Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway (original) (raw)
Abstract
RAD53 and MEC1 are essential genes required for the transcriptional and cell cycle responses to DNA damage and DNA replication blocks. We have examined the essential function of these genes and found that their lethality but not their checkpoint defects can be suppressed by increased expression of genes encoding ribonucleotide reductase. Analysis of viable null alleles revealed that Mec1 plays a greater role in response to inhibition of DNA synthesis than Rad53. The loss of survival in mec1 and rad53 null or point mutants in response to transient inhibition of DNA synthesis is not a result of inappropriate anaphase entry but primarily to an inability to complete chromosome replication. We propose that this checkpoint pathway plays an important role in the maintenance of DNA synthetic capabilities when DNA replication is stressed.
Keywords: DNA replication, S-phase, checkpoint pathway, ribonucleotide reductase, nucleotide levels
The fidelity of DNA replication is critical to the proper duplication of a cell. Not only must cells replicate chromosomes, they must do so with great accuracy; without stretches of unreplicated DNA, without gaps, without replicational slippage in repetitive regions, without recombination causing rearrangements, and without breaks. S phase, the period of the cell cycle during which DNA is replicated, is a period of great vulnerability for a cell. Many complicated processes are undertaken during S phase, including the complete unwinding and replication of enormously complex DNA molecules, and chances for cataclysmic error are high. Interference with DNA replication by DNA damage, nucleotide depletion or imbalance, or polymerase malfunction can lead to a number of deleterious events, including increased mutagenesis, chromosome instability, gene amplification, microsatellite instability, and hyper-recombination (Loeb and Kunkel 1982). Each of these events can have severe consequences for an organism, including cell death, birth defects, and cancer. A number of factors cooperate to ensure the fidelity of DNA replication. These include processivity factors, proofreading functions, mismatch repair proteins, a variety of DNA repair activities, and regulatory pathways that sense DNA damage and replicational stress (Loeb and Kunkel 1982). For example, in response to DNA damage and DNA replicational interference, cells induce the transcription of genes that enhance repair capacities and arrest cell cycle progression to provide time for these repair processes to occur (for review, see Elledge 1996). This ensures that DNA replication and segregation—the critical events that allow genetic damage to become irreversibly inherited—are delayed until optimal repair can be achieved. In eukaryotes, these regulatory pathways are called checkpoints.
Checkpoint pathways ensure the proper order and timing of cell cycle events, and compromising these pathways contributes to genomic instability and cancer. The outline of the DNA damage response checkpoint pathway in mammals is emerging. ATM (ataxia telangiectasia mutated), a central player, is a member of the lipid kinase family of proteins and is likely a transducer of a DNA damage signal (for review, see Elledge 1996). ATM controls the timely activation of p53, a transcription factor that activates transcription of the cdk inhibitor p21 (Kastan et al. 1992). Cells defective for any of these genes show a defect in G1 arrest in response to DNA damage, and ATM mutants are also defective in G2 arrest and display radioresistant DNA synthesis. The roles of p53 and ATM in tumorigenesis underscore the importance of the DNA damage response to organismal homeostasis. In the case of ATM, there are additional phenotypes that include specific neural degeneration (Friedberg et al. 1995; Meyn 1995). Recently, an additional mammalian checkpoint gene encoding a protein kinase, Chk1, has been identified (Flaggs et al. 1997; Sanchez et al. 1997). Mammalian Chk1 is phosphorylated in response to DNA damage and is capable of phosphorylating Cdc25C on an inhibitory serine residue (Peng et al. 1997; Sanchez et al. 1997). The fission yeast Chk1 homolog acts downstream of the ATM homolog Rad3 (Walworth et al. 1993; Ford et al. 1994; Carr et al. 1995, Walworth and Bernards 1996; Furnari et al. 1997).
In the budding yeast Saccharomyces ceriviseae a number of genes have been identified that control the ability of cells to arrest the cell cycle and/or activate the transcriptional response. Upstream regulators involved in early steps in this pathway include RAD9, RAD17, RAD24, and MEC3, which are required for cell cycle arrest in G1 and G2 in response to DNA damage. POL2, encoding DNA polymerase 2, DPB11, and RFC5 are upstream components of the cell cycle arrest and transcription pathways that respond to replication blocks (Elledge 1996). Checkpoint signal transducers include MEC1 and RAD53, which are required for the S-phase checkpoint as well as the transcriptional and G1 and G2 arrest responses to DNA damage (Allen et al. 1994; Kato and Ogawa 1994; Weinert et al. 1994). DUN1, which encodes a protein kinase that is activated in response to DNA damage and replication blocks in a _MEC1_- and _RAD53_-dependent manner (Allen et al. 1994), is necessary for the transcriptional response (Zhou and Elledge 1993) and plays a partial role in the G2 arrest in response to DNA damage (Pati et al. 1997). MEC1 belongs to the same subfamily of proteins as ATM, underscoring the evolutionary conservation of this pathway (Greenwell et al. 1995; Morrow et al. 1995). MEC1 and TEL1 regulate the phosphorylation of the Rad53p kinase in response to DNA damage and replication blocks (Sanchez et al. 1996; Sun et al. 1996).
Whereas MEC1 and RAD53 control both the transcriptional and cell cycle responses to DNA damage and replication blocks, it is not clear whether these are the only roles these proteins carry out or whether these proteins play equivalent roles in these responses. In addition, the issue of whether these genes coordinate DNA replication and mitosis in an unperturbed cycle or only in response to replicational stress remains to be resolved. Both genes are essential for viability, perhaps suggesting a role for the checkpoint in each cell cycle, but to date their essential roles have remained obscure. In this study we sought to determine the essential functions of RAD53 and MEC1 by isolation of dosage suppressors of the null allele of rad53. We have discovered that increasing dNTP synthetic capacity can suppress both rad53 and mec1 null alleles. Furthermore, the primary lethal defect in these mutant strains in response to nucleotide depletion is not mitotic entry but a profound defect in the ability to finish chromosomal replication. We propose that one of the roles of this checkpoint pathway is the stabilization of replication structures under conditions of replication inhibition.
Results
RNR1 overexpression suppresses Δrad53 and Δmec1 lethality
To investigate the essential function of the S-phase checkpoint, we selected dosage suppressors of the lethality associated with a deletion of RAD53. A TRP1 2μ S. cerevisiae cDNA library under control of the GAL1 promoter (Mulligan and Elledge 1994) was constructed in λTRP, converted to plasmid form by cre–lox automatic subcloning (Elledge et al. 1991) and used to transform a rad53 null strain, Y324, being kept alive by RAD53 on a URA3 CEN plasmid, pJA92 (Allen et al. 1994). Transformants were selected on synthetic complete medium lacking tryptophan (SC − Trp), with galactose as a carbon source to induce cDNA expression, and replica plated onto the same medium containing 5-fluoro-orotic acid (5-FOA) to select for strains able to grow in the absence of pJA92. We subsequently examined the ability of these 5-FOAr transformants to grow with glucose as the carbon source. Because _GAL_-driven RAD53 is capable of sustaining cell growth under repressed conditions (glucose), choosing only clones that exhibited partial galactose dependence eliminated both the RAD53 background and any plasmid-independent extragenic suppressors. Twelve clones were at least partially dependent on galactose for suppression of Δrad53. These plasmids were sequenced and the identities of the encoded genes are listed in Table 1, along with the efficiency with which they suppress the growth defect of rad53 deletion mutants. We called those genes SRL, for suppressors of r ad53 lethality. A variety of genes are capable of suppressing Δrad53 to varying extents, including a number of transcription factors, both positive and negative. Those suppressors are likely to rescue the lethality indirectly, through effects on the transcription of other genes. Two suppressors are putative 26S proteasome components and are also likely to be indirect suppressors that act by changing the stability of other proteins that suppress the lethality of the rad53 deletion. Other suppressors consist of a protein kinase (MCK1), a putative chaperone (PDR13), and the regulatory subunit of ribonucleoside diphosphate reductase (RNR1). The remainder, designated SRL1, SRL2, and SRL3, show no similarity to other proteins in the database.
Table 1.
Summary of rad53 and mec1 deletion suppressors
| ORF name | Gene name | Function | Strength of suppressiona of | ORF size (nt) | Portion cloned (nt)b | |
|---|---|---|---|---|---|---|
| rad53 | mec1 | |||||
| YBR112c | SSN6/CYC8/CRT8 | transcriptional repressor | weak | poor | 2898 | 1–600 |
| YDR173c | ARGR3/ARG82 | transcriptional repressor/activator | good | weak | 1065 | entire |
| YER070w | RNR1/CRT7 | ribonucleoside diphosphate reductase | strong | strong | 2664 | 66–end |
| YHR064c | PDR13 | drug resistance, Hsp70 family | weak | weak | 1716 | 82–end |
| YJL110c | GZF3/NIL2 | transcriptional repressor | weak | poor | 1653 | 637–end |
| YKR091w | SRL3 | weak | poor | 456 | entire | |
| YLR082c | SRL2 | good | strong | 1176 | entire | |
| YNL307c | MCK1 | meiotic protein kinase | poor | poor | 1125 | entire |
| YOR247w | SRL1 | weak | poor | 630 | entire | |
| YOR259c | RPT4/SUG2 | SPB duplication, 26S proteasome | good | poor | 1311 | 35–end |
| YOR261c | RPN8 | 26S proteasome | good | poor | 1014 | 76–880 |
| YPL129w | ANC1/TFG3 | transcription factor | poor | weak | 731 | 161–end |
RNR1 overexpression suppresses mec1, indicating a common essential function for RAD53 and MEC1
Because RAD53 and MEC1 operate in the same checkpoint pathway (Sanchez et al. 1996; Sun et al. 1996), it is possible that they are essential for the same reason. In an effort to determine whether these genes have the same essential function, we examined the SRL genes for their ability to suppress Δmec1 lethality. Most of the suppressors were capable of suppressing the mec1 deletion mutant, albeit poorly. Only one plasmid was able to efficiently suppress both the rad53 and mec1 deletion mutants (Table 1). This plasmid contained the RNR1 gene encoding a predicted protein product starting with amino acid 22 of Rnr1 and continuing to the end of the 888-amino-acid protein. RNR1 was also shown to suppress the lethality of the Δmec1Δtel1 and Δmec1Δrad53 double mutants (data not shown). RNR1 is an essential gene that encodes the large subunit of ribonucleoside diphosphate reductase (RNR), the rate-limiting enzyme of deoxyribonucleotide synthesis and the target of the DNA synthesis inhibitor hydroxyurea (HU). RNR1 is both inducible by DNA damage and tightly cell cycle regulated (Elledge and Davis 1990). A gene encoding an alternative large subunit of Rnr, RNR3, is a target gene of the DNA damage and replication interference response pathways and is 80% identical to RNR1 at the amino acid level. We found that full-length RNR1 and RNR3 are both able to efficiently suppress Δrad53 and Δmec1 when expressed from the constitutive GAP promoter on a 2μ plasmid (p_GAP–RNR1_, p_GAP–RNR3_; Fig. 1A).
