Gi protein activation in intact cells involves subunit rearrangement rather than dissociation (original) (raw)
Abstract
G protein-coupled receptors transduce diverse extracellular signals, such as neurotransmitters, hormones, chemokines, and sensory stimuli, into intracellular responses through activation of heterotrimeric G proteins. G proteins play critical roles in determining specificity and kinetics of subsequent biological responses by modulation of effector proteins. We have developed a fluorescence resonance energy transfer (FRET)-based assay to directly measure mammalian G protein activation in intact cells and found that Gi proteins activate within 1-2 s, which is considerably slower than activation kinetics of the receptors themselves. More importantly, FRET measurements demonstrated that Gαi- and Gβγ-subunits do not dissociate during activation, as has been previously postulated. Based on FRET measurements between Gαi-yellow fluorescent protein and Gβγ-subunits that were fused to cyan fluorescent protein at various positions, we conclude that, instead, G protein subunits undergo a molecular rearrangement during activation. The detection of a persistent heterotrimeric composition during G protein activation will impact the understanding of how G proteins achieve subtype-selective coupling to effectors. This finding will be of particular interest for unraveling Gβγ-induced signaling pathways.
A variety of physiological signals such as neurotransmitters, hormones, and light are detected by members of the seven transmembrane domain receptor family. These G protein-coupled receptors (GPCRs) activate G proteins by promoting binding of GTP in exchange for GDP. Both, Gα and Gβγ-subunits of activated G proteins can regulate downstream effectors such as adenylyl cyclases, phospholipases, or ion channels. Based on biochemical experiments and structural studies, it is known that conformational rearrangements in the “switch regions” of the α-subunits on GTP binding weaken the interaction with Gβγ-subunits (1-3). It is generally assumed that the reduced affinity of Gα-GTP for Gβγ causes G proteins to dissociate into Gα-GTP and a Gβγ complex. Reassociation is then assumed to occur on hydrolysis of the GTP bound to Gα, which can be accelerated by RGS proteins (4-6). However, this model fails to explain well established phenomena in G protein-mediated signaling. Most importantly, a major open question is how Gβγ effectors, such as G protein-activated inwardly rectifying K+ (GIRK) channel, are selectively regulated through specific Gα subtypes despite the lack of subtype selectivity for Gβγ subtypes (7-10). In attempts to answer this central question, it has been hypothesized that selectivity may be caused by scaffolding of G proteins and effectors, either by direct binding of Gα to its effector (11), or by temporally and spatially restricting G protein activity (12, 13).
The possibility to investigate protein-protein interactions in living cells by using fluorescence resonance energy transfer (FRET) between recombinant fluorescent proteins (14), has recently led to new insights in temporal signaling properties of G protein effectors (15-17) as well as temporal aspects of GPCR activation (18). Most of these studies make use of variants of GFP, which are fused to the protein of interest (14). By using FRET between cyan fluorescent protein (CFP)- and yellow fluorescent protein (YFP)-labeled Gβγ-and Gα-subunits in Dictyostelium, Devreotes and coworkers (19) reported receptor-mediated dissociation of G proteins of the Gαs family. In the present study, we studied the kinetics and spatial pattern of mammalian heterotrimeric Gi proteins in living cells by using a similar approach. We found, in contradiction to the current prevailing model, that heterotrimeric G proteins of the Gi family undergo a conformational rearrangement during activation without subunit dissociation.
Experimental Procedures
Molecular Biology and Cell Culture. Site-directed mutagenesis was performed on the cDNAs of rat Gαi1 and human Gβ1 and Gγ2. The cDNA encoding the enhanced YFP F46L (20) was inserted between position 91 and 92 of Ptx-insensitive C351I mutants of Gαi1 (Gai1-1-87-MGRL_ EFMV-YFP-LYSS_KIDF-Gai1-96-353) (21). CFP 1-239 was fused either to the N terminus of Gβ1 (_CFP_-ELYK_G-S_MSEL-Gβ1) or to the C terminus of Gγ2 (Gγ2-CAIL_SR_ MVSK-CFP). Constructions were performed by conventional PCR and verified by sequencing. G protein cDNAs were cloned into pCDNA3 (Invitrogen). CFP-N-Gγ2 was kindly provided by S. Ikeda (Guthrie Research Institute, Sayre, PA) (22). Transient transfections of HEK cells were performed by using effectene, according to the manufacturers protocol (Qiagen), by using (μg cDNA/5-cm dish: Gαi1, 1.8; Gβ1, 0.5; Gγ2, 0.2; α2A-adrenergic receptor (AR), 0.5 and GIRK1/4, 0.4 (bicistronic vector kindly provided by L. Y. Jan, University of California, San Francisco).
Electrophysiology. GIRK currents activated by fluorescent G protein subunits were measured as described (23), except that, in the present study, an Axopatch 200B and CLAMPEX software (Axon Instruments, Foster City, CA) was used. Experimental conditions including external and internal solutions were the same as described (24).
External Buffer. The following was used as external buffer: 124 mM NaCl/20 mM KCl/10 mM Hepes/2 mM CaCl2/1 mM MgCl2, pH 7.3.
