Direct observation of multiple misfolding pathways in a single prion protein molecule (original) (raw)
Abstract
Protein misfolding is a ubiquitous phenomenon associated with a wide range of diseases. Single-molecule approaches offer a powerful tool for deciphering the mechanisms of misfolding by measuring the conformational fluctuations of a protein with high sensitivity. We applied single-molecule force spectroscopy to observe directly the misfolding of the prion protein PrP, a protein notable for having an infectious misfolded state that is able to propagate by recruiting natively folded PrP. By measuring folding trajectories of single PrP molecules held under tension in a high-resolution optical trap, we found that the native folding pathway involves only two states, without evidence for partially folded intermediates that have been proposed to mediate misfolding. Instead, frequent but fleeting transitions were observed into off-pathway intermediates. Three different misfolding pathways were detected, all starting from the unfolded state. Remarkably, the misfolding rate was even higher than the rate for native folding. A mutant PrP with higher aggregation propensity showed increased occupancy of some of the misfolded states, suggesting these states may act as intermediates during aggregation. These measurements of individual misfolding trajectories demonstrate the power of single-molecule approaches for characterizing misfolding directly by mapping out nonnative folding pathways.
Keywords: protein folding, optical tweezers
Protein folding involves a stochastic search through the configurational energy landscape of the protein to find the native structure. Although most proteins have evolved to fold efficiently into a unique native structure, misfolding (the formation of nonnative structures) occurs frequently in vivo (1). Cellular processes act to mitigate the effects of misfolding, e.g., by preventing misfolding from occurring with the help of molecular chaperones or by removing misfolded proteins once they have formed through the action of the proteasome (1, 2). Misfolded proteins that escape such quality-control pathways, however, can lead to a wide range of diseases, such as Alzheimer’s, Parkinson’s, and the prion disorders (3).
Biophysical studies of protein misfolding show that it is a very complex process (3). The many different conformations involved, the numerous alternative pathways, and the likely importance of rare or transient states all pose key technical challenges for characterizing misfolding mechanisms. Single-molecule (SM) spectroscopic approaches are well suited to overcome these challenges: Not only are they well established for studying protein folding mechanisms, but they can directly characterize distinct subpopulations, map out folding pathways, and observe rare or transient states (4). However, SM spectroscopy has not yet been widely applied to characterize protein misfolding. Misfolding has been observed in a few protein constructs using both force (5–7) and fluorescence spectroscopies (8), and the formation and growth of aggregates has been monitored with fluorescence spectroscopy (9–11), but the network of pathways available for misfolding has only begun to be mapped out in any detail (12) and not yet for any disease-related protein.
Here we describe a SM force spectroscopy study of misfolding in the prion protein PrP, a highly conserved membrane-associated protein notable for its ability to misfold into a conformation that is infectious. The infectious form, denoted PrPSc, recruits natively folded PrP, denoted PrPC, to form additional PrPSc, thereby permitting transmission between individuals and species (13, 14). PrP has been studied extensively to elucidate the mechanisms for misfolding and conversion. PrPC has a structured C-terminal domain containing three α-helices and two short β-strands (Fig. 1A and Fig. S1) and an unstructured N-terminal domain (15–17). In contrast, PrPSc is rich in β-sheets (18). Although its structure remains unknown, several models have been proposed for the amyloid fibers into which it aggregates (19–21). Infectivity is believed to arise from oligomeric PrPSc (22), with the dominant model for conversion of PrPC to PrPSc involving seeded nucleation (14): Fluctuations of PrPC monomers produce a rare, nonnative conformation able to form an ordered, misfolded oligomer that then recruits and stabilizes additional monomers. Partially folded intermediates of PrP have long been proposed to play a key role in this process (23), as in protein aggregation more generally (3), but evidence for their existence is conflicting: Some studies indicate only two-state folding (24), whereas others suggest the presence of an intermediate (25–28).
Fig. 1.

Force spectroscopy measurements of PrP show apparent two-state folding. (A) SHaPrP(90–231) labeled at both termini with Cys residues was attached to sulfhydryl-labeled DNA strands bound to beads held in optical traps. The extension of the molecule held under tension by the traps was measured as the protein unfolded or refolded. (B) FECs of PrP. The handles stretch as the force rises monotonically until PrP unfolds at approximately 10 pN, causing a discrete jump in the extension and force (black). By overlaying 100 unfolding FECs (red), the contour length change is found from WLC fits to the folded (orange) and unfolded (cyan) states to be the value expected for unfolding of the native state. (C) With the force held constant at 9.1 pN by a passive force clamp, the extension jumps between two values corresponding to the unfolded state (U) at higher extension and natively folded state (N) at lower extension.
