Progressive Multifocal Leukoencephalopathy Complicating HIV-1 Infection (original) (raw)
. Author manuscript; available in PMC: 2010 Oct 1.
Abstract
Progressive multifocal leukoencephalopathy (PML) caused by the polyomavirus, JC virus (JCV), is one of the most dreaded complications of HIV-1 infection. Unlike other opportunistic infections, PML may present while blood CD4+ T cells remain above AIDS-defining levels and while patients receive combined antiretroviral therapy (cART), either shortly after starting or, more rarely, during chronic successful treatment. PML can be suspected by typical presentation with focal neurological deficits and corresponding demyelinating lesions at magnetic resonance imaging (MRI), while definitive diagnosis requires identification of JCV in cerebrospinal fluid (CSF) or brain tissue. While there is no specific treatment, reversal of immunosuppression by cART leads to clinical and MRI stabilization in 50-60% of PML patients and JCV clearance from CSF. A proportion of cART-treated patients develop inflammatory lesions, which may either accompany a favorable outcome or associate with clinical worsening. The reasons for variability in PML natural history and treatment responses are largely undefined, and more specific and rational approaches to management are sorely needed.
Keywords: HIV-1, progressive multifocal leukoencephalopathy, PML, JC virus, JCV, brain, central nervous system, CNS, antiretroviral treatment, ART, cART, HAART
Introduction
Progressive multifocal leukoencephalopathy (PML) is an infection of the central nervous system (CNS) by the polyoma virus, JC virus (JCV), that destroys oligodendrocytes and their myelin processes. Outside of the context of HIV-1 infection, PML is rare and develops as a complication of conditions with compromised immunity, such as hematologic malignancies and immunesuppressive treatments [1, 2]. Interest in PML has recently increased by its presentation in patients receiving humanized antibody-based immunomodulatory treatments, such as natalizumab [3, 4], efalizumab [5] and, possibly, rituximab [6]. However, it is in HIV-1 infection that PML remains most common and its biology, diagnosis and treatment issues have been most extensively studied.
In this review we summarize the epidemiology, biology and clinical presentation of HIV-1-related PML, and discuss current views on management, emphasizing areas in need of better information and understanding.
Epidemiology of HIV-1-related PML
Antiretroviral treatment has reduced the incidence of PML in HIV-1 infection and its mortality. Before the advent of combined antiretroviral therapy (cART), PML developed in 3-7% of HIV-1-infected patients [7, 8] and comprised up to 18% of fatal CNS diseases [9]. This frequency has decreased substantially in the current treatment era, though not to the same extent as other CNS opportunistic infections (CNS-OIs) [10, 11]. In the Eurosida cohort, PML incidence decreased from 0.7 per person year of follow-up in 1994 to 0.07 in 2001-2002, with the lowest annual decrease of incidence, of 31%, among all CNS-OIs [10]. Unlike the other major CNS-OIs, PML may develop in patients with blood CD4+ T-cell counts above 200 cells/μL, in patients initiating cART, and, more rarely, even in stably treated patients with full viral suppression [12, 13].
PML survival has increased substantially over the last 10 years, from 0-30% at 1 year in the pre-cART period to 38-62% following cART introduction [12-16]. However, this increase appears to be the smallest among all AIDS-defining diseases [11, 17, 18]. In the Antiretroviral Therapy Cohort Collaboration, mortality for PML was indeed 10-fold higher compared to all AIDS-related diseases [17], and, in a 2005 French survey, PML accounted for 14% of all AIDS-related deaths, second only to non-Hodgkin's lymphoma [18].
Pathogenesis of JCV Infection and PML
JCV is a small, ubiquitous DNA virus belonging to the family of human polyomaviruses (Figure 1). JCV infection is common worldwide. Both inhalation and ingestion of contaminated water or food have been suggested as major modes of human transmission [20-22]. The seroprevalence of JCV among healthy persons varies among studies, likely depending on the technique employed. In a large British survey, the overall seroprevalence was 35% by hemoagglutination inhibition assay against VP1, with figures rising with age - from 11% in children below 5 years to 50% at 60-69 years [23]. Figures were higher in other studies using the more sensitive enzyme immunoassays [24-26], with an IgG seroprevalence of 58% in a recent Swiss study on 400 healthy blood donors, also showing an increasing trend with age [26].
Figure 1.

Schematic representation of the JCV genome, composed by double-stranded circular DNA, 5,130 base pairs long - according to the JCV Mad-1 strain [24], and organized in three functional regions: regulatory, early and late. The regulatory region (RR, or non coding control region, NCCR) is a non-coding region that contains the origin of replication, viral promoter-enhancing sequences and binding sites for cell transcription factors. The early genes encode the regulatory proteins large T antigen (T-Ag) that regulates early gene transcription, initiates viral DNA replication and activates transcription of late proteins and small t antigens (t-Ag). The late genes code for the major viral capsid protein-1 (VP-1) that mediates cell attachment, and is likely the main target of both humoral and cellular immune responses, the minor capsid proteins VP-2 and VP-3, and the agnoprotein (agno).
Following primary infection, JCV establishes a persistent infection. There is evidence supporting both the urinary tract and the bone marrow as potential peripheral sites of this persistence. JCV is shed in urine of approximately 30% of immunologically normal individuals [26-28], and, recently, viral DNA has been demonstrated by polymerase chain reaction (PCR) in bone marrow of 13% of HIV-negative and 47% of HIV-1-positive patients without PML [29]. The CNS has also been suggested as a possible site of JCV persistence. The virus might reach the CNS at the time of primary infection or during its peripheral persistence phase and establish latent or low level benign CNS infection. In support of this hypothesis, PCR studies have shown JCV DNA in the brain of patients without PML [30, 31], including, recently, detection of JCV DNA fragments in oligodendrocytes and astrocytes [31]. However, efforts to identify viral proteins or DNA by in-situ techniques have generally been unsuccessful [31-33].
A proportion of patients with PML have detectable JCV DNA in plasma (see below) and the analysis of the highly variable regulatory region of JCV usually shows similar, highly-rearranged sequences in CSF and blood, but not in urine, of PML patients [34-36]. These observations support the hematogenous spread of JCV to the CNS from bone marrow or a related source at the time of PML, although, theoretically, they are also compatible with virus reactivation in the CNS and subsequent release into blood.
JCV-specific immunity is likely critical in controlling persistent JCV infection. CD4 cell responses have been detected in healthy subjects who excrete JCV in urine [37], and JCV-speci?c cytotoxic T lymphocytes were demonstrated in 73% of JCV-seropositive, immunocompetent subjects [38]. Conversely, the loss of the immune control against JCV is certainly key in favoring viral reactivation, either in periphery or in the CNS, and allowing viral propagation in the CNS, lytic infection of oligodendrocytes and brain disease. The importance of host defenses is clear from both the context of PML, with immune defects, and its remission after reversion of immune defect [39], most notably by cART [12-16]. Indeed, PML remission after initiation of cART is often associated with restoration of JCV-specific CD4 and CD8 T-cell and B-cell responses in blood [37, 40, 41] and CSF [42, 43].
Neuropathology of PML
The PML brain shows multiple areas of demyelination, varying in size and stage of evolution. Initial foci of demyelination expand and may coalesce into larger areas that in advanced cases can evolve into cavitary necrosis. All CNS regions may be involved, although spinal cord lesions are rare [44].
Oligodendrocytes sustain productive-lytic infection accompanied by characteristic histopathology (Figure 2). Their nuclei become enlarged and densely basophilic, filled with eosinophilic inclusions, and stain immunocytochemically and by in situ hybridization for JCV gene proteins and nucleic acid. Astrocytes may also appear enlarged, sometimes with multiple or multilobate hyperchromatic nuclei, at times resembling neoplastic cells – the so-called “bizarre” astrocytes [1], and also harbor JCV gene products. Foamy macrophages, though not specific for PML, are commonly present and are a response to myelin breakdown. While clinically and pathologically less conspicuous, JCV has also been identified in cerebellar granule cells, either within the context of PML, or in isolation in patients presenting with ataxia and cerebellar atrophy [45]. Thus, while usually considered as sparing neurons, JCV can also cause an infectious cerebellar degeneration in the context of immunosuppression related to its tropism for this particular class of neurons.
Figure 2.