Fluorescence Measurements. Fluorescence microscopy was performed as described (18) by placing cells plated on coverslips on a Zeiss inverted microscope (Axiovert 200) with an oil-immersion ×63 objective and a dual-emission photometric system and a polychrome IV (both TILL Photonics, Planegg, Germany). To minimize photobleaching, the illumination time was set to <40 ms applied with a frequency between 1 and 20 Hz, which depended on agonist concentration. FRET ratios were measured as the ratio of the YFP and CFP emission, FYFP/FCFP, where FYFP and FCFP are the (corrected) emission intensities at 535 ± 15 and 480 ± 20 nm (beam splitter DCLP 505 nm) on excitation at 436 ± 10 nm (beam splitter DCLP 460 nm). The YFP emission on excitation at 480 nm was recorded at the beginning of each experiment to subtract direct excitation of YFP (YFP emission at 436-nm excitation/YFP emission at 480-nm excitation was 0.06). Spillover of CFP into the 535-nm channel (spillover of YFP into the 480-nm channel was negligible) was subtracted to give a corrected FRET ratio of FYFP/FCFP.
FRET between CFP and YFP was also determined by donor dequenching after acceptor photobleaching (APB) (5-min illumination at 480 nm). To do so, FCFP was recorded (436-nm excitation) followed by the APB protocol, and, subsequently, FCFP was recorded again, and the increase of FCFP after APB of YFP was determined. Direct bleaching of CFP by the APB procedure was determined in cells expressing CFP-N-Gγ2 alone (3.5%) and all data were corrected for this effect. To determine agonist-induced changes in FRET, cells were continuously superfused with external buffer (see above) and agonist was applied by using a rapid superfusion device (18).
origin (Microcal, Amherst, MA) was used for data analysis and statistics (summarized data were expressed as mean ± SEM).
FRET Imaging. Images of CFP and YFP emission were recorded simultaneously by using a similar procedure, as described above, and equipped for dual-emission imaging (beam splitter: 505 DCXR; emission filters: 535 ± 20 and 480 ± 15 nm), by using a Cool Snap HQ charge-coupled device camera (Visitron Systems, Puchheim, Germany). Illumination time was set to 100 ms. Bleedthrough of CFP into the 535-nm channel and direct excitation of YFP by 436-nm excitation were subtracted. For image acquisition and analysis, METAFLUOR and METAMORPH (Visitron Systems) software were used.
Results
FRET Between Fluorescent G Protein Subunits in Intact Cells. To study G protein subunit interactions in intact cells, we used a FRET-based approach, where we made use of fusion proteins between G protein subunits and GFP-derived fluorescent proteins. Enhanced YFP (F46L) (20) was inserted into the αAB-loop within the α-helical domain of the Gαi1-subunit, a domain that has been used previously to insert various sequences into Gα-subunits (19, 25, 26). The fluorescent Gαi-subunit was in addition mutated in position C351I to make it insensitive to PTX (21). To detect FRET between Gαi-YFP and Gβγ, we cotransfected cells with Gβ1γ2-subunits that had been fused to CFP on the N terminus of Gβ1 (CFP-N-Gb1). First, we tested for functional interaction of CFP-N-Gβ1 with Gγ2 and found plasma membrane targeting of the fluorescent Gβγ-subunit, only in the context of a cotransfected Gγ-subunit (data not shown). The expression levels of the fluorescent G proteins were substantially lower than endogenous nonfluorescent G protein expression as determined by immunoblotting and fluorescence intensity (data not shown). To detect FRET between Gαi-YFP and CFP-N-Gβ1, ratiometric measurements of the spillover-corrected YFP/CFP fluorescence in response to 436-nm excitation (FYFP/FCFP; ref. 15) were recorded by using FRET imaging (Fig. 1_B_ and ref. 18). On excitation at 436 nm, CFP fluorescence was localized partially to intracellular structures and partially to the plasma membrane (Fig. 1_B_ Left). Gαi-YFP fluorescence (excitation: 480 nm) was found exclusively at the plasma membrane (Fig. 1_B_ Center). FRET between CFP-N-Gβ1 and Gαi-YFP was solely detected in places where Gαi-YFP was expressed (Fig. 1_B_ Right). In addition, FRET was also measured by dequenching the fluorescence donor (CFP) after ABP (YFP) (Fig. 1_C_ and refs. 17 and 27). The detection of FRET between Gαi-YFP and CFP-N-Gb1Gγ2 by means of two independent methods indicated a heterotrimeric formation of these fluorescent G protein subunits.
Fig. 1.

Increase in FRET on activation of fluorescent G protein subunits. A structural model of a heterotrimeric Gi protein is depicted, and insertion sites for YFP and CFP are indicated. (B) Fluorescence microscopy images of HEK cells transiently transfected with Gαi-YFP, CFP-N-Gβ1, and Gγ2 were recorded by using either 436-nm (Left and Right) and 500-nm excitation (Center) and emission filters for CFP (Left) or YFP (Center and Right). FRET was determined by using donor dequenching after APB (YFP) (C) of a representative cell expressing fluorescent G proteins as described in B. NA-induced activation of coexpressed α2A-AR resulted in a rapid and reversible increase in the emission ratio of YFP and CFP on 436-nm excitation (D).