Despite significant advances in the characterization of PrP, misfolding and conversion still remain poorly understood, in part because they are difficult to observe directly. There have been few SM studies of prions to date: Yeast prion structural dynamics were studied with SM fluorescence (29), and the structural interactions within prion amyloid fibers were probed by force spectroscopy (30, 31), but the properties of individual molecules of PrP—especially their ability to form nonnative structures—have not yet been investigated. To observe misfolding directly in single PrP molecules, we attached kilobase-long DNA handles to the protease-resistant fragment (residues 90–231) of Syrian hamster PrP (SHaPrP), and then bound the handles specifically to polystyrene beads held in optical tweezers (Fig. 1A). Using the tweezers to apply denaturing tension to a single PrP molecule, we monitored the resulting dynamic structural changes in the protein by measuring the end-to-end extension of the molecule with high spatial and temporal resolution.
Results
The extension of the PrP constructs was first measured while moving the traps apart at a constant rate to ramp up the force, creating force-extension curves (FECs). The extension increased monotonically with force as the handles were stretched (Fig. 1B) until the protein unfolded at approximately 10 pN, causing an abrupt increase in extension and concomitant drop in force indicative of an apparently two-state process (4). Refolding curves, where the force was ramped down, also showed two-state behavior (Fig. S2). The same two-state behavior, without any distinguishable subpopulations (e.g., different contour length changes or unfolding forces), was displayed by 3,250 FECs measured on seven molecules. The contour length change, Δ_L_ c, determined from worm-like chain (WLC) fits to the FECs (Fig. 1B), was Δ_L_ c = 34.1 ± 0.4 nm (all uncertainties represent the standard error on the mean). The number of amino acids unfolded, _n_aa, was calculated from 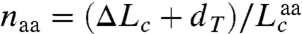 , where d T is the distance between the termini of the structured domain and
, where d T is the distance between the termini of the structured domain and 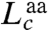 is the contour length per amino acid (Supporting Information). Given d T = 3.1 nm from the NMR structure (15) and the crystallographic value
is the contour length per amino acid (Supporting Information). Given d T = 3.1 nm from the NMR structure (15) and the crystallographic value  (32), we found _n_aa = 103 ± 1, consistent with complete unfolding and refolding of the 104-aa native structure (Fig. S1).
(32), we found _n_aa = 103 ± 1, consistent with complete unfolding and refolding of the 104-aa native structure (Fig. S1).
FECs probe the folding out of equilibrium, due to the changing force. To investigate the folding under equilibrium conditions, we also measured the extension of the molecule as a function of time while the force was held constant using a passive force clamp (33). The extension jumped between two values corresponding to natively folded (N) and unfolded (U) PrP as in the FECs, spending very little time between (Fig. 1C). No intermediates are immediately obvious in these data, despite the proven effectiveness of constant-force measurements for detecting them (34–37). Although the observation of two-state folding agrees with some previous ensemble measurements (24), others have inferred the existence of an intermediate (25–28). To search for direct evidence of an intermediate that might be too rare or short lived to be readily apparent at first glance, we examined extensive constant-force records (several hours in total) measured at high bandwidth. A passive force clamp was essential here, to avoid artifactual transients from feedback loop closure (Fig. S3). Because these measurements were made under equilibrium conditions, the protein must have sampled all possible transitions between different conformations. Any intermediate state, I, on the native folding pathway should thus have been seen not only as a step between U and N when the molecule unfolded or refolded but also as transient excursions from U and, separately, from N (Fig. S4).
We first searched for I during the brief time spent moving between N and U. We aligned all transitions on their midpoints [Fig. 2A (red)] by fitting them to the logistic function (Fig. S5), and then averaged them to reduce noise: 3,364 unfolding transitions were aligned and averaged [Fig. 2A (black)], as were 3,318 folding transitions [Fig. 2B (red)]. Because an intermediate state would slow down the transition from N to U (or U to N) compared to a simple two-state process, we next compared the measured transitions to the signal produced by a single step-like motion in the trap, as would be expected for two-state folding. To do this, the motion of a reference construct containing only the DNA handles (no protein) was measured as the trap was moved abruptly (< 1 μs) by 20 nm, equal to the U–N extension change. Aligning and averaging 200 such response curves as above [Fig. 2B (blue)], we observed that the folding and unfolding transitions [Fig. 2B (red and black, respectively)] have precisely the same shape as the instrument response to a step function, confirming that there is no intermediate. To determine the measurement resolution and set an upper limit on the lifetime of any putative state I, τ I, we used the transfer function describing the instrument response (Fig. S6) to simulate the shape of the average transition that would have resulted from an intermediate located at an extension Δ_x_UI from the unfolded state (Fig. S7_A_), as described in Supporting Information. The results are illustrated in Fig. 2C for Δ_x_UI = 7 nm and τ I = 0–100 μs. Multiple simulations using different Δ_x_UI and τ I values (Fig. S7) indicate that any obligate on-pathway intermediate must have a lifetime under tension of less than approximately 50–100 μs.