Histopathological findings of PML**. a.** Early PML: white matter vacuolization, infected oligodendrocytes (arrows) with two to three times enlarged and basophilic nuclei; no inflammatory infiltrate (Hematoxylin-eosin, O.M. 20x). b. Demyelinating PML: foamy macrophages engulfing myelin debris (*), scattered inflammatory cells (**) and infected oligodendrocytes (arrow) with enlarged basophilic nuclei (Hematoxylin-eosin, O.M. 20x). c. An enlarged, bizarre-appearing astrocyte (arrow) with atypical multilobated nuclei and altered chromatin (Hematoxylin-eosin, O.M. 40x). d. In situ hybridization with JCV-specific probe, showing JCV DNA as brown nuclear staining of an infected oligodendrocyte (ISH, JC Virus BioProbe Labeled Probe, Enzo Diagnostics, hematoxilin counterstaining, O.M. 100x). Kindly provided by dr. Manuela Nebuloni.
One of the classical features of PML is the paucity or absence inflammation [1]. However, inflammatory forms of PML have been observed with increasing frequency following the introduction of cART. These are characterized by either diffuse or focal perivascular mononuclear infiltrates comprised mostly of CD8+ T lymphocytes and monocyte/macrophages (Figure 3); B lymphocytes, plasma cells and CD4+ T-cells are found only in small numbers [46-49]. This inflammatory PML may be the initial presenting disease phenotype or, more frequently, evolve after initiation of cART (see below).
Figure 3.
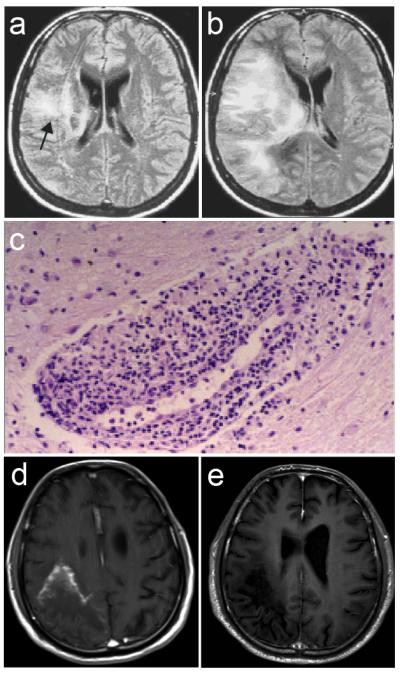
MRI and histopathology findings in two cases of inflammatory PML. Case 1 (a-c). a. At PML diagnosis in a cART-naive patient: FLAIR hyperintense lesion in the right temporal lobe extending to the periventricular white matter (CD4+ cells 4/μL, plasma HIV-1 RNA 200,000 c/mL); b. After 11 weeks of cART: increased lesion size with extension to frontal and occipital lobes and mass effect, associated with neurological worsening (CD4+ cells 64/μL, plasma HIV-1 RNA 235 c/mL). c. Biopsy of PML lesion after 15 weeks of cART: perivascular lymphomonocytic infiltration (Hematoxylin-eosin, O.M. 20x). Kindly provided by dr. Pilar Miralles. Case 2 (d, e). d. Six months after onset of PML symptoms and 4 months after cART initiation in a cART-naive patient: Gd-T1 large hypointense lesion involving the right parietal and occipital lobes with marked peripheral contrast enhancement; patient was tetraparetic and aphasic (CD4+ cells: 79 c/μL; plasma HIV-1 RNA: <50 c/mL). e. Two weeks later, following intravenous methylprednisone (1 g per day for 5 days, tapered with oral prednisone): disappearance of the enhancement, associated with frank clinical improvement (patient could walk and speak).
Clinical Manifestations of PML
Classically, PML presents insidiously with focal neurological deficits that vary depending on lesion localization. Thus, there may be hemisensory defects or hemiparesis (following involvement of parietal or frontal lobes), visual field loss and hemianopsia (occipital lobes or optic radiations), aphasia (language-dominant hemisphere), or ataxia and dysmetria (cerebellar hemispheres and peduncles). These often begin as ‘partial’ deficits that worsen with time (for example, arm weakness evolving to hemiparesis), reflecting the spread of individual lesions concentrically or along white matter tracts. Approximately 20 percent of patients develop seizures, in association with lesions adjacent to the cortex [50]. While cognitive deficits are reported in up to one third of the cases, isolated dementia without motor, sensory or visual deficits is rare [51, 52]. Headache and fever are usually absent.
Before the introduction of cART, PML was almost invariably fatal. Median survival from the diagnosis was only of a few months, although continuous progression over one year or more was occasionally observed among patients with relatively high CD4 T-cell counts [53]. In cART-treated patients, disease stabilization is observed in approximately half of the cases (see below).
Diagnosis of PML
Establishing a definitive diagnosis of PML is not only useful for clinical studies, but important for individual patient management. This is necessary in atypical cases, and, in the more typical cases, it prevents the need to revisit diagnosis in the face of disease progression, helps physicians to proceed rapidly and with certainty in therapy, and guides prognosis for patient and family decisions. One can consider three ‘stages’ to diagnosis of PML: clinical suspicion, radiological identification and etiological confirmation by CSF or tissue analysis (Table 1)[54-56]. The first of these relies on the character and temporal evolution of focal neurological symptoms and signs as outlined above, along with the setting of disease susceptibility.
Table 1.
Diagnosic criteria for PML
| Definite (etiological) diagnosis: CSF-confirmed PML: Clinical and MRI findings consistent with PML and Evidence of JCV DNA in CSF Tissue-confirmed PML:Evidence of PML neuropathology in brain tissues (biopsy or autopsy) with JCV DNA or protein detected by in situ techniques. |
|---|
| Presumptive (clinical) diagnosis: Evidence of typical clinical and MRI findings and Brain biopsy and lumbar puncture either not performed or JCV DNA not detected in CSF. |
Neuroimaging
The second stage in diagnosis entails detection and characterization of brain lesions by neuroimaging, preferably magnetic resonance imaging (MRI). MRI shows characteristic white matter lesions in brain areas corresponding to the clinical deficits. Because lesions involve demyelination, they are usually hyperintense on T2-weighted and FLAIR MRI sequences, but also hypointense on T1-weighted sequences, indicating white matter destruction. The latter helps distinguish PML from other pathologies, primarily HIV-1 encephalopathy, with more diffuse central white matter changes that are not detected on T1 sequences. While lesions can develop in any part of the brain, including the deep grey matter, they are most common in the subcortical white matter, the white matter of the cerebellar peduncles or hemispheres and in the brain stem (Figure 4). In the classical, non-inflammatory form, there is either no or only minimal contrast-enhancement, and no mass effect, unlike cerebral toxoplasmosis and primary CNS lymphoma [57, 58]. Inflammatory forms may be defined radiologically by the presence of contrast enhancement, edema, mass effect or displacement of normal structures, at times making distinction from the other opportunistic infections more difficult (Figure 3)[46, 59, 60].
Figure 4.

MRI appearance of active PML lesions (a,d,g, T2; b,e,h, FLAIR; c,f,,i, Gd-T1 axial sequences). In all cases the lesions are hyperintense (white) in T2 and FLAIR sequences and hypointense (dark) on T1 sequences, showing no enhancement after Gd administration. a-c. Hemispheric localization: large white matter signal alteration in the left frontal lobe extending to the corpus callosum and left deep white matter, with additional FLAIR/T2 high intensity signal alterations in the right frontal and temporal lobes. d-f. Cerebellar localization: large signal alterations of the left middle cerebellar peduncle and hemisphere. g-i. Brain stem localization: focal signal alterations of the left bulb (arrow) and cerebellar hemispheres.
Newer imaging techniques may provide additional diagnostic information. By diffusion-weighted imaging (DWI) sequences, which read the degree of diffusion of water molecules in a lesion area, more recent lesions and the advancing edge of established lesions show high intensity signal, reflecting active infection and cell swelling (low water diffusion). In contrast, older lesions and the center of large lesions exhibit low intensity signal, marking areas of necrosis and reparative gliosis (high water diffusion)(Figure 5)[61-63]. The degree of diffusion can also be expressed numerically using the Apparent Diffusion Coefficient (ADC) maps (Figure 5).
Figure 5.

Active PML lesions at Diffusion Weighted Imaging (DWI), Apparent Diffusion Coefficient (ADC) map and Magnetic Resonance Spectroscopy (MRS)(same case of Figure 4, 1a-c). a. DWI, hyperintense signal (white) of the periphery and low intensity signal (dark) of the centre of the lesion. b. ADC map, hyperintense signal (white) of the centre and hypointensity (dark) of the lesion periphery. c. MRS, in correspondence of the centre of the lesion, increased choline (Cho) and creatine (Cr) and relative ratio (Cho/Cr), marked reduction of N-acetyl-aspartate (NAA) and lipid (lip) peak.