Gi Proteins Do Not Dissociate During Activation in Living Cells. By using cells transfected with Gαi-YFP, CFP-N-Gb1Gγ2 and α2A-ARs we tested whether a change in FRET on receptor stimulation with noradrenaline (NA) was detectable by means of time-resolved FRET ratio measurements. Based on the common notion that G protein α- and βγ-subunits dissociate on activation, an agonistinduced decrease of the FRET signal was expected (19). To detect changes in FRET with high temporal resolution in single cells, we illuminated cells for 20 ms with 10 Hz at 436 nm and simultaneously measured the emission of CFP and YFP and the ratio of FYFP/FCFP (Fig. 1_D_). In contrast to the concept of subunit dissociation, we observed an increase in FRET during stimulation of α2A-ARs. After superfusion of cells with 1 μM NA, the FRET signal rapidly increased with a half-time of ≈1 s and decreased after withdrawal of the agonist with a half-time of ≈38 s (Fig. 1_D_). The time course of the agonist-induced increase of FRET and the decrease in FRET after withdrawal of agonist closely resembled the time course of receptor-induced activation of atrial IKACh or heterologously expressed GIRK currents (refs. 23 and 24 and Fig. 5). The increase in FRET on agonist exposure reflects most likely a decrease in the distance between YFP and CFP attached to Gα- and Gβγ-subunits. Therefore, we conclude that during activation in intact cells a molecular rearrangement of the heterotrimeric Gi protein subunits rather than subunit dissociation occurs.
Fig. 5.

Coupling of fluorescent G protein subunits to GIRK channels. HEK cells expressing PTX-insensitive Gαi-YFP, Gβ1, CFP-N-Gγ2 (A), α2A-AR and GIRK1/4 channels were subsequently to 3-5 h of pretreatment with 50 ng/ml PTX subjected to whole-cell patch-clamp recording. The response to superfusion with 10 μM NA of both simultaneously recorded photometric FRET detection (red) and whole-cell GIRK currents (black) are illustrated as representative experiments (A). To compare kinetics of the FRET and GIRK current response, GIRK currents were normalized to the maximal response and traces were scaled to the maximal response (A). Gβγ-subunits N-terminally tagged with CFP (of either the Gβ1- or Gγ2-subunit) activated basal (agonist-independent) GIRK currents when expressed without Gαi-subunits (B and C). Currents carried by GIRK channels in the absence of agonist were determined by transient application of 200μMBa2+, and subsequently currents induced in response to α2A-AR stimulation were measured (B, representative current recordings; C, summarized data of basal GIRK current densities determined in 8-10 experiments per condition). GIRK current activation was mediated by G proteins composed of Gαi-YFP, CFP-N-Gβ1, and Gγ2 (D and E). Cells were transfected with these G protein subunits as well as with α2A-AR and GIRK1/4 channel subunits, and NA-induced whole-cell GIRK currents were recorded in an inward direction at -90 mV (D, representative current traces; E, summarized current densities).
To further test this hypothesis, we used a second fluorescent Gβγ-subunit in which the N terminus of Gγ2 was fused to a CFP molecule (CFP-N-Gγ2). Functional expression and plasma membrane targeting of CFP-N-Gγ2 have been described previously (22). As depicted in the structural model (Fig. 1_A_), the N termini of Gβ-and Gγ-subunits are in close proximity (1). Accordingly, in FRET measurements analogous to those described in Fig. 1_D_ substituting CFP-N-Gb1Gγ2 by Gb1CFP-N-Gγ2 we again observed an agonistinduced increase in FRET between fluorescent Gα- and Gβγ-subunits (Fig. 2_A_). This increase in FRET again indicates a decrease in the distance between CFP and YFP and is incompatible with a complete dissociation of Gαi- and Gβγ-subunits during activation. These results rather suggested that during activation the region close to the N termini of Gβ- and Gγ-subunits move closer toward the αAB-loop of Gαi1 (Fig. 1_A_).
Fig. 2.

FRET uncovers molecular switch function of heterotrimeric Gi proteins. HEK cells transfected with Gαi-YFP and Gβ1- and a Gγ2-subunit N-terminally fused to CFP (compare Fig. 1 A) exhibited an increase in FRET on α2A-AR-mediated activation (A). Fusion of the CFP to the C terminus of the Gγ2-subunit induced a decrease in FRET on activation in cells expressing Gαi-YFP and Gβ1 (B). By using FRET imaging, the NA-induced decrease in FRET was found to occur primarily at the plasma membrane (C).
Activation Induces Intersubunit Rearrangement Rather than Dissociation of Gi. To further support this hypothesis, we searched for a third reference point on Gβγ-subunits that could be labeled with CFP and is localized at a site most distant to the N-terminal region of Gβγ. The rationale was that placing CFP on the opposite side would cause a decrease in FRET if the effects are caused by a rearrangement of Gβγ relative to Gαi. The C terminus on the Gγ2-subunit qualified for such a reference point. Therefore, HEK cells were transfected similarly as described above with α2A-AR, Gαi-YFP, Gβ1, and a Gγ2-subunit C-terminally fused to CFP (Gγ2-C-CFP) and subjected to FRET measurements.
