Bayesian Phylogenetics with BEAUti and the BEAST 1.7 (original) (raw)
Abstract
Computational evolutionary biology, statistical phylogenetics and coalescent-based population genetics are becoming increasingly central to the analysis and understanding of molecular sequence data. We present the Bayesian Evolutionary Analysis by Sampling Trees (BEAST) software package version 1.7, which implements a family of Markov chain Monte Carlo (MCMC) algorithms for Bayesian phylogenetic inference, divergence time dating, coalescent analysis, phylogeography and related molecular evolutionary analyses. This package includes an enhanced graphical user interface program called Bayesian Evolutionary Analysis Utility (BEAUti) that enables access to advanced models for molecular sequence and phenotypic trait evolution that were previously available to developers only. The package also provides new tools for visualizing and summarizing multispecies coalescent and phylogeographic analyses. BEAUti and BEAST 1.7 are open source under the GNU lesser general public license and available at http://beast-mcmc.googlecode.com and http://beast.bio.ed.ac.uk
Key words: Bayesian phylogenetics, evolution, phylogenetics, molecular evolution, coalescent theory
Introduction
Molecular sequences, morphological measurements, geographic distributions and fossil remains all provide a wealth of potential information about the evolutionary history of life on Earth, the dynamics of ancient and modern biological populations, and the emergence and spread of infectious diseases. One of the challenges of modern Evolutionary Biology is the integration of these different data sources to address evolutionary hypotheses over the full range of spatial and temporal scales. The field is witnessing a transition to an increasingly quantitative science. This transformation began first through an explosion of molecular sequence data with the parallel development of mathematical and computational tools for their analysis. However, increasingly this transformation can be observed in other aspects of Evolutionary Biology where large global databases of complementary sources of information, such as fossils, geographical distributions and population history are being curated and made publicly available.
Software Advances
Here, we present a major new version of the molecular evolutionary software package Bayesian Evolutionary Analysis by Sampling Trees (BEAST), updated to version 1.7, and representing a significant software advance over that previously described (Drummond and Rambaut 2007). Alongside the primary analysis engine in BEAST, this package also includes a suite of utilities for specifying the analysis design, processing output files and summarizing and visualizing the results. Taken together, these programs enable Bayesian inference of molecular sequences with an emphasis on time-structured evolutionary models including phylodynamic models, divergence time estimates, multiloci demographic models, gene/species–tree inference, a range of spatial phylogeographic analyses and discrete and continuous trait evolution. Implementing Markov chain Monte Carlo (MCMC) algorithms to perform these inferences, the package is intended and used for rigorous statistical inference and hypothesis testing of evolutionary models with joint inference of phylogeny. It is also possible to constrain portions of the phylogenetic model space to known values, including the tree topology, and perform conditional inference if required.
User Interface
One area of significant improvement since the last release publication is in the analysis construction and model specification tool called Bayesian Evolutionary Analysis Utility (BEAUti). This acts as the graphical user interface (GUI) for BEAST and allows the user to import data, select models, choose prior distributions on individual parameters and specify the settings for the MCMC sampler (fig. 1). Although the BEAST model specification format (a standard XML format structured text file) allows for great flexibility in the construction of complex evolutionary models, the constraints of a GUI unavoidably restrict the scope of the researcher to a prespecified set of models and combinations, hiding many advanced inference models. Working directly within the BEAST XML input format, on the other hand, represents a high barrier to the accessibility of BEAST and incurs significant risk of inadvertent errors being introduced into the model. We have concentrated development efforts on BEAUti to provide greater flexibility in model specification while still maintaining the benefits of a visual, table-based representation of the model and automatic generation of BEAST XML files. Improvements to BEAUti provide support for multiple data partitions in a joint analysis and the input of fossil calibration and trait information.
FIG. 1.

BEAUti GUI for importing data and specifying the evolutionary model.
Heterogeneous Data
Multiple data partitions may reflect separate loci for simultaneous inference of genealogies and species trees (Heled and Drummond 2010) and stochastic ancestral recombination graph reconstruction (Bloomquist and Suchard 2010) or the growing wealth of nonsequence data and their respective substitution models. These latter data and models include microsatellite markers (Wu and Drummond 2011), phenotypic traits under a multistate stochastic Dollo process (Alekseyenko et al. 2008), discretized geographic diffusion (Lemey et al. 2009), and multivariate continuous relaxed random walks (Lemey et al. 2010).
We also ease the use of a growing number of tree prior specifications. These include the extended Bayesian skyline model (Heled and Drummond 2008) for multilocus data, the flexible Gaussian Markov random field skyride model (Minin et al. 2008), and birth–death models of speciation (Stadler 2010).