Figure 1.
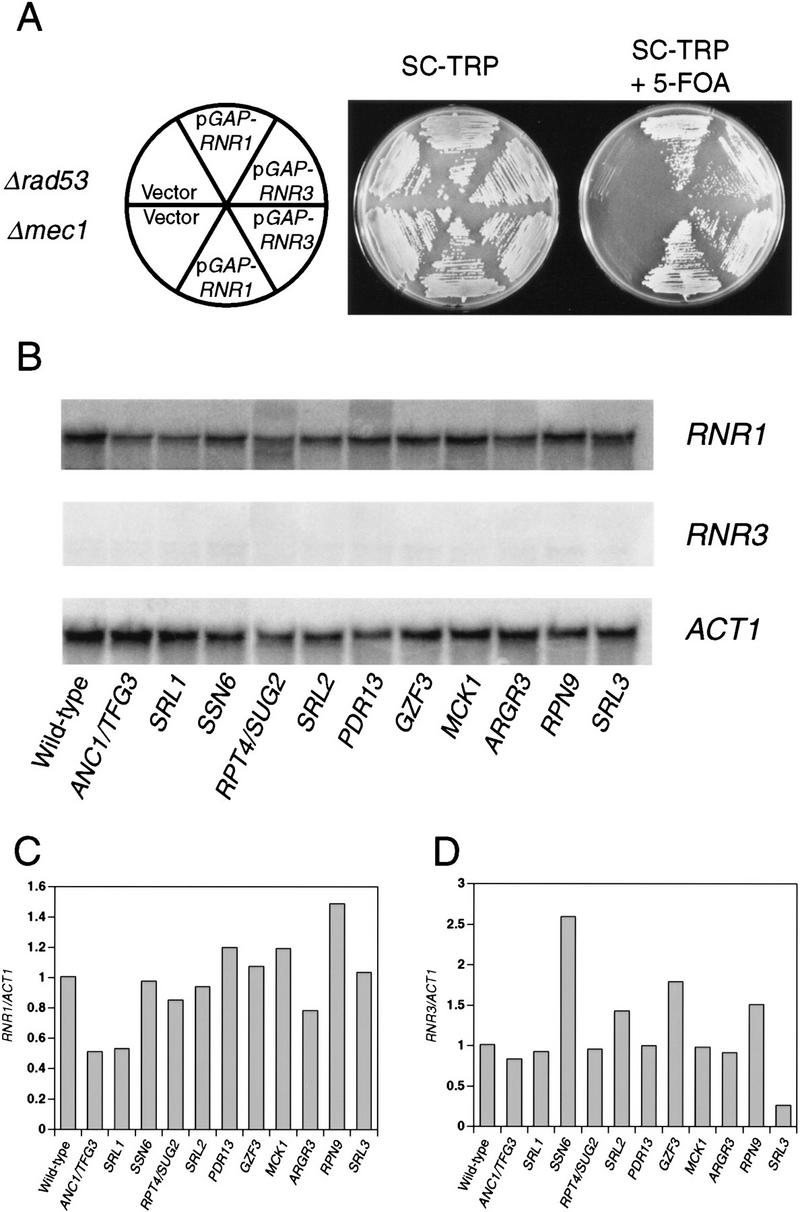
Suppression of null alleles of rad53 and mec1 by overproduction of RNR1 and other genes. (A) RNR suppression of Δrad53 and Δmec1. Y601, a Δrad53 mutant containing a wild-type copy of RAD53 on a URA3 plasmid, and Y602, a Δmec1 mutant containing a wild-type copy of MEC1 on a URA3 plasmid, were transformed with a TRP plasmid carrying _GAP_-controlled RNR1 (pBAD70) or RNR3 (pBAD79), or empty vector (pBAD54) as indicated. These transformants were struck onto SC − Trp and SC − Trp + 5-FOA to assess the ability of the null alleles to grow in the presence of the RNR expression plasmids. (B) RNR1 and RNR3 levels in suppressed Δrad53 strains. Y81 (wild-type) and Y324 (Δrad53) strains containing the indicated suppressors were grown to log phase in YPGal at 30°C. Total RNA was prepared and Northern blot analysis was performed using RNR1 (top)-, RNR3 (middle)-, or ACT1 (bottom)-specific probes (see Materials and Methods). (C, D) PhosphorImager quantitation of the Northern blots presented in B. The amount of RNR1 (C) and RNR3 (D) transcript was first normalized to the amount of ACT1 transcript present in each strain and then to the amount of RNR1 or RNR3 present in wild-type cells.
Low levels of ectopic RNR1 can suppress lethality
To examine whether up-regulation of RNR1 or RNR3 was the mechanism through which the other suppressors functioned, Northern analysis was performed on total RNA isolated from asynchronously growing cultures of each suppressed Δrad53 strain. There were no large increases in either RNR1 or RNR3 mRNA levels between wild-type cells and the suppressors (Fig. 1B–D), with the exception of Ssn6. There is a three-fold increase in RNR3 expression when truncated Ssn6 protein is expressed. RNR3 is negatively regulated by SSN6 (Zhou and Elledge 1992); therefore, this truncated Ssn6 might be acting as a dominant-negative mutant. The more general failure to detect strong differences in RNR transcription does not completely rule out altered RNR expression as a mechanism of suppression because very low amounts of exogenously supplied Rnr1 are still capable of suppressing Δrad53. For example, RNR1 under GAL1 control can still suppress when grown on glucose (data not shown). Additional support comes from the fact that one additional copy of the RNR1 gene under its own promoter is capable of efficient suppression, indicating that a twofold increase in RNR1 gene dosage is sufficient for suppression.
Mec1 has a greater role than Rad53 in response to genotoxic stress
Mec1 and Rad53 are both required for the transcriptional and cell cycle arrest responses to DNA damage and replication blocks. However, it was unclear whether they were equivalent in these functions because only hypomorphic alleles could be compared because of their essential nature. Having common suppressors of mec1 and rad53 null mutations allowed us to examine the phenotypes associated with a complete loss of function. In addition to defects in cell cycle arrest and transcriptional responses, previously isolated point mutants of RAD53 and MEC1 show a high degree of sensitivity to UV and ionizing radiation, radiomimetic drugs, and HU. Δrad53 + p_GAP–RNR1_ cells show the same degree of sensitivity to HU and UV irradiation as rad53-21 point mutants (Fig. 2A,B, circles). In addition, analysis of spindle elongation in α-factor-synchronized rad53-21 and Δrad53 cells released into media containing HU indicated that both of these alleles confer equivalent defects in the S-phase checkpoint (Fig. 2C,D, circles). The rad53 null mutant actually exhibits a slower rate of accumulation of anaphase-like spindles, but this parallels the slower rate of budding that is also observed under these conditions (Fig. 2C, circles). Thus, although RNR1 suppresses the lethality of Δrad53, it is unable to suppress the checkpoint and DNA damage sensitivity associated with loss of Rad53 function. This suggests that RNR1 overexpression allows rad53 (and mec1) null cells to tolerate an altered cellular physiology, rather than restoring function to the MEC1 RAD53 pathway.
Figure 2.


Characterization of checkpoint deficiency of rad53 and mec1 null mutants. (A) Viability in HU of mec1 and rad53 null mutants compared to point mutants. Asynchronously growing log phase cultures were treated with 0.2 m HU. Aliquots were removed at timed intervals to determine cell number and to score for viable colony-forming units on YPD plates. The strains used were Y80 (wild type, ♦), Y301 (rad53-21, •), Y603 (Δrad53 + pGAP–RNR1, ○), Y604 (mec1-21, █), and Y605 (Δmec1 + pGAP–RNR1, □). (B) UV sensitivity of mec1 and rad53 null mutants compared to point mutants. The same strains as in A were grown asynchronously to log phase at 30°C and plated onto YPD. The plates were irradiated at 0, 20, or 40 J/m2, and surviving colony-forming units were calculated. (C) Budding profiles of checkpoint mutants in HU following release from an α-factor block. Log-phase yeast cultures were incubated at 30°C in YPD supplemented with 10 μg/ml α-factor for 3 hr. To release from the block, cultures were washed into YPD lacking α-factor but containing 0.2 m HU, and aliquots were removed at timed intervals and scored for the presence of a bud. The strains used were Y580 (RAD+ MEC+ TRP1::GAP–RNR1, ♦), Y301 (rad53-21, •), Y606 (Δrad53 TRP1::GAP–RNR1, ○), and Y581 (Δmec1 TRP1::GAP-RNR1, □). (D) Kinetics of spindle elongation of checkpoint mutants in HU following release from α-factor. Samples were taken from the same experiment as in C and stained with anti-α-tubulin antibodies. Cells were scored for the presence of an elongated mitotic spindle by indirect immunofluorescence.
Δmec1 + p_GAP–RNR1_ cells are also defective in the response to DNA damage and replication blocks but more so than the mec1-21 point mutant, suggesting that mec1-21 is still partially competent in some of its responses. When the mec1 and rad53 null strains are compared, it is clear that the Δmec1 mutant is significantly more UV- and HU-sensitive (Fig. 2A,B). This indicates that MEC1 has a greater role in response to DNA damage than does RAD53, which is consistent with the fact that Rad53 is downstream of Mec1 in the pathway and indicates that MEC1 has functions in addition to its regulation of Rad53. However, inappropriate spindle elongation in the presence of HU by the mec1 null mutant occurs to the same extent as the rad53 null mutant (Fig. 2D). This suggests that the greater degree of lethality experienced by the mec1 null mutant in HU may be independent of the defect in preventing anaphase entry. The possibility that it is an event other than aberrant spindle elongation that commits checkpoint-defective cells to death is addressed further below.