Single-cell fluorescence measurements detected robust FRET between Gαi1-YFP and Gγ2-C-CFP, indicating formation of complexes (Fig. 2 B and C). With these constructs, agonist-mediated activation of G proteins led to a 30-35% decrease in FRET (Fig. 2_B_). Again, the kinetics of this response were compatible with G protein activation and deactivation: rapid activation (t1/2on = 0.69 ± 0.03s and slow deactivation t1/2off = 29.7 ± 2s, n = 7).
As detected by fluorescence imaging, fusion of CFP to the C terminus of Gγ2 led to a diffuse cytosolic staining, even in the presence of coexpressed Gβ1, presumably because of the missing lipid anchor (see below). However, as demonstrated by FRET imaging, Gγ2-C-CFP was able to functionally interact with Gαi-YFP, and this interaction was restricted to the plasma membrane, where Gαi-YFP was predominately localized (Fig. 2_C_ Left). On receptor stimulation with NA, there was a substantial decrease in plasma membrane-localized FRET (Center), which recovered after withdrawal of NA (Right). Taken together, these results suggest that a molecular rearrangement of the heterotrimer occurred on activation, which leads to a decrease of the distance between the N terminus of Gβ relative to the αAB-loop of Gαi, and,at the same time, to an increase in the distance between the C terminus of the Gγ2-subunit and the αAB-loop of Gαi.
To validate the FRET-pair-dependent differential changes in FRET during G protein activation, we measured FRET not only by means of dual emission FRET ratio recording but also by using donor dequenching after APB as a second independent method to determine FRET. As summarized in Fig. 3, FRET intensities measured in the absence of agonist depended on the FRET pair expressed. Independent of the FRET method used, Gαi-YFP exhibited strongest FRET when coexpressed with CFP-N-Gb1Gγ2, whereas the combination with Gb1CFP-N-Gγ2 or Gb1Gγ2-C-CFP exhibited equal lower FRET intensities (Fig. 3 A and C). More importantly, the directions of the agonist-induced changes in FRET were solely dependent on the FRET pair used and similar changes in FRET were observed with both methods (compare Fig. 3 B and D). However, substantial FRET remained after activation of G proteins composed of Gαi-YFP and Gb1Gγ2-C-CFP. To test whether this residual FRET was due to a significant pool of G proteins nonsusceptible to α2A-AR induced activation, 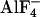 was applied during stimulation of the cells with NA (Fig. 3_F_). No significant
was applied during stimulation of the cells with NA (Fig. 3_F_). No significant 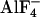 -induced decrease in FRET was observed in the presence of NA [FYFP/FCFP = 0.242 ± 0.021, compared with FYFP/FCFP = 0.247 ± 0.024 with NA alone (n = 9)].
-induced decrease in FRET was observed in the presence of NA [FYFP/FCFP = 0.242 ± 0.021, compared with FYFP/FCFP = 0.247 ± 0.024 with NA alone (n = 9)]. 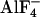 application before NA treatment activated fluorescent G proteins to a similar extent compared with NA, but with slower kinetics (Fig. 3_E_).
application before NA treatment activated fluorescent G proteins to a similar extent compared with NA, but with slower kinetics (Fig. 3_E_).
Fig. 3.

FRET determined by two independent methods. Summarized data for FRET ratio measurements (A and B) and FRET determined by donor dequenching after APB (C and D) are illustrated for all three G protein FRET pairs. Agonist-mediated changes (10 μM NA-induced change of initial FRET values) were measured either by using FRET ratio measurements (B) or by donor dequenching after APB (D). We note that, to avoid negative FRET values of the ratiometrically determined FRET, the lowest measured bleed-through of CFP to the YFP channel was used to correct YFP channel data (A). To compare the fraction of fluorescent G proteins susceptible for activation by α2A-AR to the total pool of fluorescent G proteins, cells expressing α2A-AR, Gαi-YFP, Gβ1, and Gγ2-C-CFP were exposed either before (E) or after (F) application of NA (1 μM) to 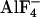 (10 mM NaF and 50 μM AlCl3).
(10 mM NaF and 50 μM AlCl3).
Functionality of Fluorescent G Protein Subunits. GFP variants are compact, but, rather large fluorophores, and therefore might disturb the function of the protein to which they are fused. Obviously, it is critically important to test the functionality of the fluorescent protein fusions. As FRET was detected by two independent methods, these fluorescent subunits formed heterotrimeric G proteins, indicating proper folding and heterotrimeric assembly (Fig. 3). Gαi-YFP was targeted exclusively to the plasma membrane (Fig. 1_B_), whereas Gβ1 and Gγ2 fused to CFP at their N termini exhibited plasma membrane staining but also intracellular staining (Fig. 1_B_ and data not shown). In contrast, Gβγ comprising Gγ2-C-CFP were primarily expressed in the cytosol, unless Gαi-subunits where coexpressed (data not shown). The defective membrane targeting of Gγ2-C-CFP was not unexpected, because fusion of CFP to the C terminus of Gγ2 removed the CAAX lipid modification signal from the C terminus of the resulting fusion protein (28).