Multispecies Coalescent
Discordance between individual gene trees that share a phylogenetic history results from incomplete lineage sorting and becomes increasingly likely when times between speciation events are short compared with species' population sizes. We provide a fully Bayesian implementation of the multispecies coalescent that improves the accuracy and precision of species tree reconstruction (Heled and Drummond 2010) and divergence time estimation (McCormack et al. 2011).
Phenotypic Trait Analysis
For trait inference including phylogeography, we now provide several tools for mapping posterior distributions of trees onto higher dimensional or geographics maps for both interactive exploration and better visualization (Bielejec et al. 2011). These tools interface with GoogleEarth via keyhole markup language and enable users to generate animations of evolutionary processes through time and real space; see http://www.phylogeography.org for several examples.
Molecular Clocks
We have refined the relaxed clock models to allow more than one branch to have the same rate value to remove anticorrelation. In practice this will only have any appreciable impact on trees that have a small number of branches (< 15 taxa). An efficient implementation of the relaxed clock models that facilitates calculation of Bayes Factors for model selection and model averaging of several clock models has also be developed (Li and Drummond, 2012). Further, we provide a new random local clock (RLC) model (Drummond and Suchard 2010), in which all possible local clock configurations and a strict clock are nested, providing a convenient model to test for a strict clock. Heled and Drummond (2011) begins to investigate alternative approaches to the calibration of tree priors with fossil and geological evidence and this area of research is still in its infancy. Often, uncertainty exists in the age of viral RNA/DNA or ancient DNA samples and these can now be incorporated (Shapiro et al. 2011), along with models for sequence damage and error (Rambaut et al. 2009).
Performance
Finally, to exploit high-performance computing, BEAST 1.7 integrates with and provides a GUI interface to configure the BEAGLE library (Ayres et al. 2011) that utilizes multicore processors, vectorization and massively parallel graphics processors to substantially decrease BEAST run-times (Suchard and Rambaut 2009).
Examples
Figure 2 presents a reconstruction of the gene tree relating 13 species of Darwin's finches from a 2,065-bp partial nucleotide alignment of the mitochondrial control region and cytochrome b genes (Sato et al. 1999) and five continuously measured phenotypic traits of the corresponding species (Sulloway 1982). In performing this simultaneous inference, we exploit the RLC model (Drummond and Suchard 2010) and find evidence for one suggestive rate change (Bayes factor in favor of the RLC over a strict clock = 2.3) in the lineage leading to the Cocos Island Finch, Pinaroloxias inornata. Multivariate Brownian trait diffusion shows strong correlation between wing and tarsus length and between bill depth and gonys length. Posterior trait prediction at any point along the history is possible and, currently unique to BEAST, comparative method inference is performed jointly with phylogenetic inference.
FIG. 2.

Simultaneous phylogenetic and phenotypic trait reconstruction of Darwin's finches. Plotted are the maximum clade credibility tree and posterior estimate of the trait correlation matrix. We annotate the tree with estimates of selected posterior clade support values and the one significant nucleotide substitution local clock (in red) and the branches scale in expected substitutions per site. We depict correlation coefficients through their bivariate ellipse sizes, where more highly correlated phenotypes return narrower ellipses.
Our second example demonstrates the application of the multispecies coalescent model (*BEAST) to a 1,165-bp fragment of the mitochondrial genome sequenced from 16 Darwin's finches representing four species (Geospiza fortis, G. magnirostris, Camarhynchus parvulus, and Certhidea olivacea). Figure 3 shows 1) a representative gene tree and 2) the two species trees with highest posterior probability. The 99% credible set for the species tree contains 3 of the 15 possible tree topologies: 65.8% (((F, M),P), O); 17.2% ((F, M),(P, O)); 16.5% (((F, M),O), P). This uncertainty in the species tree arises despite overwhelming support for Certhidea olivacea and Camarhynchus parvulus as the nested outgroup species according to the gene tree (fig. 3a), due to the possibility of incomplete lineage sorting in the deeper branches of the gene tree. The possibility of incomplete lineage sorting can be appreciated in figure 3c, in which a representative gene tree is embedded inside the most probable species tree topology for this data, showing extensive incomplete lineage sorting in the Geospiza clade and also depicting the reason that species trees necessarily have (sometimes much) younger divergence times than the corresponding gene tree might suggest. This example demonstrates that even for single-gene analyses, the multispecies coalescent can provide 1) important insight into the potential for incomplete lineage sorting, 2) more accurate assessment of uncertainty in the species tree estimate and 3) better estimates of species divergence times.
FIG. 3.