Fig. 2.

On-pathway intermediates are not observed. (A) To search for on-pathway intermediates, 100 separate transitions between folded (N) and unfolded (U) states taken from a single constant-force record were aligned on the midpoint of the transitions (red) and averaged. The average of 3,364 transitions is shown in black. (B) The average of the unfolding transitions (black) and the time-reversed average of 3,318 folding transitions (red) are identical within experimental uncertainty, and the same as the average instrumental response function measured with a reference construct lacking protein (blue). An intermediate would cause the transitions to differ from the instrument response. Error bars: standard error. (C) The average signal expected for an obligate intermediate located 7 nm from U was simulated using the instrument response function for various lifetimes τ I. Comparing the measured average (black) to the simulated signals (yellow red, cyan, purple: τ I = 0, 20, 50 and 100 μs, respectively) placed an upper limit of τ I < 50–100 μs for any intermediate.
We also searched for transient excursions into a possible intermediate coming from and returning to N, another type of event that must occur at equilibrium if I is on-pathway (Fig. S4). No transitions from N to any state other than U could be discerned directly from the constant-force records (Fig. 3). To determine the maximum possible occupancy of such a state consistent with our data, we measured the point-spread function (PSF) of the trap (the distribution of extensions expected due to fluctuations when measuring a fixed contour length), using the reference construct lacking protein (Fig. 4A). We then binned the extensions in the low-extension (nominally folded) state of the PrP trajectories and fit the resulting histogram to the PSF (Fig. 4B), looking for peaks corresponding to states with different extension. From the lack of peaks in the residual to these fits (Fig. S8), we estimate that any short-lived on-pathway intermediate must be occupied less than 0.001% of the time (Supporting Information). Because tension increases the lifetime and occupancy of extended states exponentially (38), any on-pathway intermediate would have to be exceedingly rare or short-lived at zero force.
Fig. 3.

Constant-force records reveal misfolded states. (A) High-resolution extension records at a constant force of 9.5 pN reveal frequent transient spikes down from state U, reaching two different extension values that represent off-pathway intermediates (labeled M1, M2). (Inset) Magnified view of data in blue square. (B) Transitions from U to a longer-lived off-pathway state (M3) are observed very rarely. Force = 9.2 pN.
Fig. 4.

Misfolded states form via the unfolded state only. (A) A histogram of the extension from a constant-force record of the reference construct lacking protein (Upper, gray) shows the PSF of the trap fit to Eq. S1 for the PSF (cyan). Counting noise in the residual (Lower, gray) from the histogram binning is smoothed in a 2.5-nm window (black). (B) A histogram of the native-state extension at constant force (Upper, red) fits well to the measured PSF of the trap (cyan). The residual (Lower, pink) is smoothed as above (red) and is featureless, showing that N does not fold/unfold into anything other than U. (C) Fitting the unfolded-state extension histogram (Upper, red) to the PSF (cyan) leaves a significant residual (Lower, pink). Fitting the peak in the smoothed residual (Lower, red) to the PSF (blue) again leaves a significant residual (purple), which is well fit by a third PSF (gray); the three PSFs together fit the full histogram completely (black). The residual fits yield the extension and occupancy of the misfolded states M1 and M2, which are entered only from U.