Probing the essential function of Rad53 and Mec1
To examine the possibility that the lethal defect in Δrad53 and Δmec1 mutants during an otherwise normal cell cycle is low or aberrant RNR1 expression, we measured the accumulation of endogenous RNR1 mRNA after release from an α-factor block in strains deleted for mec1 containing additional RNR1 under GAP1 control (TRP1::GAP–RNR1) (see Materials and Methods). To specifically detect endogenous RNR1 mRNA, we used a probe specific for the 3′-untranslated region of the RNR1 gene that was absent in the TRP1::GAP–RNR1 expression cassette. Endogenous RNR1 expression in a population of Δmec1 TRP1::GAP–RNR1 cells synchronously moving through the cell cycle was compared with that of a MEC1 TRP1::GAP–RNR1 strain. Although the mutant accumulates appreciable amounts of RNR1 transcript, that accumulation is delayed and occurs at a slower rate than that of wild type (Fig. 3B). By the time RNR1 levels start to decline in Δmec1 TRP1::GAP–RNR1, there is approximately a 15 to 20 minute difference between it and wild type. A similar phenomenon is observed in Δrad53 TRP1::GAP–RNR1 cells (Fig. 3A). To determine whether these differences were due to a defect in RNR1 expression in the mutants or to a general cell cycle perturbation, we examined three other indicators of cell cycle progression. Figure 3C shows the expression profile of CLN2 mRNA out of α-factor arrest. Like RNR1, CLN2 expression in the Δmec1 TRP1::GAP–RNR1 strain is delayed relative to MEC1 TRP1::GAP–RNR1 cells, with the peak occurring ∼15 minutes later. The budding profile of the mec1 null mutant also shows a delay (Figs. 3D and 2C), indicating a delayed passage through start after α-factor arrest. Finally, the FACS profiles (Fig. 3E) clearly show that the mutant cells enter S phase later than, and persist in S phase longer than, the control cells. These results demonstrate that the MEC1 pathway plays a complex role in the cell cycle, affecting several aspects of cell cycle regulation. However, whereas the regulation of RNR1 is altered, it appears to be a secondary effect of a general cell cycle perturbation and not a specific target of the MEC1/RAD53 pathway. If the apparent delay and reduced expression of RNR1 was not an artifact of general cell cycle perturbation, then RNR1 levels should also be lower in asynchronous cultures. RNR1 appears to be expressed at wild-type levels in asynchronous cultures of rad53 and mec1 null mutants kept alive with RNR3 (Fig. 3F), supporting the notion that the altered RNR1 expression in the synchrony experiment is simply a reflection of the slower kinetics of cell cycle progression.
Figure 3.




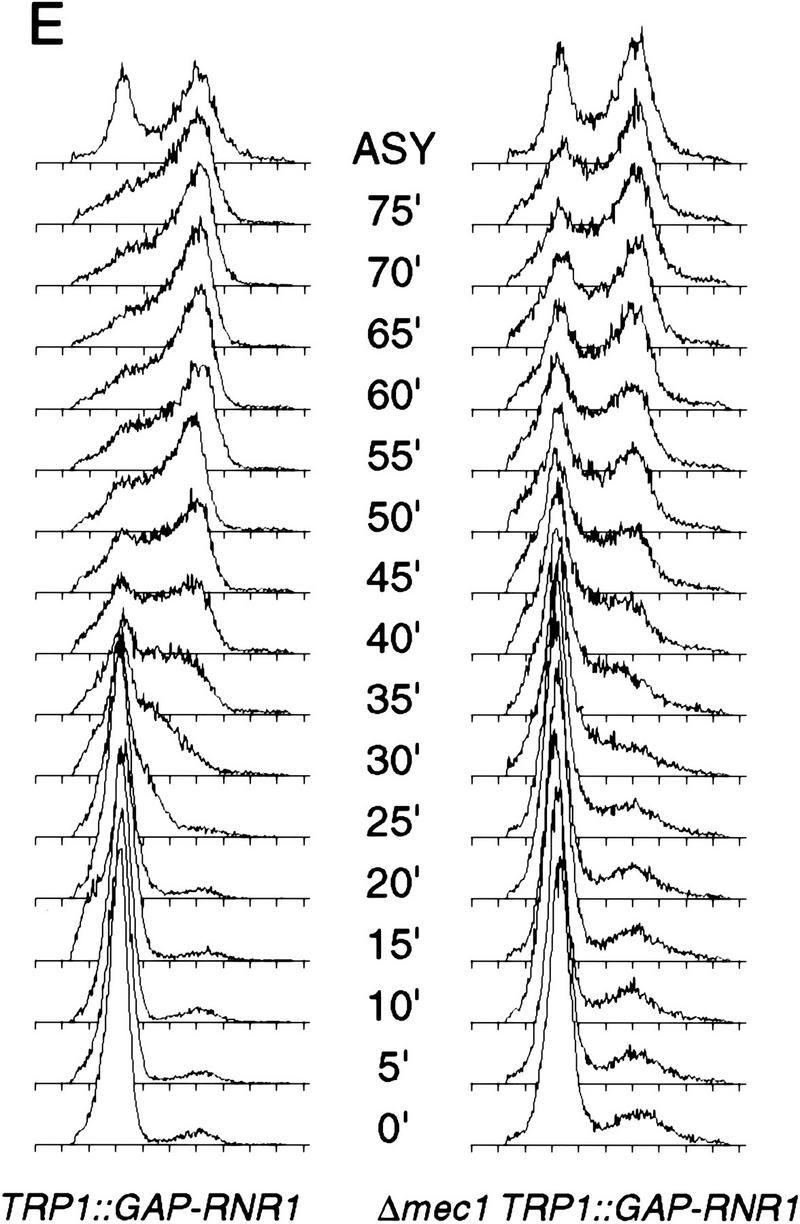
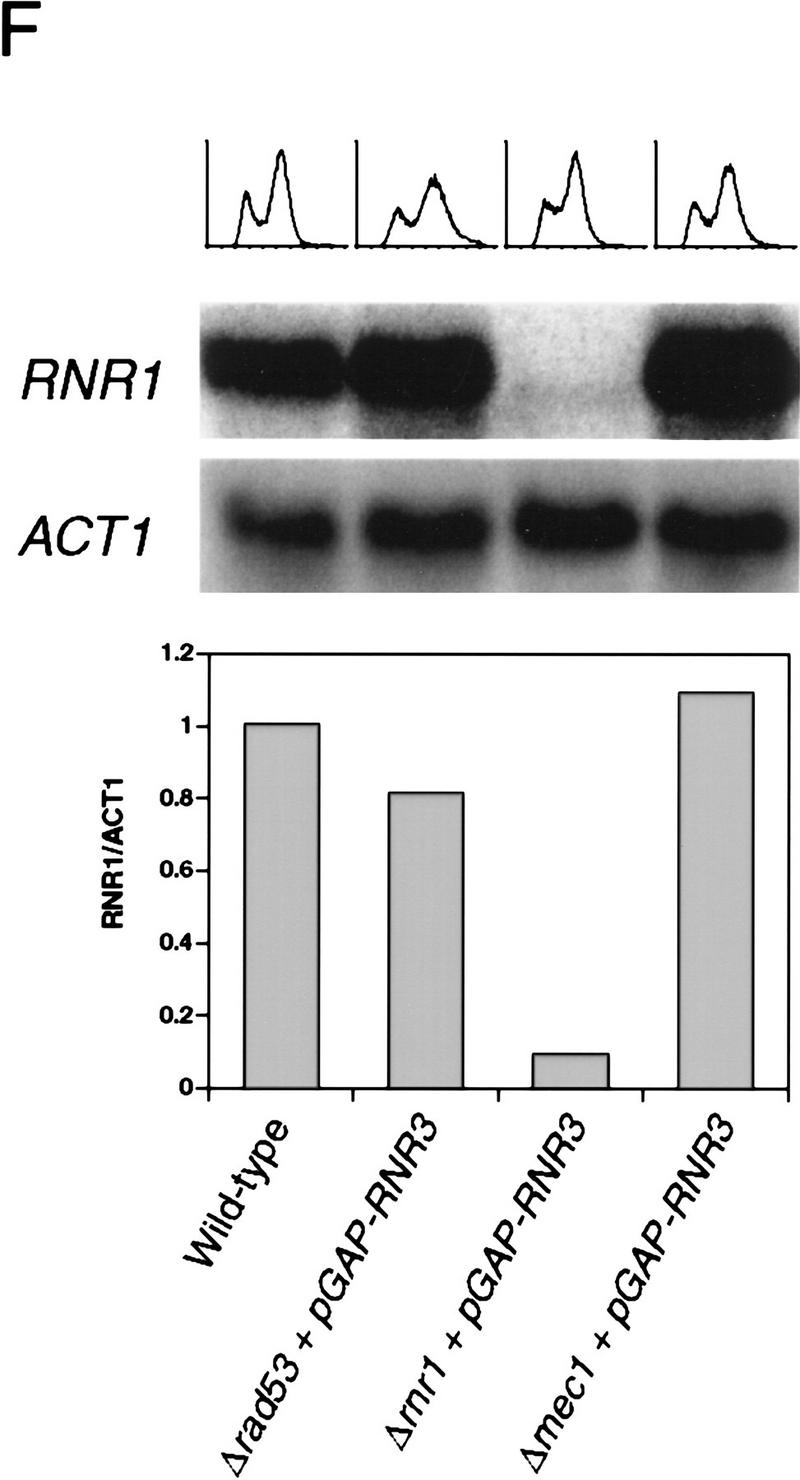
Kinetics of cell cycle events in rad53 and mec1 deletion mutants. (A) Accumulation of endogenous RNR1 mRNA in a rad53 deletion mutant. Y607 (RAD+ TRP1::GAP–RNR1, ♦) and Y606 (Δrad53 TRP1::GAP–RNR1, ○) were grown at 30°C to log phase and arrested with 10 μg/ml α-factor for 3 hr. Upon release into YPD, aliquots were taken and total RNA was prepared and blotted. The blot was probed with DNA specific to the endogenous RNR1 transcript and also to ACT1 for normalizing to the total amount of RNA in each lane (see Materials and Methods). Quantitation was performed using ImageQuant and the values obtained for each time point were plotted as a function of minutes after α-factor release. (B–E) The data presented in parts B–E all come from the same experiment and employed strains Y580 (MEC+ TRP1::GAP–RNR1, ♦) and Y581 (Δmec1 TRP1::GAP–RNR1, □). (B) Accumulation of endogenous RNR1 mRNA in the mec1 null. RNA was harvested, blotted, and probed and quantitated as in A. (C) Accumulation of CLN2 mRNA in the mec1 deletion mutant. The blot used in B was stripped and reprobed with DNA specific to the CLN2 transcript (see Materials and Methods). (D) Budding profile of the mec1 null mutant. A small aliquot of the cells used in B and C was retained for visual analysis of bud growth. The data are represented as the percentage of the total cells that have elaborated a bud at the indicated times. (E) DNA content of Δmec1 cells as they progress through the cell cycle upon release from an α-factor block. A portion of each aliquot used in parts (B–D) was stained with propidium iodide and analyzed by flow cytometry (see Materials and Methods). (F) Overall RNR1 mRNA levels in asynchronously growing rad53 and mec1 null mutants suppressed by high copy RNR3. Strains were grown to log phase at 30°C in YPD. Total RNA was purified from harvested cells, blotted, and probed with DNA specific for RNR1 and ACT1, as noted. Abundance of RNR1 transcript was calculated as noted in A and B and is represented in the bar graph below the autoradiograms. Above each lane in the autoradiograms the FACS profile of each strain is placed at the time the cells were harvested, indicating that there is a similar cell cycle distribution between them and validating the comparison of mRNA levels. The strains employed were Y692 (TRP+ MEC+), Y608 (Δrad53 + p_GAP–RNR3_), Y609 (Δrnr1 + p_GAP–RNR3_), and Y610 (Δmec1 + p_GAP–RNR3_). Y609 is a deletion of RNR1 that is suppressed by overexpression of RNR3. This provides a control for the specificity of the RNR1 probe used in this experiment.