All FRET pairs of fluorescent G protein subunits exhibited a fast change in FRET in response to stimulation of α2A-AR (Figs. 1_D_ and 2 A and B). This finding indicates a functional interaction of the fluorescent G proteins with the activating receptor. To test more specifically whether the interaction of the receptor and the G protein mutants was altered, we used a cell line stably expressing α2A-AR at 2 pmol/mg membrane protein and coexpressed FRET pairs of fluorescent G proteins. The identical cell line has been previously characterized (23) with regard to the coupling to GIRK channels through endogenous G proteins. Concentration-response curves of NA-induced FRET changes between Gαi-YFP and either Gγ2-C-CFP- or CFP-N-Gβ1-subunits were measured by using a fast superfusion system (ref. 18 and Fig. 4 A and B). Concentration-response curves of G protein activation could be reliably measured by FRET and the resulting curves were superimposable with a curve previously obtained (23) by recording G protein-mediated GIRK channel activation (Fig. 4_C_). These results suggested that the fluorescent G proteins are fully functional with respect to the interaction with GPCRs.
Fig. 4.

Concentration-response curves for α2A-AR-mediated G protein activation. HEK cells stably expressing 2 pmol α2A-AR per mg membrane protein were transiently transfected with heterotrimeric Gi proteins composed of Gαi-YFP, CFP-N-Gβ1, and Gγ2 (A and C, black squares) or Gαi-YFP, Gβ1, and Gγ2-C-CFP (B and C, red circles). NA-induced changes in the FRET signal were recorded in dependence of agonist concentration by using FRET ratio measurements. To compare coupling of the receptor to fluorescent G proteins with its coupling to endogenous G proteins, concentration-response curves of the FRET signals were compared with previously published data (23) regarding GIRK channel activation measured in the identical cell line (C, open squares). Summarized data of five to eight experiments were fitted with sigmoidal curves resulting in the following parameters: Gαi-YFP, Gβ1, and Gγ2-C-CFP: 13.7 nM/1.0; Gαi-YFP, CFP-N-Gβ1, and Gγ2: 12.3 nM/1.56; GIRK currents: 9.4 nM/0.78 (EC50/Hill coefficient).
To test the functionality of the fluorescent G proteins regarding their coupling to downstream effectors, we first tested whether fluorescent G proteins can activate coexpressed GIRK channels (Fig. 5). HEK cells stably expressing GIRK1 and GIRK4 channel subunits were transiently transfected with cDNAs encoding for Gαi-YFP, Gβ1, CFP-N-Gγ2, and α2A-AR. Three to six hours before current recordings they were preincubated with 50 ng/ml pertussis toxin (PTX) (Fig. 5_A_) to eliminate signal transduction through endogenous Gi/o proteins. Under these conditions, only cells expressing PTX-resistant Gαi-YFP gave rise to agonist-activated GIRK currents (Fig. 5_A_), indicating that Gαi-YFP can indeed couple to GIRK channels. Simultaneous recording of GIRK currents and G protein-derived FRET signals revealed a close temporal relationship between G protein activation and GIRK current activation. Similar results were obtained with CFP-N-Gβ1 (data not shown). In contrast to the N-terminally tagged Gβ-orGγ-subunits, the C-terminally tagged Gγ2-C-CFP-subunit failed to activate, but rather, inhibited GIRK currents (Fig. 6, which is published as supporting information on the PNAS web site). An important contact site on the Gβ-subunit that is required for Gβγ-mediated GIRK channel activation has been mapped to a region close to the C terminus of the Gγ-subunit (29). Therefore, the failure of Gγ2-C-CFP-containing Gβγ-subunits to activate GIRK channels is expected.
Gβγ-Subunits Containing CFP-N-Gγ2 or CFP-N-Gβ2 Activate GIRK Channels. Gβγ-subunits when coexpressed without Gα-subunits are known to constitutively activate GIRK currents (24, 30). To test for functional interaction of fluorescent Gβγ-subunits, HEK cells were transiently transfected with α2A-AR GIRK1/4 channel subunits and with or without Gβγ-subunits composed of either WT Gβ1γ2 or Gβ1 plus CFP-N-Gγ2 or CFP-N-Gβ1 plus Gγ2. Whole-cell GIRK currents were measured by means of conventional patchclamp recording in the inward direction. To do so in the context of an EK of approximately -50 mV, the membrane potential was set to -90 mV. To determine basal GIRK current density, the current amplitude that was blocked by 200 μM Ba2+ (30) was normalized to the cell capacity (Fig. 5 B and C). Cells that were cotransfected with any of the three Gβγ combinations exhibited significantly increased basal GIRK current amplitudes in the absence of receptor stimulation (Fig. 5_C_).
We questioned how to test whether GIRK channels could be activated through heterotrimeric G proteins composed of both Gαi-YFP and CFP-N-Gβ1. Knowing that both endogenous Gβγ-and Gα-subunits are limiting for maximal receptor-mediated activation of GIRK channels in these cells (Fig. 5 B and C and ref. 24), we tested whether in the presence of fluorescent Gαi-YFP coexpression of fluorescent Gβγ would lead to further increase in current density. To do so, GIRK channel- and α2A AR-expressing cells were transfected with Gαi-YFP, and with or without CFP-NGβ1 and Gγ2, and were subjected to whole-cell current analysis. NA-activated GIRK currents in the presence of fluorescent Gβγ-subunits to more than twice of the amplitude (Fig. 5 D and E) of cells expressing no exogenous Gβγ-subunits, indicating a functional signaling pathway from receptors to heterotrimeric G proteins composed of Gαi-YFP and CFP-N-Gβ1 and Gγ2 to GIRK1/4 channels.