With proton MR spectroscopy (MRS), PML lesions usually show elevated choline (Cho) or Cho/creatine ratio, related to demyelination, and decreased N-acetyl-aspartate (NAA), or NAA/creatine ratio, reflecting neuronal and axonal damage. The NAA/creatine ratio is usually lower in PML than in other white matter lesions, and it may help differentiate PML from HIV-1 encephalopathy. Lactate and lipid signals, likely reflecting necrosis, are also frequently elevated (Figure 5)[64, 65].
Virological confirmation
Etiological diagnosis of PML usually relies first on detection of JCV DNA in CSF by PCR (Table 1). Among HIV-1-infected, cART untreated patients with neurological diseases, the diagnostic sensitivity of this technique was of 72–92% and specificity of 92–100% [66]. Thus, a positive result is regarded as diagnostic in the appropriate clinical context. Because the rate of JCV DNA detection increases with progression of PML, lumbar puncture is usually repeated if initial PCR analysis is negative but suspicion remains high. However, the likelihood of detecting JCV in CSF may be reduced - down to 58% - in patients presenting with PML in the setting of cART [67]. The level of JCV DNA in CSF varies largely among PML patients, between the lower limit of detection - usually around 102 copies/mL - to over 107 copies/mL [67]. In untreated patients, the magnitude of JCV DNA load in CSF added additional prognostic information, with higher copy numbers predicting shorter survival [68, 69]. However, the correlation with survival is less clear for those who develop PML while on cART in whom high copy number is not always unfavorable [69, 70]. In addition, patients with lower CD4 T-cell counts generally exhibit higher CSF JCV DNA levels [69].
When efforts to identify JCV DNA in the CSF fail, brain biopsy is required to achieve an etiological diagnosis of PML. PML is identified by the characteristic tissue histopathology described above, and virological confirmation is obtained by identification of JCV proteins by immunohistochemistry, JCV DNA by in situ nucleic acid hybridization or JC virions by electron microscopy [1, 33, 71](Figure 2, Table 1).
Other laboratory assessments
By PCR, JCV DNA can also be detected in blood, both in mononuclear cells (PBMC) and plasma. JCV DNA has been found in PBMC of 10-60% of PML patients but also in up to 20% of immunocompromised controls [72-74]; copy numbers were generally below 103 per 106 cells but did not differ between patients and controls [74]. In plasma, JCV is detected in 16-40% of patients with PML, but only occasionally in controls without PML [3, 73, 75]; the viral load seems to vary widely in PML patients (from 102 to 106 copies/mL), although is most often low, i.e., below 103 copies/mL in controls [3](and personal observation). These figures indicate that JCV DNA detection in blood is unlikely to provide a sensitive and specific diagnostic tool for PML, though more systematic study is required in light of the wide range of published results. In urine, JCV DNA is detected in approximately one-third of both healthy and immunocompromised individuals without PML [26-28, 73, 76, 77], at a wide range of copy number, and thus is not useful in PML diagnosis.
Because of the high seroprevalence of JCV, the presence of JCV-specific serum IgG is unlikely to be useful in PML diagnosis. However, demonstration of JCV-specific intrathecal antibody synthesis might provide additional diagnostic support. Intrathecal production of antibodies to JCV-VP1 was observed in 76% of PML cases but in only 11% of HIV-1-infected controls without PML, with a significantly higher antibody specificity index in the former [78].
JCV-specific T-cell immunity has been assessed in PML by different technical approaches, including T-cell lymphoproliferation (studying the CD4 cell responses), cytotoxic T cell (CTL) chromium release and CD8+ tetramer identification [37, 40, 79], as well as by functional cytokine secretion and Elispot assays, measuring both CD4 and CD8 responses [41, 80]. At the time of diagnosis, most of the patients with HIV-1-related PML show low, most often undetectable, responses [37, 40, 41]. Although the diagnostic sensitivity and specificity of these assays has not been formally assessed, the presence of responses in some PML patients and, conversely, lack of responses in patients without the disease, suggest that these assessments are unlikely to become diagnostically useful.
Classification of HIV-1-associated PML, including definition of inflammatory forms
To provide a subject designation for planning and evaluating treatment interventions, we have developed a classification system of HIV-1-associated PML, which uses two principal components: cART context and immuno-virological response, and inflammatory features (Table 2). Inflammation characterizes the cART-associated Immune Reconstitution Inflammatory Syndrome (IRIS), and the presence of inflammation in PML justifies the combined term PML-IRIS [81]. By definition, the general term, IRIS, comprises three elements: 1. immune-reconstitution, meaning a decrease of plasma HIV-1 RNA with or without an increase of CD4 T cells temporarily associated with the start of cART; 2. tissue inflammation; and, 3. clinical disease or worsening that would not be expected from the natural course of the disease [82, 83]. In PML, this occurs in two settings. The first is when symptomatic PML is treated with cART and inflammation develops in relation to the existing PML lesions (paradoxical IRIS)[84]. In this setting, it is important to distinguish a favorable immune reconstitution, associated with clinical stabilization, from harmful IRIS associated with clinical worsening [48]. The second setting is when patients manifest PML after initiating cART and an inflammatory picture is found on MRI (unmasking IRIS)[84].
Table 2.
Classification of HIV-1-associated PML and Treatment Recommendations
| Category | Intervention |
|---|---|
| By cART context | |
| A. Off cART (naive or cART interruption) | Initiate cART |
| B. Recently started cART (< 6 months) | Continue cART |
| C. On chronic cART, HIV-1 suppressed * | Continue cART, consider cART intensification |
| D. On chronic cART, HIV-1 viremic * | Genotype-guided cART optimization . |
| By presence/absence of inflammatory features† | |
| 1. ‘Classical’ (non-inflammatory) PML | No corticosteroids |
| 2. Inflammatory PML | Consider initiation of corticosteroids in patients with worsening of symptoms or signs related to inflammation, continue cART |
Complicating this picture, asymptomatic patients initiating cART may develop PML after a few weeks of therapy, but without showing the characteristic inflammatory picture on MRI [12, 81]. The mechanism underlying PML development in these cases is unknown, but a ‘provocative effect’ of cART on JCV replication is invoked. These forms might be termed immune reconstitution (IR)-PML, but without the immunopathological connotation of the inflammatory syndrome terminology.
Thus, there is a spectrum of PML with respect to setting of incidence that includes both unmasking PML-IRIS and IR-PML in asymptomatic patients following initiation of cART; and both paradoxical PML-IRIS and PML with therapeutically useful and clinically benign inflammation when PML is treated with cART. It will be useful to further characterize these states with respect to JCV replication and immune responses, imaging characteristics and other biomarkers to more clearly guide clinicians in treatment approaches.
Treatment of PML
The current main approach to treatment of HIV-1-related PML involves cART with the objective of reversing the immunological defect that interferes with the normal host response to JCV. This is thus an ‘indirect’ approach to PML treatment, but it is the only one that has proven effective. While a number of antiviral and/or immunomodulant agents have been proposed or used as more specific PML treatments, none has proven effective after greater scrutiny (Table 3).
Table 3.
Antiviral and Immunomodulatory Treatments used in PML
| Drug | Rationale for use | Highest level of clinical evidence (number of treated patients) | Efficacy | References |
|---|---|---|---|---|
| Cytarabine (ARA-C) | Inhibits JCV replication in vitro [85]; early reports of clinical benefit. | Clinical trial of i.v. ARA-C vs. i.t. ARA-C vs. no ARA-C (n=57) | No benefit | 86 |
| Cidofovir (CDV) | Inhibits replication of mouse polyomavirus and SV40 in vitro [87] | Meta-analysis of 5 observational studies of CDV + cART vs. cART alone and one pilot study of CDV + cART (n=370) | No benefit | 70 |
| Topotecan | The analog camptothecin inhibits JCV replication in vitro [88] | Clinical uncontrolled trial (n=12) | Possible benefit; high toxicity | 89 |
| α-Interferon | Antiviral and immunomodulant activity | Retrospective analysis of observational study (n=97) | No benefit | 90 |
| β-interferon | Antiviral and immunomodulant activity | Anecdotes (n=2) | Possible benefit in one case * | 91, 92 |
| 5-HT2a antagonists | Block JCV cell entry in vitro [93] | Anecdotes (n=8) | Possible benefit in most of reported cases * | 94-98 |
| Interleukin-2 | Immunomodulant activity | Anecdotes (n=3) | Probable benefit | 99-101 |
Very recently, in vitro screening of a drug library of over thousand molecules, either already approved or under clinical trial, led to the identification of several drugs active against JCV [102]. One of these, the anti-malarial drug, mefloquine, which has favorable pharmacokinetic properties and brain penetration, has recently been brought into a phase I/II clinical trial evaluating its efficacy in decreasing CSF JCV DNA level. The trial is currently under way [103]. It is to be hoped that this is not only effective, but also leads the way to a greater effort in drug discovery and therapeutic trial for PML.