RNR1 overproduction does not enhance the rate of DNA replication
Because low levels of additional RNR1 expression are capable of suppressing the lethality of mec1 and rad53 mutants, we entertained two general hypotheses for how this suppression might work. The first is based on the assumption that because MEC1 and RAD53 coordinate S-phase completion and mitosis under certain circumstances, their loss may allow S phase and mitosis to occur based on their natural timing, akin to a race between S phase completion and mitotic onset. Thus, by adding additional nucleotides S phase may be shortened to the point where it is completed prior to a lethal mitosis. The second hypothesis is that the MEC1/RAD53 pathway provides a function other than cell cycle coordination, such that the loss of Rad53 and Mec1 creates a special nucleotide stress or a greater sensitivity to normal nucleotide levels—levels that may be suboptimal for DNA polymerization or fork stability. Because rad53 mutants are sensitive to low nucleotide levels, we know that nucleotide depletion is toxic. Although the HU sensitivity is generally assumed to be due to inappropriate mitotic entry, this has not been rigorously demonstrated and other explanations exist. For example, nucleotide depletion sensitivity could result from the occasional disassembly of a paused replication complex searching for nucleotides, and MEC1/RAD53 might help to restore the function of these (transiently) nucleotide-starved complexes. Providing additional nucleotides in the form of RNR overexpression might prevent this stress from occurring. In both hypotheses, RNR1 overexpression suppresses by providing extra dNTPs; in the first case, the dNTPs would suppress by accelerating the rate of S-phase completion, whereas in the second case they would suppress by preventing a cataclysmic response to perceived nucleotide depletion by reversing that depletion.
To test the first hypothesis, we examined whether S phase was shorter in wild-type cells overproducing RNR1 under GAP control. Cells were arrested in G1 with α-factor, released from the block, and their DNA content was measured by FACS analysis at 2 min intervals. Although a very small effect cannot be ruled out, the overexpression of RNR1 had no apparent effect on the timing of S-phase completion or the overall rate of DNA synthesis (Fig. 4).
Figure 4.
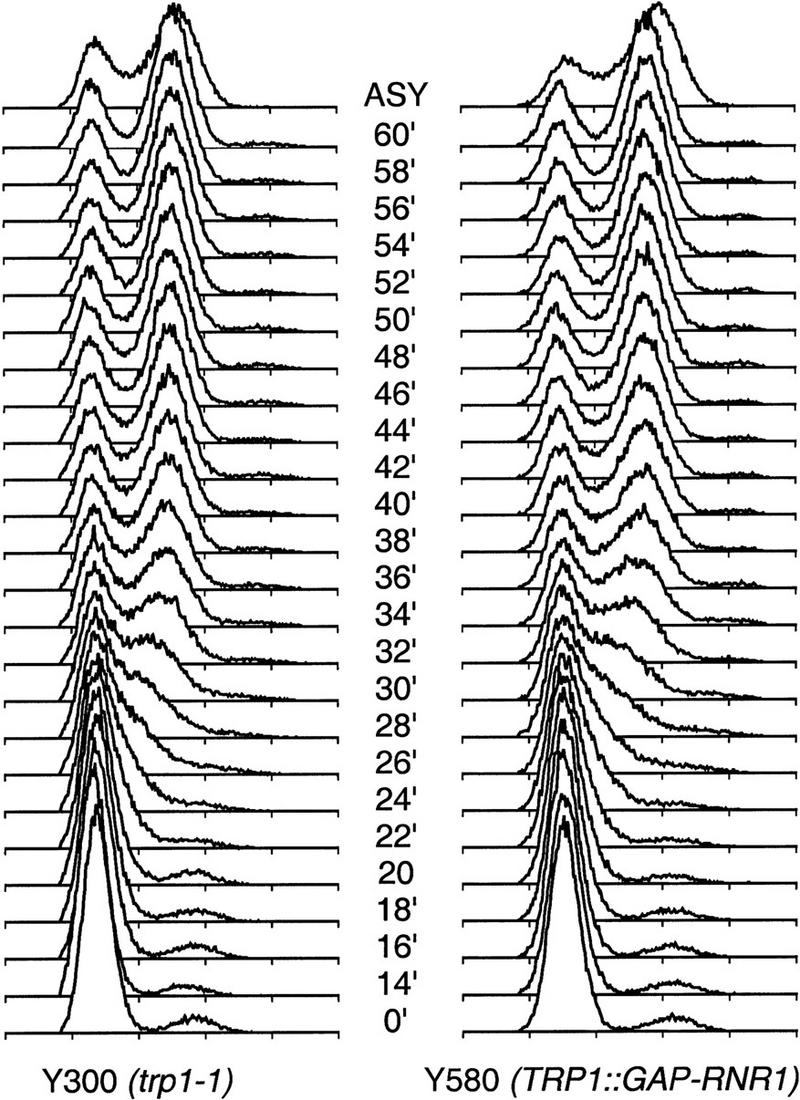
RNR1 overproduction does not accelerate progression though S phase. DNA replication timing of Y300 (wild type, trp1-1) and Y580 (TRP1::GAP–RNR1) strains is shown. Cells were grown to log phase at 30°C and arrested with 10 μg/ml α-factor for 3 hr. Upon release from the α-factor block into YPD, samples were taken at close intervals and stained with propidium iodide and analyzed by flow cytometry for the purpose of detecting subtle differences in the rate of replication due to RNR1 overproduction.
Delaying mitosis cannot rescue the lethality of mec1 and rad53 null mutants
If the outcome of a race between S phase and mitosis determines lethality, the result could be influenced not only by making S phase happen faster but also by delaying mitosis. To test this we examined the effects of agents capable of delaying mitosis on mec1 and rad53 mutants. We germinated spores from a Δrad53::HIS3/RAD53 heterozygous diploid on media containing sublethal amounts of benomyl (15 μg/ml), which delays mitosis through activation of the mitotic spindle assembly checkpoint (Elledge 1996). No His+ colonies were viable under these conditions. We also streaked rad53 null cells containing RAD53 on a URA3 CEN plasmid (pJA92) onto media containing 5-FOA and 15 μg/ml benomyl but observed no increase in the appearance of 5-FOAr colonies relative to the absence of benomyl. mec1 and rad53 null mutants are extremely sensitive to low HU levels on plates. We identified the minimal concentration of HU that blocked growth on plates (5 mm) and attempted, unsuccessfully, to suppress the lethality of either mutant with 15 μg/ml benomyl.
Because survival was measured as growth on plates in previous experiments, the concentration of microtubule inhibitors employed was necessarily not sufficient to completely block mitosis, and low levels of suppression might be obscured as a result. To examine this more thoroughly, we tested the ability of a sustained mitotic block to allow rad53 mutants to recover from a transient HU block (Fig. 5A). rad53-21 mutants were released from a G1 block into media containing 0.25 m HU. After 30 min, the HU was washed out and the cells were resuspended in media containing 80 μg/ml benomyl with no HU, and viability was measured over time. Blocking mitosis with benomyl was unable to restore any measure of viability. The inability of benomyl to rescue either the lethality of the null, or the sensitivity of either the null or the point mutant to HU, suggests that the lethal event may be the same in each case (the consequence of nucleotide depletion) and unrelated to whether or not cells are allowed to proceed into mitosis. This is consistent with the fact that in rad53 and mec1 null mutants, loss of viability in HU does not correlate with the degree of spindle elongation (Fig. 2A,D).
Figure 5.

Inability of a microtubule inhibitor to suppress the lethality of rad53 mutants transiently exposed to HU. (A) Sensitivity of rad53-21 to HU in the presence of benomyl. A rad53-21 strain, Y301, was released from α-factor arrest into 0.25 m HU for 30 min. Following this transient incubation the culture was maintained in 80 μg/ml benomyl, and timed aliquots were plated onto YPD for measurement of viable colony-forming units. (B) FACS analysis of Y301 (rad53-21) and Y300 (wild-type) cultures that had been transiently treated with HU. Wild-type and rad53-21 cultures were released from the G1 block into either 0.25 m HU for 30 min or medium lacking HU, as indicated. At 30 min after α-factor release, cells were washed and transferred into YPD containing 80 μg/ml benomyl. Progress through S phase was monitored by FACS at the indicated time points.
rad53 mutants fail to complete DNA replication after a transient replication block
As the cause of lethality in null mutants and HU-treated null and point mutants does not appear to be solely due to the relative timing of S phase and mitosis, it is likely that in rad53 and mec1 null cells a lethal event is occurring that commits the cells to death regardless of the timing of the subsequent mitosis. As we described earlier, one such event could be defective DNA replication caused by a condition of nucleotide depletion. To determine whether mutant cells transiently arrested with HU did in fact have difficulty finishing DNA replication after removal of the replication block, we examined DNA content in rad53-21 and wild-type cells under these conditions. Although the rad53 mutant showed a delay in replicating its DNA relative to wild-type cells transiently treated with HU, it eventually accumulated with an approximately G2 DNA content (Fig. 5B), indicating that it recovered the ability to produce sufficient dNTP levels to replicate a genome’s worth of DNA.