Discussion
Direct measurement of mammalian G protein activity has not been possible so far. By using a FRET approach between genetically encoded YFP- and CFP-fluorescent G protein subunits, we were able to monitor G protein activity with high temporal resolution in intact cells. Although GFP-fusion proteins have been successfully tested for a large variety of proteins (14), attachment of a large fluorophore to a protein of interest always requires a detailed test to decide whether a given protein remains still functional. The three CFP-labeled Gβγ-subunits used in the present study exhibited functional interaction with Gαi-YFP-subunits because FRET was detected for each potential FRET pair (Figs. 1, 2, 3) by using two different methods to detect FRET. In addition, the subcellular localization pattern of the expressed fluorescent G protein subunits was as expected. All G protein subunit-FRET pairs that are described in the present study remained fully functional with respect to coupling to GPCRs. Indeed, EC50 values obtained from concentration-response curves of α2A-AR-induced G protein activation were not distinguishable if measured by using FRET assays (for Gαi-YFP/CFP-N-Gβ1 or for Gαi-YFPGγ2-C-CFP) or measured indirectly by recording GIRK channel activity (Fig. 4 and ref. 23). Therefore, the developed FRET assay seems to be well suited to study GPCR activity.
The present study is the first, to our knowledge, to actually measure G protein activation in intact cells with millisecond time resolution, and we found that α2A-AR-mediated G protein activation is complete within 1-2 s. The comparison of these data with the speed of α2A-AR activation (18) uncovers the fact that GPCR activation occurs at least five times faster than G protein activation. This result suggests that receptor and G protein are not organized as a single switch unit, but rather, are loosely coupled. Vigorous tests on the functionality of the studied fluorescent G protein subunits to couple to GIRK channels have demonstrated that selective activation of PTX-insensitive Gαi1-YFP led to GIRK current activation with similar kinetics as did stimulation of endogenous PTX-sensitive subunits (Fig. 5_A_ and ref. 23). Similarly, coupling of N-terminally tagged Gβγ subunits (either CFP-N-Gγ2 or CFP-N-Gβ1) to GIRK channels was normal (Fig. 5 B and C and ref. 22). An important point for future studies designed to dissect kinetics regarding the coupling between G protein and Gβγ effectors such as GIRK- or N-type Ca channels, receptor-mediated signals were transduced through G proteins composed of Gαi-YFP- and CFP-N-Gβ1g2-subunits (Fig. 5 D and E) to activate GIRK channels.
The most striking finding in the present study is the fact that in living cells Gαi- and Gβγ-subunits, in contrast to the generally accepted model, did not dissociate during activation, because Gαi-YFP and CFP-N-Gβ1γ2 exhibited an increase in FRET during activation (Figs. 1 and 2_A_). Similarly, FRET between Gαi-YFP and Gβ1CFP-N-Gγ2 increased during activation of α2A-AR, suggesting a decrease in distance of the fluorophores during activation. In contrast, the distance between Gαi-YFP and Gβ1Gγ2-C-CFP increased during activation (reflected as a decrease in FRET; Fig. 2 B and C). However, even during maximal activation by 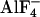 , a substantial FRET was detectable, suggesting that again, activated subunits of Gi proteins do not dissociate in intact cells. These data suggest the following model: Gi proteins exist as heterotrimers independent of the activation state. On GDP to GTP exchange on the Gαi-subunit and associated conformational changes on the Gαi, Gβγ stay complexed with Gαi but undergo a intermolecular rearrangement or switch that moves the N terminus of Gβ and Gγ closer to the αAB-loop of Gαi, and at the same time increases the distance of this position to the C terminus of Gγ. Alternatively, whether the FRET changes reflected a tilt or change in mobility of the fluorophores rather than a distance change, the observed FRET changes could indicate a rotational movement of the two subunits relative to each other, but certainly would rule out a complete dissociation of the Gαi- and Gβγ-subunits during activation. The fact that GIRK channels could be activated by heterotrimeric Gi proteins comprised of both YFP-tagged Gαi and CFP-tagged Gβγ (Fig. 5 D and E), indicates that the intermolecular rearrangement of Gα and Gβγ during activation opens up GIRK channel-activating interaction sites on Gβγ. These interaction sites have been mapped to a region on the Gβ-propeller that is quite distant from the N terminus of either Gβ or Gγ and overlap with the interaction site of Gβ with GDP-bound Gαi (7). However, these GIRK channel-interaction sites are in close proximity to the C terminus of Gγ. Therefore, it was not surprising that Gβγ comprising Gγ2-C-CFP did not activate, but rather, inhibited GIRK channels.