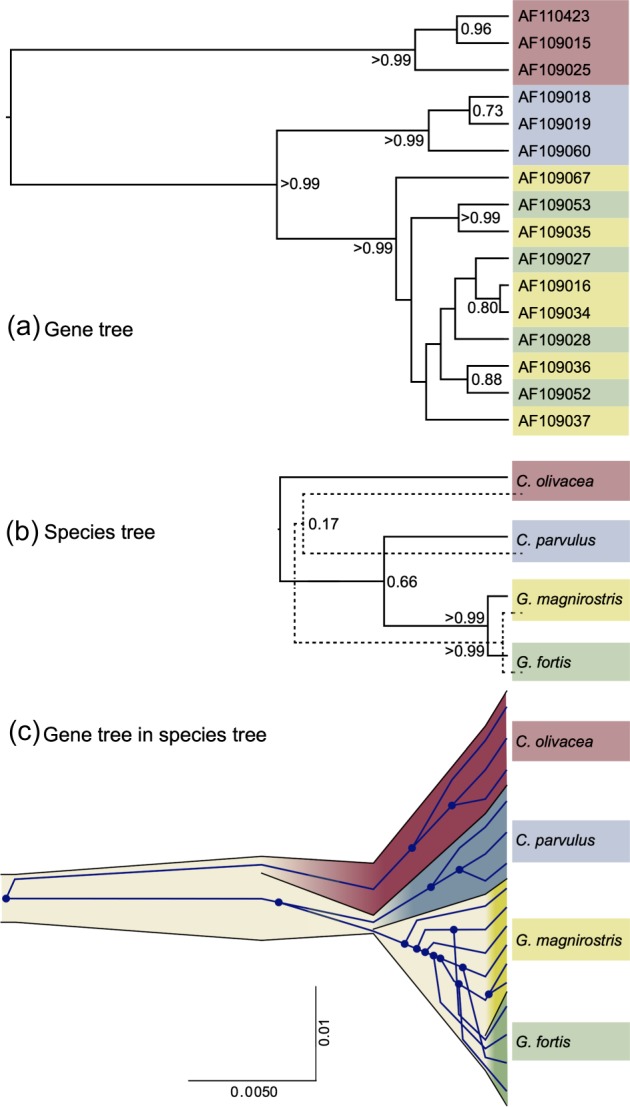
(a) Representative gene tree of mitochondrial DNA fragment from 16 Darwin's finches of four species (Geospiza fortis, G. magnirostris, Camarhynchus parvulus, and Certhidea olivacea). Nodes that have posterior clade probabilities of greater than 0.5 are labeled with their posterior clade probability. (b) The two most probable species trees (solid line represents most probable species tree; dashed line is second most probable). (c) Gene tree embedded in a point estimate of the species tree, including divergence times and effective population sizes. The x axis is divergence time in units of substitutions per site and the y axis is proportional to effective population size.
Availability and Future Directions
We make the BEAST package available in both executable and source code forms. BEAST requires Java version 1.5 or greater and executables for Windows, Mac OS and Linux platforms are located at http://beast.bio.ed.ac.uk which serves as the main page for the package. This page also links to a sizable list of self-contained step-by-step tutorials covering basic to advance usage of BEAST. Popular tutorials describe how to use BEAST to infer population dynamics and phylogeographic processes and walk users all the way through to generating a range of graphical summaries of their results.
GoogleCode houses the BEAST's version-controlled source code at http://beast-mcmc.googlecode.com and links to two GoogleGroup discussion groups related to BEAST. The first is the “beast-users” group (http://groups.google.com/group/beast-users) with over 1,500 members. At the time of writing, 47 developers belong to the “beast-dev” group that facilitates BEAST development across three continents.
Future development directions for BEAUti and BEAST focus on easing the user experience in several ways. These include in fitting hierarchical phylogenetics models (Suchard et al. 2003) that commonly arise in studies of intrahost viral evolution, in exploiting MarkovJump methods (Minin and Suchard 2008; O'Brien et al. 2009) for computationally efficient and robust estimation of complex evolutionary processes under simple models, and in specifying phylogeographic models (Lemey et al. 2009, 2010) in a convenient geographical user interface.
Supplementary Material
Supplementary Data
Acknowledgments
We thank the National Evolutionary Synthesis Center for sponsoring a working group (Software for Bayesian Evolutionary Analysis) that facilitated the development of BEAST version 1.7. We would also like to thank the many developers and contributors to BEAST, including: Alex Alekseyenko, Trevor Bedford, Erik Bloomquist, Joseph Heled, Sebastian Hoehna, Philippe Lemey, Sibon Li, Gerton Lunter, Sidney Markowitz, Vladimir Minin, Michael Defoin Platel, Oliver Pybus, Beth Shapiro and Chieh-Hsi Wu. This work was supported in part by funding from the Marsden Trust, National Science Foundation (DMS 0856099), National Institute of Health (R01 GM086887, R01 HG006139), The Royal Society of London, Biotechnology and Biological Sciences Research Council (BB/H011285/1) and the Wellcome Trust (WT092807MA).
