Diamond Blackfan anaemia: Results of an international clinical consensus conference. Br. J. Haematol. 2008;142:859–876. doi: 10.1111/j.1365-2141.2008.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ball S.E., McGuckin C.P., Jenkins G., Gordon-Smith E.C. Diamond-Blackfan anaemia in the U.K.: Analysis of 80 cases from a 20-year birth cohort. Br. J. Haematol. 1996;94:645–653. doi: 10.1046/j.1365-2141.1996.d01-1839.x. [DOI] [PubMed] [Google Scholar]
- 11.Campagnoli M.F., Garelli E., Quarello P., Carando A., Varotto S., Nobili B., Longoni D., Pecile V., Zecca M., Dufour C. Molecular basis of Diamond-Blackfan anemia: New findings from the Italian registry and a review of the literature. Haematologica. 2004;89:480–489. [PubMed] [Google Scholar]
- 12.Draptchinskaia N., Gustavsson P., Andersson B., Pettersson M., Willig T.N., Dianzani I., Ball S., Tchernia G., Klar J., Matsson H. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 1999;21:169–175. doi: 10.1038/5951. [DOI] [PubMed] [Google Scholar]
- 13.Gazda H.T., Grabowska A., Merida-Long L.B., Latawiec E., Schneider H.E., Lipton J.M., Vlachos A., Atsidaftos E., Ball S.E., Orfali K.A. Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am. J. Hum. Genet. 2006;79:1110–1118. doi: 10.1086/510020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cmejla R., Cmejlova J., Handrkova H., Petrak J., Pospisilova D. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum. Mutat. 2007;28:1178–1182. doi: 10.1002/humu.20608. [DOI] [PubMed] [Google Scholar]
- 15.Farrar J.E., Nater M., Caywood E., McDevitt M.A., Kowalski J., Takemoto C.M., Talbot C.C., Jr., Meltzer P., Esposito D., Beggs A.H. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood. 2008;112:1582–1592. doi: 10.1182/blood-2008-02-140012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gazda H.T., Sheen M.R., Vlachos A., Choesmel V., O'Donohue M.F., Schneider H., Darras N., Hasman C., Sieff C.A., Newburger P.E. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am. J. Hum. Genet. 2008;83:769–780. doi: 10.1016/j.ajhg.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Léger-Silvestre I., Caffrey J.M., Dawaliby R., Alvarez-Arias D.A., Gas N., Bertolone S.J., Gleizes P.E., Ellis S.R. Specific role for yeast homologs of the diamond blackfan anemia associated Rps19 protein in ribosome synthesis. J. Biol. Chem. 2005;280:38177–38185. doi: 10.1074/jbc.M506916200. [DOI] [PubMed] [Google Scholar]
- 18.Choesmel V., Bacqueville D., Rouquette J., Noaillac-Depeyre J., Fribourg S., Crétien A., Leblanc T., Tchernia G., Da Costa L., Gleizes P.E. Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood. 2007;109:1275–1283. doi: 10.1182/blood-2006-07-038372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flygare J., Aspesi A., Bailey J.C., Miyake K., Caffrey J.M., Karlsson S., Ellis S.R. Human RPS19, the gene mutated in Diamond-Blackfan anemia, encodes a ribosomal protein required for the maturation of 40S ribosomal subunits. Blood. 2007;109:980–986. doi: 10.1182/blood-2006-07-038232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Idol R.A., Robledo S., Du H.Y., Crimmins D.L., Wilson D.B., Ladenson J.H., Bessler M., Mason P.J. Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defects in 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells Mol. Dis. 2007;39:35–43. doi: 10.1016/j.bcmd.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 21.Choesmel V., Fribourg S., Aguissa-Touré A.H., Pinaud N., Legrand P., Gazda H.T., Gleizes P.E. Mutation of ribosomal protein RPS24 in Diamond-Blackfan anemia results in a ribosome biogenesis disorder. Hum. Mol. Genet. 2008;17:1253–1263. doi: 10.1093/hmg/ddn015. [DOI] [PubMed] [Google Scholar]
- 22.Perdahl E.B., Naprstek B.L., Wallace W.C., Lipton J.M. Erythroid failure in Diamond-Blackfan anemia is characterized by apoptosis. Blood. 1994;83:645–650. [PubMed] [Google Scholar]
- 23.Miyake K., Utsugisawa T., Flygare J., Kiefer T., Hamaguchi I., Richter J., Karlsson S. Ribosomal protein S19 deficiency leads to reduced proliferation and increased apoptosis but does not affect terminal erythroid differentiation in a cell line model of Diamond-Blackfan anemia. Stem Cells. 2008;26:323–329. doi: 10.1634/stemcells.2007-0569. [DOI] [PubMed] [Google Scholar]
- 24.Danilova N., Sakamoto K.M., Lin S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 2008;112:5228–5237. doi: 10.1182/blood-2008-01-132290. [DOI] [PubMed] [Google Scholar]
- 25.McGowan K.A., Li J.Z., Park C.Y., Beaudry V., Tabor H.K., Sabnis A.J., Zhang W., Fuchs H., de Angelis M.H., Myers R.M. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat. Genet. 2008;40:963–970. doi: 10.1038/ng.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gazda H.T., Zhong R., Long L., Niewiadomska E., Lipton J.M., Ploszynska A., Zaucha J.M., Vlachos A., Atsidaftos E., Viskochil D.H. RNA and protein evidence for haplo-insufficiency in Diamond-Blackfan anaemia patients with RPS19 mutations. Br. J. Haematol. 2004;127:105–113. doi: 10.1111/j.1365-2141.2004.05152.x. [DOI] [PubMed] [Google Scholar]
- 27.Quarello P., Garelli E., Carando A., Brusco A., Calabrese R., Dufour C., Longoni D., Misuraca A., Vinti L., Aspesi A. Diamond-Blackfan anemia: Genotype-phenotype correlation in Italian patients with RPL5 and RPL11 mutations. Haematologica. 2009 doi: 10.3324/haematol.2009.011783. Published online September 22, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Table S1