, a substantial FRET was detectable, suggesting that again, activated subunits of Gi proteins do not dissociate in intact cells. These data suggest the following model: Gi proteins exist as heterotrimers independent of the activation state. On GDP to GTP exchange on the Gαi-subunit and associated conformational changes on the Gαi, Gβγ stay complexed with Gαi but undergo a intermolecular rearrangement or switch that moves the N terminus of Gβ and Gγ closer to the αAB-loop of Gαi, and at the same time increases the distance of this position to the C terminus of Gγ. Alternatively, whether the FRET changes reflected a tilt or change in mobility of the fluorophores rather than a distance change, the observed FRET changes could indicate a rotational movement of the two subunits relative to each other, but certainly would rule out a complete dissociation of the Gαi- and Gβγ-subunits during activation. The fact that GIRK channels could be activated by heterotrimeric Gi proteins comprised of both YFP-tagged Gαi and CFP-tagged Gβγ (Fig. 5 D and E), indicates that the intermolecular rearrangement of Gα and Gβγ during activation opens up GIRK channel-activating interaction sites on Gβγ. These interaction sites have been mapped to a region on the Gβ-propeller that is quite distant from the N terminus of either Gβ or Gγ and overlap with the interaction site of Gβ with GDP-bound Gαi (7). However, these GIRK channel-interaction sites are in close proximity to the C terminus of Gγ. Therefore, it was not surprising that Gβγ comprising Gγ2-C-CFP did not activate, but rather, inhibited GIRK channels.
The result that Gi proteins did not dissociate in living cells contrasts with many biochemical studies (7, 31), and most surprisingly, with the only FRET study published on G proteins so far (19). What might be the difference? Deverotes and coworkers (19) used the same approach as was used in the present study and even labeled their G protein subunits in very similar positions with identical fluorophores, but still reported a robust decrease in FRET on G protein activation in Dictyostelium. The most likely explanation for the contradictory results might be the difference in the G protein used. The G protein used in the Dictyostelium study is homologous to Gs, and is thus quite different in the primary structure as compared with mammalian Gαi used in our study. We like to speculate, that G protein subtypes, specifically Gi and Gs, differentially undergo either subunit dissociation (Gs) or an intersubunit rearrangement (Gi) during activation. Initial FRET experiments in our laboratory designed to answer this question indicated that mammalian Gαs similarly to Gαs from Dictyostelium undergoes subunit dissociation, however Gαs-YFP exhibited dramatically impaired functionality (J. P. Vilardaga and M.B., unpublished results), limiting the significance of these experiments.
Based mainly on indirect evidence, it has been proposed by many groups that Gα contributes to the specificity of Gβγ signaling, because, despite the lack of a robust Gβγ subtype selectivity of Gβγ effectors, Gβγ-mediated responses are sensitive to PTX in primary tissues (32). So far, the most appealing hypothesis explaining how to achieve Gα-subtype-specific Gβγ responses is the subcellular complexing of effectors and heterotrimeric G proteins (11, 12). This result might be achieved either by a physical interaction of the heterotrimer with its Gβγ effector (11) or by a functional complexing, such as kinetic scaffolding (12). Heterotrimeric assembly of Gi proteins independent of the activation state of the G proteins, as discovered in the present study, would serve as a perfect scaffold to link Gβγ effectors to specific G protein subtypes, particularly in the context of additional Gα contact sites on the Gβγ effector (11). Future studies are needed to address, which G protein subtypes do or do not undergo dissociation on activation in intact cells, and what physiological consequences are associated with the specific G protein assembly during activation.
Supplementary Material
Supporting Figure
Acknowledgments
We thank Drs. S.R. Ikeda and V. Ruiz-Velasco for providing cDNAs for CFP-N-Gγ2, Drs. L. Hein and J. P. Vilardaga for project discussions and for critically reading the manuscript, and J. Humrich for performing immunoblots for control purposes. This work was supported by a Leibniz award (Deutsche Forschungsgemeinschaft) (to M.J.L.).
Abbreviations: FRET, fluorescence resonance energy transfer; GPCR, G protein-coupled receptor; YFP, yellow fluorescent protein; CFP, cyan fluorescent protein; APB, acceptor photobleaching; AR, adrenergic receptor; NA, noradrenaline; PTX, pertussis toxin; GIRK, G protein-activated inwardly rectifying K+.