Combination Antiretroviral Therapy (cART)
Approximately half of HIV-1-infected PML patients receiving cART experience an arrest of disease progression [12-16, 70]. Unfortunately, neurological deficits frequently persist because of irreparable loss of brain tissue, and only a minority of patients experience functional improvement. The outcome of PML is not readily predicted at the onset or in the early stages of the disease. In some case series, poorer prognosis was associated with lower CD4+ T cell counts, higher plasma HIV-1 RNA levels, higher CSF JCV DNA level at the time of PML diagnosis, or lesion localization to the brainstem [13-16, 70, 104]. None of these variables, however, was definitely shown to strongly predict outcome.
In clinical practice, treatment strategies depend on the patient's cART status and response (Table 2). Untreated patients should start cART immediately. Similarly, treatment should be changed to an effective regimen in treated patients with continued HIV-1 viremia [56]. More problematic is treating patients who present with PML while taking cART and with suppressed HIV-1 viremia. A recent prospective study showed survival benefit in PML patients treated with four classes of cART, including enfuvirtide [105], suggesting that cART intensification might be considered in patients with undetectable plasma HIV-1. Longer PML survival has also been associated with higher CNS penetration-effectiveness score of antiretroviral regimens [106, 107]. It can be speculated that fast and effective control of HIV-1 replication in the CNS might affect JCV activity by inhibiting HIV-1 transactivation of JCV early genes [108, 109] or through unknown mechanisms associated with a reduction of intrathecal immuneactivation [110]. These issues related to antiretroviral intensification and choice of individual anti-HIV drugs clearly need further study.
As discussed above, cART-treated patients may show an exuberant inflammatory response related to the PML lesions, i.e., IRIS-PML. In HIV-1-associated IRIS outside the PML setting, corticosteroids are used to ameliorate local inflammatory reactions [82, 83]. In PML patients who respond favorably to cART, contrast enhancement or edema may not require steroid treatment if the patient is clinically stable or improving. However, in those with progressing clinical deficits and signs of inflammatory disease, corticosteroid treatment appears justified. There is little published information to guide their initiation, dosage and duration. In a recent review of 12 cases receiving steroids, a trend was observed between good PML outcome and earlier initiation or longer duration of treatment [81]. High doses of intravenous methyl-prednisolone (500-1000 mg per day for 3-5 days), oral prednisone (1-2 mg/Kg per day) or equivalent doses of oral or intravenous dexamethasone, have improved neurological function and reduced MRI signs of inflammation in individual cases of inflammatory PML [47, 49, 81](and personal observation, Figure 4).
Monitoring PML Response to Treatment
Both for clinical trials and individual patient management, methods and strategies that enable real-time monitoring of the effects of treatment and distinguish active/progressing from inactive/stabilized disease or disease remission are essential. Currently, this is achieved by the combination of neurological assessment, conventional MRI, and JCV DNA measurement in CSF. This approach is useful to show disease stabilization or progression over a period of weeks to months, but measures that could define the direction of disease evolution and treatment effects within a shorter time frame still need to be developed and clinically validated. Because of the frequently rapid evolution of PML, clinical assessment every 2-4 weeks and MRI and CSF examination every 4-8 weeks appear reasonable options with today's methods. Additional MRI evaluation can be recommended in case of rapid clinical deterioration, particularly when inflammatory changes associated with immune reconstitution are suspected. Clinical measures of functional change, such as the expanded disability status scale (EDSS) can help to objectively identify clinical stabilization or improvement. The MRI picture is considered inactive when there are no new lesions, and already established lesions show no increase in volume, FLAIR/T2 hyperintensity and T1 hypointensity. Preliminary observations have shown that DWI disappearance of the high-signal intensity rim at the lesion periphery, accompanying disease stabilization, may precede changes revealed by T2/FLAIR images [61, 63]. Disease stabilization is also associated with decreasing Cho/Cr at MRS, increasing NAA/Cr and myoinositol (mI)/Cr ratio, the latter signaling glial activation, and resolution of the lipid and lactate signals [64, 111].
Virological remission is defined by clearance of JCV DNA in CSF [47, 69, 112], likely reflecting suppression of viral replication in brain lesions. The dynamics of decay varies, within a time frame of weeks to months, depending on initial level and, likely, the degree and dynamics of immunological restoration in individual cases [69]. Clearance of JCV DNA can be associated with clinical stabilization but transitory extension of the signal abnormalities at MRI (Figure 6)[42, 60], which might reflect the host inflammatory response (edema that is hyperintense in T2 and FLAIR) rather than actual extension of the front of myelin loss.
Figure 6.

Sequential MRI in a case with favorable outcome following cART initiation (a-e, FLAIR; f-j, Gd-T1 axial sequences). a,f. At PML diagnosis: focal signal alteration of the frontal right white matter (a) with no mass effect or contrast enhancement (f); patient had motor deficit of left arm and leg; CD4+ cells: 495/μL, plasma HIV-1 RNA 262,000 c/mL, CSF JCV DNA 10,792 c/mL; cART was started immediately. b,g. Two months later: increase of both volume and FLAIR hyperintensity of the right frontal lesion (b), which shows T1 hypointensity and mild enhancement after Gd administration (g); neurological status was improved; CD4+ cells 619/μL, plasma HIV-1 RNA 4198 c/mL, CSF JCV-DNA 335 c/mL. c,g. Six months after diagnosis: additional extension of the right lesion to the adjacent areas and new onset of small hyperintensity in the left frontal white matter (c), with increased spot-like enhancement of the right lesion after Gd administration (h); neurological condition remained stable and improved from diagnosis; CD4+ cells 804 c/mL, plasma HIV-1 RNA <50 c/mL, CSF JCV DNA <100 c/mL. d,i. Twelve months after diagnosis: reduced lesion volume (d) and markedly reduced, but still evidence of, contrast enhancement of the right frontal lesion (i); enlarged subarachnoid spaces in proximity to the right frontal lesion (d), due to focal atrophy; CD4+ cells 1252/μL, plasma HIV-1 RNA <50 c/mL. e,j. Twenty months after diagnosis: further volume reduction (e) and no longer evidence of contrast enhancement of the right frontal lesion (j); further increase of the focal atrophy; CD4+ cells 1516/μL, plasma HIV-1 RNA <50 c/mL.
Assessment of JCV-specific immunity is also likely to provide a useful tool for real-time monitoring the effects of treatment. By both CD4 T-cell lymphoproliferation and studies of CD8 T-cell activity against the JCV VP-1 protein or peptides, PML patients who survived PML showed significant anti-JCV responses both at diagnosis and during follow-up, as opposed to patients with poor prognosis [37, 40, 41]. Also systemic humoral immune responses, as assessed by ELISA at time of diagnosis, were significantly greater in survivors compared to non-survivors [41] and an increased JCV-specific intrathecal antibody index was shown in a small group of cART-treated patients undergoing PML remission, in parallel with JCV DNA decay in CSF [42].
Conclusions
PML remains an important complication of HIV-1 infection. It should be promptly considered in any HIV-1-infected patients presenting with insidious onset of focal neurological symptoms, both cART untreated or treated, and especially in those recently starting cART. If the diagnosis of PML is achieved, effective cART needs be immediately instituted or continued. More generally, there is a need for clearer understanding of its pathogenesis and for development of more effective therapies, in order to diminish the impact of this disease. We need to better understand the factors involved in JCV infection and host responses that convert a persistent, but seemingly benign, infection to progressive focal encephalitis, and to exploit this understanding to predict susceptibility and develop early diagnostic and therapeutic interventions. We need to better understand of host immune responses to JCV and to define differences between helpful and immunopathological reactions underlying the opposing outcomes of remission and PML-IRIS. Finally, we need to develop specific therapies for JCV and PML to supplement the effects of cART and better methods of monitoring therapeutic responses in a shorter time frame.
Acknowledgements
We would like to thank Dr. Manuela Nebuloni, Department of Pathology, Luigi Sacco Hospital, University of Milan, Italy, and Dr. Pilar Miralles, Hospital Universitario Gregorio Marañón, Madrid, Spain, for help with iconography.
Financial Support
IJK is supported in part by NIH grant R01 NS041198 and 047029, and K24 NS 060950
JMM is supported in part by the ‘Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III, Madrid (Spain)’, Spanish Network for AIDS Research (RIS; ISCIII-RETIC RD06/006) and the Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona (Spain).