Whereas no intermediates were observed on the native pathway, the search for rare and short-lived events did reveal several states with extensions between U and N that were only entered and exited via U and hence must be off-pathway. They were generally very short-lived (approximately 1 ms or less), appearing as “spikes” pointing down from U in the constant-force extension records (Fig. 3A, Inset). These spikes are not simply part of the extension distribution for U: In contrast to the native state distribution (Fig. 4B), a histogram of the data in the high-extension (nominally unfolded) state of the constant-force trajectory (Fig. 4C) reveals a small residual peak between N and U after subtracting the PSF, indicating the presence of extra states. Two additional states are needed to fit the residual: The first, M1, is located 7.1 ± 0.4 nm from U and is occupied 0.6 ± 0.1% of the time; the second, M2, is 10.5 ± 0.5 nm from U and occupied 0.11 ± 0.02% of the time. Very infrequently (approximately 6 hr-1, occupancy approximately 0.04%), an additional state (denoted M3 in Fig. 3B) was observed 4.9 ± 0.2 nm from U, with a lifetime of approximately 50–100 ms. From the extensions and occupancies of these states at different forces (Supporting Information), we find that M1 has approximately 40–50 aa folded and is 3 ± 1 kcal/mol more stable than U at zero force, M2 has 60–70 aa folded and is 5 ± 2 kcal/mol more stable than U, and M3 has 30–40 aa folded and is approximately as stable as U. Because these states do not lead to the natively folded structure, they must represent misfolded conformations of PrP.
To investigate whether these nonnative states are related in some way to aggregation, we measured the folding of a mutant SHaPrP, C179A/C214A. This mutant aggregates much more readily than wild-type PrP (Fig. S9) and can form β-rich oligomers (39). Because the geometry of the SM assay isolates a single protein molecule, however, aggregation is prevented during the SM measurements, allowing us to probe how the misfolding differs in a monomer that would otherwise aggregate rapidly. FECs (Fig. S10) revealed two states with Δ_L_ c = 34 ± 1 nm, the value expected for the native structure, indicating that the native fold forms when the mutant is prevented from aggregating. Similar to the wild-type protein, misfolded states were observed in extension records at constant force (Fig. 5A), at the same extensions as M1–M3 and with similar kinetic properties. These states were therefore identified as states M1–M3. Notably, however, the occupancy of M1 was increased four- to fivefold and that of M2 by two- to threefold in the mutant as compared to wild-type PrP (Fig. 5 B and C), indicating that these states were, respectively, 1 and 0.5 kcal/mol more stable in the mutant (Fig. S11).
Fig. 5.

Mutant PrP shows increased misfolding. (A) Constant-force records of the C179A/C214A mutant show transient misfolding at the same extensions as M1, M2, and M3 for wild-type PrP, with similar lifetimes. Force = 9.2 pN (Left), 9.7 pN (Right). (B) The native-state histogram (Upper, red) is well fit by the trap PSF (blue). The residual to this fit (Lower, pink) is featureless after smoothing (red) to remove counting noise. (C) The unfolded-state histogram (Upper, red) has a prominent shoulder. Fitting the peak and shoulder to one PSF for U (blue) and another at the location of M1 (cyan) leaves a significant peak in the smoothed residual (Lower, red) at the location of M2, which is well fit by a third PSF (black). M1 and M2 are several times more prevalent in the mutant than the wild-type.
Discussion
These results offer another perspective on the complex structural dynamics of protein misfolding, through direct observation of the conformational fluctuations of single PrP molecules. The picture of the network of folding pathways that emerges for PrP (Fig. 6) reveals some surprising details that shed light on PrP misfolding. First, we find that native folding is a two-state process. We see no evidence for any partially folded intermediate on the native pathway, such as hypothesized to mediate misfolding (23), despite previous reports (25–28). We cannot completely rule out on-pathway intermediates, because force probes are not sensitive to structural changes that leave the molecular extension unchanged (as in a proposed intermediate where helix 1 restructures (40)); the chemical denaturants and low pH used in most misfolding studies might also produce intermediates not observable under the conditions here. Nevertheless, partially native intermediates in which the structure changes near the termini, such as proposals with a detached β-strand 1 (41) or selectively disordered helices 2 and 3 (28), are inconsistent with our observations.
Fig. 6.

Schematic diagram of folding/misfolding pathways. SHaPrP does not pass through an intermediate on the native pathway, but the unfolded state leads to three different misfolded states at varied rates. Folding and misfolding rates at approximately 9 pN are indicated. U = unfolded, I = intermediate, N = native, _M_1–_M_3 = misfolded.
Instead of misfolding proceeding through an on-pathway intermediate, we find that nonnative structures are only accessible from the unfolded state. Indeed, there is not just one misfolding pathway but rather three of them, leading to different nonnative structures. Remarkably, PrP explores these misfolding pathways more frequently than it does the native pathway: The rates for formation of M1 and M2 estimated from the constant-force trajectories, approximately 15 s-1 and 3 s-1, respectively (Supporting Information), are considerably higher than the rate for native folding at the same force, approximately 1 s-1. The relative rates imply that the vast majority (> 90%) of all attempts at structure formation lead to nonnative structures rather than the native state, although the resulting misfolded states are not very stable for isolated PrP molecules under these conditions and are thus rarely occupied. One type of misfolding we did not observe, however, despite the amyloid-forming propensity of residues 106–126 (42), was structure within the natively unstructured N terminus of the protein: Even transient structure formation by the N terminus while the C-terminal domain was folded would have produced a detectable peak at short extensions in the residual to the N-state extension histogram fit (Figs. 4B and 5B).