Because FACS analysis cannot determine to what extent mitochondrial DNA contributes to the amount of G2 DNA observed in this experiment, we performed a similar experiment in ρ0 rad53-21 mutant strains (Fig. 6A). ρ0 rad53-21 mutants were released from α-factor into 0.2 m HU and 10 μg/ml nocodazole, the HU was washed away after 1 hr, and samples were analyzed for DNA content for up to 3 hr (Fig. 6B, bottom). Under transient HU-treatment conditions that resulted in 75% lethality (Fig. 6A), we observed the same accumulation of apparent G2 DNA content as in the ρ+ strains (cf. Figs. 6B and 5B). The control experiment in the absence of HU (Figs. 5B and 6B, top) indicates that the effect is specific to HU. The observed delay in replication in rad53-21 mutants was not unexpected because rad53 mutants are unable to induce expression of the RNR1, RNR2, RNR3, and RNR4 genes to quickly increase nucleotide biosynthetic capacity (Allen et al. 1994; Huang and Elledge 1997). Alternatively, the delay could be due to the presence of lesions that occur in the transiently nucleotide-starved cells (e.g., stalled replication complexes or abandoned replication forks) that persist and impede the function of the active replication complexes that subsequently encounter them. These data confirm that rad53-21 cells are delayed but not deficient in restoring DNA synthetic capability after transient HU treatment. However, the cells are clearly dying, and forestalling mitosis with microtubule destabilizing drugs has no effect on this.
Figure 6.

Inability of ρ0 rad53-21 mutants to complete chromosomal replication after a transient HU treatment. Y623 (wild-type ρ0) and Y624 (rad53-21 ρ0) cells were arrested in α-factor for 3 hr and washed into YPD media containing either 10 μg/ml nocodazole or 0.2 m HU and nocodazole. After a 60-min incubation, cells were washed and resuspended into YPD medium containing 10 μg/ml nocodazole only and monitored for viability (A), DNA content (B), and chromosome integrity (C,D). (A) Sensitivity of rad53-21 (•) to transient HU treatment in the presence of nocodazole. Wild-type (♦) is shown for comparison. (B) Flow cytometric analysis of the DNA content of wild-type and rad53-21 strains. Transiently HU-treated cultures are shown at bottom, with the asterisk (*) indicating the time at which the cells were washed out of HU. (Top) Cultures released from α-factor into nocodazole only. (C) CHEF gel of chromosomes from wild-type (left) and rad53-21 (right) strains transiently treated with HU. The vertical bar over each lane indicates time points at which HU was present (shaded bars) or had been washed out (open bars). The two chromosomes that were used in part (D) are indicated. (D) Quantitation of replication of chromosomes from wild-type [ρ0 Chr A (♦) and ρ0 Chr B (⋄)] and rad53-21 [rad53-21 ρ0 Chr A (•) and rad53-21 ρ0 Chr B (○)] cultures that had been transiently treated with HU and resolved by CHEF in C. The two chromosomes examined are indicated in C. The amount of fully duplicated chromosomes in the rad53 mutants precisely correlates with the percentage survival. Intensities of the bands were quantitated using NIH Image software and plotted as a function of time after release from α-factor and plotted in arbitrary units.
FACS analysis measures only bulk DNA content, and it cannot determine whether a small percentage of the DNA is unreplicated or, in the case of the previous experiment, whether the apparently replicated chromosomes at the end of the experiment are intact. To examine the integrity of chromosome structure, we employed pulsed-field gel electrophoretic (PFGE) analysis. Incompletely replicated chromosomes fail to enter a pulsed-field gel because of the presence of forks and replication bubbles that impede migration (Hennessy et al. 1991). Chromosomal DNA was prepared from the cultures of wild-type ρ0 and rad53-21 ρ0 mutant cells that had been treated transiently with HU and kept in the presence of nocodazole. At timed intervals, DNA from these cells was prepared and examined by PFGE (Fig. 6C) and quantitated densitometrically (Fig. 6D) (see Materials and Methods). Transient HU treatment delayed the re-entry of chromosomes from wild-type cells, consistent with the kinetics observed by FACS analysis. In contrast, chromosomes from the rad53 mutant never re-entered the gel, even during a 6-hr mitotic block. Similar results were obtained with mec1 mutants (data not shown). Quantitation of the intensities of two chromosome bands, designated A and B, shows that wild-type chromosomes double in intensity from 150 min, indicating completed replication. rad53 chromosomes reappear at 180 min at half the original intensity, indicating that a quarter of the population has properly completed DNA synthesis, consistent with the survival data. This indicates that in addition to experiencing a significant delay in the recovery of bulk DNA synthetic capacity, when the rad53 mutant’s chromosomes do eventually become apparently fully replicated (by FACS analysis), they have a profoundly abnormal structure (by PFGE).
Genetic interactions between the checkpoint and origin initiation machinery
We have described defective DNA replication as a consequence of transient nucleotide depletion in checkpoint mutants. Because checkpoint null mutants can be suppressed by increasing nucleotide biosynthetic capacity, it is likely that the null mutants experience a nucleotide depletion and die for the same reason as hypomorphic mutants that experience a transient nucleotide depletion. Therefore, an important issue is the nature of the perceived nucleotide depletion in checkpoint null cells. These mutants could be sensitive to the normal dNTP levels present in each cell cycle, or alternatively, the absence of the checkpoint could create a nucleotide depletion to which the cells cannot subsequently respond. In the latter case, the mechanism could be a direct failure to up-regulate RNR activity or an indirect consequence of a failure to properly regulate the nucleotide consumption of other cellular machinery. While investigating the genetic interactions between checkpoint mutants and origin-firing mutants, we have uncovered support for the idea that timing of origin firing may contribute to the nucleotide depletion that kills checkpoint null mutants. The temperature-sensitive origin firing mutant orc2-1 (Liang et al. 1995) displays an extended duration of S phase upon release from an α-factor arrest, even at the permissive temperature (data not shown). To determine whether this might be mimicking the effect that HU has on S phase, we constructed orc2-1 mec1-21 and orc2-1 rad53-21 double mutants. Surprisingly, both double mutants are viable, suggesting that the lengthened S phase in orc2-1 is a qualitatively different phenomenon than that caused by HU treatment, which kills these checkpoint mutants. Even more startling is the fact that the mec1-21 mutation, but not the rad53-21 mutation, can suppress the temperature sensitivity of orc2-1 (Fig. 7A) at 30°C. This observation suggests that the checkpoint pathway is acting antagonistically to the origin-firing defect of orc2-1.
Figure 7.


Genetic interactions between mec1 mutants and origin-firing mutants. (A) Suppression of orc2-1 by the mec1-21 mutation. Y300 (wild-type), Y604 (mec1-21), Y611 (orc2-1), and Y612 (orc2-1 mec1-21) cultures were grown to log phase in YPD at 24°C. Serial dilutions of 105, 104, 103, and 102 cells were spotted onto YPD plates at either 24°C (left) or 30°C (right). (B) Suppression of Δmec1 by dbf4-1. Representative Δmec1 dbf4-1 double mutants (Y613–Y616) containing MEC1 on a URA3 CEN plasmid (pBAD45) were struck to 5-FOA plates to identify suppressors of the mec1 null mutation. The wild-type and Δmec1 controls that were used in this experiment were isolated from the same cross as the double mutants.
The suppression of orc2 by a mec1 mutation bears on the essential function of the DNA replication checkpoint because if there is an antagonistic interplay between checkpoint genes and origin-firing genes at the level of origin firing, then it could be that inappropriate origin firing in checkpoint null mutants creates a nucleotide depletion that commits the cells to lethality. If true, then origin firing mutants might be expected to abrogate this effect and suppress the lethality of checkpoint null mutants. The concept that the checkpoint and the origin-firing machinery specifically interact with each other is further supported by recent work (Santocanale and Diffley 1998, and pers. comm.) indicating that the timing of origin firing is negatively regulated by the DNA replication checkpoint pathway. To further explore this idea we examined interactions between the checkpoint pathway and the Dbf4/Cdc7 complex, a protein kinase that is required for origin initiation (Jackson et al. 1993). We tested dbf4-1 and cdc7-1 mutants for suppression of Δrad53 and Δmec1 by isolating double mutants that contained the wild-type alleles of RAD53 or MEC1 on a URA3 plasmid. These strains were struck onto plates containing 5-FOA to assess their ability to grow in the absence of checkpoint gene product. We found that Δmec1 but not Δrad53 was suppressible by dbf4-1 and cdc7-1 (Fig. 7B, data not shown), supporting the plausibility of this idea. Why mec1 and not rad53 mutants would exhibit these interactions with origin firing mutants is not clear, but the explanation may lie in the additional functions of Mec1 somehow impinging on these events or in a more complex relationship between origin firing and checkpoint function, as detailed in the Discussion.
Discussion
Cell cycle checkpoints have been thought of primarily as surveillance mechanisms that respond to aberrations in cellular structures, such as DNA damage or replication blocks, and prevent catastrophic cell cycle transitions. Unlike the checkpoint genes specific for DNA damage, those involved in the DNA replication checkpoint are essential for viability. The fact that all known replication interference checkpoint genes in S. cerevisiae are essential is an indication either that events occurring during the course of a normal cell cycle require the coordinating activities of this checkpoint or that the DNA replication checkpoint genes have activities in addition to the cell cycle coordination traditionally thought to be their primary function (Weinert and Hartwell 1988). We investigated this poorly understood aspect of checkpoint function by performing a high copy suppressor screen of the lethal rad53 null mutation. We found that overproduction of RNR1 eliminated the requirement for both MEC1 and RAD53, indicating an interaction between nucleotide levels and checkpoint function even in the absence of nucleotide-depleting drugs. We also determined that lethality caused by nucleotide stress in checkpoint-deficient cells can be attributed to failure of replication structures to completely recover from the immediate effects of nucleotide depletion, suggesting that replicational stress due to suboptimal nucleotide levels may occur during a normal cell cycle.
Functional distinction between MEC1 and RAD53
The mec1 and rad53 alleles that were previously available for study were necessarily hypomorphic and not complete loss-of-function alleles. This has made determination of the relative roles played by each in the checkpoint pathway impossible to definitively establish. The existence of a common suppressor allows a direct comparison of the two null mutants with existing hypomorphic alleles and with each other. The UV and HU sensitivities of the mec1-21 mutant are much less severe than the mec1 null mutant, indicating that the mec1-21 allele retains significant residual function. The UV and HU sensitivities of the rad53-21 and rad53 null mutants are very similar. Furthermore, the kinetics and extent of spindle elongation in HU-treated rad53-21 mutants are essentially indistinguishable from that of both rad53 and mec1 null mutants after general cell cycle perturbations are taken into account, indicating that rad53-21 can be considered to be nearly completely defective for the cell cycle delay function.