Footnotes
Search strategy and selection criteria
Data for this review were identified by searches of PubMed, and references from relevant articles. No date restrictions were set in the search. We also reviewed abstracts from major meetings on HIV-1 infection and neurology during the past three years. Search terms were ‘progressive multifocal leukoencephalopathy’ and ‘JC virus’. English language papers only were reviewed.
Conflict of Interest
PC is a member of the steering committee and site investigator of the Biogen Idec-sponsored trial of mefloquine for PML, has received research support from Biogen Idec and Biogen Dompè, and honoraria from Abbott, Biogen Idec, Biogen Dompè, Glaxo Smith Kline and Pfizer.
IJK is a site investigator in the Biogen Idec-sponsored trial of mefloquine for PML and has received research support from Biogen Idec and honoraria from Merck-Serono, Bristol-Myers-Squibb, Ono pharmaceuticals, Antisense and Alnylam.
SG has received honoraria from Biogen Dompè.
JMM has received honoraria and/or research grants from Abbott, Boehringer-Ingelheim, Bristol-Myers Squibb, Cubist, Gilead Sciences, Glaxo Smith Kline, Novartis, Pfizer, Roche and Theravance.
RWP is a site investigator in the Biogen Idec-sponsored trial of mefloquine for PML and has received research support from Biogen Idec and Merck and Co.
References
- 1.Richardson EP., Jr. Progressive multifocal leukoencephalopathy. N Engl J Med. 1961;265:815–23. doi: 10.1056/NEJM196110262651701. [DOI] [PubMed] [Google Scholar]
- 2.Garcia-Suarez J, de Miguel D, Krsnik I, Banas H, Arribas I, Burgaleta C. Changes in the natural history of progressive multifocal leukoencephalopathy in HIV-negative lymphoproliferative disorders: impact of novel therapies. Am J Hematol. 2005;80:271–81. doi: 10.1002/ajh.20492. [DOI] [PubMed] [Google Scholar]
- 3.Kappos L, Bates D, Hartung HP, et al. Natalizumab treatment for multiple sclerosis: recommendations for patient selection and monitoring. Lancet Neurol. 2007;6:431–41. doi: 10.1016/S1474-4422(07)70078-9. [DOI] [PubMed] [Google Scholar]
- 4.Hartung HP. New cases of progressive multifocal leukoencephalopathy after treatment with natalizumab. Lancet Neurol. 2009;8:28–31. doi: 10.1016/S1474-4422(08)70281-3. [DOI] [PubMed] [Google Scholar]
- 5.Pugashetti R, Koo J. Efalizumab discontinuation: a practical strategy. J Dermatolog Treat. 2009;20:132–6. doi: 10.1080/09546630902984596. [DOI] [PubMed] [Google Scholar]
- 7.Petito CK, Cho ES, Lemann W, Navia BA, Price RW. Neuropathology of acquired immunodeficiency syndrome (AIDS): an autopsy review. Journal Neuropathol Experimental Neurol. 1986;45:635–46. doi: 10.1097/00005072-198611000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Lang W, Miklossy J, Deruaz JP, et al. Neuropathology of the acquired immune deficiency syndrome (AIDS): a report of 135 consecutive autopsy cases from Switzerland. Acta Neuropathol (Berl) 1989;77:379–90. doi: 10.1007/BF00687372. [DOI] [PubMed] [Google Scholar]
- 9.Cinque P, Vago L, Dahl H, et al. Polymerase chain reaction on cerebrospinal fluid for diagnosis of virus-associated opportunistic diseases of the central nervous system in HIV-infected patients. AIDS. 1996;10:951–8. doi: 10.1097/00002030-199610090-00004. [DOI] [PubMed] [Google Scholar]
- 10.d'Arminio Monforte A, Cinque P, Mocroft A, et al. Changing incidence of central nervous system diseases in the EuroSIDA cohort. Ann Neurol. 2004;55:320–8. doi: 10.1002/ana.10827. [DOI] [PubMed] [Google Scholar]
- 11.Grabar S, Lanoy E, Allavena C, et al. Causes of the first AIDS-defining illness and subsequent survival before and after the advent of combined antiretroviral therapy. HIV Med. 2008;9:246–56. doi: 10.1111/j.1468-1293.2008.00554.x. [DOI] [PubMed] [Google Scholar]
- 12.Cinque P, Bossolasco S, Brambilla AM, et al. The effect of highly active antiretroviral therapy-induced immune reconstitution on development and outcome of progressive multifocal leukoencephalopathy: study of 43 cases with review of the literature. Journal Neurovirol. 2003;9(Suppl 1):73–80. doi: 10.1080/13550280390195351. [DOI] [PubMed] [Google Scholar]
- 13.Falco V, Olmo M, del Saz SV, et al. Influence of HAART on the clinical course of HIV-1-infected patients with progressive multifocal leukoencephalopathy: results of an observational multicenter study. J Acquir Immune Defic Syndr Hum Retrovirol. 2008;49:26–31. doi: 10.1097/QAI.0b013e31817bec64. [DOI] [PubMed] [Google Scholar]
- 14.Clifford DB, Yiannoutsos C, Glicksman M, et al. HAART improves prognosis in HIV-associated progressive multifocal leukoencephalopathy. Neurology. 1999;52:623–5. doi: 10.1212/wnl.52.3.623. [DOI] [PubMed] [Google Scholar]
- 15.Berenguer J, Miralles P, Arrizabalaga J, et al. Clinical course and prognostic factors of progressive multifocal leukoencephalopathy in patients treated with highly active antiretroviral therapy. Clin Infect Dis. 2003;36:1047–52. doi: 10.1086/374048. [DOI] [PubMed] [Google Scholar]
- 16.Khanna N, Elzi L, Mueller NJ, et al. Incidence and outcome of progressive multifocal leukoencephalopathy over 20 years of the Swiss HIV Cohort Study. Clin Infect Dis. 2009;48:1459–66. doi: 10.1086/598335. [DOI] [PubMed] [Google Scholar]
- 17.Mocroft A, Sterne JA, Egger M, et al. Variable impact on mortality of AIDS-defining events diagnosed during combination antiretroviral therapy: not all AIDS-defining conditions are created equal. Clin Infect Dis. 2009;48:1138–51. doi: 10.1086/597468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewden C, May T, Rosenthal E, et al. Changes in causes of death among adults infected by HIV between 2000 and 2005: The “Mortalite 2000 and 2005” surveys (ANRS EN19 and Mortavic) J Acquir Immune Defic Syndr Hum Retrovirol. 2008;48:590–8. doi: 10.1097/QAI.0b013e31817efb54. [DOI] [PubMed] [Google Scholar]
- 19.Frisque RJ, Bream GL, Cannella MT. Human polyomavirus JC virus genome. J Virol. 1984;51:458–69. doi: 10.1128/jvi.51.2.458-469.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bofill-Mas S, Formiga-Cruz M, Clemente-Casares P, Calafell F, Girones R. Potential transmission of human polyomaviruses through the gastrointestinal tract after exposure to virions or viral DNA. J Virol. 2001;75:10290–9. doi: 10.1128/JVI.75.21.10290-10299.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monaco MC, Jensen PN, Hou J, Durham LC, Major EO. Detection of JC virus DNA in human tonsil tissue: evidence for site of initial viral infection. J Virol. 1998;72:9918–23. doi: 10.1128/jvi.72.12.9918-9923.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sabath BF, Major EO. Traffic of JC virus from sites of initial infection to the brain: the path to progressive multifocal leukoencephalopathy. J Infect Dis. 2002;186(Suppl 2):S180–6. doi: 10.1086/344280. [DOI] [PubMed] [Google Scholar]
- 23.Knowles WA, Pipkin P, Andrews N, et al. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J Med Virol. 2003;71:115–23. doi: 10.1002/jmv.10450. [DOI] [PubMed] [Google Scholar]
- 24.Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamilton RS, Gravell M, Major EO. Comparison of antibody titers determined by hemagglutination inhibition and enzyme immunoassay for JC virus and BK virus. J Clin Microbiol. 2000;38:105–9. doi: 10.1128/jcm.38.1.105-109.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Egli A, Infanti L, Dumoulin A, et al. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis. 2009;199:837–46. doi: 10.1086/597126. [DOI] [PubMed] [Google Scholar]