Our results suggest that the key state for misfolding may be the unfolded state (43), rather than the native state or an on-pathway intermediate. PrP could be unfolded in vivo during translocation or retro-translocation across the endoplasmic reticulum (ER) membrane (44), providing opportunities to misfold both in the ER and the cytosol. Interestingly, our measurements were made at neutral pH, similar to conditions in the cytosol, ER, and extracellular space. Previous work in vitro found that PrP does not readily misfold at neutral pH, but it does at low pH (45), supporting the hypothesis that PrPSc develops in endosomes (13). The misfolded states we found in single PrP molecules are so rarely occupied that they would be unlikely to be detected by ensemble methods, but the high misfolding rates we observe clearly indicate that PrP does indeed readily misfold at neutral pH.
The behavior of the C179A/C214A mutant provides a first look at how the observed misfolding pathways relate to aggregate formation. The β-rich oligomers that this mutant can form are similar to isoforms that have been investigated as possible intermediates for PrPSc conversion (45). The fact that the misfolded states M1 and M2 are stabilized in the mutant suggests that they could act as intermediates leading to oligomerization, with the mutation driving increased aggregation via enhanced occupancy of the misfolded intermediates. The existence of different misfolding pathways might possibly relate to the ability of PrP to form different oligomeric structures (46, 47), but additional measurements will clearly be needed to establish such a link. Additional studies will also be needed to address the question of how the misfolding of isolated PrP molecules relates to PrPSc formation, e.g., by exploring the effects of mutations enhancing pathogenicity (48), probing the effects of different pH conditions, and observing aggregate formation directly. If M1–M3 are involved, however, the need to completely unfold the native state could support models where the C-terminal domain is significantly restructured in PrPSc (19), although partially native structures (20, 21) might still form (despite being unstable in monomeric PrP) if they were stabilized during refolding into an amyloid.
A key feature of this study is the ability to observe very short-lived conformational fluctuations directly within the folding trajectories of single protein molecules, enabled by high time resolution and the capacity to resolve states with extremely low occupancies. The ability to map out the network of pathways that compete with native structure formation provides a powerful platform for investigating the molecular mechanisms of protein misfolding. Applying this approach to other proteins involved in misfolding disorders should provide additional insight both into what features drive the unique behavior of PrP and into what commonalities exist between misfolding mechanisms for different proteins.
Methods
Sample Preparation.
Truncated wild-type SHaPrP(90–232) was cloned into the pET-15b plasmid between the XhoI and EcoRI sites. N- and C-terminal Cys residues were introduced by mutating Ser residues in the thrombin cleavage site and the prion site S232, respectively. The 19-kDa, His-tagged SHaPrP was expressed in Escherichia coli BL21(DE3), then purified similarly to a previous protocol (49). PrP was refolded on a nickel-nitriloacetic acid column after purification, with native folding confirmed by circular dichroism spectroscopy (50). The C179A/C214A mutations were introduced by site-directed mutagenesis, and the mutant was expressed and purified similarly (39). DNA handles were attached to the protein similarly to a previous protocol (51): Refolded PrP was reduced with tris(2-carboxyethyl)phosphine (TCEP), activated with 2,2′-dithiodipyridine, then reacted with sulfhydryl-labeled DNA handles prepared by PCR (one 798 bp, labeled by biotin, the other 1,261 bp, labeled with digoxigenin). Reference constructs consisting only of the DNA handles without protein were made by creating a disulfide bond between handles.
PrP–DNA and reference constructs were incubated at approximately 100 pM with 250 pM polystyrene beads (600 nm diameter labeled with avidin, 800 nm diameter labeled with antidigoxigenin), to form dumbbells (52). Dumbbells were diluted to approximately 500 fM in 50 mM MOPS, pH 7.0, with 200 mM KCl and oxygen scavenging system (8 mU/μL glucose oxidase, 20 mU/μL catalase, 0.01% wt/vol D-glucose), before insertion into a sample cell for the optical trap.
Measurements and Analysis.