The major point of similarity between the mec1 and rad53 null strains is the fact that even moderate RNR1 overproduction can efficiently suppress them both. Furthermore, a rad53 mec1 double null mutant is also easily suppressible by RNR1 (data not shown). This indicates that the essential functions of both genes are the same. Moreover, using the common suppressor approach we can state unequivocally that there is a functional distinction between RAD53 and MEC1 observable at the level of sensitivity to UV irradiation and HU treatment, with MEC1 contributing more to resistance than RAD53. Given that the kinetics of anaphase entry of rad53 and mec1 null mutants in the presence of HU are very similar to each other, we believe that the actual cell cycle regulatory functions of the two gene products are therefore also similar but that MEC1 has additional roles required for recovery from replicational stress. This is also consistent with the fact that MEC1 acts upstream of RAD53 in the checkpoint pathway and is required for its phosphorylation in response to DNA damage and replication blocks.
What is the essential function of the S-phase checkpoint?
Whereas RAD53 and MEC1 are essential genes in S. cerevisiae, their homologs in Schizosaccharomyces pombe, cds1+ and rad3+, respectively, are not (Al-Khodairy et al. 1994; Murakami and Okayama 1995). The _MEC1_-related gene ATM is also dispensable for cell growth in humans and mice (Barlow et al. 1996; Elson et al. 1996; Xu et al. 1996). This suggests that the essential natures of MEC1 and RAD53 are reflections of a checkpoint requirement that manifests in every cell cycle in S. cerevisiae. Our findings that RNR1 and RNR3, the rate-limiting regulatory subunits of ribonucleotide reductase, are dosage suppressors of the lethality of the mec1 and rad53 null mutations support this idea and indicate that the essential function of these genes involves maintaining an adequate nucleotide supply, as opposed to responding to some kind of DNA damage. The fact that low amounts of exogenously supplied RNR1 can efficiently suppress lethality suggests that the defect responsible for lethality is just below the threshold for survival. However, RNR1 can do little to overcome the effects of exposure to the RNR inhibitor HU, which requires full activation of the checkpoint for a prolonged period of time.
dNTPs levels are highly regulated (for review, see Elledge et al. 1992). The mRNA for RNR1 is tightly cell cycle regulated, the mRNAs for all four RNR genes are inducible in response to DNA damage and replication blocks, the substrate specificity of the reductase is modulated by particular dNTPs to ensure an equal supply of all four dNTPs, and dATP feedback inhibits the overall activity of the enzyme to prevent excessive build up of dNTPs. An important question is why the levels of dNTPs in mec1 and rad53 mutants are insufficient for survival. One possibility is that mec1 and rad53 cells are simply more sensitive to normal levels of nucleotides. Perhaps nucleotide levels are normally maintained at a level that is limiting for polymerase function. In vitro it has been shown that high nucleotide levels lead to increased misincorporation rates because proofreading mechanisms have less time to function before the next nucleotide is inserted (Fersht 1979). Thus, it is possible that normal in vivo nucleotide levels cause polymerase pausing in a state that is deleterious in the absence of the replication stress response pathway. A second possibility is that the checkpoint has a direct role in up-regulating dNTP synthesis during S phase such that the loss of checkpoint function would actually cause a nucleotide depletion to which it then would not be able to respond. RAD53 does regulate the transcription of RNR1, RNR2, RNR3, and RNR4 in response to HU treatment and DNA damage; however, the viable rad53-21 allele is completely defective for this transcriptional regulation (Allen et al. 1994; Huang and Elledge 1997), suggesting that this function is not specifically lacking in null mutants. If up-regulation of nucleotide synthesis is regulated by the checkpoint, the defect is not at the level of RNR1 accumulation because RNR1 levels appear to be normal in the null mutants. Furthermore, overproduction of RNR2 and RNR4 fail to suppress rad53 lethality (data not shown). A third possibility is that in the absence of the checkpoint, a secondary event causes a more rapid consumption of dNTPs such that their levels are lower than normal, mimicking HU treatment. This, together with an inability to respond to such a nucleotide depletion, however transient, could cause lethality.
Currently we cannot distinguish between the three models presented in the preceding paragraph. However, the third model, indirect nucleotide depletion as a secondary effect of checkpoint deficiency, has recently gained support. The firing of late replication origins is advanced in rad53 and mec1 mutants (Santocanale and Diffley 1998, and pers. comm.). Consistent with this observation, we found that the mec1-21 point mutant suppresses the temperature sensitivity of mutations in ORC2, a gene required for origin recognition and firing. Normally at the G1-S transition, up-regulation of ribonucleotide reductase and the triggering of replication origins occur by separate but parallel regulatory networks. Yet the activation of replication complexes and the dNTP supply must be coordinated because firing of origins with insufficient nucleotide levels would cause a condition of effective nucleotide deprivation. The S-phase checkpoint pathway may provide this coordination. Failure to do so would result in premature or excessive origin firing as observed in mec1 and rad53 mutants. The presence of more origins replicating DNA at the same time might consume nucleotides faster than they can be synthesized, leading to DNA replicative stress, a checkpoint requiring situation. RNR1 overexpression could alleviate this problem without restoring checkpoint function. We tested this by artificially slowing down origin firing in checkpoint mutant backgrounds using temperature-sensitive dbf4-1, cdc7-1, and orc2-1 mutants. Although these mutants were unable to suppress the lethality of rad53 null mutants, we have found that mutations in dbf4 and cdc7 can suppress the mec1 null mutant. The inability to suppress the rad53 null mutation might indicate a novel role for RAD53 relative to MEC1, or a possible redundancy in RAD53 regulation. We have shown previously that TEL1, a MEC1 homolog, can activate Rad53 to a limted degree (Sanchez et al. 1996). Thus, it is possible that a rad53 null mutant could have a more severe defect than a mec1 null mutant under certain circumstances. In addition, it is possible that dbf4 mutants can suppress the lethality of rad53 null mutations but that the double mutant then dies because of a condition unique to the rad53 null mutation. In support of such a possibility we have observed that dbf4-1 rad53-21 and cdc7-1 rad53-21 double mutants are inviable (B.A. Desany and S.J. Elledge, unpubl.).
The genetic interactions between the checkpoint and origin initiation pathways support the notion that the MEC1/RAD53 pathway is acting antagonistically to the origin firing machinery for the purpose of maintaining coordination between the initiation of DNA replication and the nucleotide supply. Furthermore, we believe that the simplest interpretation of our data is that in the absence of the checkpoint pathway, nucleotide levels become limiting either by increased consumption due to increased origin-firing or by an unknown mechanism, and this situation, together with the absence of the ability to properly respond to nucleotide depletion, results in lethality.
What is responsible for lethality in the presence of HU?
Replication checkpoint-defective cells die rapidly when exposed to HU, and inappropriate spindle elongation has been thought to be responsible for this lethality. However, microtubule-inhibiting drugs are incapable of rescuing either the lethality of the mec1 and rad53 null mutants or the HU sensitivity of the point mutants. Additionally, the spindle elongation defects of the mec1 and rad53 null mutants are similar to each other, whereas their sensitivities to HU are significantly different. We interpret this to indicate that spindle elongation, rather than being the sole lethal event in these cells, is being misregulated independently of another event that is irreversibly committing cells to death. This is similar to the results obtained in S. pombe with mutations in cds1, the gene related most closely to RAD53. cds1 mutants die in response to HU treatment but do not appear to enter mitosis prematurely (Murakami and Okayama 1995; Lindsay et al. 1998). Similar results were obtained with hus1 mutants (Enoch et al. 1992). Although there was no attempt to artificially delay mitotic entry to rescue the lethality in those experiments, it is likely that these mutants are dying for the same reasons as rad53 mutants in HU. Our experiments show that rad53 mutant cells have a reduced ability to synthesize intact chromosomes following transient nucleotide depletion. This is not due to an inability to resume dNTP production because bulk DNA synthesis resumes after the block is removed, albeit with slower kinetics than wild type. Whether the structures that prevent chromosome migration in pulsed field gels are normal replicational intermediates that persist much longer than usual, such as replication forks, or are structurally aberrant in some way because of errors resulting from stalled polymerases is not clear. Stalled replication complexes could occasionally disintegrate and require checkpoint-mediated restoration. Alternatively, the collapse of complexes on converging forks could leave lethal gaps of unreplicated DNA. Aberrant DNA repair could also lead to defective chromosomal structure. Although it is not known whether the MEC1/RAD53 pathway directly controls repair processes, it is clear that HU causes damage because rad51 and rad52 mutants are very sensitive to HU (Allen et al. 1994).
Taken together, our results suggest that inviability of rad53 and mec1 null mutations is not due to premature mitotic entry but to an inability to survive with the existing nucleotide levels present in those mutants. Furthermore, our results indicate that the lethality resulting from limiting nucleotides is not purely a cell cycle transition phenomenon but is due instead to the profound inability of these mutants to properly carry out chromosomal replication after transient nucleotide depletion. Although this defect could be caused by misregulation of an as yet unappreciated aspect of cell cycle coordination distinct from anaphase commitment, it is clearly not the onset of anaphase that is causing lethality in these mutants because preventing anaphase cannot restore viability after a transient replication block. We favor the model that the checkpoint pathway is more than a cell cycle response. The fact that mec1 and rad53 null mutants appear to be equally checkpoint defective but have significantly different sensitivities to DNA-damage and replication-blocking agents suggests that this pathway controls repair activities in addition to coordination of cell cycle transitions. In this light, these pathways should be considered to be DNA-damage and DNA replication-block stress-response pathways as opposed to solely concerning themselves with cell cycle transitions.
Materials and methods
Yeast growth conditions
Yeast cells were grown at 30°C unless indicated otherwise. Rich and SC medium was formulated according to Kaiser et al. (1994). The carbon source was glucose, unless indicated, in which case the glucose was replaced by galactose. Where indicated, 5-FOA was used at 0.1%, and benomyl in solid media was used at 15 μg/ml.