- 27.Markowitz RB, Thompson HC, Mueller JF, Cohen JA, Dynan WS. Incidence of BK virus and JC virus viruria in human immunodeficiency virus-infected and -uninfected subjects. J Infect Dis. 1993;167:13–20. doi: 10.1093/infdis/167.1.13. [DOI] [PubMed] [Google Scholar]
- 28.Lednicky JA, Vilchez RA, Keitel WA, et al. Polyomavirus JCV excretion and genotype analysis in HIV-infected patients receiving highly active antiretroviral therapy. AIDS. 2003;17:801–7. doi: 10.1097/00002030-200304110-00004. [DOI] [PubMed] [Google Scholar]
- 29.Tan CS, Dezube BJ, Bhargava P, et al. Detection of JC virus DNA and proteins in the bone marrow of HIV-positive and HIV-negative patients: implications for viral latency and neurotropic transformation. J Infect Dis. 2009;199:881–8. doi: 10.1086/597117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White FA, 3rd, Ishaq M, Stoner GL, Frisque RJ. JC virus DNA is present in many human brain samples from patients without progressive multifocal leukoencephalopathy. J Virol. 1992;66:5726–34. doi: 10.1128/jvi.66.10.5726-5734.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perez-Liz G, Del Valle L, Gentilella A, Croul S, Khalili K. Detection of JC virus DNA fragments but not proteins in normal brain tissue. Ann Neurol. 2008 doi: 10.1002/ana.21443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vago L, Cinque P, Sala E, et al. JCV-DNA and BKV-DNA in the CNS tissue and CSF of AIDS patients and normal subjects. Study of 41 cases and review of the literature. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;12:139–46. doi: 10.1097/00042560-199606010-00006. [DOI] [PubMed] [Google Scholar]
- 33.Procop GW, Beck RC, Pettay JD, et al. JC virus chromogenic in situ hybridization in brain biopsies from patients with and without PML. Diagn Mol Pathol. 2006;15:70–3. doi: 10.1097/00019606-200606000-00002. [DOI] [PubMed] [Google Scholar]
- 34.Ciappi S, Azzi A, De Santis R, et al. Archetypal and rearranged sequences of human polyomavirus JC transcription control region in peripheral blood leukocytes and in cerebrospinal fluid. J Gen Virol. 1999;80:1017–23. doi: 10.1099/0022-1317-80-4-1017. [DOI] [PubMed] [Google Scholar]
- 35.Fedele CG, Ciardi MR, Delia S, et al. Identical rearranged forms of JC polyomavirus transcriptional control region in plasma and cerebrospinal fluid of acquired immunodeficiency syndrome patients with progressive multifocal leukoencephalopathy. J Neurovirol. 2003;9:551–8. doi: 10.1080/13550280390241188. [DOI] [PubMed] [Google Scholar]
- 36.Pfister LA, Letvin NL, Koralnik IJ. JC virus regulatory region tandem repeats in plasma and central nervous system isolates correlate with poor clinical outcome in patients with progressive multifocal leukoencephalopathy. J Virol. 2001;75:5672–6. doi: 10.1128/JVI.75.12.5672-5676.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gasnault J, Kahraman M, de Goer de Herve MG, Durali D, Delfraissy JF, Taoufik Y. Critical role of JC virus-specific CD4 T-cell responses in preventing progressive multifocal leukoencephalopathy. AIDS. 2003;17:1443–9. doi: 10.1097/00002030-200307040-00004. [DOI] [PubMed] [Google Scholar]
- 38.Du Pasquier RA, Schmitz JE, Jean-Jacques J, et al. Detection of JC virus-specific cytotoxic T lymphocytes in healthy individuals. J Virol. 2004;78:10206–10. doi: 10.1128/JVI.78.18.10206-10210.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Price RW, Nielsen S, Horten B, Rubino M, Padgett B, Walker D. Progressive multifocal leukoencephalopathy: a burnt-out case. Ann Neurol. 1983;13:485–90. doi: 10.1002/ana.410130503. [DOI] [PubMed] [Google Scholar]
- 40.Du Pasquier RA, Kuroda MJ, Zheng Y, Jean-Jacques J, Letvin NL, Koralnik IJ. A prospective study demonstrates an association between JC virus-specific cytotoxic T lymphocytes and the early control of progressive multifocal leukoencephalopathy. Brain. 2004;127:1970–8. doi: 10.1093/brain/awh215. [DOI] [PubMed] [Google Scholar]
- 41.Khanna N, Wolbers M, Mueller NJ, et al. JC virus-specific immune responses in human immunodeficiency virus type 1 patients with progressive multifocal leukoencephalopathy. J Virol. 2009;83:4404–11. doi: 10.1128/JVI.02657-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giudici B, Vaz B, Bossolasco S, et al. Highly active antiretroviral therapy and progressive multifocal leukoencephalopathy: effects on cerebrospinal fluid markers of JC virus replication and immune response. Clin Infect Dis. 2000;30:95–9. doi: 10.1086/313598. [DOI] [PubMed] [Google Scholar]
- 43.Du Pasquier RA, Autissier P, Zheng Y, Jean-Jacques J, Koralnik IJ. Presence of JC virus-specific CTL in the cerebrospinal fluid of PML patients: rationale for immune-based therapeutic strategies. AIDS. 2005;19:2069–76. doi: 10.1097/01.aids.0000194804.97164.86. [DOI] [PubMed] [Google Scholar]
- 44.Bernal-Cano F, Joseph JT, Koralnik IJ. Spinal cord lesions of progressive multifocal leukoencephalopathy in an AIDS patient. J Neurovirol. 2007;13:474–6. doi: 10.1080/13550280701469178. [DOI] [PubMed] [Google Scholar]
- 45.Wuthrich C, Cheng YM, Joseph JT, et al. Frequent infection of cerebellar granule cell neurons by polyomavirus JC in progressive multifocal leukoencephalopathy. J Neuropatho Exp Neurol. 2009;68:15–25. doi: 10.1097/NEN.0b013e3181912570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang D, Cossoy M, Li M, Choi D, Taege A, Staugaitis SM, Rehm S, Ransohoff RM. Inflammatory progressive multifocal leukoencephalopathy in human immunodeficiency virus-negative patients. Ann Neurol. 2007 Jul;62(1):34–9. doi: 10.1002/ana.21085. [DOI] [PubMed] [Google Scholar]
- 47.Miralles P, Berenguer J, Lacruz C, et al. Inflammatory reactions in progressive multifocal leukoencephalopathy after highly active antiretroviral therapy. AIDS. 2001;15:1900–2. doi: 10.1097/00002030-200109280-00028. [DOI] [PubMed] [Google Scholar]
- 48.Vendrely A, Bienvenu B, Gasnault J, Thiebault JB, Salmon D, Gray F. Fulminant inflammatory leukoencephalopathy associated with HAART-induced immune restoration in AIDS-related progressive multifocal leukoencephalopathy. Acta Neuropathol (Berl) 2005;109:449–55. doi: 10.1007/s00401-005-0983-y. [DOI] [PubMed] [Google Scholar]
- 49.Martinez JV, Mazziotti JV, Efron ED, et al. Immune reconstitution inflammatory syndrome associated with PML in AIDS: a treatable disorder. Neurology. 2006;67:1692–4. doi: 10.1212/01.wnl.0000242728.26433.12. [DOI] [PubMed] [Google Scholar]
- 50.Lima MA, Drislane FW, Koralnik IJ. Seizures and their outcome in progressive multifocal leukoencephalopathy. Neurology. 2006;66:262–4. doi: 10.1212/01.wnl.0000194227.16696.11. [DOI] [PubMed] [Google Scholar]
- 51.Berger JR, Pall L, Lanska D, Whiteman M. Progressive multifocal leukoencephalopathy in patients with HIV infection. J Neurovirol. 1998;4:59–68. doi: 10.3109/13550289809113482. [DOI] [PubMed] [Google Scholar]
- 52.Zunt JR, Tu RK, Anderson DM, Copass MC, Marra CM. Progressive multifocal leukoencephalopathy presenting as human immunodeficiency virus type 1 (HIV)-associated dementia. Neurology. 1997;49:263–5. doi: 10.1212/wnl.49.1.263. [DOI] [PubMed] [Google Scholar]
- 53.Berger JR, Levy RM, Flomenhoft D, Dobbs M. Predictive factors for prolonged survival in acquired immunodeficiency syndrome-associated progressive multifocal leukoencephalopathy. Ann Neurol. 1998;44:341–9. doi: 10.1002/ana.410440309. [DOI] [PubMed] [Google Scholar]
- 54.Cinque P, Koralnik IJ, Clifford DB. The evolving face of human immunodeficiency virus-related progressive multifocal leukoencephalopathy: defining a consensus terminology. J Neurovirol. 2003;9(Suppl 1):88–92. doi: 10.1080/13550280390195298. [DOI] [PubMed] [Google Scholar]