References
- 1.Wall, M. A., Coleman, D. E., Lee, E., Iniguez-Lluhi, J. A., Posner, B. A., Gilman, A. G. & Sprang, S. R. (1995) Cell 83**,** 1047-1058. [DOI] [PubMed] [Google Scholar]
- 2.Lambright, D. G., Sondek, J., Bohm, A., Skiba, N. P., Hamm, H. E. & Sigler, P. B. (1996) Nature 379**,** 311-319. [DOI] [PubMed] [Google Scholar]
- 3.Mixon, M. B., Lee, E., Coleman, D. E., Berghuis, A. M., Gilman, A. G. & Sprang, S. R. (1995) Science 270**,** 954-960. [DOI] [PubMed] [Google Scholar]
- 4.Doupnik, C. A., Davidson, N., Lester, H. A. & Kofuji, P. (1997) Proc. Natl. Acad. Sci. USA 94**,** 10461-10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watson, N., Linder, M. E., Druey, K. M., Kehrl, J. H. & Blumer, K. J. (1996) Nature 383**,** 172-175. [DOI] [PubMed] [Google Scholar]
- 6.Druey, K. M., Blumer, K. J., Kang, V. H. & Kehrl, J. H. (1996) Nature 379**,** 742-746. [DOI] [PubMed] [Google Scholar]
- 7.Hamm, H. E. (1998) J. Biol. Chem. 273**,** 669-672. [DOI] [PubMed] [Google Scholar]
- 8.Wickman, K. D., Iniguez-Lluhl, J. A., Davenport, P. A., Taussig, R., Krapivinsky, G. B., Linder, M. E., Gilman, A. G. & Clapham, D. E. (1994) Nature 368**,** 255-257. [DOI] [PubMed] [Google Scholar]
- 9.Ruiz-Velasco, V. & Ikeda, S. R. (2000) J. Neurosci. 20**,** 2183-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hou, Y., Chang, V., Capper, A. B., Taussig, R. & Gautam, N. (2001) J. Biol. Chem. 276**,** 19982-19988. [DOI] [PubMed] [Google Scholar]
- 11.Peleg, S., Varon, D., Ivanina, T., Dessauer, C. W. & Dascal, N. (2002) Neuron 33**,** 87-99. [DOI] [PubMed] [Google Scholar]
- 12.Zhong, H., Wade, S. M., Woolf, P. J., Linderman, J. J., Traynor, J. R. & Neubig, R. R. (2003) J. Biol. Chem. 278**,** 7278-7284. [DOI] [PubMed] [Google Scholar]
- 13.Dunphy, J. T., Greentree, W. K. & Linder, M. E. (2001) J. Biol. Chem. 276**,** 43300-43304. [DOI] [PubMed] [Google Scholar]
- 14.Tsien, R. Y. (1998) Annu. Rev. Biochem. 67**,** 509-544. [DOI] [PubMed] [Google Scholar]
- 15.Erickson, M. G., Alseikhan, B. A., Peterson, B. Z. & Yue, D. T. (2001) Neuron 31**,** 973-985. [DOI] [PubMed] [Google Scholar]
- 16.Zaccolo, M. & Pozzan, T. (2002) Science 295**,** 1711-1715. [DOI] [PubMed] [Google Scholar]
- 17.Riven, I., Kalmanzon, E., Segev, L. & Reuveny, E. (2003) Neuron 38**,** 225-235. [DOI] [PubMed] [Google Scholar]
- 18.Vilardaga, J. P., Bünemann, M., Krasel, C., Castro, M. & Lohse, M. J. (2003) Nat. Biotechnol. 21**,** 807-812. [DOI] [PubMed] [Google Scholar]
- 19.Janetopoulos, C., Jin, T. & Devreotes, P. (2001) Science 291**,** 2408-2411. [DOI] [PubMed] [Google Scholar]
- 20.Nagai, T., Ibata, K., Park, E. S., Kubota, M., Mikoshiba, K. & Miyawaki, A. (2002) Nat. Biotechnol. 20**,** 87-90.11753368 [Google Scholar]
- 21.Wise, A., Watson-Koken, M. A., Rees, S., Lee, M. & Milligan, G. (1997) Biochem. J. 321**,** 721-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruiz-Velasco, V. & Ikeda, S. R. (2001) J. Physiol. (London) 537**,** 679-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bünemann, M., Bucheler, M. M., Philipp, M., Lohse, M. J. & Hein, L. (2001) J. Biol. Chem. 276**,** 47512-47517. [DOI] [PubMed] [Google Scholar]
- 24.Hommers, L. G., Lohse, M. J. & Bünemann, M. (2003) J. Biol. Chem. 278**,** 1037-1043. [DOI] [PubMed] [Google Scholar]
- 25.Yu, J. Z. & Rasenick, M. M. (2002) Mol. Pharmacol. 61**,** 352-359. [DOI] [PubMed] [Google Scholar]
- 26.Hughes, T. E., Zhang, H., Logothetis, D. E. & Berlot, C. H. (2001) J. Biol. Chem. 276**,** 4227-4235. [DOI] [PubMed] [Google Scholar]
- 27.Miyawaki, A., Griesbeck, O., Heim, R. & Tsien, R. Y. (1999) Proc. Natl. Acad. Sci. USA 96**,** 2135-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muntz, K. H., Sternweis, P. C., Gilman, A. G. & Mumby, S. M. (1992) Mol. Biol. Cell 3**,** 49-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ford, C. E., Skiba, N. P., Bae, H., Daaka, Y., Reuveny, E., Shekter, L. R., Rosal, R., Weng, G., Yang, C. S., Iyengar, R., et al. (1998) Science 280**,** 1271-1274. [DOI] [PubMed] [Google Scholar]
- 30.Bünemann, M. & Hosey, M. M. (1998) J. Biol. Chem. 273**,** 31186-31190. [DOI] [PubMed] [Google Scholar]
- 31.Hepler, J. R. & Gilman, A. G. (1992) Trends Biochem. Sci. 17**,** 383-387. [DOI] [PubMed] [Google Scholar]
- 32.Albert, P. R. & Robillard, L. (2002) Cell Signalling 14**,** 407-418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Figure