Isolation of SRL genes
Strain Y324 (see text and Table 2) was grown in YPD and transformed with a 2μ TRP1 S. cerevisiae cDNA library (ATTC nos. 87288 and 47059) using the lithium acetate method. Transformants were plated on SC − Trp GAL (containing galactose) and replica-plated to SC − Trp GAL supplemented with 5-FOA. Positive clones were tested for their ability to grow on SC − Trp supplemented with 100 mm HU. Negatives were then struck to either YPD or YPD with the glucose replaced by galactose (YPGal). Clones that displayed any degree of galactose-dependent growth were tested for repeatability by plasmid rescue and retransformation of Y324, followed by verification of 5-FOA resistance. These final positive clones were christened SRL genes.
Table 2.
Strains and plasmids used in this study
| Strain | Genotype | Source |
|---|---|---|
| Y81 | _MAT_α trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-100 | Allen et al. (1994) |
| Y300 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-100 | Allen et al. (1994) |
| Y301 | as Y300 rad53-21 | Allen et al. (1994) |
| Y312 | as Y323 Δrad53::HIS3/RAD53 | Allen et al. (1994) |
| Y323 | MATa/α trp1-1/trp1-1 ura3-1/ura3-1 his3-11,15/his3-11,15 leu2-3,112/leu2-3,112 ade2-1/ade2-1 can 1-100/can 1-100 | Allen et al. (1994) |
| Y324 | as Y81 Δrad53::HIS3 + pJA92 | Allen et al. (1994) |
| Y580 | as Y300 TRP1::GAP–RNR1 | this study |
| Y581 | as Y300 Δmec1::HIS3 TRP1::GAP–RNR1 | this study |
| Y601 | as Y300 Δrad53::HIS3 + pJA92 | this study |
| Y602 | as Y300 Δmec1::HIS3 + pBAD45 | this study |
| Y603 | as Y300 Δrad53::HIS3 + pBAD70 | this study |
| Y604 | as Y300 mec1-21 | this study |
| Y605 | as Y300 Δmec1::HIS3 + pBAD70 | this study |
| Y606 | as Y300 Δrad53::HIS3 TRP1::GAP–RNR1 | this study |
| Y607 | as Y300 TRP1::GAP–RNR1 | this study |
| Y608 | as Y300 Δrad53::HIS3 + pBAD79 | this study |
| Y609 | as Y300 Δrnr::HIS3 + pBAD79 | this study |
| Y610 | as Y300 Δmec1::HIS3 + pBAD79 | this study |
| Y611 | as Y300 orc2-1 | this study |
| Y612 | as Y300 orc2-1 mec1-21 | this study |
| Y613–616 | as Y300 dbf4-1 Δmec1::HIS3 + pBAD45 | this study |
| Y617 | as Y323 Δmec1::HIS3/MEC1 | this study |
| Y618 | as Y323 Δrad53::HIS3/RAD53 TRP1::GAP–RNR1/trp1-1 | this study |
| Y619 | as Y323 Δmec1::HIS3/MEC1 TRP1::GAP–RNR1/trp1-1 | this study |
| Y620 | as Y81 mec1-21 | this study |
| Y621 | as Y323 orc2-1/ORC2 mec1-21/MEC1 | this study |
| Y622 | as Y323 dbf4-1/DBF4 Δmec1::HIS3/MEC1 | this study |
| Y623 | as Y300 ρ0_HIS3_ | this study |
| Y624 | as Y301 ρ0_HIS3_ | this study |
| Y692 | as Y300 TRP+ | this study |
| YCH266 | as Y81 dbf4-1 | C. Hardy (Washington University, St. Louis, MO) |
| Plasmid | Relevant markers | |
| pAB23BXN | Apr 2μ URA3 GAP promoter | T. Brake (Chiron Corporation, Emeryville, CA) |
| pTRP | Apr TRP1 2μ GAL promoter | Mulligan and Elledge (1994) |
| pJA50 | Apr Knr HIS3 | Allen and Elledge (1994) |
| pJA92 | Apr URA3 CEN4 RAD53 | Allen et al. (1994) |
| pSAD3-3B | Apr CEN4 TRP1 MEC1 | this study |
| pWJ87 | Apr CEN4 TRP1 Δmec1::HIS3 | this study |
| pJR1267 | Apr URA3 orc2-1 | C. Fox and J. Rine (University of California, Berkeley) |
| pSE734 | Apr RNR3 | Elledge and Davis (1990) |
| pSE757 | _Apr_2μ TRP1 RNR1 | Elledge and Davis (1990) |
| pBAD40 | Apr CEN4 URA3 | this study |
| pBAD45 | Apr URA3 CEN4 MEC1 | this study |
| pBAD49 | Apr RNR1 PCR product | this study |
| pBAD54 | Apr TRP1 2μ GAP promoter | this study |
| pBAD58 | Apr RNR3 PCR product | this study |
| pBAD62 | Apr RNR1 ORF | this study |
| pBAD70 | Apr TRP1 2μ GAP–RNR1 | this study |
| pBAD74 | Apr RNR3 ORF | this study |
| pBAD79 | Apr TRP1 2μ GAP–RNR3 | this study |
| pBAD114 | Apr TRP1 GAP–RNR1 | this study |
RNA purification and Northern blotting
RNA purification and Northern blotting were performed as described (Navas et al. 1995). For detection of the endogenous RNR1 transcript in the presence of exogenously provided RNR1, we used a _Hin_dIII–_Spe_I fragment as a probe corresponding to nucleotides 2642–3317 of the 3′ end of the RNR1 transcript. These sequences are not present on the exogenous RNR1 expression constructs. For detection of CLN2 mRNA, we probed using a _Sty_I fragment of CLN2 comprising nucleotides 460–1541 of the 1638 nucleotide ORF.
Quantitation of bands was performed by exposing the blots to a Storage Phosphor Screen (Molecular Dynamics, Sunnyvale, CA) and using ImageQuant software to quantitate the band intensities. In all cases, the lane background was subtracted from each band prior to normalization to the loading control (ACT1).
HU- and UV-killing assays
For HU killing, cultures were grown to log phase in YPD, whereupon the medium was replaced with YPD + 0.2 m HU (unless indicated otherwise), and aliquots were removed and plated on YPD at timed intervals and allowed to grow for several days at 30°C. For UV killing, cells were grown to log phase in YPD, plated on YPD, and irradiated (Stratagene UV Stratalinker 1800) with 0, 20, or 40 J/m2 prior to incubation at 30°C.
Synchronization of cells in G1 phase
Strains were grown to log phase in YPD (pH 3.9), treated with 10 μg/ml α-factor for 1.5 hr, and supplemented with an additional 5 μg/ml α-factor for another 1.5 hr. Cells were then centrifuged and resuspended in YPD containing the 0.2 m HU, 0.25 m HU, 80 μg/ml benomyl, and/or 10 μg/ml nocodazole as indicated in the individual experiments.
Staining of cells for microtubule visualization
Cells were fixed by the addition of 5% formaldehyde to growing cultures and allowed to stand for at least 4 hr at 4°C. Cells were washed in PBS, and microtubules were immunostained using the antitubulin antibody YOL1/34 and a FITC-conjugated secondary antibody as described (Allen et al. 1994).
FACS analysis
The amount of 250 μl of cell culture (∼1.5 × 106 to 4 × 106 cells) was added directly to 1 ml of ethanol and allowed to stand 1 hr for fixation. Cells were washed once with 70% ethanol and once with FACS buffer (0.2 m Tris at pH 7.5, 20 mm EDTA). In a volume of 100 μl of FACS buffer, cells were treated with 1 mg/ml RNase A at 37°C for 2 hr. Cells were then washed in PBS, treated with 5 μg/ml propidium iodide in a final volume of 1 ml of PBS, and analyzed for fluorescence content using a Coulter model Epics XL-MCL. The DNA content of ∼30,000 cells was determined for each sample.
PFGE of replication intermediates
α-Factor-arrested ρ0 strains were released into YPD containing 0.2 m HU and 10 μg/ml nocodazole for 60 min; cells were spun down, washed, and resuspended in YPD containing 10 μg/ml nocodazole. Cells from different time points during and after HU treatment were fixed in 70% ethanol overnight. These were subsequently resuspended in 0.5 m EDTA, 1.2 m sorbitol, and 1 m Tris (pH 7.5). Chromosome plugs were prepared following a rapid two-step protocol without use of proteinase K (Johnston 1994). Each 75 μl plug contained 4.5 × 106 cells. PFGE was carried out in a Bio-Rad DR II apparatus for 24 hr, at 200 V. Switching was done every 60 sec for the first 15 hr, and every 90 sec for the last 9 hr. Chromosomes were visualized with ethidium bromide. The gel was photographed and chromosome band intensities were quantitated using NIH Image software.
Strain and plasmid construction
The source of the MEC1 gene was pSAD3-3B, which is a 9.5-kb fragment of the MEC1 genomic locus cloned into pRS414 (Sikorski and Hieter 1989). pBAD45 contains the 7.7-kb _Sac_I _MEC1_-containing fragment from pSAD3-3B cloned into the _Sac_I site of pBAD40, which is a derivative of pRS416 (Sikorski and Hieter 1989) deleted between the _Not_I and _Sal_I sites. pBAD54 is a GAP promoter expression vector made by cloning the GAP expression cassette, containing the GAP promoter and GAP terminator flanking a multicloning site, as a _Bam_HI fragment from pAB23BXN into the _Bam_HI site of YEplac112 (Gietz and Sugino 1988).
The RNR1 and RNR3 ORFs were cloned by PCR and subcloned into pBS II KS(−) to make pBAD49 and pBAD58. The ends of each ORF were sequenced to verify lack of mutation, and the central parts of each ORF were replaced by the corresponding fragment from a functional genomic clone. For RNR1 this was a _Bst_EII–_Xba_I fragment from pSE757 generating pBAD62, and for RNR3 it was a _Bst_EII–_Hin_dIII fragment from pSE734 generating pBAD74. pBAD70 was made by subcloning the RNR1 ORF as a _Xho_I–_Not_I fragment from pBAD62 into _Xho_I–_Not_I-digested pBAD54. pBAD79 was made by subcloning the RNR3 ORF as a Psp1406I(T4-filled in)–_Not_I fragment from pBAD74 into pBAD54 that had been cut with _Xho_I and T4-filled in and subsequently cut again with _Not_I.
The RAD53 gene knockout has been described previously (Allen et al. 1994). MEC1 was knocked out by replacing a 7.5-kb _Bam_HI fragment from pSAD3-3B with the Bam_HI fragment from pJA50 containing the HIS3 gene and a kanamycin resistance gene from Tn_5 to form pWJ87. This removes all but the amino-terminal 33 amino acids from the MEC1 ORF. The 4.4-kb _Sac_I fragment from pWJ87 containing the Δmec1::HIS3 deletion construct was transformed into Y323 to generate a diploid heterozygous for the mec1 knockout Y617.