- 55.Portegies P, Solod L, Cinque P, et al. Guidelines for the diagnosis and management of neurological complications of HIV infection. Eur J Neurol. 2004;11:297–304. doi: 10.1111/j.1468-1331.2004.00856.x. [DOI] [PubMed] [Google Scholar]
- 56.Kaplan JE, Benson C, Holmes KH, Brooks JT, Pau A, Masur H. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep. 2009;58:1–207. quiz CE1-4. [PubMed] [Google Scholar]
- 57.Manzardo C, Del Mar Ortega M, Sued O, Garcia F, Moreno A, Miro JM. Central nervous system opportunistic infections in developed countries in the highly active antiretroviral therapy era. J Neurovirol. 2005;11(Suppl 3):72–82. doi: 10.1080/13550280500513846. [DOI] [PubMed] [Google Scholar]
- 58.Skiest DJ. Focal neurological disease in patients with acquired immunodeficiency syndrome. Clin Infect Dis. 2002;34:103–15. doi: 10.1086/324350. [DOI] [PubMed] [Google Scholar]
- 59.Post MJ, Yiannoutsos C, Simpson D, et al. Progressive multifocal leukoencephalopathy in AIDS: are there any MR findings useful to patient management and predictive of patient survival? AIDS Clinical Trials Group, 243 Team. AJNR Am J Neuroradiol. 1999;20:1896–906. [PMC free article] [PubMed] [Google Scholar]
- 60.Thurnher MM, Post MJ, Rieger A, Kleibl-Popov C, Loewe C, Schindler E. Initial and follow-up MR imaging findings in AIDS-related progressive multifocal leukoencephalopathy treated with highly active antiretroviral therapy. AJNR Am J Neuroradiol. 2001;22:977–84. [PMC free article] [PubMed] [Google Scholar]
- 61.Mader I, Herrlinger U, Klose U, Schmidt F, Kuker W. Progressive multifocal leukoencephalopathy: analysis of lesion development with diffusion-weighted MRI. Neuroradiology. 2003;45:717–21. doi: 10.1007/s00234-003-0966-4. [DOI] [PubMed] [Google Scholar]
- 62.Bergui M, Bradac GB, Oguz KK, et al. Progressive multifocal leukoencephalopathy: diffusion-weighted imaging and pathological correlations. Neuroradiology. 2004;46:22–5. doi: 10.1007/s00234-003-1115-9. [DOI] [PubMed] [Google Scholar]
- 63.Usiskin SI, Bainbridge A, Miller RF, Jager HR. Progressive multifocal leukoencephalopathy: serial high-b-value diffusion-weighted MR imaging and apparent diffusion coefficient measurements to assess response to highly active antiretroviral therapy. AJNR Am J Neuroradiol. 2007;28:285–6. [PMC free article] [PubMed] [Google Scholar]
- 64.Chang L, Ernst T, Tornatore C, et al. Metabolite abnormalities in progressive multifocal leukoencephalopathy by proton magnetic resonance spectroscopy. Neurology. 1997;48:836–45. doi: 10.1212/wnl.48.4.836. [DOI] [PubMed] [Google Scholar]
- 65.Simone IL, Federico F, Tortorella C, et al. Localised 1H-MR spectroscopy for metabolic characterisation of diffuse and focal brain lesions in patients infected with HIV. J Neurol Neurosurg Psychiatry. 1998;64:516–23. doi: 10.1136/jnnp.64.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cinque P, Scarpellini P, Vago L, Linde A, Lazzarin A. Diagnosis of central nervous system complications in HIV-infected patients: cerebrospinal fluid analysis by the polymerase chain reaction. AIDS. 1997;11:1–17. doi: 10.1097/00002030-199701000-00003. [DOI] [PubMed] [Google Scholar]
- 67.Marzocchetti A, Di Giambenedetto S, Cingolani A, Ammassari A, Cauda R, De Luca A. Reduced rate of diagnostic positive detection of JC virus DNA in cerebrospinal fluid in cases of suspected progressive multifocal leukoencephalopathy in the era of potent antiretroviral therapy. J Clin Microbiol. 2005;43:4175–7. doi: 10.1128/JCM.43.8.4175-4177.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yiannoutsos CT, Major EO, Curfman B, et al. Relation of JC virus DNA in the cerebrospinal fluid to survival in acquired immunodeficiency syndrome patients with biopsy-proven progressive multifocal leukoencephalopathy. Ann Neurol. 1999;45:816–21. doi: 10.1002/1531-8249(199906)45:6<816::aid-ana21>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 69.Bossolasco S, Calori G, Moretti F, et al. Prognostic significance of JC virus DNA levels in cerebrospinal fluid of patients with HIV-associated progressive multifocal leukoencephalopathy. Clin Infect Dis. 2005;40:738–44. doi: 10.1086/427698. [DOI] [PubMed] [Google Scholar]
- 70.De Luca A, Ammassari A, Pezzotti P, et al. Cidofovir in addition to antiretroviral treatment is not effective for AIDS-associated progressive multifocal leukoencephalopathy: a multicohort analysis. AIDS. 2008;22:1759–67. doi: 10.1097/QAD.0b013e32830a5043. [DOI] [PubMed] [Google Scholar]
- 71.Jochum W, Weber T, Frye S, Hunsmann G, Luke W, Aguzzi A. Detection of JC virus by anti-VP1 immunohistochemistry in brains with progressive multifocal leukoencephalopathy. Acta Neuropathol (Berl) 1997;94:226–31. doi: 10.1007/s004010050697. [DOI] [PubMed] [Google Scholar]
- 72.Tornatore C, Berger JR, Houff SA, et al. Detection of JC virus DNA in peripheral lymphocytes from patients with and without progressive multifocal leukoencephalopathy. Ann Neurol. 1992;31:454–62. doi: 10.1002/ana.410310426. [DOI] [PubMed] [Google Scholar]
- 73.Koralnik IJ, Boden D, Mai VX, Lord CI, Letvin NL. JC virus DNA load in patients with and without progressive multifocal leukoencephalopathy. Neurology. 1999;52:253–60. doi: 10.1212/wnl.52.2.253. [DOI] [PubMed] [Google Scholar]
- 74.Andreoletti L, Lescieux A, Lambert V, et al. Semiquantitative detection of JCV-DNA in peripheral blood leukocytes from HIV-1-infected patients with or without progressive multifocal leukoencephalopathy. J Med Virol. 2002;66:1–7. doi: 10.1002/jmv.2103. [DOI] [PubMed] [Google Scholar]
- 75.Dubois V, Moret H, Lafon ME, et al. Prevalence of JC virus viraemia in HIV-infected patients with or without neurological disorders: a prospective study. J Neurovirol. 1998;4:539–44. doi: 10.3109/13550289809113498. [DOI] [PubMed] [Google Scholar]
- 76.Shah KV, Daniel RW, Strickler HD, Goedert JJ. Investigation of human urine for genomic sequences of the primate polyomaviruses simian virus 40, BK virus, and JC virus. J Infect Dis. 1997;176:1618–21. doi: 10.1086/517340. [DOI] [PubMed] [Google Scholar]
- 77.Knowles WA, Pillay D, Johnson MA, Hand JF, Brown DW. Prevalence of long-term BK and JC excretion in HIV-infected adults and lack of correlation with serological markers. J Med Virol. 1999;59:474–9. [PubMed] [Google Scholar]
- 78.Weber T, Trebst C, Frye S, et al. Analysis of the systemic and intrathecal humoral immune response in progressive multifocal leukoencephalopathy. J Infect Dis. 1997;176:250–4. doi: 10.1086/514032. [DOI] [PubMed] [Google Scholar]
- 79.Weber F, Goldmann C, Kramer M, et al. Cellular and humoral immune response in progressive multifocal leukoencephalopathy. Ann Neurol. 2001;49:636–42. [PubMed] [Google Scholar]
- 80.Binggeli S, Egli A, Schaub S, et al. Polyomavirus BK-specific cellular immune response to VP1 and large T-antigen in kidney transplant recipients. Am J Transplant. 2007;7:1131–9. doi: 10.1111/j.1600-6143.2007.01754.x. [DOI] [PubMed] [Google Scholar]
- 81.Tan K, Roda R, Ostrow L, McArthur J, Nath A. PML-IRIS in patients with HIV infection: clinical manifestations and treatment with steroids. Neurology. 2009;72:1458–64. doi: 10.1212/01.wnl.0000343510.08643.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shelburne SA, 3rd, Hamill RJ, Rodriguez-Barradas MC, et al. Immune reconstitution inflammatory syndrome: emergence of a unique syndrome during highly active antiretroviral therapy. Medicine. 2002;81:213–27. doi: 10.1097/00005792-200205000-00005. [DOI] [PubMed] [Google Scholar]