Samples were measured in a custom dual-beam optical trap described previously (37). FECs were measured by moving the traps apart in 1–2 nm steps at pulling rates of 20–230 nm/s. Data were sampled at 20 kHz, filtered online with an eight-pole 10 kHz Bessel filter, and averaged over each step. Trap stiffness, calibrated as described previously (53), was 0.3 and 0.9 pN/nm. Multiple FECs from a given molecule were aligned offline to correct for instrumental drift (52). Aligned data were fit to an extensible worm-like chain model (54) parameterized by L p (polymer persistence length), L c (contour length), and K (elastic modulus). Two WLCs in series were used (37): one for the handles and one for the amino acids. We treated the handle L c, L p, and K as free parameters for fitting the folded branch of the FECs but thereafter as fixed parameters for fitting the unfolded branch. L p and K for the unfolded amino acids were also treated as fixed parameters when fitting the unfolded branch of the FECs: L p = 0.65 nm (55) and K = 2,000 pN. Hence the latter fit involved only a single free parameter, the unfolded amino acid contour length. The crystallographic contour length of an amino acid,  (32), was used to convert Δ_L_ c into _n_aa.
(32), was used to convert Δ_L_ c into _n_aa.
Constant-force data were measured with a passive force clamp (33). Measurements were sampled at 50 kHz and filtered online with a 25 kHz eight-pole Bessel filter or sampled at 20 kHz and filtered at 10 kHz. The stiffness of the trap used to measure force was 0.3 pN/nm. Data were median-filtered offline in a 0–2 ms window depending on the application: 2 ms for separating U and N (as in Fig. 1C), 0.5 ms for extension histograms (as in Fig. 4), and unfiltered for measuring the transition between U and N (as in Fig. 2). Histograms were binned in 0.05 nm increments. All offline data analysis used custom software in Igor Pro (Wavemetrics).
Additional details are provided in Supporting Information.
Supplementary Material
Supporting Information
ACKNOWLEDGMENTS.
We thank V. Semenchenko and D. Wishart for providing cells expressing SHaPrP(90–231); D. Foster and M. Belov for assistance with instrumentation development; and members of the Woodside lab and Prion Structure & Dynamics collaboration for helpful discussions. We thank PrioNet Canada, the Alberta Prion Research Institute, the _nano_Works program of Alberta Innovates (AI), and the National Institute for Nanotechnology for financial support. X.L. and A.S. were supported by fellowships from AI Health Solutions and AI Technology Solutions, respectively.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 2.Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11:777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- 3.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 4.Borgia A, Williams PM, Clarke J. Single-molecule studies of protein folding. Annu Rev Biochem. 2008;77:101–125. doi: 10.1146/annurev.biochem.77.060706.093102. [DOI] [PubMed] [Google Scholar]
- 5.Bechtluft P, et al. Direct observation of chaperone-induced changes in a protein folding pathway. Science. 2007;318:1458–1461. doi: 10.1126/science.1144972. [DOI] [PubMed] [Google Scholar]
- 6.Oberhauser AF, Marszalek PE, Carrion-Vazquez M, Fernandez JM. Single protein misfolding events captured by atomic force microscopy. Nat Struct Biol. 1999;6:1025–1028. doi: 10.1038/14907. [DOI] [PubMed] [Google Scholar]
- 7.Yu J, Malkova S, Lyubchenko YL. Alpha-Synuclein misfolding: Single molecule AFM force spectroscopy study. J Mol Biol. 2008;384:992–1001. doi: 10.1016/j.jmb.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Borgia MB, et al. Single-molecule fluorescence reveals sequence-specific misfolding in multidomain proteins. Nature. 2011;474:662–665. doi: 10.1038/nature10099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hillger F, Nettels D, Dorsch S, Schuler B. Detection and analysis of protein aggregation with confocal single molecule fluorescence spectroscopy. J Fluoresc. 2007;17:759–765. doi: 10.1007/s10895-007-0187-z. [DOI] [PubMed] [Google Scholar]
- 10.Nath S, Meuvis J, Hendrix J, Carl SA, Engelborghs Y. Early aggregation steps in alpha-synuclein as measured by FCS and FRET: Evidence for a contagious conformational change. Biophys J. 2010;98:1302–1311. doi: 10.1016/j.bpj.2009.12.4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orte A, et al. Direct characterization of amyloidogenic oligomers by single-molecule fluorescence. Proc Natl Acad Sci USA. 2008;105:14424–14429. doi: 10.1073/pnas.0803086105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stigler J, Ziegler F, Gieseke A, Gebhardt JC, Rief M. The complex folding network of single calmodulin molecules. Science. 2011;334:512–516. doi: 10.1126/science.1207598. [DOI] [PubMed] [Google Scholar]