The TRP1::GAP–RNR1 expression cassette was created by subcloning a _Pst_I–_Sac_I fragment from pBAD70 into _Pst_I–_Sac_I-digested pRS404 to create pBAD114. rad53 and mec1 null mutants suppressed by this GAP–RNR1 expression cassette were generated as follows. pBAD114 was linearized within the TRP1 gene and transformed into Y312 and Y617 to create Y618 and Y619, and correct integration was confirmed by Southern blotting. Y618 was sporulated and Y606 and Y607 were recovered. Y619 was sporulated to obtain Y580 and Y581.
The temperature-sensitive orc2-1 mutant Y611 was generated by looping the orc2-1 allele from the _URA3_-integrating plasmid pJR1267 into Y300. We then selected transformants for 5-FOA resistance and screened them for temperature sensitivity. Y612 was made by crossing Y611 with Y620 and sporulating and dissecting the resulting diploid Y621. Y613, Y614, Y615, and Y616 are four spores of identical genotype that were isolated from the diploid Y622, which was in turn created by a mating between Y602 and YCH266.
Y623 and Y624, His+ ρ0 derivatives of Y300 and Y301, respectively, were generated by serial culturing in minimal media containing ethidium bromide, as described in Fox et al. (1991).
Acknowledgments
We thank Ted Weinert, J. Diffley for sharing unpublished data, W. Shoeber for FACS analysis, B. Sclafani, C. Hardy, C. Fox, and J. Rine. We also thank J. Diller for discussions, and members of the Elledge laboratory for comments, helpful discussions, and/or reagents. This work was supported by National Institutes of Health grant GM44664 to S.J.E. S.J.E. is an investigator of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL selledge@bcm.tmc.edu; FAX (713) 798-8717.
References
- Al-Khodairy F, Fotou E, Sheldrick KS, Griffiths DJF, Lehmann AR, Carr AM. Identification and characterization of new elements involved in checkpoint and feedback controls in fission yeast. Mol Biol Cell. 1994;5:147–160. doi: 10.1091/mbc.5.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JB, Elledge SJ. A family of vectors that facilitate transposon and insertional mutagenesis of cloned genes in yeast. Yeast. 1994;10:1267–1272. doi: 10.1002/yea.320101003. [DOI] [PubMed] [Google Scholar]
- Allen JB, Zhou Z, Siede W, Friedberg EC, Elledge SJ. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes & Dev. 1994;8:2416–2428. doi: 10.1101/gad.8.20.2401. [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Carr AM, Moudjou M, Bentley NJ, Hagan IM. The chk1 pathway is required to prevent mitosis following cell-cycle arrest at ‘start’. Curr Biol. 1995;5:1179–1190. doi: 10.1016/s0960-9822(95)00234-x. [DOI] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Elledge SJ, Davis RW. Two genes differentially regulated in the cell cycle and by DNA-damaging agents encode alternative regulatory subunits of ribonucleotide reductase. Genes & Dev. 1990;4:740–751. doi: 10.1101/gad.4.5.740. [DOI] [PubMed] [Google Scholar]
- Elledge SJ, Mulligan J, Ramer S, Spottswood M, Davis RW. Lambda YES: A multifunctional cDNA expression vector for the isolation of genes by complementation of yeast and E. coli mutations. Proc Natl Acad Sci. 1991;88:1731–1734. doi: 10.1073/pnas.88.5.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ, Zhou Z, Allen JB. Ribonucleotide reductase: Regulation, regulation, regulation. Trends Biochem Sci. 1992;17:119–123. doi: 10.1016/0968-0004(92)90249-9. [DOI] [PubMed] [Google Scholar]
- Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, Leder P. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci. 1996;93:13084–13089. doi: 10.1073/pnas.93.23.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch T, Carr A, Nurse P. Fission yeast genes involved in coupling mitosis to completion of DNA replication. Genes & Dev. 1992;6:2035–2046. doi: 10.1101/gad.6.11.2035. [DOI] [PubMed] [Google Scholar]
- Fersht AR. Fidelity of replication of phage phi X174 DNA by DNA polymerase III holoenzyme: Spontaneous mutation by misincorporation. Proc Natl Acad Sci. 1979;76:4946–4950. doi: 10.1073/pnas.76.10.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaggs G, Plug AW, Dunks KM, Mundt KE, Ford JC, Quiggle MR, Taylor EM, Westphal CH, Ashley T, Hoekstra MF, Carr AM. Atm-dependent interactions of a mammalian chk1 homolog with meiotic chromosomes. Curr Biol. 1997;7:977–986. doi: 10.1016/s0960-9822(06)00417-9. [DOI] [PubMed] [Google Scholar]
- Ford JC, Al-Khodairy F, Fotou E, Sheldrick KS, Griffiths DJ, Carr AM. 14-3-3 protein homologs required for the DNA damage checkpoint in fission yeast. Science. 1994;265:533–555. doi: 10.1126/science.8036497. [DOI] [PubMed] [Google Scholar]
- Fox TD, Folley LS, Mulero JJ, McMullin TW, Thorsness PE, Hedin LO, Costanzo MC. Analysis and manipulation of yeast mitochondrial genes. Methods Enzymol. 1991;194:149–165. doi: 10.1016/0076-6879(91)94013-3. [DOI] [PubMed] [Google Scholar]
- Friedberg E C, Walker GC, Siede W. DNA repair and mutagenesis. Washington D.C.: ASM Press; 1995. [Google Scholar]
- Furnari B, Rhind N, Russell P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science. 1997;277:1495–1497. doi: 10.1126/science.277.5331.1495. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD. TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell. 1995;82:823–829. doi: 10.1016/0092-8674(95)90479-4. [DOI] [PubMed] [Google Scholar]
- Hennessy KM, Lee A, Chen E, Botstein D. A group of interacting yeast DNA replication genes. Genes & Dev. 1991;5:958–969. doi: 10.1101/gad.5.6.958. [DOI] [PubMed] [Google Scholar]
- Huang M, Elledge SJ. Identification of RNR4, encoding a second essential small subunit of ribonucleotide reductase in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:6105–6113. doi: 10.1128/mcb.17.10.6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Pahl PM, Harrison K, Rosamond J, Sclafani RA. Cell cycle regulation of the yeast Cdc7 protein kinase by association with the Dbf4 protein. Mol Cell Biol. 1993;13:2899–1908. doi: 10.1128/mcb.13.5.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JR. Pulsed field gel electrophoresis. In: Johnston JR, editor. Molecular genetics of yeast: A practical approach. Oxford, UK: Oxford University Press; 1994. pp. 83–95. [Google Scholar]
- Kaiser C, Michaelis S, Mitchell A. Methods in yeast genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fournace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in Ataxia-Telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Kato R, Ogawa H. An essential gene, ESR1, is required for mitotic cell growth, DNA repair and meiotic recombination in Saccharomyces cerevisiae. Nucleic Acids Res. 1994;22:3104–3112. doi: 10.1093/nar/22.15.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Weinreich M, Stillman B. ORC and Cdc6p interact and determine the frequency of initiation of DNA replication in the genome. Cell. 1995;81:667–676. doi: 10.1016/0092-8674(95)90528-6. [DOI] [PubMed] [Google Scholar]
- Lindsay HD, Griffiths DJ, Edwards RJ, Christensen PU, Murray JM, Osman F, Walworth N, Carr AM. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes & Dev. 1998;12:382–395. doi: 10.1101/gad.12.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb LA, Kunkel TA. Fidelity of DNA synthesis. Annu Rev Biochem. 1982;51:429–457. doi: 10.1146/annurev.bi.51.070182.002241. [DOI] [PubMed] [Google Scholar]
- Meyn MS. Ataxia-telangiectasia and cellular responses to DNA damage. Cancer Res. 1995;55:5991–6001. [PubMed] [Google Scholar]
- Morrow DM, Tagle DA, Shiloh Y, Collins FS, Hieter P. TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell. 1995;82:831–840. doi: 10.1016/0092-8674(95)90480-8. [DOI] [PubMed] [Google Scholar]
- Mulligan JT, Elledge SJ. The construction and use of cDNA libraries for genetic selections. In: Johnston JR, editor. Molecular genetics of yeast: A practical approach. Oxford, UK: Oxford University Press; 1994. pp. 65–81. [Google Scholar]
- Murakami H, Okayama H. A kinase from fission yeast responsible for blocking mitosis in S phase. Nature. 1995;374:817–819. doi: 10.1038/374817a0. [DOI] [PubMed] [Google Scholar]
- Navas TA, Zhou Z, Elledge SJ. DNA polymerase ε links the DNA replicational machinery to the S phase checkpoint. Cell. 1995;80:29–39. doi: 10.1016/0092-8674(95)90448-4. [DOI] [PubMed] [Google Scholar]
- Pati D, Keller C, Groudine M, Plon SE. Reconstitution of a MEC1-independent checkpoint in yeast by expression of a novel human fork head cDNA. Mol Cell Biol. 1997;17:3037–3046. doi: 10.1128/mcb.17.6.3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones W, Liu Q, Wang B, Elledge SJ. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thomas RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–1501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- Santocanale, S. and J.F.X. Diffley. 1998. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature (in press). [DOI] [PubMed]
- Sikorski RS, Hieter P. A system of shuttle vectors and host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Fay DS, Marini F, Stern DF. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes & Dev. 1996;10:395–406. doi: 10.1101/gad.10.4.395. [DOI] [PubMed] [Google Scholar]
- Walworth N, Bernards R. rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science. 1996;271:353–356. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- Walworth N, Davey S, Beach D. Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature. 1993;363:368–371. doi: 10.1038/363368a0. [DOI] [PubMed] [Google Scholar]
- Weinert TA, Hartwell LH. Cell cycle arrest of cdc mutants and specificity of the RADX9, checkpoint. Genetics. 1993;134:63–80. doi: 10.1093/genetics/134.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert TA, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes & Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes & Dev. 1996;10:2411–2422. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Elledge SJ. Isolation of crt mutants constitutive for transcription of the DNA damage inducible gene RNR3 in Saccharomyces cerevisiae. Genetics. 1992;131:851–866. doi: 10.1093/genetics/131.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— DUN1 encodes a protein kinase that controls the DNA damage response in yeast. Cell. 1993;75:1119–1127. doi: 10.1016/0092-8674(93)90321-g. [DOI] [PubMed] [Google Scholar]