- 83.French MA, Price P, Stone SF. Immune restoration disease after antiretroviral therapy. AIDS. 2004;18:1615–27. doi: 10.1097/01.aids.0000131375.21070.06. [DOI] [PubMed] [Google Scholar]
- 84.French MA. HIV/AIDS: immune reconstitution inflammatory syndrome: a reappraisal. Clin Infect Dis. 2009;48:101–7. doi: 10.1086/595006. [DOI] [PubMed] [Google Scholar]
- 85.Hou J, Major EO. The efficacy of nucleoside analogs against JC virus multiplication in a persistently infected human fetal brain cell line. J Neurovirol. 1998;4:451–6. doi: 10.3109/13550289809114545. [DOI] [PubMed] [Google Scholar]
- 86.Hall CD, Dafni U, Simpson D, et al. Failure of cytarabine in progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. AIDS Clinical Trials Group 243 Team. N Engl J Med. 1998;338:1345–51. doi: 10.1056/NEJM199805073381903. [DOI] [PubMed] [Google Scholar]
- 87.Andrei G, Snoeck R, Vandeputte M, De Clercq E. Activities of various compounds against murine and primate polyomaviruses. Antimicrob Agents Chemother. 1997;41:587–93. doi: 10.1128/aac.41.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kerr DA, Chang CF, Gordon J, Bjornsti MA, Khalili K. Inhibition of human neurotropic virus (JCV) DNA replication in glial cells by camptothecin. Virology. 1993;196:612–8. doi: 10.1006/viro.1993.1517. [DOI] [PubMed] [Google Scholar]
- 89.Royal W, 3rd, Dupont B, McGuire D, et al. Topotecan in the treatment of acquired immunodeficiency syndrome-related progressive multifocal leukoencephalopathy. J Neurovirol. 2003;9:411–9. doi: 10.1080/13550280390201740. [DOI] [PubMed] [Google Scholar]
- 90.Geschwind MD, Skolasky RI, Royal WS, McArthur JC. The relative contributions of HAART and alpha-interferon for therapy of progressive multifocal leukoencephalopathy in AIDS. J Neurovirol. 2001;7:353–7. doi: 10.1080/13550280152537238. [DOI] [PubMed] [Google Scholar]
- 91.Tashiro K, Doi S, Moriwaka F, Maruo Y, Nomura M. Progressive multifocal leucoencephalopathy with magnetic resonance imaging verification and therapeutic trials with interferon. J Neurol. 1987;234:427–9. doi: 10.1007/BF00314091. [DOI] [PubMed] [Google Scholar]
- 92.Nath A, Venkataramana A, Reich DS, Cortese I, Major EO. Progression of progressive multifocal leukoencephalopathy despite treatment with beta-interferon. Neurology. 2006;66:149–50. doi: 10.1212/01.wnl.0000191322.93310.a1. [DOI] [PubMed] [Google Scholar]
- 93.Elphick GF, Querbes W, Jordan JA, et al. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306:1380–3. doi: 10.1126/science.1103492. [DOI] [PubMed] [Google Scholar]
- 94.Lima MA, Auriel E, Wuthrich C, Borenstein NM, Koralnik IJ. Progressive multifocal leukoencephalopathy as a complication of hepatitis C virus treatment in an HIV-negative patient. Clin Infect Dis. 2005;41:417–9. doi: 10.1086/431769. [DOI] [PubMed] [Google Scholar]
- 95.Vulliemoz S, Lurati-Ruiz F, Borruat FX, et al. Favourable outcome of progressive multifocal leucoencephalopathy in two patients with dermatomyositis. J Neurol Neurosurg Psychiatry. 2006;77:1079–82. doi: 10.1136/jnnp.2006.092353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Focosi D, Fazzi R, Montanaro D, Emdin M, Petrini M. Progressive multifocal leukoencephalopathy in a haploidentical stem cell transplant recipient: a clinical, neuroradiological and virological response after treatment with risperidone. Antiviral Res. 2007;74:156–8. doi: 10.1016/j.antiviral.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 97.Verma S, Cikurel K, Koralnik IJ, et al. Mirtazapine in progressive multifocal leukoencephalopathy associated with polycythemia vera. J Infect Dis. 2007;196:709–11. doi: 10.1086/520514. [DOI] [PubMed] [Google Scholar]
- 98.Cettomai D, McArthur JC. Mirtazapine use in human immunodeficiency virus-infected patients with progressive multifocal leukoencephalopathy. Arch Neurol. 2009;66:255–8. doi: 10.1001/archneurol.2008.557. [DOI] [PubMed] [Google Scholar]
- 99.Przepiorka D, Jaeckle KA, Birdwell RR, et al. Successful treatment of progressive multifocal leukoencephalopathy with low-dose interleukin-2. Bone Marrow Transplant. 1997;20:983–7. doi: 10.1038/sj.bmt.1701010. [DOI] [PubMed] [Google Scholar]
- 100.Buckanovich RJ, Liu G, Stricker C, et al. Nonmyeloablative allogeneic stem cell transplantation for refractory Hodgkin's lymphoma complicated by interleukin-2 responsive progressive multifocal leukoencephalopathy. Ann Hematol. 2002;81:410–3. doi: 10.1007/s00277-002-0481-4. [DOI] [PubMed] [Google Scholar]
- 101.Kunschner L, Scott TF. Sustained recovery of progressive multifocal leukoencephalopathy after treatment with IL-2. Neurology. 2005;65:1510. doi: 10.1212/01.wnl.0000183064.10227.b5. [DOI] [PubMed] [Google Scholar]
- 102.Brickelmaier M, Lugovskoy A, Kartikeyan R, Reviriego-Mendoza MM, Allaire N, Simon K, Frisque RJ, Gorelik L. Identification and characterization of mefloquine efficacy against JC virus in vitro. Antimicrob Agents Chemother. 2009;53(1):840–9. doi: 10.1128/AAC.01614-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gasnault J, Taoufik Y, Goujard C, et al. Prolonged survival without neurological improvement in patients with AIDS-related progressive multifocal leukoencephalopathy on potent combined antiretroviral therapy. J Neurovirol. 1999;5:421–9. doi: 10.3109/13550289909029483. [DOI] [PubMed] [Google Scholar]
- 105.Gasnault J, Chavez HH, Dorofeev E, et al. Acceleration of Immune Recovery on Intensified ART Improves Survival in Patients with AIDS-related Progressive Multifocal Leukoencephalopathy: Preliminary Reports of the ANRS 125 Trial (abstr); Presented at the 14th Conference on Retroviruses and Opportunistic Infections; Los Angeles, CA. 2007 Febr 25-27. [Google Scholar]
- 106.Letendre S, Marquie-Beck J, Capparelli E, et al. Validation of the CNS Penetration-Effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch Neurol. 2008;65:65–70. doi: 10.1001/archneurol.2007.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gasnault J, Lanoy E, Bentata M, Guiguet M, Costagliola D. Intracerebral Penetrating ART Are More Efficient on Survival of HIV+ Patients with Progressive Multifocal Leucoencephalopathy (ANRS CO4 - FHDH)(abstr); Presented at the 15th Conference on Retroviruses and opportunistic infections; Boston, MA, USA. February 3-6, 2008. [Google Scholar]
- 108.Tada H, Rappaport J, Lashgari M, Amini S, Wong-Staal F, Khalili K. Trans-activation of the JC virus late promoter by the tat protein of type 1 human immunodeficiency virus in glial cells. Proc Natl Acad Sci USA. 1990;87:3479–83. doi: 10.1073/pnas.87.9.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Berger JR, Chauhan A, Galey D, Nath A. Epidemiological evidence and molecular basis of interactions between HIV and JC virus. J Neurovirol. 2001;7:329–38. doi: 10.1080/13550280152537193. [DOI] [PubMed] [Google Scholar]
- 110.Sinclair E, Ronquillo R, Lollo N, et al. Antiretroviral treatment effect on immune activation reduces cerebrospinal fluid HIV-1 infection. J Acquir Immune Defic Syndr Hum Retrovirol. 2008;47:544–52. doi: 10.1097/QAI.0b013e318162754f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Katz-Brull R, Lenkinski RE, Du Pasquier RA, Koralnik IJ. Elevation of myoinositol is associated with disease containment in progressive multifocal leukoencephalopathy. Neurology. 2004;63:897–900. doi: 10.1212/01.wnl.0000137420.58346.9f. [DOI] [PubMed] [Google Scholar]
- 112.Taoufik Y, Gasnault J, Karaterki A, et al. Prognostic value of JC virus load in cerebrospinal fluid of patients with progressive multifocal leukoencephalopathy. J Infect Dis. 1998;178:1816–20. doi: 10.1086/314496. [DOI] [PubMed] [Google Scholar]