- 13.Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: Oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem. 2009;78:177–204. doi: 10.1146/annurev.biochem.78.082907.145410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cobb NJ, Surewicz WK. Prion diseases and their biochemical mechanisms. Biochemistry. 2009;48:2574–2585. doi: 10.1021/bi900108v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.James TL, et al. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc Natl Acad Sci USA. 1997;94:10086–10091. doi: 10.1073/pnas.94.19.10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lysek DA, et al. Prion protein NMR structures of cats, dogs, pigs, and sheep. Proc Natl Acad Sci USA. 2005;102:640–645. doi: 10.1073/pnas.0408937102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zahn R, et al. NMR solution structure of the human prion protein. Proc Natl Acad Sci USA. 2000;97:145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan KM, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cobb NJ, Sonnichsen FD, McHaourab H, Surewicz WK. Molecular architecture of human prion protein amyloid: A parallel, in-register beta-structure. Proc Natl Acad Sci USA. 2007;104:18946–18951. doi: 10.1073/pnas.0706522104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeMarco ML, Daggett V. From conversion to aggregation: Protofibril formation of the prion protein. Proc Natl Acad Sci USA. 2004;101:2293–2298. doi: 10.1073/pnas.0307178101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc Natl Acad Sci USA. 2004;101:8342–8347. doi: 10.1073/pnas.0402254101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silveira JR, et al. The most infectious prion protein particles. Nature. 2005;437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen FE, et al. Structural clues to prion replication. Science. 1994;264:530–531. doi: 10.1126/science.7909169. [DOI] [PubMed] [Google Scholar]
- 24.Wildegger G, Liemann S, Glockshuber R. Extremely rapid folding of the C-terminal domain of the prion protein without kinetic intermediates. Nat Struct Biol. 1999;6:550–553. doi: 10.1038/9323. [DOI] [PubMed] [Google Scholar]
- 25.Apetri AC, Maki K, Roder H, Surewicz WK. Early intermediate in human prion protein folding as evidenced by ultrarapid mixing experiments. J Am Chem Soc. 2006;128:11673–11678. doi: 10.1021/ja063880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hart T, et al. Folding kinetics of the human prion protein probed by temperature jump. Proc Natl Acad Sci USA. 2009;106:5651–5656. doi: 10.1073/pnas.0811457106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenkins DC, Sylvester ID, Pinheiro TJ. The elusive intermediate on the folding pathway of the prion protein. FEBS J. 2008;275:1323–1335. doi: 10.1111/j.1742-4658.2008.06293.x. [DOI] [PubMed] [Google Scholar]
- 28.Kuwata K, et al. Locally disordered conformer of the hamster prion protein: A crucial intermediate to PrPSc? Biochemistry. 2002;41:12277–12283. doi: 10.1021/bi026129y. [DOI] [PubMed] [Google Scholar]
- 29.Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc Natl Acad Sci USA. 2007;104:2649–2654. doi: 10.1073/pnas.0611503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong J, Castro CE, Boyce MC, Lang MJ, Lindquist S. Optical trapping with high forces reveals unexpected behaviors of prion fibrils. Nat Struct Mol Biol. 2010;17:1422–1430. doi: 10.1038/nsmb.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganchev DN, Cobb NJ, Surewicz K, Surewicz WK. Nanomechanical properties of human prion protein amyloid as probed by force spectroscopy. Biophys J. 2008;95:2909–2915. doi: 10.1529/biophysj.108.133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pauling L, Corey RB. The pleated sheet, a new layer configuration of polypeptide chains. Proc Natl Acad Sci USA. 1951;37:251–256. doi: 10.1073/pnas.37.5.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenleaf WJ, Woodside MT, Abbondanzieri EA, Block SM. Passive all-optical force clamp for high-resolution laser trapping. Phys Rev Lett. 2005;95:208102. doi: 10.1103/PhysRevLett.95.208102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cecconi C, Shank EA, Bustamante C, Marqusee S. Direct observation of the three-state folding of a single protein molecule. Science. 2005;309:2057–2060. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 35.Gebhardt JC, Bornschlogl T, Rief M. Full distance-resolved folding energy landscape of one single protein molecule. Proc Natl Acad Sci USA. 2010;107:2013–2018. doi: 10.1073/pnas.0909854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woodside MT, et al. Direct measurement of the full, sequence-dependent folding landscape of a nucleic acid. Science. 2006;314:1001–1004. doi: 10.1126/science.1133601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neupane K, Yu H, Foster DAN, Wang F, Woodside MT. Single-molecule force spectroscopy of the add adenine riboswitch relates folding to regulatory mechanism. Nucleic Acids Res. 2011;39:7677–7687. doi: 10.1093/nar/gkr305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bustamante C, Chemla YR, Forde NR, Izhaky D. Mechanical processes in biochemistry. Annu Rev Biochem. 2004;73:705–748. doi: 10.1146/annurev.biochem.72.121801.161542. [DOI] [PubMed] [Google Scholar]
- 39.Maiti NR, Surewicz WK. The role of disulfide bridge in the folding and stability of the recombinant human prion protein. J Biol Chem. 2001;276:2427–2431. doi: 10.1074/jbc.M007862200. [DOI] [PubMed] [Google Scholar]
- 40.De Simone A, Zagari A, Derreumaux P. Structural and hydration properties of the partially unfolded states of the prion protein. Biophys J. 2007;93:1284–1292. doi: 10.1529/biophysj.107.108613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hosszu LL, et al. Definable equilibrium states in the folding of human prion protein. Biochemistry. 2005;44:16649–16657. doi: 10.1021/bi051277k. [DOI] [PubMed] [Google Scholar]
- 42.Kuwata K, et al. NMR-detected hydrogen exchange and molecular dynamics simulations provide structural insight into fibril formation of prion protein fragment 106–126. Proc Natl Acad Sci USA. 2003;100:14790–14795. doi: 10.1073/pnas.2433563100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hosszu LL, et al. Structural mobility of the human prion protein probed by backbone hydrogen exchange. Nat Struct Biol. 1999;6:740–743. doi: 10.1038/11507. [DOI] [PubMed] [Google Scholar]
- 44.Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science. 2002;298:1781–1785. doi: 10.1126/science.1073725. [DOI] [PubMed] [Google Scholar]
- 45.Bjorndahl TC, et al. Detailed biophysical characterization of the acid-induced PrP(c) to PrP(beta) conversion process. Biochemistry. 2011;50:1162–1173. doi: 10.1021/bi101435c. [DOI] [PubMed] [Google Scholar]
- 46.Chakroun N, et al. The oligomerization properties of prion protein are restricted to the H2H3 domain. FASEB J. 2010;24:3222–3231. doi: 10.1096/fj.09-153924. [DOI] [PubMed] [Google Scholar]
- 47.Gerber R, Tahiri-Alaoui A, Hore PJ, James W. Oligomerization of the human prion protein proceeds via a molten globule intermediate. J Biol Chem. 2007;282:6300–6307. doi: 10.1074/jbc.M608926200. [DOI] [PubMed] [Google Scholar]
- 48.van der Kamp MW, Daggett V. The consequences of pathogenic mutations to the human prion protein. Protein Eng Des Sel. 2009;22:461–468. doi: 10.1093/protein/gzp039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zahn R, von Schroetter C, Wuthrich K. Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett. 1997;417:400–404. doi: 10.1016/s0014-5793(97)01330-6. [DOI] [PubMed] [Google Scholar]
- 50.Stockel J, Safar J, Wallace AC, Cohen FE, Prusiner SB. Prion protein selectively binds copper(II) ions. Biochemistry. 1998;37:7185–7193. doi: 10.1021/bi972827k. [DOI] [PubMed] [Google Scholar]
- 51.Cecconi C, Shank EA, Dahlquist FW, Marqusee S, Bustamante C. Protein-DNA chimeras for single molecule mechanical folding studies with the optical tweezers. Eur Biophys J. 2008;37:729–738. doi: 10.1007/s00249-007-0247-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greenleaf WJ, Frieda KL, Foster DA, Woodside MT, Block SM. Direct observation of hierarchical folding in single riboswitch aptamers. Science. 2008;319:630–633. doi: 10.1126/science.1151298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neuman KC, Block SM. Optical trapping. Rev Sci Instrum. 2004;75:2787–2809. doi: 10.1063/1.1785844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang MD, Yin H, Landick R, Gelles J, Block SM. Stretching DNA with optical tweezers. Biophys J. 1997;72:1335–1346. doi: 10.1016/S0006-3495(97)78780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shank EA, Cecconi C, Dill JW, Marqusee S, Bustamante C. The folding cooperativity of a protein is controlled by its chain topology. Nature. 2010;465:637–640. doi: 10.1038/nature09021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information