References
- Alekseyenko A, Lee C, Suchard M. Wagner and Dollo: a stochastic duet by composing two parsimonious solos. Syst Biol. 2008;57(5):772–784. doi: 10.1080/10635150802434394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres D, Darling A, Zwickl D, et al. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst Biol. 2011;61(1):170–173. doi: 10.1093/sysbio/syr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielejec F, Rambaut A, Suchard MA, Lemey P. Spread: spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics. 2011;27(20):2910–2. doi: 10.1093/bioinformatics/btr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomquist E, Suchard M. Unifying vertical and nonvertical evolution: a stochastic ARG-based framework. Syst Biol. 2010;59(1):27–41. doi: 10.1093/sysbio/syp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. Beast: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA. Bayesian random local clocks, or one rate to rule them all. BMC Biol. 2010;8:114. doi: 10.1186/1741-7007-8-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heled J, Drummond AJ. Bayesian inference of population size history from multiple loci. BMC Evol Biol. 2008;8:289. doi: 10.1186/1471-2148-8-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heled J, Drummond AJ. Bayesian inference of species trees from multilocus data. Mol Biol Evol. 2010;27(3):570–580. doi: 10.1093/molbev/msp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heled J, Drummond AJ. Calibrated tree priors for relaxed phylogenetics and divergence time estimation. Syst Biol. 2011;61(1):138–149. doi: 10.1093/sysbio/syr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLoS Comput Biol. 2009;5(9):e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P, Rambaut A, Welch J, Suchard M. Phylogeography takes a relaxed random walk in continuous space and time. Mol Biol Evol. 2010;27:1877–1885. doi: 10.1093/molbev/msq067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WLS, Drummond AJ. Model Averaging and Bayes Factor Calculation of Relaxed Molecular Clocks in Bayesian Phylogenetics. Mol Biol Evol. 2012;29:751–761. doi: 10.1093/molbev/msr232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JE, Heled J, Delaney KS, Peterson AT, Knowles LL. Calibrating divergence times on species trees versus gene trees: implications for speciation history of aphelocoma jays. Evolution. 2011;65:184–202. doi: 10.1111/j.1558-5646.2010.01097.x. [DOI] [PubMed] [Google Scholar]
- Minin V, Suchard M. Counting labeled transitions in continuous-time Markov models of evolution. J Math Biol. 2008;56:391–412. doi: 10.1007/s00285-007-0120-8. [DOI] [PubMed] [Google Scholar]
- Minin VN, Bloomquist EW, Suchard MA. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol Biol Evol. 2008;25:1459–1471. doi: 10.1093/molbev/msn090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien J, Minin V, Suchard M. Learning to count: robust estimates for labeled distances between molecular sequences. Mol Biol Evol. 2009;26:801–814. doi: 10.1093/molbev/msp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Ho S, Drummond AJ, Shapiro B. Accommodating the effect of ancient DNA damage on inferences of demographic histories. Mol Biol Evol. 2009;26:245–248. doi: 10.1093/molbev/msn256. [DOI] [PubMed] [Google Scholar]
- Sato A, O'hUigin C, Figueroa F, Grant P, Grant B, Tichy H, Klein J. Phylogeny of Darwin's finches as revealed by mtDNA sequences. Proc Natl Acad Sci U S A. 1999;96:5101–5106. doi: 10.1073/pnas.96.9.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro B, Ho S, Drummond AJ, Suchard M, Pybus O, Rambaut A. A Bayesian phylogenetic method to estimate unknown sequence ages. Mol Biol Evol. 2011;28:879–887. doi: 10.1093/molbev/msq262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler T. Sampling-through-time in birth–death trees. J Theor Biol. 2010;267:396–404. doi: 10.1016/j.jtbi.2010.09.010. [DOI] [PubMed] [Google Scholar]
- Suchard MA, Rambaut A. Many-core algorithms for statistical phylogenetics. Bioinformatics. 2009;25:1370–1376. doi: 10.1093/bioinformatics/btp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard MA, Kitchen CMR, Sinsheimer JS, Weiss RE. Hierarchical phylogenetic models for analyzing multipartite sequence data. Syst Biol. 2003;52:649–664. doi: 10.1080/10635150390238879. [DOI] [PubMed] [Google Scholar]
- Sulloway F. The Beagle collections of Darwin's finches (Geospizinae) Bull Br Mus. 1982;43:49–94. [Google Scholar]
- Wu C, Drummond AJ. Joint inference of microsatellite mutation models, population history and genealogies using trans-dimensional MCMC. Genetics. 2011;188:151–164. doi: 10.1534/genetics.110.125260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data